
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cyanide-catalyzed imino-Stetter reaction enables the concise total syntheses of rucaparib†
Jinjae Park and
Cheol-Hong Cheon*
and
Cheol-Hong Cheon*
Department of Chemistry Korea University 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea. E-mail: cheon@korea.ac.kr
First published on 1st August 2022
Abstract
Two routes toward the synthesis of rucaparib, an FDA-approved drug used for the treatment of ovarian and prostate cancers, have been developed from commercially available starting materials utilizing the cyanide-catalyzed imino-Stetter reaction as the key step for the construction of the indole motif bearing all the desired substituents in their correct positions. In the first-generation synthesis, meta-fluorobenzoate, the starting material currently used in the process chemistry route of rucaparib, was converted into 4,6-disubstituted 2-aminocinnamic acid derivatives (ester or amide). The cyanide-catalyzed imino-Stetter reaction of aldimines derived from the resulting 2-aminocinnamic acid derivatives and a commercially available aldehyde afforded the desired indole-3-acetic acid derivatives. The final azepinone formation completed the total synthesis of rucaparib in 27% overall yield. To resolve the issues raised in the first-generation synthesis, we further developed a second-generation synthesis of rucaparib. The Heck reaction of a commercially available ortho-iodoaniline derivative with acrylonitrile provided 4,6-disubstituted 2-aminocinnamonitrile, which was subjected to the imino-Stetter reaction with the same aldehyde to provide the desired indole-3-acetonitrile product. Subsequent construction of the azepinone scaffold completed the total synthesis of rucaparib in 59% overall yield over three separation operations. The synthetic strategy reported herein can provide a highly practical route to access rucaparib from commercially available starting materials (5.2% overall yield in the current process chemistry route vs. 59% overall yield in the second-generation synthesis).
Introduction
Rucaparib (1, Fig. 1) is a poly(ADP-ribose)polymerase (PARP) inhibitor used as an anticancer agent.1 It was approved by the Food and Drug Administration (FDA) for the treatment of ovarian and prostate cancers in 2016 and 2020, respectively.2,3 In addition, rucaparib has been, and still is, used in several advanced clinical trials for the treatment of other relevant cancers associated with PARP inhibition. To supply a sufficient quantity of rucaparib (1) for cancer treatment and its further applications, the development of a more practical synthetic route is the research of importance.4–8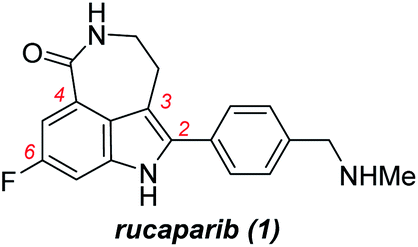 | ||
| Fig. 1 Structure of rucaparib (1). | ||
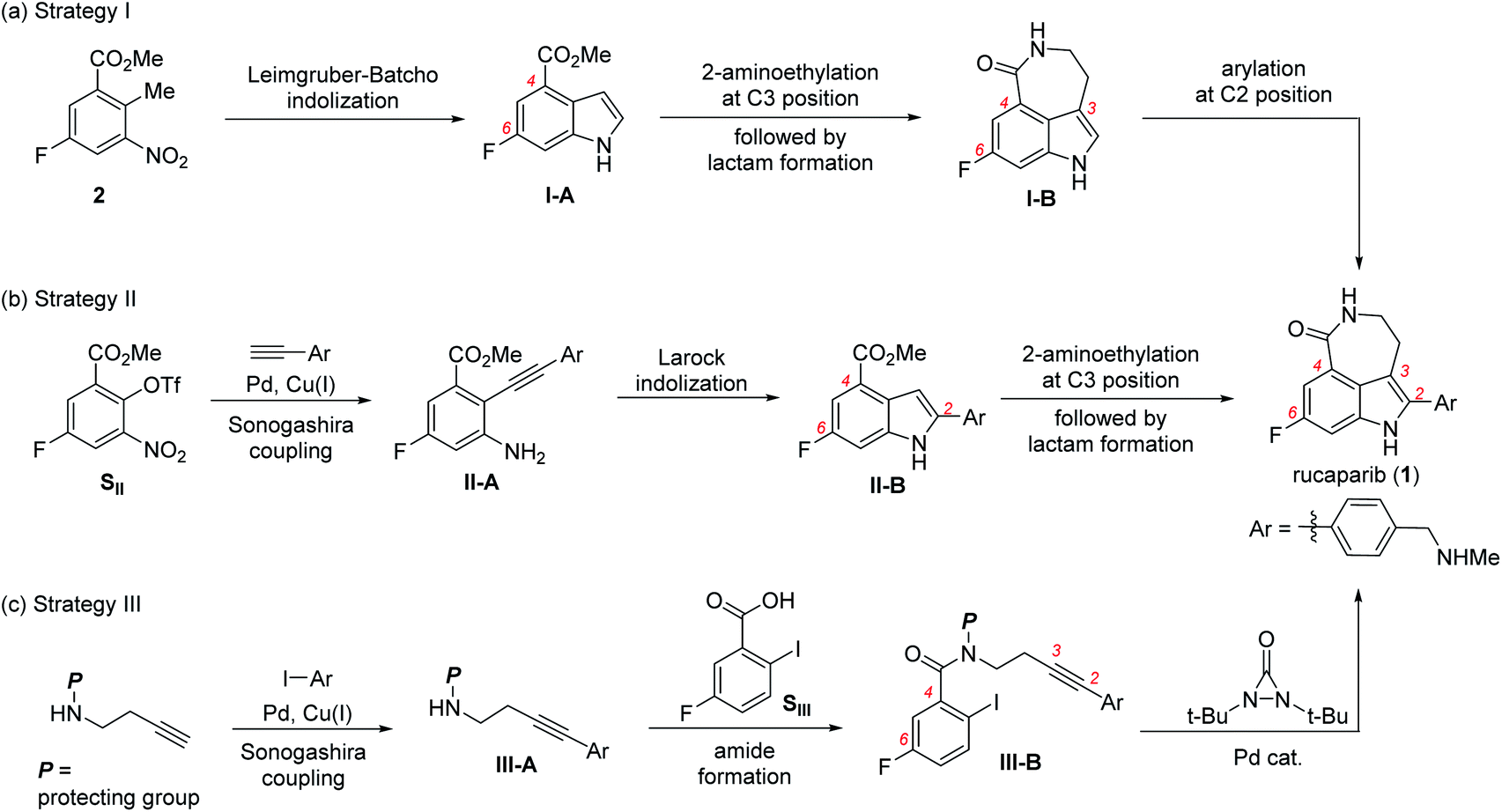
Rucaparib (1) displays unique structural features, as shown in Fig. 1. It possesses an indole subunit bearing four substituents along its periphery, and the two substituents at C3 and C4 positions form an additional seven-membered lactam ring. As indole derivatives bearing four substituents at the correct positions are not readily available, the previous syntheses of rucaparib (1) have been developed based on protocols used to construct the indole subunit with the required substituents at the right positions from available meta-fluorobenzoic acid derivatives as starting materials (Scheme 1).4–8
 | ||
| Scheme 1 Previous synthetic strategies of rucaparib (1). | ||
The first strategy involves the construction of the indole scaffold bearing two substituents at the C4 and C6 positions, followed by sequential introduction of the desired substituents at the C3 and C2 positions (Scheme 1a).5,6 In this approach, indole compound I-A bearing the fluoride and methoxycarbonyl groups at the C6 and C4 positions, respectively, was first prepared from meta-fluorobenzoate 2 via the Leimgruber–Batcho reaction. Subsequent introduction of a 2-aminoethyl group at the C3 position followed by construction of the azepinone scaffold furnished the azepinoindole intermediate I-B. Final introduction of the 4-(N-methylaminomethyl)phenyl group at the C2 position completed the total synthesis of rucaparib (1).
The second strategy utilizes the indole intermediate, bearing three substituents at the 2,4,6-positions, from which the azepinone formation would complete the synthesis of rucaparib (Scheme 1b).7
Sonogashira coupling reaction of meta-fluorobenzoate SII, generated from the corresponding phenol derivative through trifluoromethanesulfonate (triflate) formation, with arylacetylene afforded diarylacetylene intermediate II-A. Larock indolization of II-A provided 2,4,6-trisubstituted indole intermediate II-B. Subsequent construction of the azepinone scaffold through the introduction of the 2-aminoethyl group at the C3 position followed by lactam formation completed the total synthesis of compound 1.
The third strategy relies on the direct construction of an indole framework bearing four substituents at appropriate positions from a meta-fluorobenzoic acid derivative (Scheme 1c). Along this line, Zhang and co-workers recently reported a fascinating synthesis of rucaparib.8 Condensation of meta-fluorobenzoic acid SIII with amine III-A, which was prepared through Sonogashira coupling between homopropargyl amine and aryl iodide, furnished the corresponding amide III-B. Subsequent palladium-catalyzed indoloazepinone formation of III-B with N,N-di-tert-butyldiaziridone furnished rucaparib (1) after the removal of protecting groups.
Despite the development of several novel strategies for the synthesis of rucaparib (1), the current process chemistry route for rucaparib has been developed through the construction of an indole scaffold, followed by the sequential installation of substituents on the indole periphery as demonstrated in Scheme 1a.5 Consequently, the overall synthesis was not efficient, providing rucaparib (1) in low overall yield with rather lengthy synthetic sequence.9 Furthermore, to understand the metabolic pathway of rucaparib, several examples of the synthesis of rucaparib bearing a radioactive isotope have been reported recently.10 However, these radiosyntheses of rucaparib have also relied on the current process chemistry route, providing rucaparib bearing radioactive isotopes with very low yields. Thus, there is still the need for the development of a more practical synthetic route from readily available starting materials, which are possibly commercially available.
To address the issues related to the current process chemistry synthesis of rucaparib, our group commenced with a research program toward the development of a practical synthetic route for rucaparib. Consideration of the structural features of rucaparib (1), we envisioned that the development of a more efficient protocol for the preparation of an indole intermediate bearing all four substituents in their correct positions from available starting materials would provide a solution for the development of a more practical route for the synthesis of rucaparib (1). Based on this idea, very recently our group reported the total synthesis of rucaparib from commercially available starting materials using the cyanide-catalyzed imino-Stetter reaction as the key step in the synthesis of the requisite indole intermediate.11 Although this new approach provided a highly efficient synthetic route for rucaparib, we believed that it is necessary to report how we have designed and developed these synthetic routes for rucaparib utilizing the cyanide-catalyzed imino-Stetter reaction to prepare the requisite tetra-substituted indole intermediates.
Herein, we report the development of two different routes for the total synthesis of rucaparib from commercially available starting materials. In the first-generation synthesis, meta-fluorobenzoate, which is the starting material used in the current process chemistry route for rucaparib, can be converted into the desired (E)-2-aminocinnamic acid derivative via benzylic olefination with an glyoxalic acid derivative, followed by reduction of the nitro group. The cyanide-catalyzed imino-Stetter reaction of the aldimine derived from the resulting 2-aminocinnamic acid derivative and commercially available aryl aldehyde provides the desired indole-3-acetic acid derivative bearing all of the required substituents present in rucaparib in their correct positions. The final azepinone scaffold formation step between the C3 and C4 substituents on the indole scaffold completes the synthesis of rucaparib in 27% overall yield, albeit over slightly longer steps. To address the issues raised in the first-generation synthesis, we further developed a second-generation synthesis of rucaparib. The Heck reaction of a commercially available ortho-iodoaniline derivative with acrylonitrile affords 2-aminocinnamonitrile. The cyanide-catalyzed imino-Stetter reaction of the resulting 2-aminocinnamonitrile and the aldehyde provides indole-3-acetonitrile with all of the desired substituents in their correct positions. The final azepinone formation completed the synthesis of rucaparib over only three separation operations. Furthermore, we could improve the overall yield of rucaparib from 54% in the previous synthesis11 to 59% in this new synthesis through the modified protocol for the azepinone formation from the indole intermediate. Detailed studies on the preparation of 2-aminocinnamic acid derivatives, the preparation of the key indole intermediates via the imino-Stetter reaction, and azepinone scaffold formation from the indole intermediates will be described in this paper.
Results and discussion
1. First-generation synthesis of rucaparib (1)
Our synthetic plan for the first-generation synthesis of rucaparib (1) was devised from meta-fluorobenzoate 2, the starting material used in the current process chemistry route for rucaparib (Scheme 2). Rucaparib (1) could be synthesized from tetra-substituted indole intermediate 6 via the azepinone formation between the substituents at the C3 and C4 positions. Tetra-substituted indole derivative 6 would be prepared via the cyanide-catalyzed imino-Stetter reaction of the aldimine derived from 2-aminocinnamic acid derivative 4 and aryl aldehyde 5. 2-Aminocinnamic acid derivative 4 bearing the fluoride and methoxycarbonyl groups at the C4 and C6 positions, respectively, could be prepared through benzylic olefination of 2 with glyoxalic acid derivative 7, followed by the reduction of the nitro group.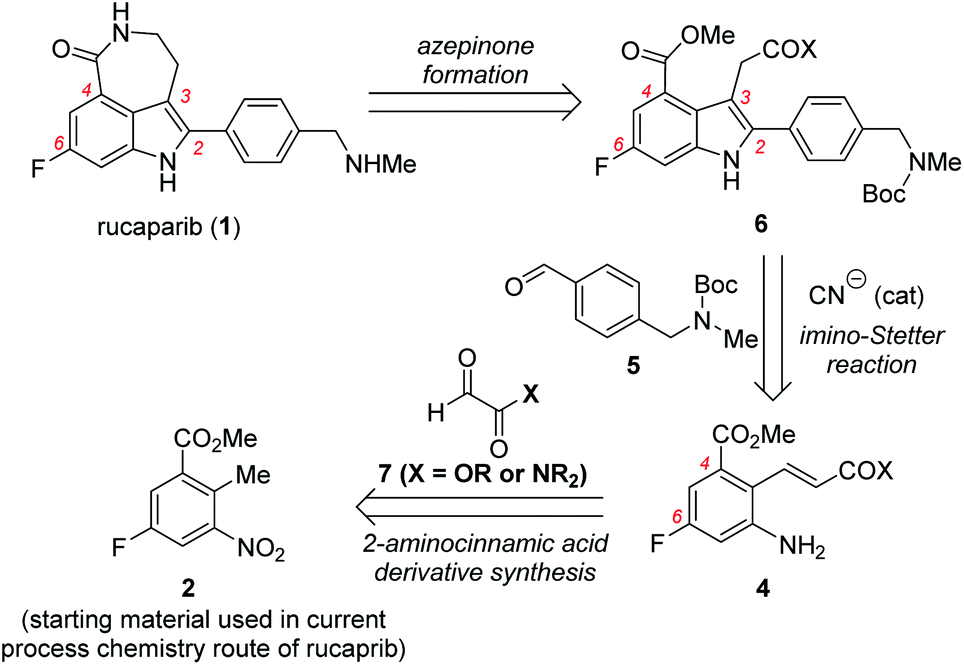 | ||
| Scheme 2 Retrosynthetic analysis of the first-generation synthesis of rucaparib (1). | ||
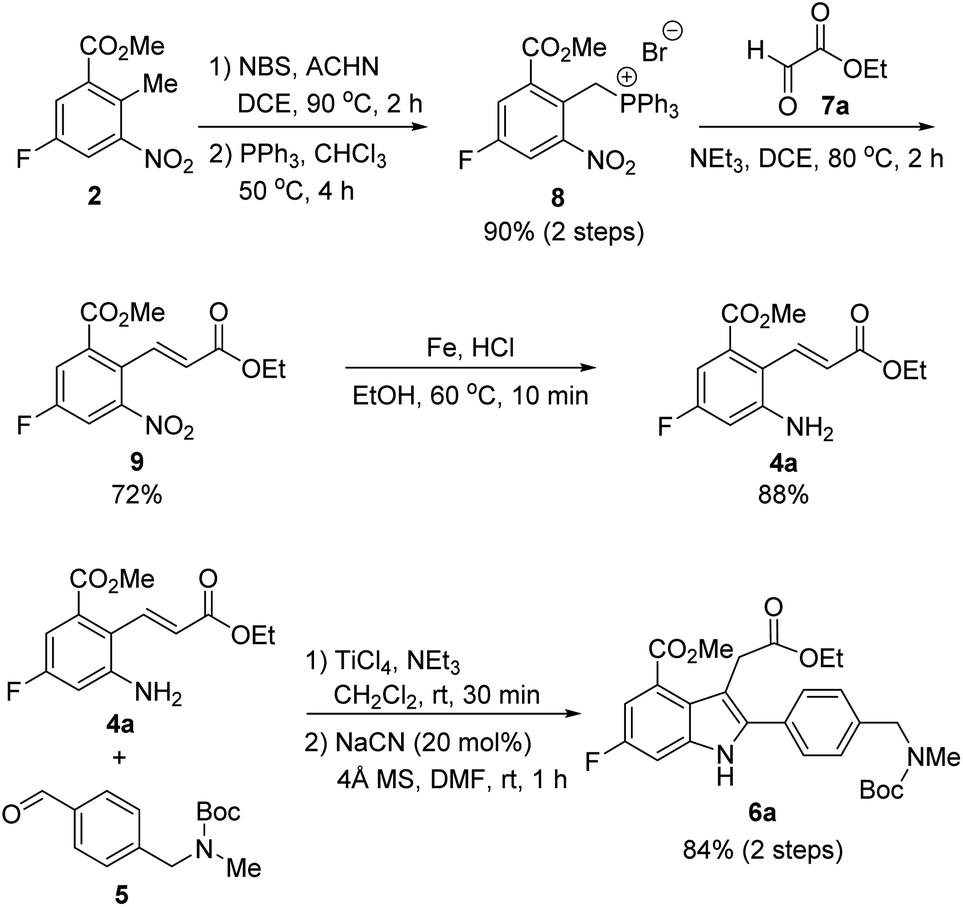
With this synthetic plan in mind, our synthesis began with the preparation of indole intermediate 6 bearing the four desired substituents via the cyanide-catalyzed imino-Stetter reaction of 2-aminocinnamic acid derivative 4 and aldehyde 5 (Scheme 3).
 | ||
| Scheme 3 Preparation of indole-3-acetate 6a. | ||
Since aldehyde 5 could be obtained from commercial suppliers, we focused on the preparation of 2-aminocinnamate 4a (X = OEt) from meta-fluorobenzoate 2. To construct the cinnamic acid moiety from 2, we chose to perform a Wittig olefination reaction with ethyl glyoxalate (7a). Treatment of 2 with N-bromosuccinimide (NBS) in the presence of a catalytic amount of 1,1′-azobis(cyclohexanecarbonitrile) (ACHN) in 1,2-dichloroethane (DCE) afforded its corresponding benzyl bromide, which was then reacted with triphenylphosphine to give the desired phosphonium salt 8 in 90% yield over two steps. The Wittig reaction of phosphonium salt 8 with glyoxalate 7a in the presence of triethylamine gave cinnamate 9 in 72% yield. Subsequent reduction of the nitro group in 9 using iron under acidic conditions afforded 2-aminocinnamate 4a in 88% yield. The condensation of 4a and 5 using TiCl4 as a dehydrating reagent furnished the desired aldimine, which was then treated with a catalytic amount of cyanide to yield the target indole intermediate 6a in 84% yield over two steps.
With indole 6a in hand, we focused on the construction of the azepinone moiety between the substituents at C3 and C4-positions to complete the total synthesis of rucaparib (1). For the successful construction of the azepinone scaffold, we needed to convert the aliphatic ester in 6a into its corresponding amine in the presence of an aromatic ester group. To achieve this, we designed a two-step sequence involving the conversion of the aliphatic ester in 6a into its corresponding amide 10, followed by the reduction of the amide group into its corresponding amine, leading to compound 11 (Scheme 4). However, there are a few anticipated challenges in this approach. First, we needed to develop (1) a chemoselective reaction of the aliphatic ester (highlighted in blue) in 6a to form an amide in the presence of the aromatic ester (highlighted in red) and (2) a chemoselective reduction of the resulting amide group (highlighted in blue) in 10 into its corresponding amine 11 in the presence of the aromatic ester moiety (highlighted in red). Second, under the reduction conditions in which amide 10 could be converted into amine 11, the resulting amine group in 11 may undergo cyclization with the adjacent aromatic ester group to furnish a seven-membered lactam 12. In this scenario, the reduction of the cyclic amide moiety (highlighted in red) in 12 must be prevented under the same reduction conditions so that unwanted cyclic amine 13 would not be generated. Thus, we had to establish conditions to convert amide 10 into amine 11 or lactam 12 without the formation of cyclic amine 13.
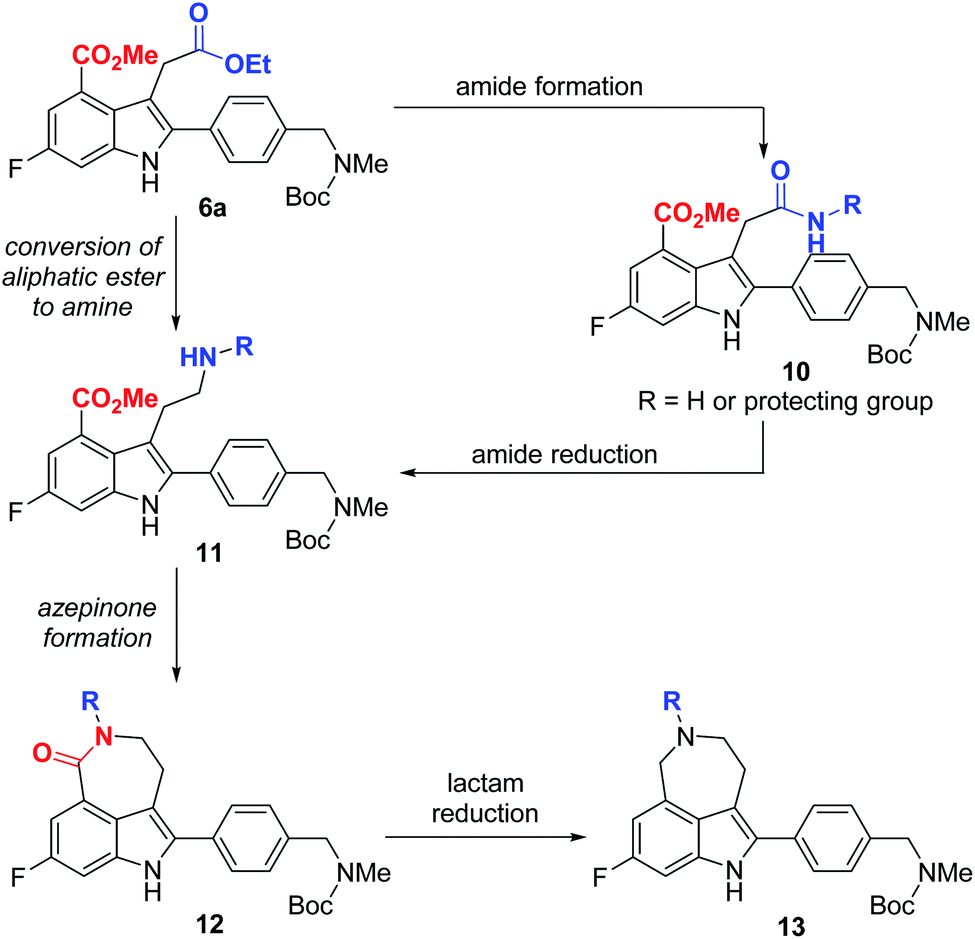 | ||
| Scheme 4 Anticipated challenges in the construction of the azepinone scaffold from compound 6a. | ||
With these chemoselectivity issues in mind, we attempted to complete the total synthesis of rucaparib (1) by constructing the azepinone moiety from 6a (Scheme 5). We first explored the chemoselective conversion of the aliphatic ester group in 6a into its corresponding amide in the presence of the aromatic ester group. As expected, the chemoselective formation of the amide from the aliphatic ester in the presence of the aromatic ester was difficult to control. When 6a was treated with para-methoxybenzyl (PMB) amine in the presence of trimethylaluminum,15 the chemoselective conversion of the aliphatic ester into its corresponding secondary amide proceeded with very poor selectivity with monoamide 6b and diamide 14 being obtained in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Despite the poor chemoselectivity in the amide formation step, the two resulting amides (6b and 14) could be separable. Thus, we further explored the chemoselective reduction of the amide group in 6b into its corresponding amine in the presence of the ester group.
:1 ratio. Despite the poor chemoselectivity in the amide formation step, the two resulting amides (6b and 14) could be separable. Thus, we further explored the chemoselective reduction of the amide group in 6b into its corresponding amine in the presence of the ester group.
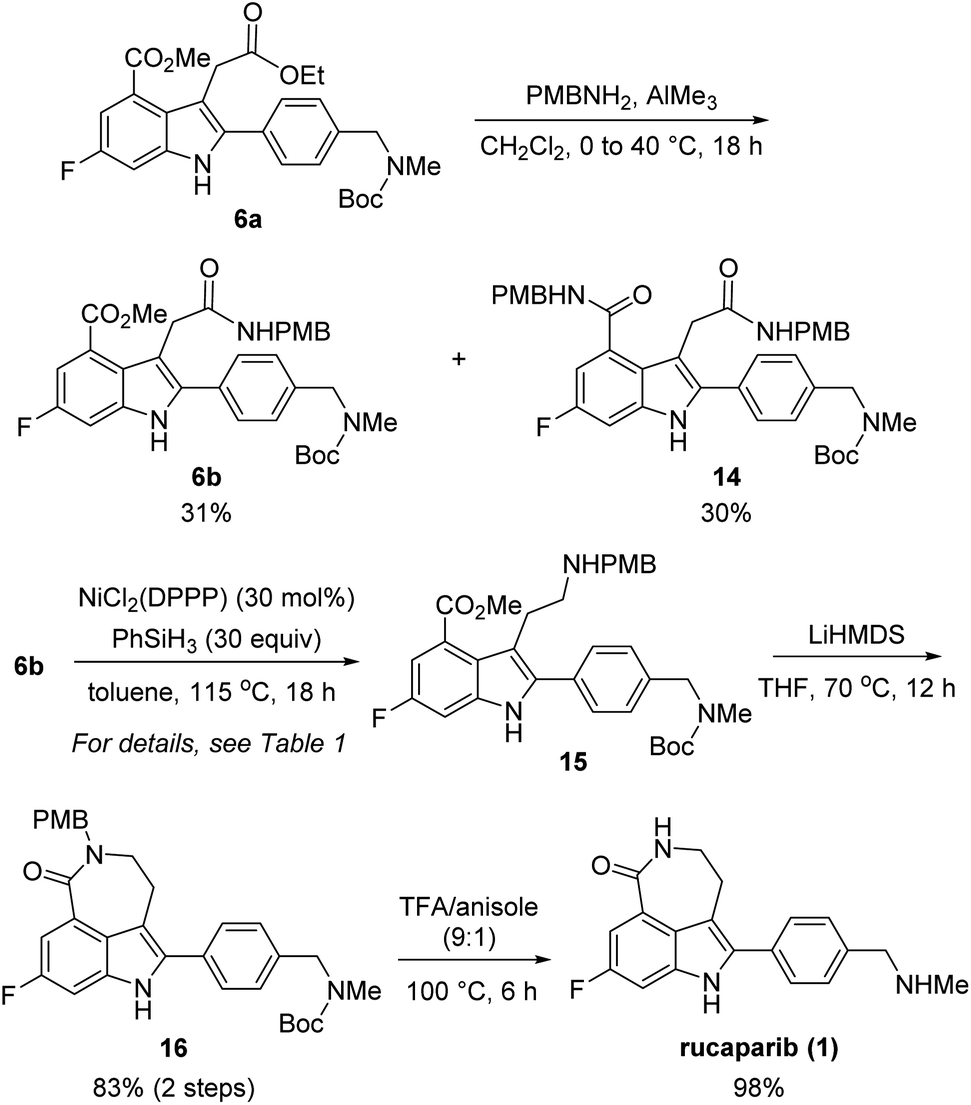 | ||
| Scheme 5 End game to the first-generation synthesis of rucaparib (1). | ||
However, the chemoselective reduction of the amide group of 6b was also found to be very challenging. After extensively investigating the reaction conditions,16,17 we conducted the chemoselective reduction of the amide using silane as the reducing reagent in the presence of a nickel catalyst (Table 1).18,19 Interestingly, the efficiency of the reduction reaction was significantly increased when using an additional ligand on the nickel catalyst.20 The amide could not be reduced with NiCl2 (entry 1), whereas the reaction using NiCl2(dme) (dme = 1,2-dimethoxyethylene) as the catalyst gave aminoester 15 in 27% yield without the formation of any side products (entry 2). However, the yield of 15 did not improve, even after extending the reaction time. We further examined other ligands to improve the reactivity of the catalyst. When NiCl2(PPh3)2 was used as the catalyst, 6b was completely consumed in 18 h. However, the desired amine 15 was obtained in a moderate yield (57%) and rather disappointingly, cyclic amine 17 was obtained as a side product without the formation of lactam 16 (entry 3).21
|
|||
|---|---|---|---|
| Entry | Ni cat. | Conversionb (%) | Ratio of 15:16:17b |
| a Reaction conditions: 6b (0.10 mmol), Ni catalyst (10 mol%), and PhSiH3 (1.0 mmol) in toluene (1.0 mL) at 115 °C for 18 h.b The conversion and the ratio of 15, 16 and 17 were determined using 1H NMR spectroscopy of the crude mixture.c Not determined.d Isolated yield of 15 after column chromatography.e 30 mol% of NiCl2(DPPP) was used. | |||
| 1 | NiCl2 | 0 | N. D.c |
| 2 | NiCl2(dme) | 42 | 1:0:0 (27%)d |
| 3 | NiCl2(PPh3)2 | 100 | 1:0:0.2 (57%)d |
| 4 | NiCl2(DPPF) | 0 | N. D.c |
| 5 | NiCl2(BINAP) | 52 | 1:0:0.1 |
| 6 | NiCl2(Xantphos) | 100 | 1:0:0.2 |
| 7 | NiCl2(DPPE) | 36 | 1:0:0 |
| 8 | NiCl2(DPPP) | 56 | 1:0:0 |
| 9e | NiCl2(DPPP) | 100 | 1:0:0 (80%)d |
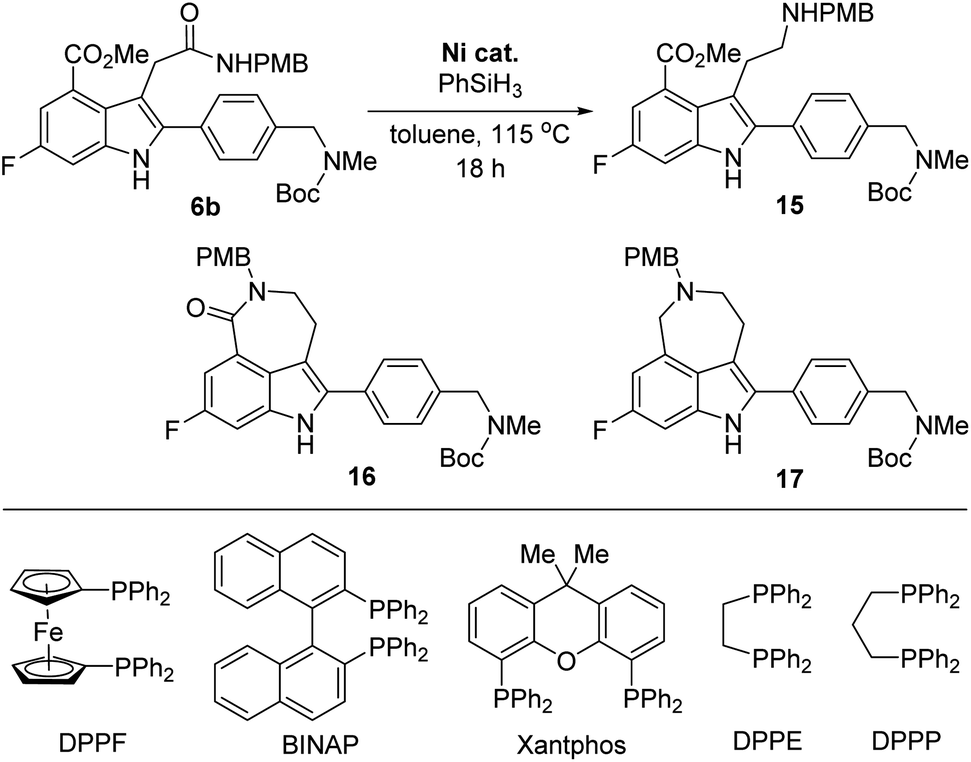
This result strongly suggested that the reduction of the cyclic amide in 16 proceeds faster than the reduction of the acyclic amide in 6b. Consequently, we must suppress the formation of lactam 16 to increase the yield of the final product, rucaparib (1). Thus, we investigated the use of a nickel catalyst bearing a bidentate diphosphine ligand in the reaction with an expectation that the bidentate ligand would decrease the Lewis acidity of the nickel catalyst, suppressing the lactamization reaction of the amine product 15 (entries 4–9).22 As expected, the bidentate phosphine ligands considerably influenced the reactivity of nickel catalysts. The nickel catalyst derived from 1,1′-bis(diphenylphosphino)ferrocene (DPPF) did not afford the desired aminoester product 15 (entry 4), while the reaction using 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) afforded 15 in a moderate yield, along with cyclic amine 17 (entry 5).
To further improve the reactivity of the nickel catalyst, we examined other bidentate ligands. Using Xantphos, a less strong σ-donor bidentate ligand, in the nickel catalyst, the reaction was completed within 18 h, but cyclic amine 17 was again obtained as a side product (entry 6). Although the reaction with a nickel complex derived from 1,2-bis(diphenylphosphino)ethane (DPPE) produced the desired product 15 in low yield, the formation of side product 17 was not observed even after a prolonged reaction time (entry 7). Fortunately, the nickel catalyst derived from 1,3-bis(diphenylphosphino)propane (DPPP) exhibited better reactivity and the desired product 15 was obtained in an improved yield with no formation of cyclic amide 16 or cyclic amine 17 (entry 8). To further improve the yield of the desired product 15, the catalyst loading was increased. To our delight, the desired product 15 was obtained in 80% yield when the reaction was carried out using 30 mol% of the catalyst, without the formation of lactam 16 and side product 17 (entry 9).
With the optimized reaction conditions for the chemoselective reduction of the amide in 6b in hand, we attempted to complete the total synthesis of rucaparib (1) (Scheme 5). The reduction of amide 6b with phenylsilane in the presence of a catalytic amount of NiCl2(DPPP) could be performed on a 1.0 mmol scale affording the desired aminoester 15,23 which was directly subjected to the cyclization step under basic conditions to yield the desired lactam 16 in 83% yield over two steps. Removal of the Boc and PMB groups with trifluoroacetic acid (TFA) in anisole provided rucaparib (1) in 98% yield. The spectroscopic data obtained for 1 were in good agreement with those reported in the literature.5–8 Overall, we completed the total synthesis of rucaparib (1) in an overall 12% yield over seven separation operations from commercially available starting materials.
Although we successfully completed the total synthesis of rucaparib (1) from commercially available starting materials, we encountered challenges in the construction of the azepinone scaffold from indole derivative 6a, particularly in regard to the chemoselective conversion of the aliphatic ester in 6a into its corresponding amide 6b in the presence of the aromatic ester. Therefore, we attempted to develop an improved synthetic route to rucaparib (1) by resolving the problems associated with the first-generation synthesis. Particularly, since we successfully developed a route to prepare rucaparib (1) from indole-3-acetamide 6b, and already demonstrated that the cyanide-catalyzed imino-Stetter reaction can be applied toward the preparation of indole-3-acetamides from aldimines derived from 2-aminocinnamamides,13 we envisioned that the synthesis of rucaparib (1) could be streamlined if indole-3-acetamide 6b was directly prepared from 2-aminocinnamamide 4b bearing a PMB group on the nitrogen atom in the amide moiety (X = NHPMB, Scheme 2) via the cyanide-catalyzed imino-Stetter reaction.
With this synthetic plan in mind, we focused on the preparation of 2-aminocinnamamide 4b from meta-fluorobenzoate 2 (Scheme 6). The Wittig reaction of phosphonium salt 8 and glyoxalamide 7b24 performed in the presence of triethylamine furnished cinnamamide 18 in 85% yield. Unexpectedly, the undesired Z-isomer of the cinnamamide product, (Z)-18, was obtained as the major product. Thus, the undesired Z-isomer must be converted into the desired E-isomer via an olefin isomerization step. We speculated that the predominant formation of the Z-isomer from the Wittig reaction may be due to the intramolecular hydrogen bonds between the N–H bond in the amide and the oxygen atoms in the ortho-substituents on the phenyl ring.25 Thus, the free N–H of the amide in (Z)-18 was first protected using tert-butoxycarbonyl (Boc) anhydride to afford N-Boc-protected (Z)-cinnamamide, (Z)-19, in 98% yield. Treatment of (Z)-19 with NBS afforded the desired E-isomer, (E)-19, in 65% yield, along with the recovered Z-isomer (Z)-19 in 15% yield.26,27 Reduction of the nitro group using iron under acidic conditions furnished amine 20. However, under the acidic reduction conditions, some of the Boc groups in 20 were cleaved, affording 2-aminocinnamamide 4b with the correct (E)-configuration. Thus, the resulting reaction mixture was further treated with TFA to yield (E)-2-aminocinnamamide 4b in 92% yield over two steps.
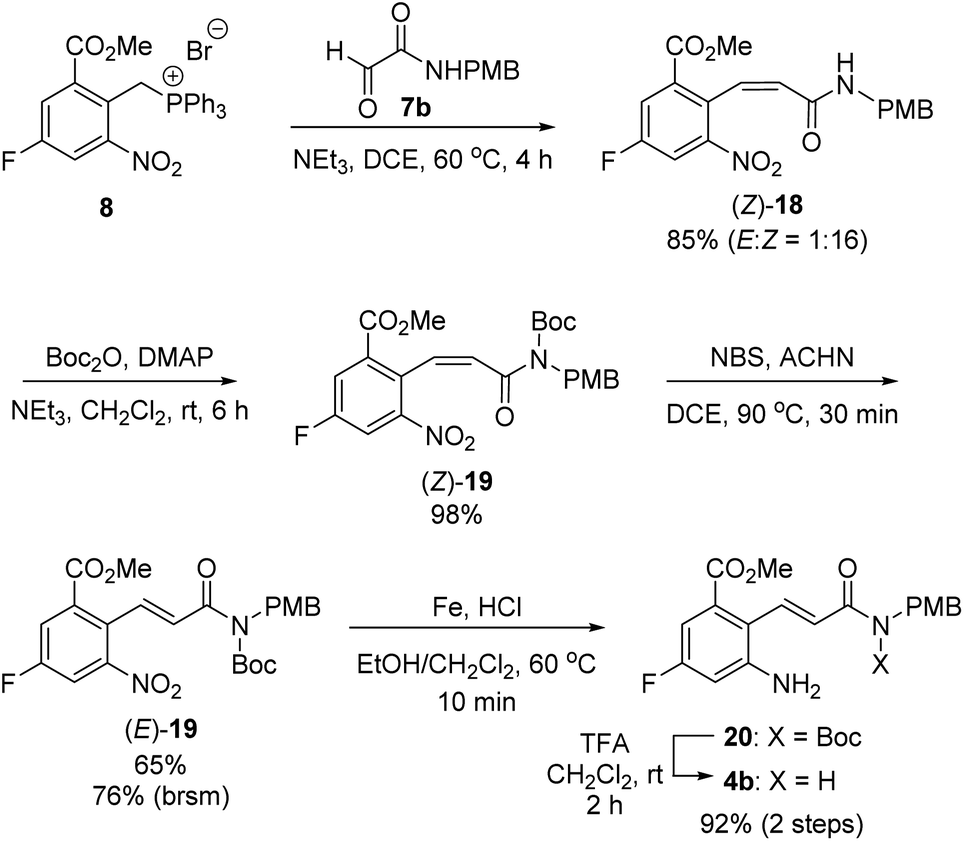 | ||
| Scheme 6 Preparation of 2-aminocinnamamide 4b. | ||
With 2-aminocinnamamide 4b in hand, we attempted to complete the total synthesis of rucaparib (1) (Scheme 7). The condensation of 4b and 5 using TiCl4 as a dehydrating reagent furnished the desired aldimine. Subsequent treatment of the resulting aldimine with a catalytic amount of cyanide provided the target tetra-substituted indole 6b in 80% yield over two steps. From indole-3-acetamide 6b, we were able to complete the total synthesis of rucaparib (1) using the procedures developed in our first-generation synthesis of rucaparib (1) (see Scheme 5). Overall, we completed the total synthesis of rucaparib in 27% overall yield over eight separation operations from commercially available starting materials.
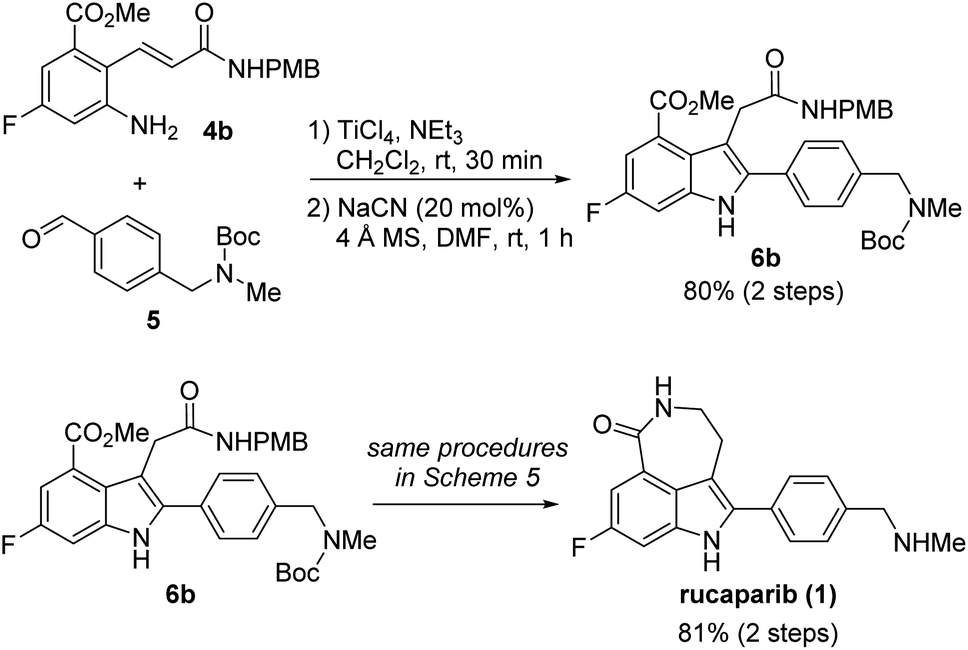 | ||
| Scheme 7 A modified first-generation synthesis of rucaparib (1). | ||
2. Second-generation synthesis of rucaparib (1)
Although we have developed a novel route for the total synthesis of rucaparib (1) with an improved yield (27% yield in our first-generation synthesis vs. 5.2% yield in the current process chemistry route) from commercially available starting materials such as meta-fluorobenzoate 2, which is the starting material used in the current process chemistry route, our first-generation synthesis of rucaparib (1) has two major drawbacks.The first is the chemoselectivity observed during the construction of the azepinone scaffold from indole-3-acetic acid derivative 6. In the first approach, the chemoselective conversion of the aliphatic ester in indole-3-acetate 6a into its corresponding amide in the presence of the aromatic ester group was problematic; both aliphatic and aromatic esters showed similar reactivity, leading to the formation of a 1:1 mixture of monoamide 6b and diamide 14. Moreover, during the construction of the azepinone scaffold from indole-3-acetamide 6b, the chemoselective reduction of the amide in the presence of the ester group present in 6b needed to be carefully controlled. Furthermore, because the cyclic amide in the azepinone scaffold of 16, generated via cyclization of the resulting amine 15, displays higher reactivity toward the reductant than the acyclic amide in 6b, comprehensive optimization of the reaction conditions was required to suppress the reduction of the lactam, preventing the unwanted formation of cyclic amine 17. The second problem was the preparation of 2-aminocinnamamide 6b from compound 2. The Wittig reaction of phosphonium salt 8 with glyoxalamide favored the formation of the unwanted Z-isomer of the cinnamamide product, (Z)-18, which required additional steps to be converted into the desired E-isomer.
Despite the issues raised in the first-generation synthesis of rucaparib (1), it was found that the cyanide-catalyzed imino-Stetter reaction was a highly reliable method for constructing the tetra-substituted indole derivative with the correct regioselectivity. Therefore, we attempted to devise a second-generation synthetic route for rucaparib (1) using the cyanide-catalyzed imino-Stetter reaction as the key step to construct the indole subunit. Since the issues mentioned above can be ascribed to the amide moiety in 6b and 18, we envisioned that the use of a different carboxylic acid moiety other than an amide could resolve these issues. Among the carboxylic acid derivatives, a nitrile group would be an ideal group for the second-generation synthesis of rucaparib (1) for the following reasons:
(1) The chemoselective the reduction of the nitrile group will be possible since there have been several reliable methods to reduce a nitrile group into its corresponding amine in the presence of an ester group reported in the literature.28,29
(2) Since the nitrile group displays different reactivity to the cyclic amide in the azepinone scaffold, which can be generated by the cyclization of the resulting amine with the adjacent ester group, the formation of the undesired cyclic amine, possibly formed by the reduction of the cyclic amide, may be avoided.
(3) The nitrile group in the 2-aminocinnamonitrile will favor the formation of the E-isomer because the nitrile group does not form favorable H-bond interactions with the ortho-substituents of the cinnamonitrile.25
(4) Very recently, our group has developed a protocol for the synthesis of 2-substituted indole-3-acetonitriles using the cyanide-catalyzed imino-Stetter reaction of aldimines derived from 2-aminocinnamonitriles and aldehydes.30
(5) Since 2-aminocinnamonitriles, one of the key building blocks for the cyanide-catalyzed imino-Stetter reaction, are prepared by the Heck reaction of an aryl halide and acrylonitrile,30 4,6-disubstituted 2-aminocinnamonitrile can be synthesized in a much more efficient manner via the Heck reaction of a suitable aryl halide (or pseudohalide) and acrylonitrile.
With these ideas in mind, the retrosynthetic analysis of the second-generation synthesis of rucaparib (1) is shown in Scheme 8.11 Rucaparib (1) could be synthesized from indole-3-acetonitrile 6c bearing all of the desired substituents at the 2,4,6-positions through the construction of the azepinone scaffold. Indole-3-acetonitrile 6c could be prepared from 2-aminocinnamonitrile 4c and aldehyde 5 via the key cyanide-catalyzed imino-Stetter reaction.30 2-Aminocinnamonitrile 4c could be prepared using the Heck reaction of an appropriate aryl halide (or pseudohalide) 21 with acrylonitrile.
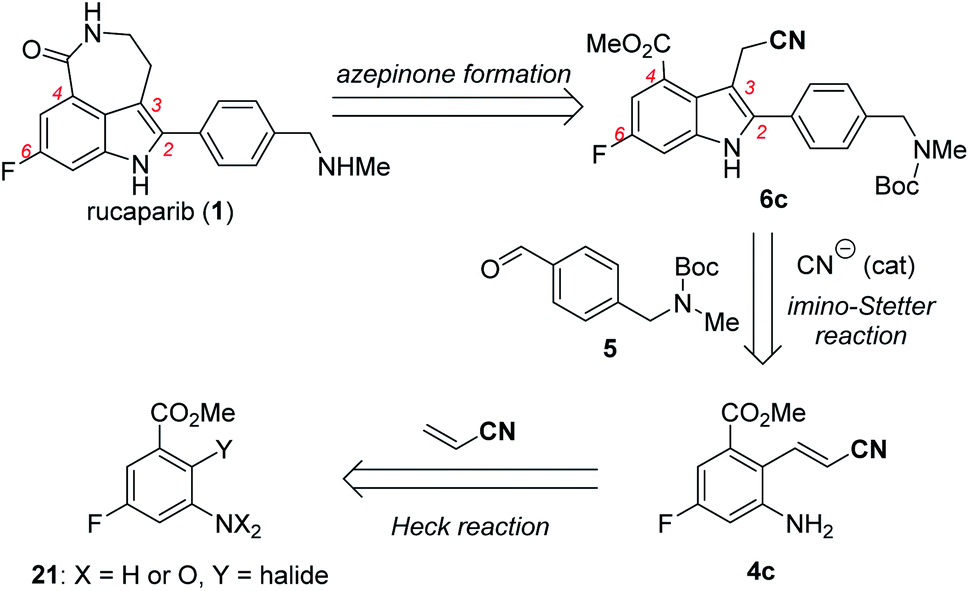 | ||
| Scheme 8 Retrosynthetic analysis of the second-generation synthesis of rucaparib (1). | ||
Our second-generation synthesis of rucaparib (1) began with the preparation of (E)-2-aminocinnamonitrile 4c via the Heck reaction of an appropriate aryl halide (or pseudohalide) with acrylonitrile (Scheme 9).31 As ortho-nitroaryl trifluoromethanesulfonate (triflate; OTf) SII was used in the previous synthesis,7 we first attempted to synthesize 2-nitrocinnamonitrile derivative 23 from aryl triflate SII via the Heck reaction with acrylonitrile. However, the reaction of SII with acrylonitrile using several different palladium catalysts did not provide the desired coupling product 23; in all cases, the triflate group in SII was hydrolyzed to form the corresponding phenol product 24 (eqn (1)). To prevent the undesired hydrolysis of the triflate group, we attempted to prepare the desired cinnamonitrile using the Heck reaction of an aryl iodide32 and acrylonitrile. Interestingly, the ortho-substituent on the iodide group significantly affected the outcome of the coupling reaction. Although ortho-nitroaryl iodide 22 did not afford the desired coupling product 23 (eqn (2)), the coupling reaction ortho-aminoaryl iodide 3 with acrylonitrile proceeded smoothly with Pd(t-Bu3P)2 as the catalyst,33 affording the coupling product 4c in 92% yield on a 10 mmol scale (eqn (3)).
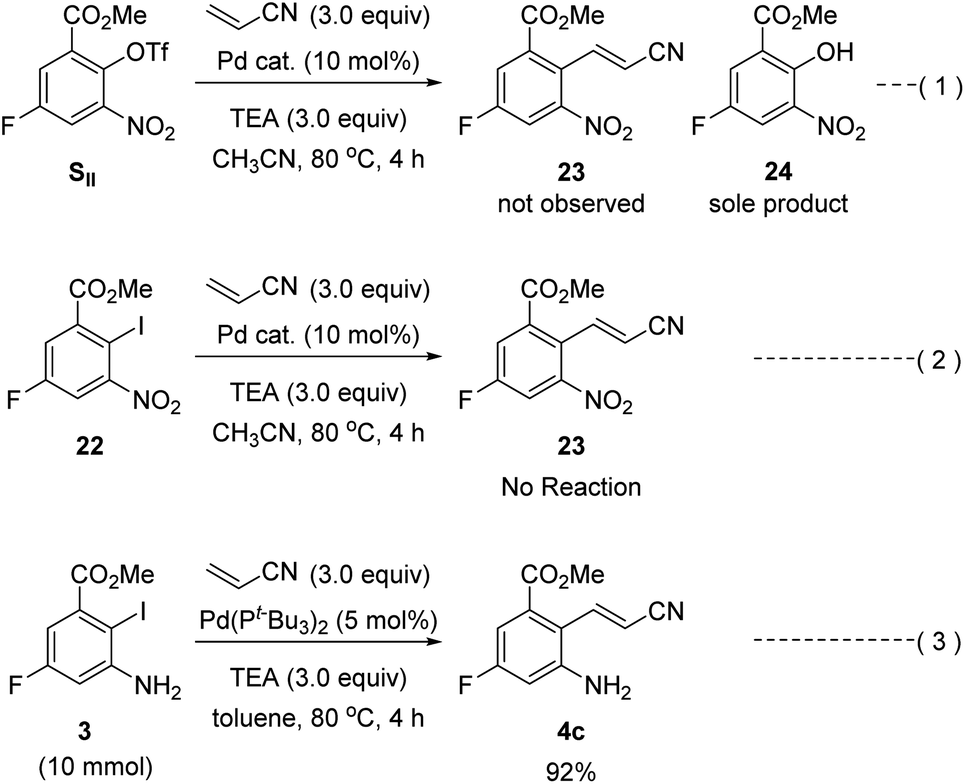 | ||
| Scheme 9 Heck reaction of ortho-aminoaryl iodide 3 or ortho-nitroaryl iodide 22 (or triflate SII) with acrylonitrile. | ||
With 2-aminocinnamonitrile 4c in hand, we focused on the preparation of indole-3-acetonitrile 6c bearing all of the desired substituents in their correct positions via the cyanide-catalyzed imino-Stetter reaction (Scheme 10a). Aldimine formation from 4c and 5 using TiCl4 as a dehydrating reagent followed by the cyanide-catalyzed imino-Stetter reaction afforded the desired indole-3-acetonitrile 6c in 80% yield on a 10 mmol scale.12–14,30
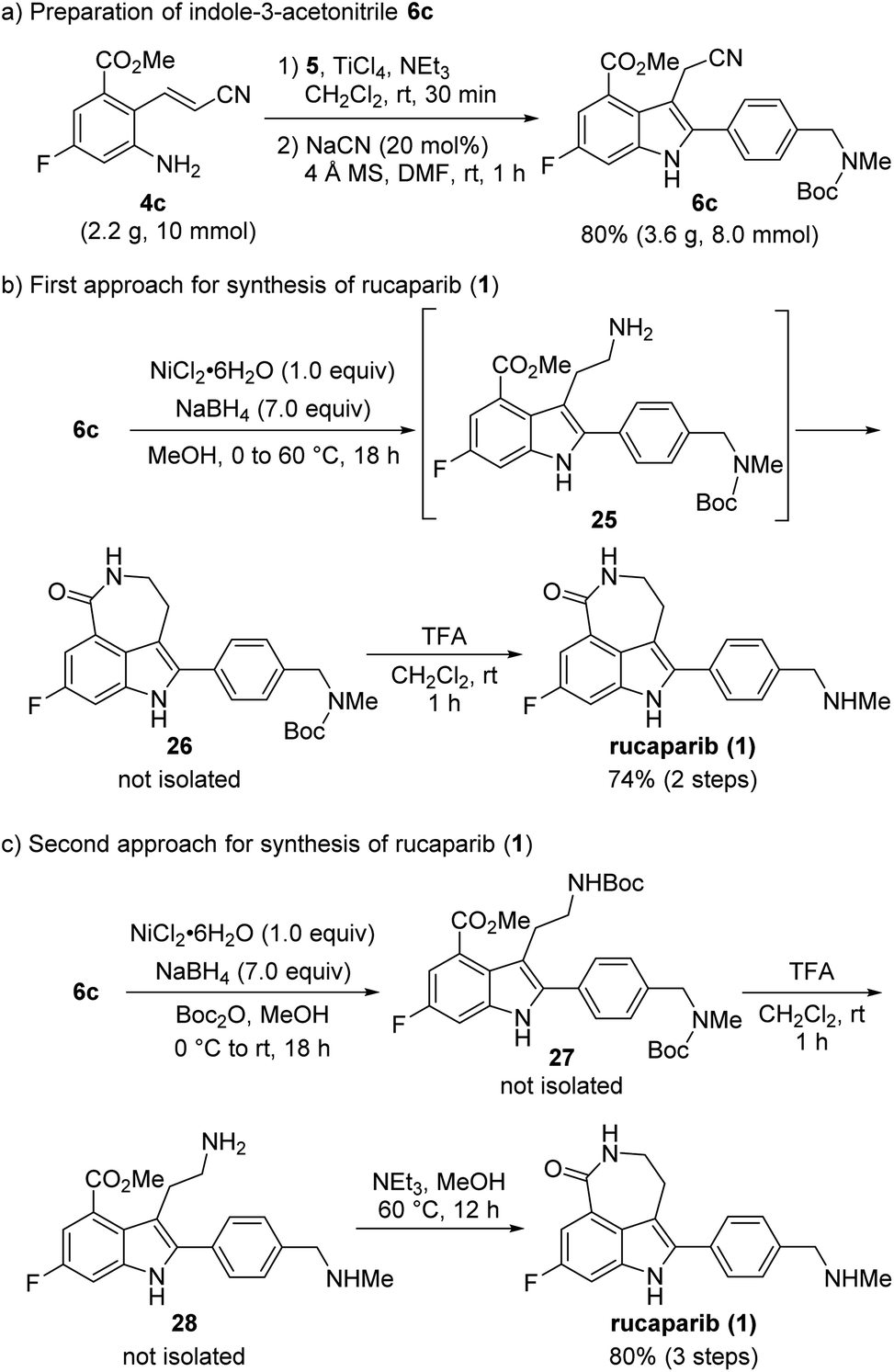 | ||
| Scheme 10 The second-generation synthesis of rucaparib (1). | ||
Our next efforts were directed toward constructing the azepinone scaffold via the reduction of the nitrile into its corresponding amine, followed by lactam formation to complete the total synthesis of rucaparib (1). Initially, the reduction of the nitrile group was attempted using hydrogenation.28 However, the nitrile group was not reduced, and the starting material 6c remained unreacted in the reaction mixture under these conditions. Thus, alternative methods to reduce the nitrile group were explored. After a quick screen of the other reducing agents available, it was found that nickel boride, which was generated in situ from NiCl2·6H2O and NaBH4, was effective for the chemoselective reduction of the nitrile group in the presence of the ester group (Scheme 10b).29 When 6c was treated with nickel boride, the nitrile group was successfully reduced into its corresponding primary amine 25, which spontaneously underwent lactamization to provide Boc-protected rucaparib 26. Removal of the Boc group upon treatment with TFA furnished rucaparib (1) in 74% yield over two steps. Spectroscopic data of the resulting rucaparib (1) were in good agreement with those reported in the literature.5–8
Although the total synthesis of rucaparib (1) was accomplished in two steps from indole-3-acetontrile 6c, the formation of a trace amount of side products was observed during the reduction of the nitrile group in 6c.34 A literature survey revealed that protection of the resulting primary amine with an electron-withdrawing group could exert a beneficial effect on the reduction of the nitrile group by preventing the formation of the unwanted side products.35 Therefore, we attempted to carry out the reduction of the nitrile group with nickel boride in the presence of Boc2O to suppress the formation of the unwanted side products (Scheme 10c).
To our delight, when indole-3-nitrile 6c was treated with nickel boride in the presence of Boc2O, Boc-protected primary amine 27 was obtained via the reduction of the nitrile group into amine 25, followed by its reaction with Boc2O. Subsequent removal of both Boc groups with TFA provided primary amine 27, which underwent the final cyclization step in the presence of an amine in methanol to provide rucaparib (1) in 80% yield from indole 6c over three steps. Overall, rucaparib (1) was obtained from commercially available starting materials in an overall 59% yield over three separation operations in our second-generation synthesis.
Conclusions
In conclusion, we developed two routes for the total synthesis of rucaparib, an FDA-approved drug used for the treatment of ovarian and prostate cancers, starting from commercially available starting materials using the cyanide-catalyzed imino-Stetter reaction as the key step to construct an indole scaffold bearing the four desired substituents in their correct positions.In the first-generation synthesis, 4,6-disubstituted 2-aminocinnamic acid derivatives (ester or amide) were prepared from the starting material currently used in the process chemistry route of rucaparib. The subsequent cyanide-catalyzed imino-Stetter reaction of the aldimine derived from the 2-aminocinnamic acid derivative and a commercially available aryl aldehyde allowed the preparation of the target tetra-substituted indole derivative. The final azepinone formation step completed the total synthesis of rucaparib in 27% overall yield.
To resolve the formation of undesired Z-isomer of 2-aminocinnamide and chemoselectivity during the azepinone formation raised in the first-generation synthesis, a second-generation total synthesis of rucaparib was developed. The Heck reaction of an ortho-iodoaniline derivative with acrylonitrile afforded the desired 2-aminocinnamonitrile. The subsequent cyanide-catalyzed imino-Stetter reaction of the resulting 2-aminocinnamonitrile and the same aldehyde afforded the desired indole-3-acetonitrile bearing all of the required substituents in their correct positions. The reduction of the nitrile group, followed by lactam formation completed the total synthesis of rucaparib in an overall 59% yield over only three separation operations.
It should be noted that our newly developed synthetic route will allow rucaparib to be prepared in a higher yield over less steps from commercially available starting materials. Further efforts to achieve a scale-up synthesis and a more practical synthesis of this important compound are currently underway in our laboratory and will be reported in the future.
Author contributions
J. P. and C.-H. C. conceived and designed the project. J. P. performed experiments and collected the data. All authors participated in discussing the data and wrote the manuscript.Conflicts of interest
The authors declare the following competing financial interest(s): a patent application for the synthesis of rucaparib has been filed (KR 10-2021-0101905; KR 10-2021-0101907).Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korean Government (NRF-2021R1A2C1012984, NRF-2021R1A5A6002803 (Center for New Directions in Organic Synthesis).Notes and references
- N. J. Curtin, Cancers, 2020, 12, 564–581 CrossRef CAS PubMed.
- S. alasubramaniam, J. A. eaver, S. Horton, L. L. Fernandes, S. Tang, H. N. Horne, J. Liu, C. Liu, S. J. Schrieber, J. Yu, P. Song, W. Pierce, K. J. Robertson, T. R. Palmby, H. J. Chiu, E. Y. Lee, R. Philip, R. Schuck, R. Charlab, A. Banerjee, X. H. Chen, X. Wang, K. B. Goldberg, R. Sridhara, G. Kim and R. F. D. A. Pazdur, Clin. Cancer Res., 2017, 23, 7165–7170 CrossRef PubMed.
- M. S. Anscher, E. Chang, X. Gao, Y. Gong, C. Weinstock, E. Bloomquist, O. Adeniyi, R. Charlab, S. Zimmerman, M. Serlemitsos-Day, Y. M. Ning, R. Mayrosh, B. Fuller, A. M. Trentacosti, P. Gallagher, K. Bijwaard, R. Philip, S. Ghosh, F. Fahnbulleh, F. Diggs, S. Arora, K. B. Goldberg, S. Tang, L. Amiri-Kordestani, R. Pazdur, A. Ibrahim and J. A. F. D. A. Beaver, Oncologist, 2021, 26, 139–146 CrossRef CAS PubMed.
- For a review, see: D. L. Hughes, Org. Process Res. Dev., 2017, 21, 1227–1244 CrossRef CAS.
- A. T. Gillmore, M. Badland, C. L. Crook, N. M. Castro, D. J. Critcher, S. J. Fussell, K. J. Jones, M. C. Jones, E. Kougoulos, J. S. Mathew, L. McMillan, J. E. Pearce, F. L. Rawlinson, A. E. Sherlock and R. Walton, Org. Process Res. Dev., 2012, 16, 1897–1904 CrossRef CAS.
- (a) S. E. Webber, S. S. Canan-Koch, J. Tikhe and L. H. Thoresene, PCT Int. Appl. WO2000042040A1, 2000; (b) D. S. Samec, J. Dogan, T. Biljan, M. M. Skugor, M. Mihovilovic, T. Mundorfer, N. Janton, M. Tuksar, S. M. Pipercic and N. Baus, PCT Int. Appl. WO2018140377A1, 2018; (c) I. Beckers, G. O'Rourke and D. D. Vos, Synthesis, 2022, 54, 334–340 CrossRef CAS.
- C. Ma, N. Nayyar and N. S. Stankovic, US Pat. US20060063926A1, 2006.
- (a) C. Cheng, X. Zuo, D. Tu, B. Wan and Y. Zhang, Org. Lett., 2020, 22, 4985–4989 CrossRef CAS PubMed; (b) Y. Zhang and C. Cheng, China Patent CN111646990A, 2020.
- In the current process chemistry route, rucaparib (1) is prepared from the available starting materials with an overall yield of 5.2% in seven separation operations. For details, see ref. 5.
- For the synthesis of rucaparib derivatives bearing radioactive isotopes, see: (a) Z. Chen, G. Destro, F. Guibbal, C. Y. Chan, B. Cornelissen and V. Gouverneur, Org. Lett., 2021, 23, 7290–7294 CrossRef CAS PubMed; (b) M. Liao, S. Watkins, E. Nash, J. Isaacson, J. Etter, J. Beltman, R. Fan, L. Shen, A. Mutlib, V. Kemeny, Z. Pápai, P. van Tilburg and J. J. Xiao, Invest. New Drugs, 2020, 38, 765–775 CrossRef CAS PubMed; (c) S. R. Alluri and P. J. Riss, ACS Chem. Neurosci., 2018, 9, 1259–1263 CrossRef CAS PubMed.
- J. Park and C.-H. Cheon, J. Org. Chem., 2022, 87, 4803–4817 CrossRef PubMed.
- For a review of the cyanide-catalyzed imino-Stetter reaction, see: E. Park, J. Jeon and C.-H. Cheon, Chem. Rec., 2018, 18, 1474–1488 CrossRef CAS PubMed.
- For seminal reports on the cyanide-catalyzed imino-Stetter reaction, see: (a) S. J. Lee, H.-A. Seo and C.-H. Cheon, Adv. Synth. Catal., 2016, 358, 1566–1570 CrossRef CAS; (b) H.-A. Seo and C.-H. Cheon, J. Org. Chem., 2016, 81, 7917–7923 CrossRef CAS PubMed.
- For selected examples of the application of the imino-Stetter reaction to the total synthesis of natural products, see: (a) S. H. Kwon, H.-A. Seo and C.-H. Cheon, Org. Lett., 2016, 18, 5280–5283 CrossRef CAS PubMed; (b) S. Lee, K.-H. Kim and C.-H. Cheon, Org. Lett., 2017, 19, 2785–2788 CrossRef CAS PubMed; (c) C. Bae, E. Park, C.-G. Cho and C.-H. Cheon, Org. Lett., 2020, 22, 2354–2358 CrossRef CAS PubMed; (d) J. Jeon, S. E. Lee and C.-H. Cheon, Org. Lett., 2021, 23, 2169–2173 CrossRef CAS PubMed.
- T. Gustafsson, F. Pontén and P. H. Seeberger, Chem. Commun., 2008, 1100–1102 RSC.
- For a review of the chemoselective reduction of carboxamides, see: A. Volkov, F. Tinnis, T. Slagbrand, P. Trillo and H. Adolfsson, Chem. Soc. Rev., 2016, 45, 6685–6697 RSC.
- We also investigated the reduction of the amide with electrophilic reducing reagents such as BH3. However, although the reduction of amide proceeded slightly faster than that of the ester, both amide and ester were reduced with this reagent. For details, see ESI.†.
- B. J. Simmons, M. Hoffmann, J. Hwang, M. K. Jackl and N. K. Garg, Org. Lett., 2017, 19, 1910–1913 CrossRef CAS PubMed.
- When other metal catalysts were used, either the reaction did not proceed or only the Boc group was removed. For an example of the reduction of amides with a metal catalyst, see: S. Das, D. Addis, K. Junge and M. Beller, Chem.–Eur. J., 2011, 17, 12186–12192 CrossRef CAS PubMed.
- S. Yu, T. Shin, M. Zhang, Y. Xia, H. Kim and S. Lee, Org. Lett., 2018, 20, 7563–7566 CrossRef CAS PubMed.
- The effect of other halide ligands on the nickel complex was also investigated. However, there were no differences in the outcome of amide reduction. The use of NiBr2(PPh3)2 afforded the desired product 15 in similar efficiency as the use of the corresponding chloride catalyst.
- For a selected review on the effect of bidentate phosphine ligands on the reactivity, see: M. N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- Although we were able to perform the synthesis of rucaparib (1) on a larger scale, we decided to carry out this transformation on a 1.0 mmol scale due to the safety issue.
- Compound 7b was prepared by the slight modification of the literature protocol, N. A. Petasis and Y. Xin, US Pat. US20040010162A1, 2004.
- For examples of favorable formation of Z-olefin via intramolecular hydrogen bonding, see: (a) S. M. Kim, D. Lee and S. H. Hong, Org. Lett., 2014, 16, 6168–6171 CrossRef CAS PubMed; (b) D. Lee, S. M. Kim, H. Hirao and S. H. Hong, Org. Lett., 2017, 19, 4734–4737 CrossRef CAS PubMed.
- M. M. Baag, A. Kar and N. P. Argade, Tetrahedron, 2003, 59, 6489–6492 CrossRef CAS.
- The olefin isomerization went to completion upon increasing the reaction time. However, formation of several unwanted side products was observed when the reaction was allowed to proceed for a prolonged time period. Thus, the reaction was stopped before completion to avoid the formation of unwanted side products.
- For examples of the reduction of a nitrile group by hydrogenation, see: (a) W. Huber, J. Am. Chem. Soc., 1944, 66, 876–879 CrossRef CAS; (b) F. E. Gould, G. S. Johnson and A. F. Ferris, J. Org. Chem., 1960, 25, 1658–1660 CrossRef CAS.
- For examples of the reduction of a nitrile group by nickel boride, see: (a) T. Satoh, S. Suzuki, Y. Suzuki, Y. Miyaji and Z. Imai, Tetrahedron Lett., 1969, 10, 4555–4558 CrossRef; (b) J. M. Khurana and G. Kukreja, Synth. Commun., 2002, 32, 1265–1269 CrossRef CAS.
- For the synthesis of 2-substituted indole-3-acetonitriles via cyanide-catalyzed imino-Stetter reaction, see: H. J. Kim and C.-H. Cheon, Asian J. Org. Chem., 2020, 9, 2103–2107 CrossRef CAS.
- For details on the results of the Heck coupling reactions, see ref. 11.
- Aryl iodides, 3 and 22, are commercially available.
- A. F. Littke and G. C. Fu, J. Am. Chem. Soc., 2001, 123, 6989–7000 CrossRef CAS PubMed.
- For examples of the formation of side products during the reduction of nitrile, see: (a) C. Wang, Z. Jia, B. Zhen and M. Han, Molecules, 2018, 23, 92–102 CrossRef PubMed; (b) D. M. Sharma and B. Punji, Adv. Synth. Catal., 2019, 361, 3930–3936 CrossRef CAS.
- S. Caddick, D. B. Judd, A. KdK. Lewis, M. T. Reich and M. R. V. Williams, Tetrahedron, 2003, 59, 5417–5423 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03619c |
| This journal is © The Royal Society of Chemistry 2022 |