
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum chemical study of the reaction paths and kinetics of acetaldehyde formation on a methanol–water ice model†
Islem Ben Chouikha *a,
Boutheïna Kerkeni*ab,
Ghofrane Ouerfelliac,
Lily Makronid and
Gunnar Nyman*e
*a,
Boutheïna Kerkeni*ab,
Ghofrane Ouerfelliac,
Lily Makronid and
Gunnar Nyman*e
aDépartement de Physique, LPMC, Faculté des Sciences de Tunis, Université de Tunis el Manar, Tunis 2092, Tunisia. E-mail: islem.benchouikha@fst.utm.tn
bISAMM, Université de la Manouba, La Manouba 2010, Tunisia. E-mail: boutheina.kerkeni@isamm.uma.tn
cTaif University, Taif, Saudi Arabia
dKey Laboratory for Macromolecular Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an, Shaanxi 710119, China
eDepartment of Chemistry and Molecular Biology, University of Gothenburg, Sweden. E-mail: nyman@chem.gu.se
First published on 29th June 2022
Abstract
Acetaldehyde (CH3CHO) is ubiquitous in interstellar space and is important for astrochemistry as it can contribute to the formation of amino acids through reaction with nitrogen containing chemical species. Quantum chemical and reaction kinetics studies are reported for acetaldehyde formation from the chemical reaction of C(3P) with a methanol molecule adsorbed at the eighth position of a cubic water cluster. We present extensive quantum chemical calculations for total spin S = 1 and S = 0. The UωB97XD/6-311++G(2d,p) model chemistry is employed to optimize the structures, compute minimum energy paths and zero-point vibrational energies of all reaction steps. For the optimized structures, the calculated energies are refined by CCSD(T) single point computations. We identify four transition states on the triplet potential energy surface (PES), and one on the singlet PES. The reaction mechanism involves the intermediate formation of CH3OCH adsorbed on the ice cluster. The rate limiting step for forming acetaldehyde is the C–O bond breaking in CH3OCH to form adsorbed CH3 and HCO. We find two positions on the reaction path where spin crossing may be possible such that acetaldehyde can form in its singlet spin state. Using variational transition-state theory with multidimensional tunnelling we provide thermal rate constants for the energetically rate limiting step for both spin states and discuss two routes to acetaldehyde formation. As expected, quantum effects are important at low temperatures.
1 Introduction
The complexity of the chemical networks that take part in the formation and fragmentation processes of interstellar species depend on the physical evolution of the star-formation cycles, such as changes in density, temperature, and spectral shape and intensity of radiation.1 The synthesis of interstellar species occurs in some cases on surfaces of grains, or via gas phase reactions, and in some other cases as a combination of these. Knowledge of the pathways that lead to the synthesis of organic molecules in space could help in understanding if such chemistry could lead to the emergence of life on Earth.2The search for the chemical routes leading to the formation of interstellar Complex Organic Molecules (COMs), viz. organic species with six or more atoms,3,4 has motivated several research groups to investigate them theoretically,5–11 observationally,1,12 and experimentally.13–15 In particular, acetaldehyde (CH3CHO) was first detected in 1973 (ref. 16) and plays a key role in the chemistry of the Interstellar Medium (ISM).17 Astronomical observations toward cold and dark clouds in star-forming regions show the abundance of acetaldehyde to be approximately ∼10−10 to 10−9 with respect to the density of H2.18–20 Acetaldehyde has been observed in the gas phase toward several environments such as translucent clouds,21 pre-stellar cores,22 the comet Hale–Bopp23 but also in interstellar ices6,7,24 and meteorites.25
Acetaldehyde is the simplest methyl-bearing aldehyde and constitutes a critical precursor to prebiotic molecules such as glycolaldehyde (CH2OHCHO)26 and acrolein (CH2CHCHO). The latter is a crucial intermediate in the prebiotic synthesis of various amino acids27 and plays an important role in astrobiology.28 For these reasons, CH3CHO is one of the most studied molecules in the ISM in recent years.
So far, formation of acetaldehyde has been investigated in the gas phase,29,30 on pure water ice models,31–33 ice containing methanol,34 and on amorphous CO-rich ice cluster models.6,7,35,36 Quantum chemical calculations on the formation of CH3CHO from the reaction between HCO and CH3 radicals have been performed by Enrique-Romero et al.31 The broken symmetry (BS) approach was not initially considered in their DFT calculations, and only CH4 + CO can be formed on an amorphous molecular water ice cluster. In gas phase, possible spontaneous products are CH3CHO, CH4 + CO, and CH3OCH depending on the initial orientation of the reagents.31 The gas phase formation of CH3CHO and CH3OCH however requires a third body or spontaneous emission of a photon in order to stabilize the molecules. These conclusions have been revoked later by Enrique-Romero et al.,33 when an unrestricted BS approach was employed. As a consequence, the authors report that in gas phase the CH3CHO and CH4 + CO channels are barrierless, while CH3OCH presents a high barrier. In contrast, on an ice model represented by a single H2O molecule, H transfer from HCO to CH3 to form CH4 + CO presented a very high barrier, while the formation of CH3CHO was found to be barrierless, as in the gas phase. Other calculations on two model clusters comprised of 18 and 33 water molecules reported similar findings.32
Synthesis of acetaldehyde in the gas phase has been studied through several mechanisms using the KIDA37 and UDfA38 databases.
None of these appear to yield sufficient acetaldehyde to explain its interstellar abundance and Vazart and co-workers5 wrote a critical review of the gas-phase formation routes of acetaldehyde invoked in the literature. The prevailing theory of the formation of COMs relies on their chemistry at the surface of warm grains. Acetaldehyde has, however, been observed at very low temperatures, hence challenging the warm scenario.
Bennett and co-workers6,7 studied synthetic routes toward acetaldehyde formation inside ice mixtures of carbon monoxide and methanol condensed at 10 K from both experimental and computational points of view.
These and other laboratory experiments provide compelling evidence that acetaldehyde can easily form in interstellar ices containing carbon monoxide and methane,6,7,35 methanol,34 ethane (CH3CH3) or ethylene (CH2CH2),39 and pure methanol ices,40 or by oxidation of ethanol (CH3CH2OH)41/2-propanol (CH3CH2OHCH3)42,43 when exposed to ionizing radiation.42
In this article, we focus on a solid-state chemistry formation mechanism that has been proposed in the literature.44 We perform new electronic structure calculations on the energetics of this mechanism. Our main purpose is to investigate the role of the spin and calculate thermal rate constants for the rate limiting step of the proposed mechanism. This information can then be used in astrochemical modelling.
We study the formation of acetaldehyde following the Eley-Rideal mechanism where gas phase C(3P) approaches a methanol molecule which is part of a model grain also composed of 7 water molecules.
The overall reaction is:
| C(3P)(gas) + 1CH3OH-7w(cluster) → 1CH3CHO(gas) + 17w(cluster), |
Singh et al.44 proposed, where we below essentially use their notation, some (involving only total spin S = 1) of the following elementary steps:
Step 1: RC → I1
| C(3P)(gas) + 1CH3OH-7w(cluster) → 3CH3OHC(ads)-7w(cluster) |
Step 2: I1 → I2
| 3CH3OHC(ads)-7w(cluster) → 3CH3OCH(ads)-7w(cluster) |
We also consider intersystem crossing and investigate formation of 1CH3OCH(ads)-7w(cluster) in Step 2. Consequently, the Step 3 below has also been studied on the singlet spin potential energy surface in this paper.
Step 3: I2 → P1
| 3CH3OCH(ads)-7w(cluster) → 3[CH3 + HCO](ads)-7w(cluster) |
I2 → P1
| 1CH3OCH(ads)-7w(cluster) → 1[CH3 + HCO](ads)-7w(cluster) |
Step 4: P1 → P2
| 1[CH3+ HCO](ads)-7w(cluster) → 1CH3CHO(ads)-7w(cluster). |
| 3[CH3 + HCO](ads)-7w(cluster) → 3CH3CHO(ads)-7w(cluster). |
We will additionally comment on:
Step 4′: P1 → P3
| 1[CH3 + HCO](ads)-7w(cluster) → 1[CH4(ads) + CO](ads)-7w(cluster). |
| 3[CH3 + HCO](ads)-7w(cluster) → 3[CH4(ads) + CO](ads)-7w(cluster). |
In Section 2 we describe the adopted computational methods for electronic structure and rate constants calculations and in Section 3 we discuss the results and their implications. Finally conclusions are given in Section 4.
2 Methodology
In the present work we employ density functional theory (DFT),45 and CCSD(T)46 to investigate acetaldehyde (Ac) formation on a model grain comprised of seven water molecules and a single methanol molecule that substitutes the 8th water molecule of a cubic cluster.All quantum chemical calculations have been performed with the Gaussian 16 software.47
2.1 Density functional theory (DFT) calculations
In our work all structures are fully optimized and the harmonic frequencies computed using DFT. Frequency calculations were performed in order to verify that all intermediates are true minima on the potential energy surface, and that all transition states exhibit a single imaginary frequency.We use the range separated UωB97XD hybrid meta-GGA exchange–correlation functional48 with its dispersion corrections which yields satisfactory accuracy for the thermochemistry, kinetics, and non-covalent interactions. Thus, it can be used to study our model system and describes accurately the interactions that play an important role in the stability of the different fragments involved in the reaction process leading to acetaldehyde.48
The split valence triple ζ Pople basis set 6-311++G(2d,p)49 includes polarisation and diffuse functions and is able to describe van der Waals interactions inside the ice model and molecular interactions with the fragments. Further, this basis set is shown to provide near experimental accuracy for many chemical reactions.50,51 We study all species in the reaction mechanism with the UωB97XD/6-311++G(2d,p) model chemistry. Gaussian 16 automatically includes an ultrafine integration grid in the DFT calculations in order to improve the accuracy of the results. The grid greatly enhances the accuracy at reasonable additional cost.
The reaction paths are computed using the intrinsic reaction coordinate (IRC) methodology52,53 to confirm the identities of the reactants and products for every transition state.
IRC calculations require initial force constants of the transition state. Then, the first and second order energy derivatives are obtained to calculate the projected harmonic vibrational frequencies along each reaction path. The Minimum Energy Paths (MEPs) for Steps 2, 3 and 4 were computed using the Page–McIver integrator with a gradient step size of 0.1 a0.
2.2 Coupled cluster calculations
In order to better describe the energetics, single point energy calculations were performed for all involved species using CCSD(T)46 on the DFT optimized geometries.As the calculations become time intensive for larger basis set choices, the same basis set was used as in the DFT calculations and this composite method will in the following be referred to as CCSD(T)//UωB97XD/6-311++G(2d,p).
2.3 Semi-classical theory of the rate constants
Rate constants for Step 3 (for both spin states S = 0, 1) in the reaction mechanism, which is energetically rate limiting, are computed over the temperature range [105–2000 K] with the general polyatomic rate constant code POLYRATE version 2010-A.54 Transition state theory is employed in various forms as described below.The connectivity between a transition state and the desired reactant or product is verified by performing intrinsic reaction coordinate calculations in both the forward and backward directions with a step size of 0.1 Bohr.
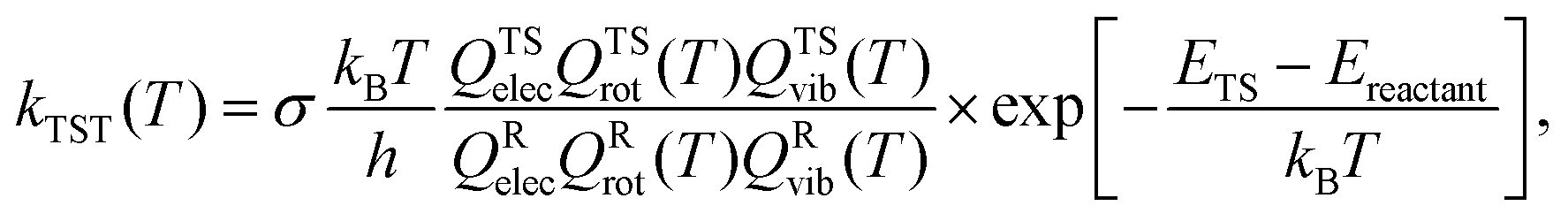 | (1) |
Note that in using a free energy of activation criterion to determine the location of the variational transition state, both entropic and energetic contributions are taken into account. We may write:
| kCVT(T) = minskG(T,s) = kG(T,s⋆,CVT(T)), | (2) |
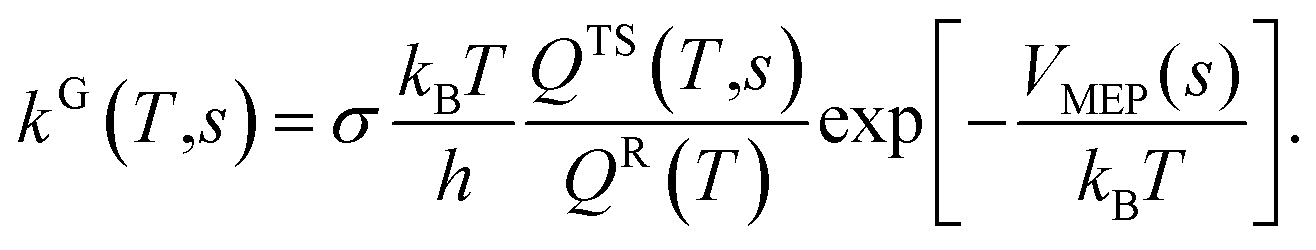 | (3) |
s = 0 at the transition state, at the reactants it is a finite negative number, and for the products a finite positive number.
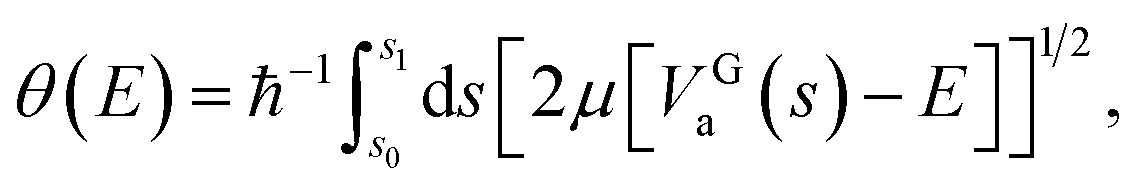 | (4) |
A tunnelling probability is calculated as
| P(E) = exp[−2θ(E)], | (5) |
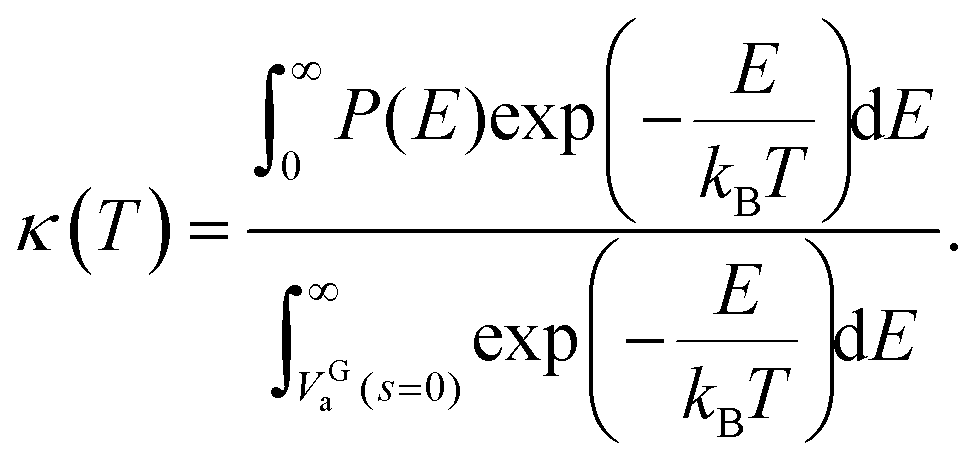 | (6) |
This κ(T) multiplies the otherwise obtained thermal rate constant to give a tunnelling corrected rate constant,
| kZCT(T) = κ(T) × kTST(T). | (7) |
This approach is called the zero curvature tunneling (ZCT) method.57
In the small curvature tunneling method (SCT),56 transmission coefficients,57,59 which include effects of the reaction-path curvature, are based on the centrifugal-dominant small-curvature semi-classical adiabatic ground-state (CD-SCSAG) approximation.56 The SCT method is known to predict reasonably accurate rate constants60–62 and is therefore widely used.
In the SCT method the effect of the reaction curvature is included by replacing the reduced mass μ by an effective mass μeff. The tunnelling probability at energy E is
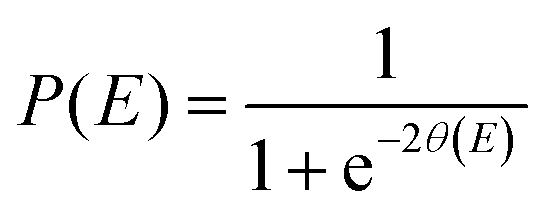 | (8) |
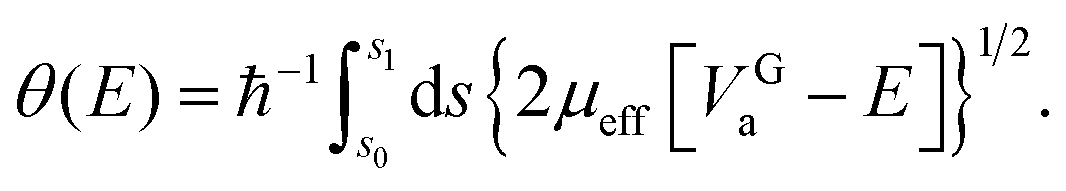 | (9) |
3 Results and discussion
In this section we first report our electronic structure calculations and then the rate constants that we have obtained for the energetically rate determining step, Step 3, on both the singlet and triplet potential energy surfaces.3.1 Electronic structure calculations
From the electronic structure calculations we computed intermediates, transition states and products as illustrated in Fig. 1 for the singlet and triplet species.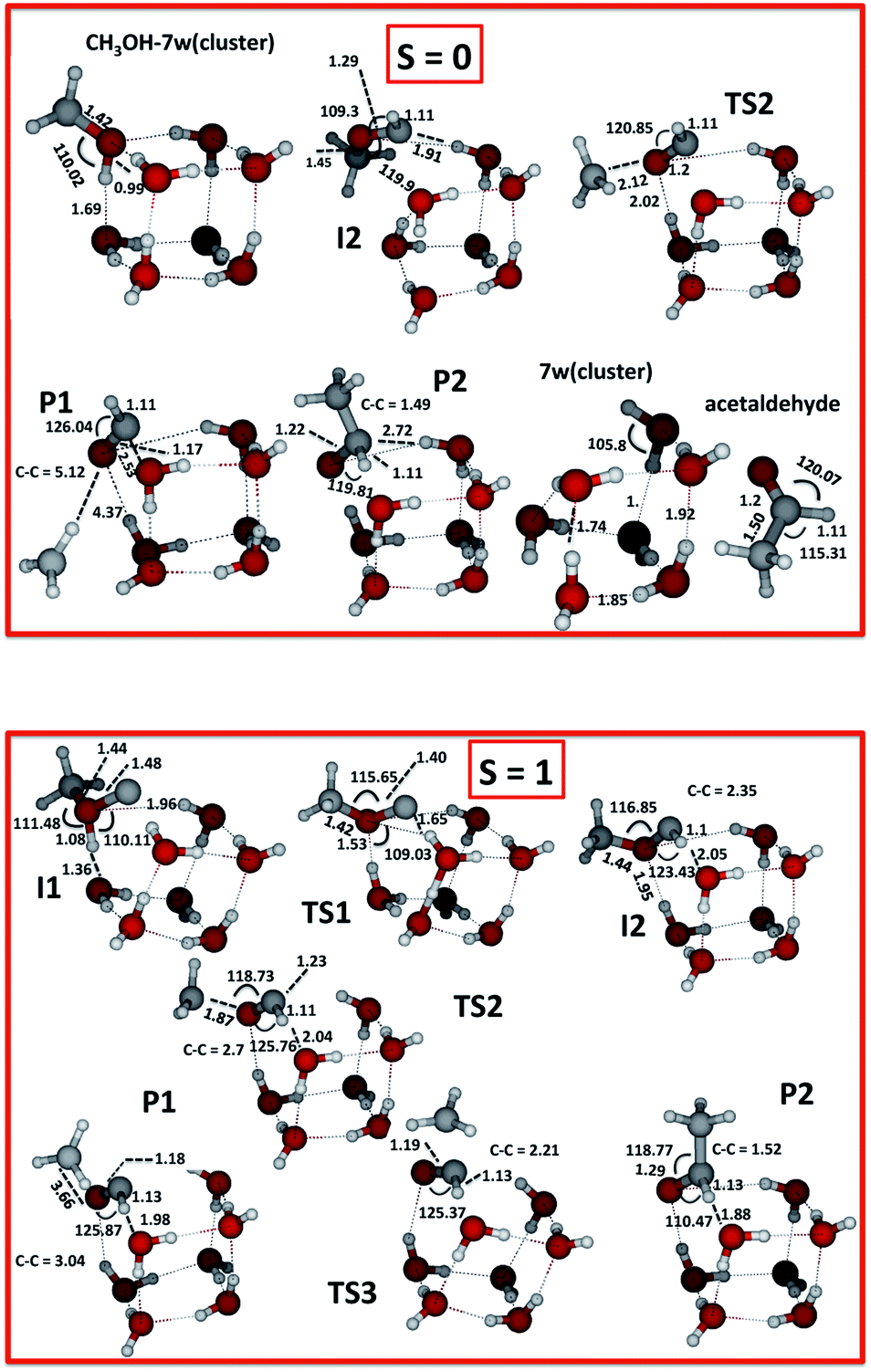 | ||
| Fig. 1 Fully optimised final geometries for all reactants, and complexes computed with UωB97XD/6-311++G(2d,p) for the two spin states S = 0 and 1. | ||
All structures are optimised at the UωB97XD/6-311++G(2d,p) level. Transition states correspond to the first-order saddle points characterized by single imaginary frequencies. For the triplet species these frequencies are 758i cm−1 for the H atom transfer in Step 2, 762i cm−1 for the C–O bond breakage in Step 3, 243i cm−1 for the C–C bond formation in Step 4, and 1445i cm−1 for the H abstraction in Step 4′.
As the carbon atom approaches the CH3OH–water complex, the formation of intermediate I1 occurs without a barrier (Step 1). This is followed by hydrogen transfer from the oxygen atom of methanol to the carbon atom via a proton relay-mechanism involving the water molecules of the ice model through the transition state TS1, to yield a more stable intermediate I2 (Step 2). Another barrier-mediated (TS2) step generates the adsorbed radicals CH3 and HCO, i.e. the P1 complex (Step 3). These first three steps may occur on the triplet PES. Spin flip could however take place close to the intermediate I2 (S = 1), thereby yielding I2 (S = 0). This would allow the reaction to continue on the singlet PES, see more below. In Step 4 acetaldehyde adsorbed on the ice (P2) forms.
We have computed the possible fragments on the singlet (S = 0) PES employing “Stable = Opt” first to ensure that we get the lowest singlet SCF solution, as we are interested in finding an SCF solution of an open shell unrestricted singlet wavefunction. For S = 0, our calculations lead to the following stationary species I2, TS2, P1, P2 and P3, as seen in Table 1. Step 3 has a transition state TS2, as can be seen in Fig. 1 and 2, and a single imaginary frequency of 464i cm−1.
| Species | Energy (UωB97XD) | ZPE (UωB97XD) | Energy + ZPE (UωB97XD) | Energy CCSD(T)//UωB97XD | Energy + ZPE CCSD(T)//UωB97XD |
|---|---|---|---|---|---|
| S = 1 | |||||
| C(3P) | −37.840178 | — | −37.840178 | −37.773973 | −37.773973 |
| I1 | −688.780084 | 0.236295 | −688.543789 | −687.548089 | −687.311794 |
| TS1 | −688.777331 | 0.230375 | −688.546956 | −687.541491 | −687.311116 |
| I2 | −688.828564 | 0.235519 | −688.593045 | −687.595794 | −687.360275 |
| TS2 | −688.801766 | 0.230675 | −688.571091 | −687.569784 | −687.339109 |
| P1 | −688.837208 | 0.227453 | −688.609755 | −687.609865 | −687.382412 |
| TS3 | −688.834115 | 0.230081 | −688.604034 | −687.601360 | −687.371279 |
| P2 | −688.862109 | 0.234261 | −688.627848 | −687.623084 | −687.388823 |
| TS4 | −688.790605 | 0.228354 | −688.562251 | −687.551798 | −687.323444 |
| P3 | −688.807343 | 0.233586 | −688.573757 | −687.571456 | −687.337870 |
| CH3CHO(gas) | −153.709122 | 0.052916 | −153.656206 | −153.399772 | −153.346856 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| S = 0 | |||||
| Ice model: CH3OH-7w | −650.894314 | 0.235504 | −650.658810 | −649.742209 | −649.506705 |
| 7w(cluster) | −535.138321 | 0.179647 | −534.958674 | −534.211967 | −534.032320 |
| I2 | −688.875062 | 0.237861 | −688.637201 | −687.644363 | −687.406502 |
| TS2 | −688.827359 | 0.229755 | −688.597604 | −687.597604 | −687.368810 |
| P1 | −688.837677 | 0.227578 | −688.610099 | −687.610245 | −687.382667 |
| P2 | −688.982611 | 0.237554 | −688.745056 | −687.750202 | −687.512648 |
| P3 | −688.973831 | 0.232680 | −688.741151 | −687.755034 | −687.522354 |
| CH3CHO(gas) | −153.829051 | 0.055686 | −153.773365 | −153.523343 | −153.467657 |
 | ||
| Fig. 2 Schematic potential energy surfaces for the complete reaction mechanism computed with UωB97XD/6-311++G(2d,p) and CCSD(T)//UωB97XD. Relative classical (top) and adiabatic (bottom) energies are compared to those from ref. 44 (blue lines). We plot our calculated values for the two spin states S = 0 (red and purple lines) and S = 1 (black and green lines). | ||
The energies of the fragments and their corresponding zero point energies are given in Table 1, for each spin state.
We provide in the ESI† the Cartesian coordinates for all fragments involved in the two spin states.
In Table 2 we collect the computed energetics for both total spin states, S = {0,1}.
| Method | UωB97XD | CCSD(T)//UωB97XD | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Reaction steps | Label | ΔV | ΔVGa | ΔE | ΔEa | ΔV | ΔVGa | ΔE | ΔEa |
| a – indicates barrierless. | |||||||||
| S = 1 | |||||||||
| RC → I1 | Step 1 | — | — | −28.61 | −28.11 | — | — | −20.02 | −19.53 |
| I1 → I2 | Step 2 | 1.727 | −1.987 | −30.42 | −30.91 | 4.14 | 0.43 | −29.93 | −30.42 |
| I2 → P1 | Step 3 | 16.82 | 13.78 | −5.42 | −10.49 | 16.32 | 13.28 | −8.83 | −13.89 |
| P1 → P2 | Step 4 | 1.94 | 3.59 | −15.63 | −11.35 | 5.34 | 6.98 | −8.29 | −4.02 |
| P1 → P3 | Step 4′ | 29.24 | 29.81 | 18.74 | 22.59 | 36.44 | 37.00 | 24.10 | 27.95 |
|
|||||||||
| S = 0 | |||||||||
| I2 → P1 | Step 3 | 29.93 | 24.85 | 23.46 | 17.01 | 28.73 | 23.55 | 21.41 | 14.96 |
| P1 → I2 | Step 3′ | 6.47 | 7.84 | −23.46 | −17.01 | 13.28 | 14.65 | −21.41 | −14.96 |
| P1 → P2 | Step 4 | — | — | −90.95 | −84.69 | — | — | −76.62 | −70.36 |
| P1 → P3 | Step 4′ | — | — | −85.44 | −82.24 | — | — | −79.65 | −76.45 |
| P2 → Ac(gas) + 7w(cluster) | — | — | 9.56 | 8.17 | — | — | 9.34 | 7.95 | |
In Fig. 2 we plot the relative classical and adiabatic energies of the reaction mechanism under consideration. We see that on the triplet PES, Step 2 can lead to the formation of intermediate I2 without a barrier to reaction on the UωB97XD/6-311++G(2d,p) vibrationally adiabatic PES while there is a small barrier of 0.43 kcal mol−1 using CCSD(T)//UωB97XD, whereas ref. 44 reports a barrier of 5 kcal mol−1.
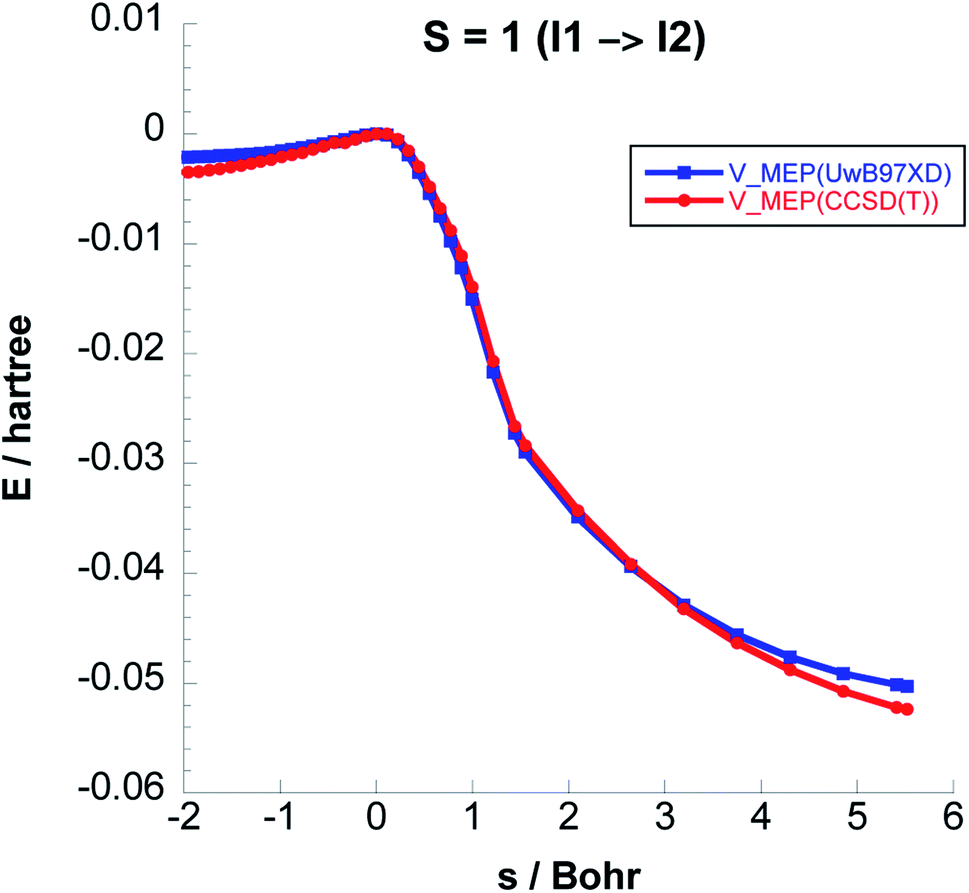
To balance the computational cost and chemical accuracy, the MEP energies were refined by performing CCSD(T)//UωB97XD/6-311++G(2d,p) single-point energy calculations on the UωB97XD/6-311++G(2d,p) IRC optimized stationary points. The comparison between the MEP energies obtained with the two levels of theory for Steps 2 & 3 can be seen in Fig. 3 and 4.
 | ||
| Fig. 3 Comparison between the classical potential energies VMEP, computed with CCSD(T)//UωB97XD/6-311++G(2d,p) and UωB97XD/6-311++G(2d,p) levels, for Step 2 on the S = 1 PES. | ||
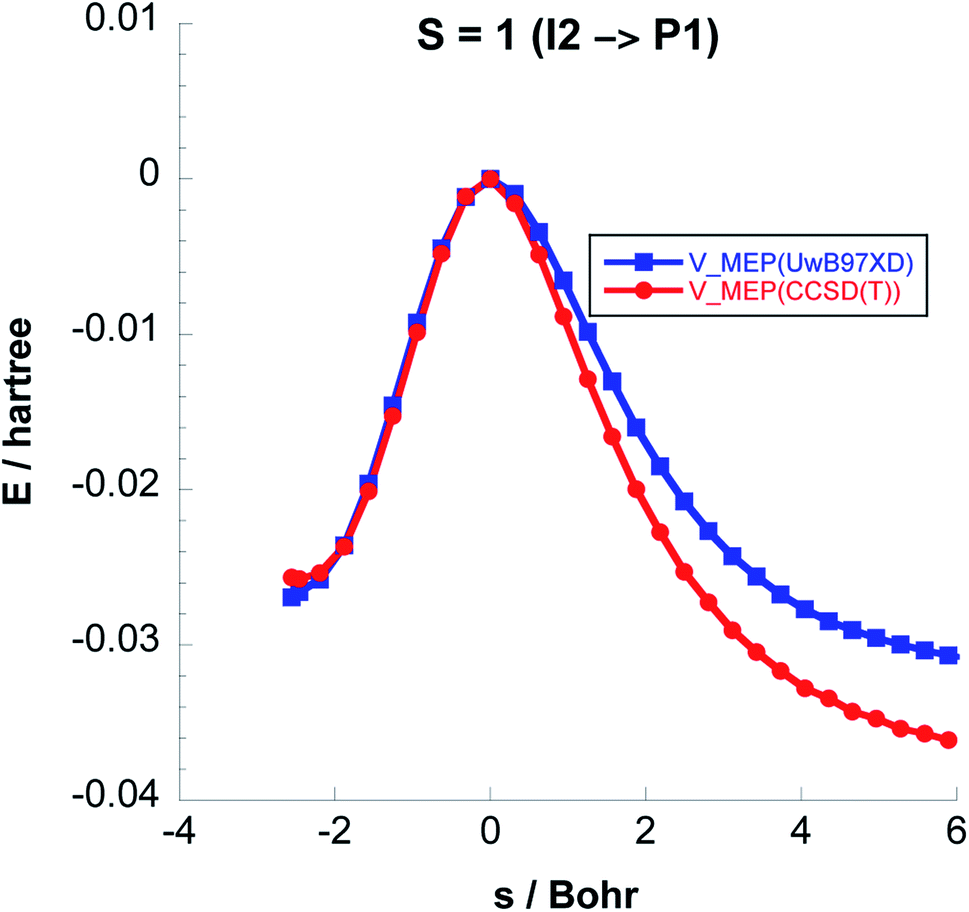 | ||
| Fig. 4 Comparison between the classical potential energies VMEP, computed with CCSD(T)//UωB97XD/6-311++G(2d,p) and UωB97XD/6-311++G(2d,p) levels, for Step 3 on the S = 1 PES. | ||
In Fig. 5 we plot the potential energy, ZPE, and vibrationally adiabatic ground state energy VGa along the minimum energy path for Step 2. The ZPE variation with respect to the reaction coordinate s exhibits a dip around the transition state, which results in that the 4.14 kcal mol−1 classical barrier is reduced to 0.43 kcal mol−1.
 | ||
| Fig. 5 Classical potential energies VMEP, ground-state vibrational adiabatic potential energy curves (VGa), and ZPE as functions of s (Bohr) at the CCSD(T)//UωB97XD/6-311++G(2d,p) level, for Step 2 at a total spin S = 1. | ||
Fig. 6 shows the reaction path pertaining to Step 3 on the triplet surface, i.e. the energetically rate determining step. The adiabatic barrier height is 13.28 kcal mol−1 for this step, which is exothermic. Formation of adsorbed acetaldehyde (Ac) through Step 4 is exothermic by −8.29 kcal mol−1 with a moderate vibrationally adiabatic barrier height of 6.98 kcal mol−1. In Fig. 7 we plot, for Step 4 the UωB97XD/6-311++G(2d,p) VMEP(s), VGa(s) and ZPE(s) where we clearly see how the variation in ZPE results in an increase of the adiabatic barrier.
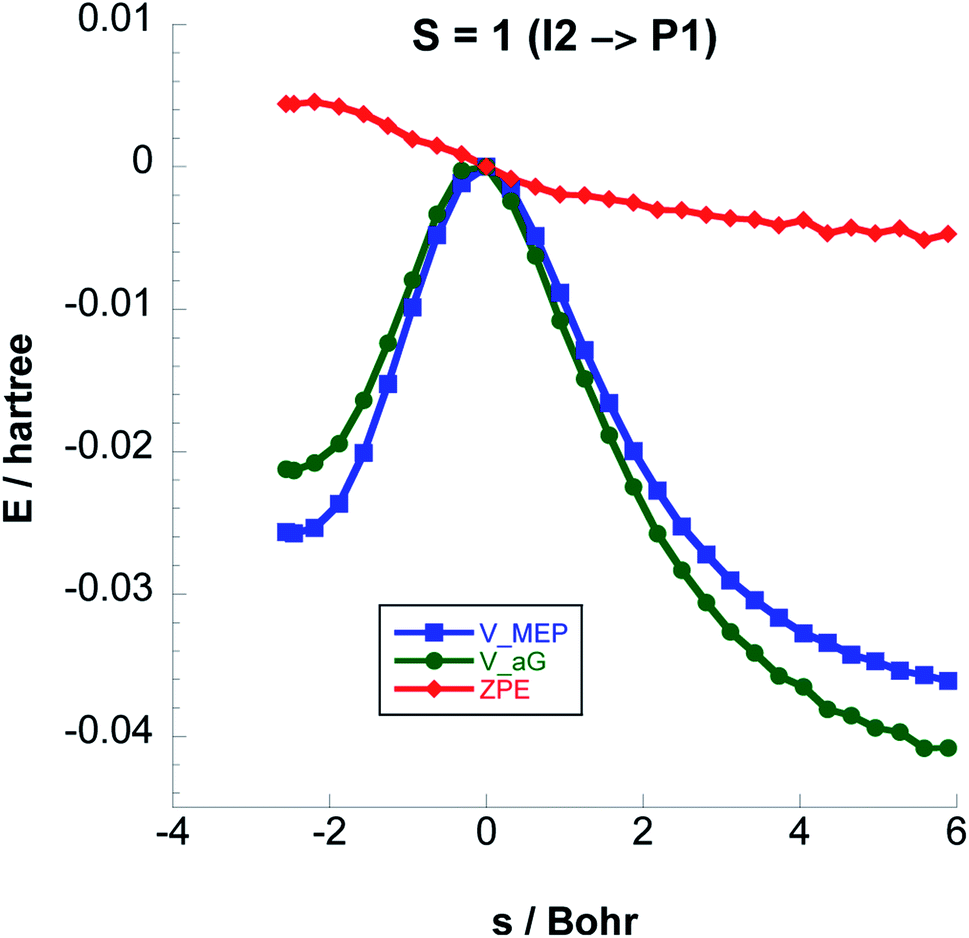 | ||
| Fig. 6 Classical potential energies VMEP, ground-state vibrational adiabatic potential energy curves (VGa), and ZPE as functions of s (Bohr) at the CCSD(T)//UωB97XD/6-311++G(2d,p) level, for Step 3 on the S = 1 PES. | ||
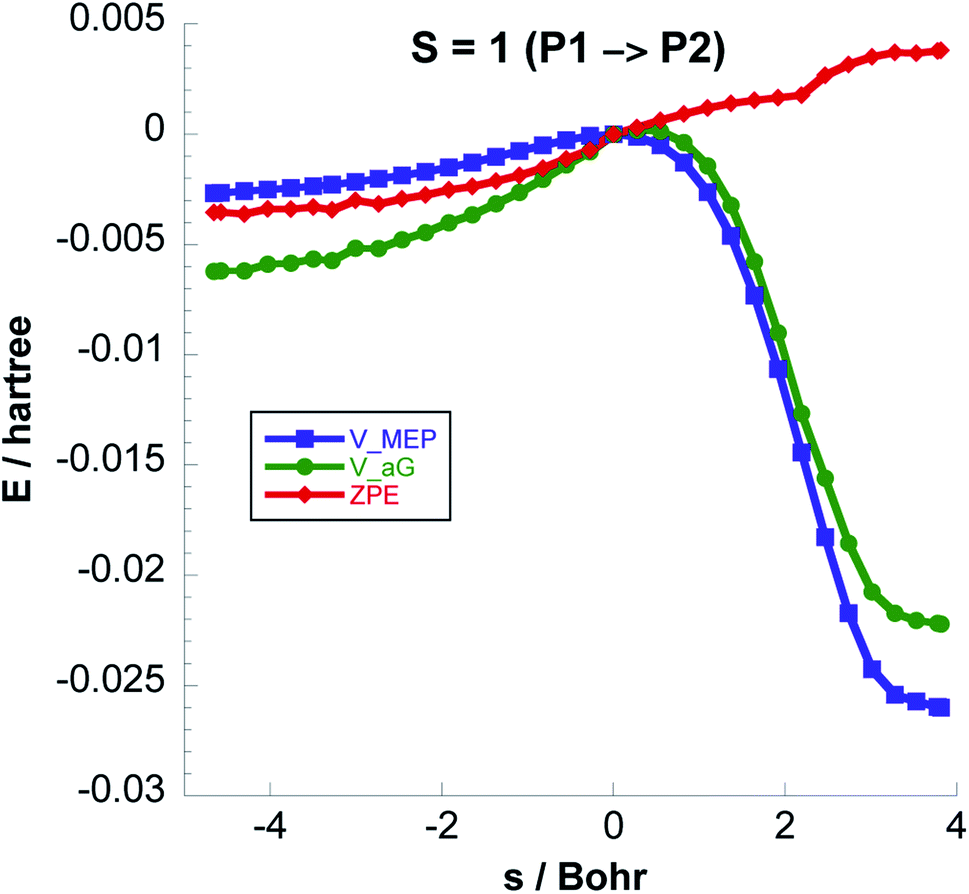 | ||
| Fig. 7 Classical potential energies VMEP, ground-state vibrational adiabatic potential energy curves (VGa), and ZPE of 3N-6 vibrational modes as functions of s (Bohr) at the UωB97XD/6-311++G(2d,p) level, for Step 4 at a total spin S = 1. | ||
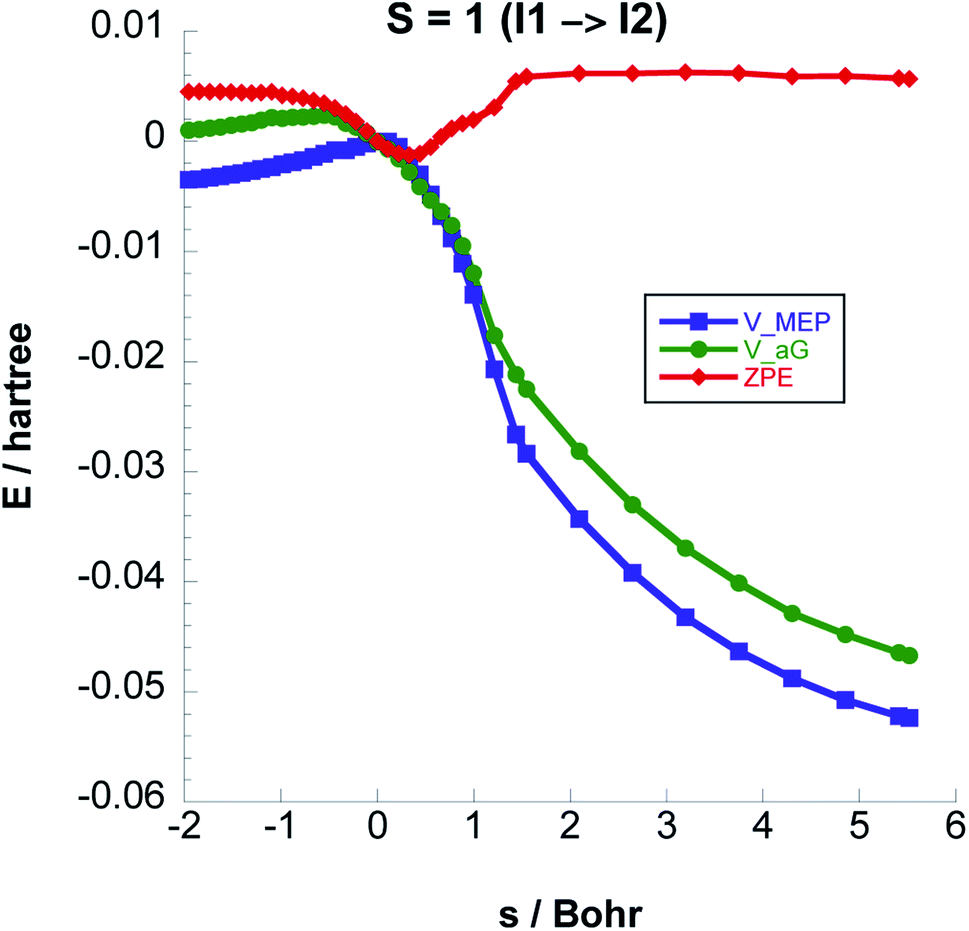
The I2 singlet state has a noticeably lower energy (see Table 1) than the triplet state. We have therefore investigated if a spin flip could happen such that I2 may form in its singlet state.
We have computed the downhill reaction path starting from the triplet I2 geometry but with a total spin S = 0. We also computed the single point energies for each geometry of this path with total spin S = 1. The energies are shown in Fig. 8. The crossing point between the two states is only about 3 kcal mol−1 above the energy of the triplet I2 geometry and is thus energetically easily accessible considering the energy released in coming from I1. The point on the IRC where the energies of triplet and singlet states are the same, indicates a possible intersystem crossing geometry such that I2 can form as a singlet.
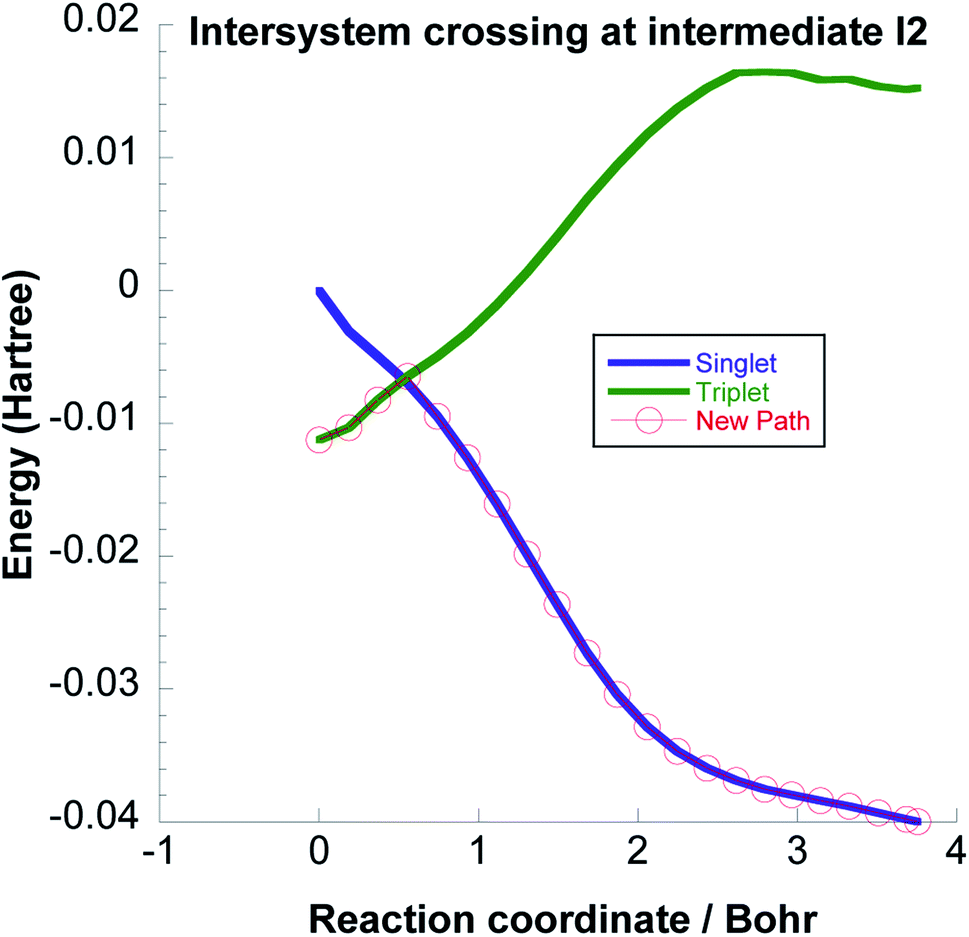 | ||
| Fig. 8 Intersystem crossing between the triplet and singlet potential energy surfaces at the intermediate I2. | ||
As a result of a spin flip close to the I2 intermediate, the next step in order to form acetaldehyde would be I2 (singlet) → P1 (singlet) instead of I2 (triplet) → P1 (triplet). In Fig. 9 we plot the classical potential energies VMEP from UωB97XD and CCSD(T)//UωB97XD, and also the ZPE for Step 3. The adiabatic barrier height for this step is 23.55 kcal mol−1 at the CCSD(T)//UωB97XD level.
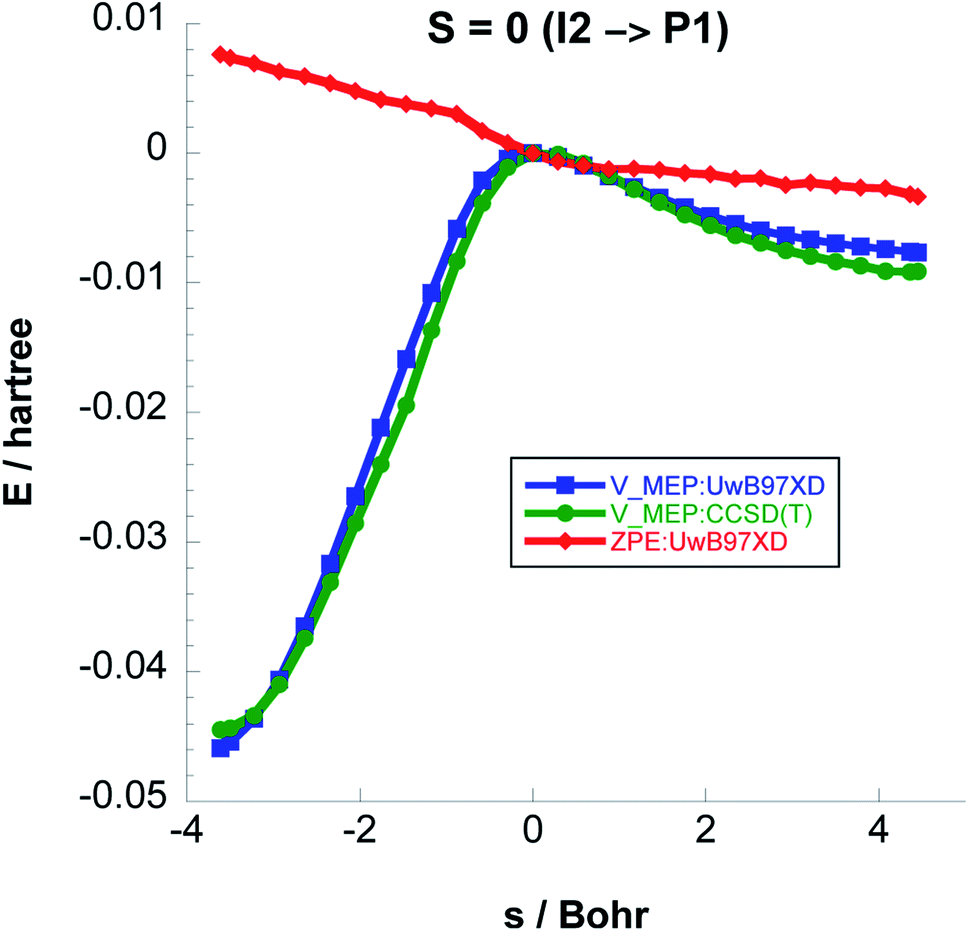 | ||
| Fig. 9 Classical potential energies VMEP, at the UωB97XD/6-311++G(2d,p) and CCSD(T)//UωB97XD/6-311++G(2d,p) levels of theory, with ZPE as functions of s (Bohr) for Step 3 on the S = 0 PES. | ||
The following step to form adsorbed acetaldehyde (step 4) is exoergic by 70.36 kcal mol−1. Desorbtion of acetaldehyde into the gas phase is endoergic by only 7.95 kcal mol−1, suggesting that the large exoergicity of step 4 would lead to fast desorption.
If there is no spin flip close to I2, P1 would form in the triplet state. Again, the singlet P1 is lower in energy and we have investigated if a spin flip could occur also here, see Fig. 10, which is energetically quite plausible. Thereafter the reaction would proceed on the singlet surface to form adsorbed acetaldehyde which is likely to easily desorb as discussed just above.
 | ||
| Fig. 10 Intersystem crossing between the triplet and singlet potential energy surfaces at the intermediate P1. | ||
For S = 0 the P1 → P2 and P1 → P3 reactions are downhill with exoergicities of −70.36 and −76.45 kcal mol−1 respectively. For the reactions on the triplet PES, only one unpaired electron (on the CH3 radical) is involved in either the C–C bond formation (to form P2) or the H-transfer (to form CH4), while the other unpaired electron (on HCO-wat radical) stays uninvolved. This is obviously different than the processes on the singlet PES where the two unpaired electrons have opposite spins and they either recombine without barrier for the (C–C) bond formation or transfer an H atom without barrier to lead to two closed shell species. For these reasons on the singlet potential energy surface P1 → P2 (Step 4) and P1 → P3 (Step 4′) are barrierless as can be seen in Fig. 2. Formation of adsorbed CO + CH4 products (P3) on the S = 1 PES has a barrier of 37 kcal mol−1 as can be seen in Table 2.
3.2 Rate constants
In this section we report our calculated rate constants for the energetically rate determining step on both the singlet and triplet potential energy surfaces. The thermal rate constants are evaluated by means of canonical variational transition-state theory, i.e. CVT, in which the flux is minimized for a canonical ensemble.64 ZCT and SCT tunneling corrections have been computed for the temperature range 105–2000 K where the fully optimised structures, energies, gradients, and force constants of points along the MEP are provided as inputs. Tables 3 and 4 summarize the TST, CVT, CVT/ZCT, and CVT/SCT, rate constants computed at various temperatures.| T (K) | TST | CVT | CVT/ZCT | CVT/SCT |
|---|---|---|---|---|
| 105 | 1.19 × 10−15 | 5.28 × 10−16 | 2.49 × 10−6 | 1.19 × 10−5 |
| 120 | 4.0 × 10−12 | 1.78 × 10−12 | 6.94 × 10−6 | 2.77 × 10−5 |
| 150 | 3.64 × 10−7 | 1.61 × 10−7 | 1.64 × 10−4 | 3.73 × 10−4 |
| 200 | 3.63 × 10−2 | 1.55 × 10−2 | 1.79 × 10−1 | 2.20 × 10−1 |
| 300 | 4.33 × 103 | 1.69 × 103 | 3.32 × 103 | 3.47× 103 |
| 400 | 1.71× 106 | 6.01 × 105 | 7.35× 105 | 7.51 × 105 |
| 500 | 6.61× 107 | 2.11 × 107 | 2.14 × 107 | 2.17 × 107 |
| 600 | 7.89 × 108 | 2.33 × 108 | 2.16× 108 | 2.18 × 108 |
| 700 | 4.76 × 109 | 1.32 × 109 | 1.16 × 109 | 1.17 × 109 |
| 800 | 1.86 × 1010 | 4.86× 109 | 4.13 × 109 | 4.15 × 109 |
| 900 | 5.41 × 1010 | 1.35 × 1010 | 1.12 × 1010 | 1.12 × 1010 |
| 1000 | 1.28 × 1011 | 3.07 × 1010 | 2.48 × 1010 | 2.48 × 1010 |
| 1200 | 4.72 × 1011 | 1.04 × 1011 | 7.58 × 1010 | 7.59 × 1010 |
| 1400 | 1.21 × 1012 | 2.44 × 1011 | 1.62 × 1011 | 1.62 × 1011 |
| 1600 | 2.46 × 1012 | 4.55 × 1011 | 2.80 × 1011 | 2.81 × 1011 |
| 1800 | 4.27 × 1012 | 7.29 × 1011 | 4.35 × 1011 | 4.35× 1011 |
| 2000 | 6.67 × 1012 | 1.06 × 1012 | 6.29 × 1011 | 6.29× 1011 |
| T (K) | TST | CVT | CVT/ZCT | CVT/SCT |
|---|---|---|---|---|
| 107 | 2.41 × 10−34 | 1.13 × 10−34 | 3.32 × 10−32 | 1.02 × 10−31 |
| 120 | 3.34 × 10−29 | 1.55 × 10−29 | 3.04 × 10−28 | 4.81 × 10−28 |
| 150 | 1.06 × 10−20 | 4.73 × 10−21 | 2.07 × 10−20 | 2.23 × 10−20 |
| 200 | 3.95 × 10−12 | 1.63 × 10−12 | 3.07 × 10−12 | 3.16 × 10−12 |
| 300 | 2.09 × 10−3 | 7.11 × 10−4 | 7.60 × 10−4 | 7.67 × 10−4 |
| 400 | 62.3 | 17.8 | 15.2 | 15.2 |
| 500 | 3.48 × 104 | 8.44 × 103 | 6.06 × 103 | 6.07 × 103 |
| 600 | 2.56 × 106 | 5.22 × 105 | 3.15 × 105 | 3.16 × 105 |
| 700 | 5.76 × 107 | 9.93 × 106 | 5.22 × 106 | 5.23 × 106 |
| 800 | 6.12 × 108 | 8.78 × 10 7 | 3.35 × 107 | 3.35 × 107 |
| 900 | 3.91 × 109 | 4.64 × 108 | 1.67 × 108 | 1.67 × 108 |
| 1000 | 1.75 × 1010 | 1.74 × 109 | 6.12 × 108 | 6.13 × 108 |
| 1200 | 1.68 × 1011 | 1.23 × 1010 | 4.22 × 109 | 4.23 × 109 |
| 1400 | 8.54 × 1011 | 4.87 × 1010 | 1.73 × 1010 | 1.73 × 1010 |
| 1600 | 2.91 × 1012 | 1.35 × 1011 | 5.13 × 1010 | 5.13 × 1010 |
| 1800 | 7.60 × 1012 | 2.98 × 1011 | 1.19 × 1011 | 1.19 × 1011 |
| 2000 | 1.64 × 1013 | 5.59 × 1011 | 2.35 × 1011 | 2.35× 1011 |
The variational effect is defined as the ratio between the TST and CVT rate constants, and the tunneling effect is the ratio between the (CVT/ZCT, CVT/SCT) and CVT rate constants. The ratio between CVT/ZCT and CVT/SCT gives information about the reaction path curvature in the vicinity of the transition state. As such, for Step 3, the CVT/SCT rate constants are predicted to be consistently larger than the CVT/ZCT ones at the same temperature.
In Table 5, we give the calculated ratios of kCVT/SCT:kCVT/ZCT, kTST:kCVT, and kCVT/SCT:kCVT at some selected temperatures, indicating the importance of the small-curvature, variational and tunneling effects.
:kCVT/ZCT, kTST:kCVT, and kCVT/SCT:kCVT ratios for a total spin S = 1, for Step 3 (I2 → P1) computed at CCSD(T)//UωB97XD/6-311++G(2d,p) level
| T (K) | kCVT/SCT:kCVT/ZCT |
kTST:kCVT |
kCVT/SCT:kCVT |
|---|---|---|---|
| 105 | 4.7791 | 2.2538 | 2.2538 × 1010 |
| 300 | 1.0452 | 2.5621 | 2.0533 |
| 500 | 1.0140 | 3.1327 | 1.0284 |
| 800 | 1.0048 | 3.8272 | 0.85391 |
| 1000 | 1.0 | 4.1694 | 0.80782 |
| 1500 | 1.0 | 5.1765 | 0.64118 |
| 2000 | 1.0 | 6.2925 | 0.59340 |
Table 5 and Fig. 11 show that tunneling totally dominates at the lowest temperature, but quickly becomes insignificant with increasing temperature, such that reflection by the barrier becomes more important. We also see that the variational effects affect the rate constant, particularly at high temperature.
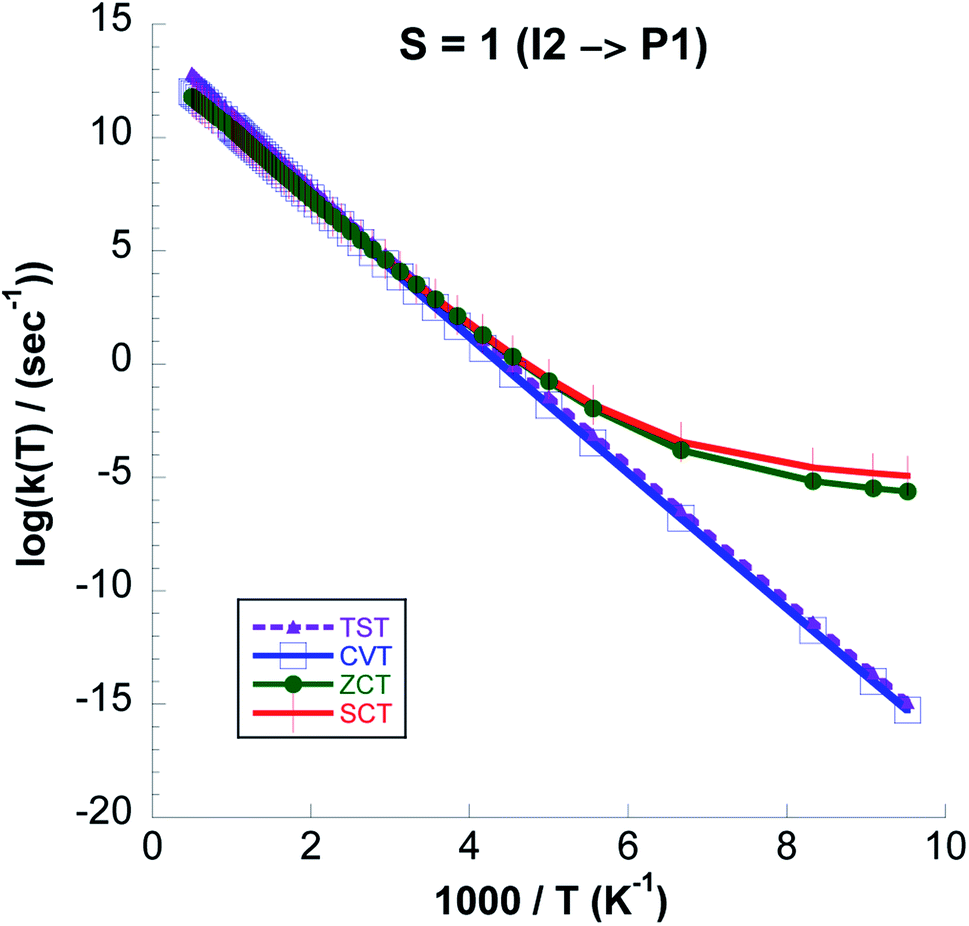 | ||
| Fig. 11 Logarithmic representation of the k(TST), k(CVT), k(CVT/ZCT) and k(CVT/SCT) rate constants computed at the CCSD(T)//UωB97XD/6-311++G(2d,p) level for the I2 → P1 reaction versus 1000/T for S = 1. | ||
From Fig. 12 where we compare (CVT/ZCT) and (CVT/SCT) rate constants computed with UωB97XD/6-311++G(2d,p) and CCSD(T)//UωB97XD/6-311++G(2d,p) on the triplet surface we see that the latter model chemistry predicts 4 orders of magnitude higher rate values due to the lower barrier. On the singlet surface, the shape of the reaction path in Fig. 9 shows a broad barrier resulting in only ∼3 orders of magnitude difference between the CVT/SCT rates and those from purely classical TST at the lowest temperature.
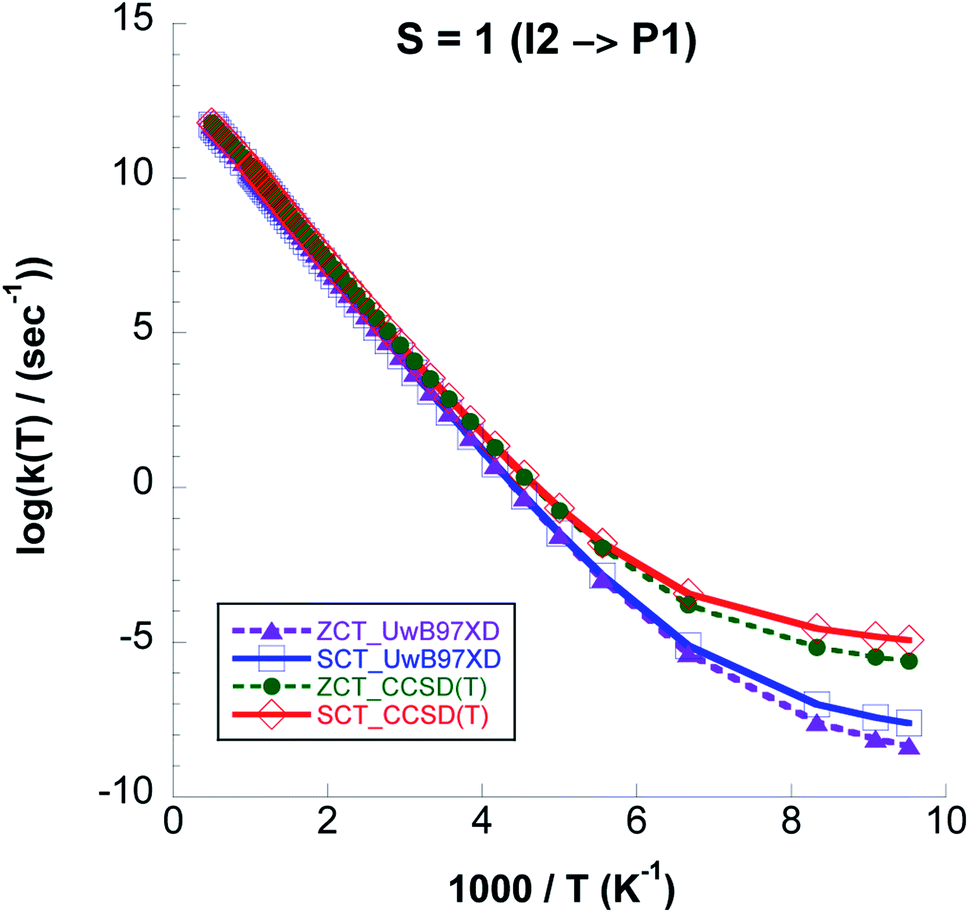 | ||
| Fig. 12 Logarithm of k(CVT/ZCT) and k(CVT/SCT) rate constants, computed with CCSD(T)//UωB97XD/6-311++G(2d,p) and UωB97XD/6-311++G(2d,p) levels, for Step 3 on the S = 1 PES. | ||
In Fig. 13 we plot rate constants computed for the I2 → P1 step and total spin S = 0. We see that quantum tunneling is only important at temperatures lower than ∼130 K. This is also seen from the kCVT/SCT:kCVT ratio in Table 6. Except for the highest temperatures the CVT/SCT rate constants on the S = 0 surface are much lower than those on the S = 1 PES. From Fig. 14 it is seen that for the whole temperature range CCSD(T)//UωB97XD/6-311++G(2d,p) rate constants are higher than those computed with UωB97XD/6-311++G(2d,p).
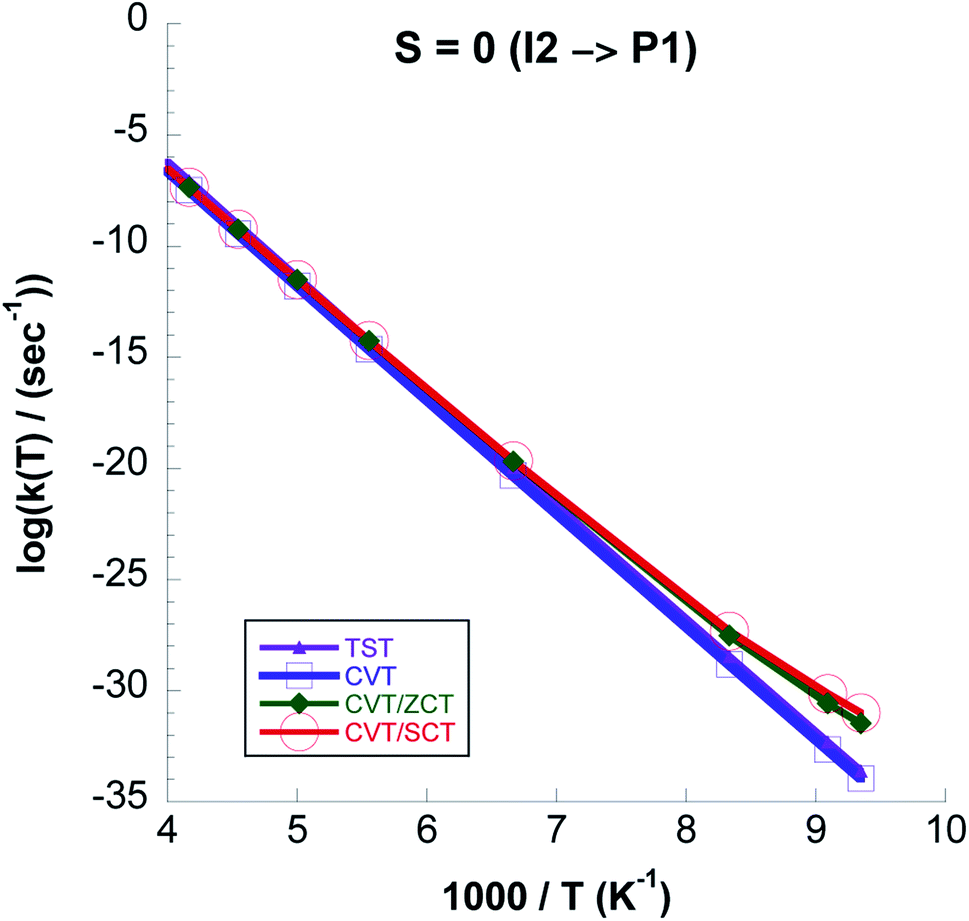 | ||
| Fig. 13 Logarithmic representation of the k(TST), k(CVT), k(CVT/ZCT) and k(CVT/SCT) rate constants computed at the CCSD(T)//UωB97XD/6-311++G(2d,p) level for the I2 → P1 reaction versus 1000/T for S = 0. | ||
:kCVT/ZCT, kTST:kCVT, and kCVT/SCT:kCVT ratios for a total spin S = 0, for Step 3 (I2 → P1) computed at CCSD(T)//UωB97XD/6-311++G(2d,p) level
| T (K) | kCVT/SCT:kCVT/ZCT |
kTST:kCVT |
kCVT/SCT:kCVT |
|---|---|---|---|
| 106 | 3.1337 | 2.1298 | 1.2840 × 103 |
| 300 | 1.0081 | 2.9478 | 1.0783 |
| 500 | 1.0020 | 4.1292 | 0.71910 |
| 800 | 1.0019 | 6.9857 | 0.38286 |
| 1000 | 1.0018 | 10.0 | 0.35188 |
| 1500 | 1.0 | 19.512 | 0.36856 |
| 2000 | 1.0 | 29.408 | 0.42130 |
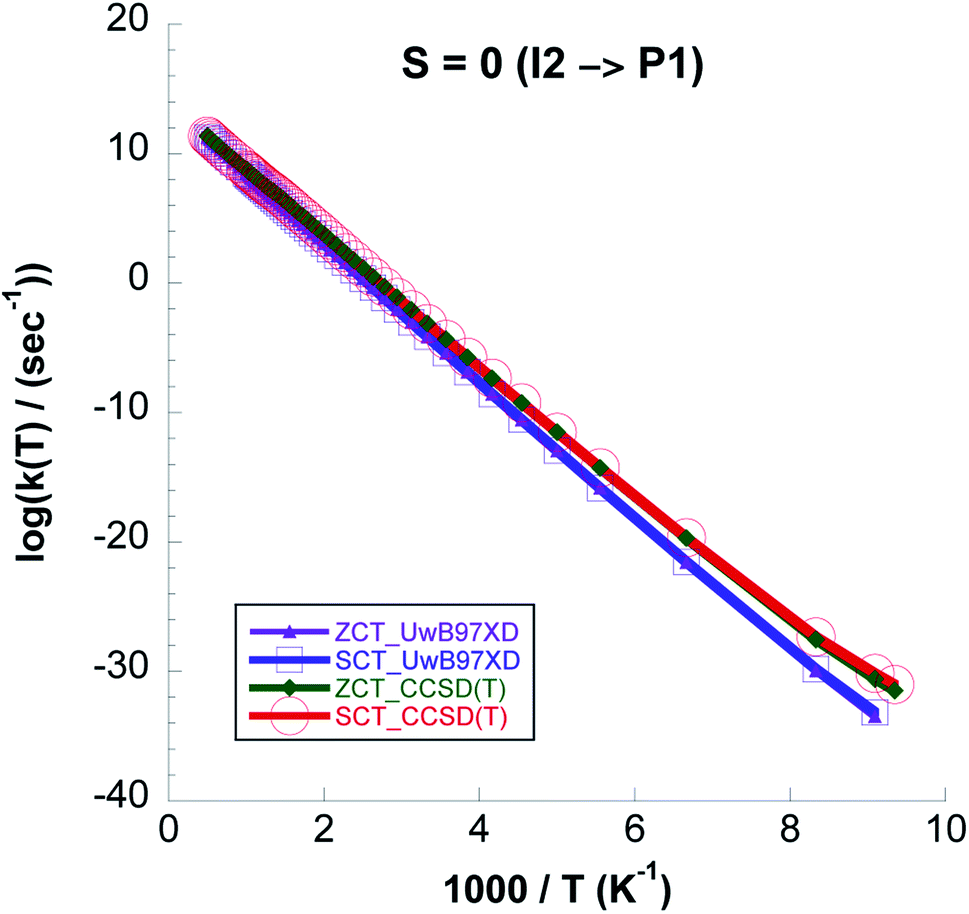 | ||
| Fig. 14 Comparison between the logarithmic representation of the k(CVT/ZCT) and k(CVT/SCT) rate constants computed with the UωB97XD/6-311++G(2d,p) and the CCSD(T)//UωB97XD/6-311++G(2d,p) level of theory for the I2 → P1 reaction versus 1000/T for S = 0. | ||
The present work aims to improve the understanding of the reaction mechanisms and kinetics of radical–radical addition on a methanol–water ice model.
The data we provide is of a major interest in regard to astrochemical models. Other molecular species in the ice composition can also be tested in the future, in order to investigate to what extent acetaldehyde formation paths can differ and how the composition of species influences the reaction mechanisms that may change in specific astronomical sources such as cold or hot molecular cores, and star forming regions.
4 Conclusions
We have investigated an Eley-Rideal-type mechanism where C(3P) reacts with CH3OH adsorbed on a water cluster. We consider the reaction mechanism on the triplet and the singlet surfaces and discuss two possible intersystem crossings. The reaction begins on the triplet surface and is downhill. It may spin flip close to the first intermediate, i.e. between TS1 and TS2. If that happens the reaction continues on the singlet PES, where however the energetically rate limiting step, I2 to P1, is much slower than on the triplet surface at interstellar temperatures.The reaction may on the other hand remain on the triplet surface, pass TS2 and close to P1 cross to the singlet surface. From P1 to P2, i.e. to form adsorbed acetaldehyde, the reaction is barrierless and exoergic by 70.36 kcal mol−1. Desorption of the acetaldehyde only requires 8 kcal mol−1 and is thus likely to happen fast given the large exoergicity of the previous step. This thus appears to be a plausible route to acetaldehyde formation in the gas phase.
We use transition state theory, with tunnelling corrections, to obtain the thermal rate constants for the energetically rate determining step of the reaction mechanism. In this step adsorbed CH3OCH breaks up to form adsorbed CH3 and HCO (on both the triplet and singlet surfaces).
Specifically, variational transition state theory calculations with zero curvature and small tunneling corrections are employed to find thermal rate constants over the temperature range 105–2000 K, relevant to a set of astrophysical environments.
The variational effect is found to be important and have a dominating effect on the obtained rate constants except at the lowest temperature. Tunneling totally dominates the rate constant at the lowest temperature but decreases quickly as the temperature increases. In fact reflection by the barrier clearly dominates over tunneling at the higher temperatures.
We also notice that the zero curvature tunneling correction works just as well as the small curvature correction except at the lowest temperature reported in Tables 5 and 6.
This study may serve as a useful investigation for further model variants to be explored computationally, and to suggest possible constraints on new experimental setups relevant to ice models to gain new impetus in the understanding of COM formation on icy mantles in general.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Professor D. G. Truhlar for providing the licenses for the Polyrate and Gaussrate programs. The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at Chalmers Centre for Computational Science and Engineering (C3SE) and partially funded by the Swedish Research Council through grant agreement no. 2018-05973. GN is also grateful to the Swedish Research Council for grant 2020-05293 which in part supported this work. BK is very grateful to Gaussian help team for useful insights into the Gaussian computations. IBC expresses her gratitude to Dr Keshav Kumar Singh for providing the Cartesian coordinates of some selected structures.References
- J. K. Jorgensen, A. Belloche and R. T. Garrod, Annu. Rev. Astron. Astrophys., 2020, 58, 727–778 CrossRef.
- J. Jortner, Phil. Trans. Biol. Sci., 2006, 361, 1877–1891 CrossRef CAS.
- E. Herbst and E. Dishoeck, Annu. Rev. Astron. Astrophys., 2009, 47, 427–480 CrossRef CAS.
- E. Herbst, Int. Rev. Phys. Chem., 2017, 36, 287–331 Search PubMed.
- F. Vazart, C. Ceccarelli, N. Balucani, E. Bianchi and D. Skouteris, Mon. Not. R. Astron. Soc., 2020, 499, 5547–5561 CrossRef CAS.
- C. J. Bennett, Y. Osamura, M. D. Lebar and R. I. Kaiser, Astrophys. J., 2005, 634, 698–711 CrossRef CAS.
- C. J. Bennett, C. S. Jamieson, Y. Osamura and R. I. Kaiser, Astrophys. J., 2005, 624, 1097–1115 CrossRef CAS.
- M. Moore and R. Hudson, Icarus, 1998, 135, 518–527 CrossRef CAS.
- J. E. Hudson, M. L. Hamilton, C. Vallance and P. W. Harland, Phys. Chem. Chem. Phys., 2003, 5, 3162–3168 RSC.
- A. J. Barnes and H. E. Hallam, Trans. Faraday Soc., 1970, 66, 1920–1931 RSC.
- V. A. Basiuk and K. Kobayashi, Int. J. Quantum Chem., 2004, 97, 713–718 CrossRef CAS.
- E. Herbst, G. Vidali and C. Ceccarelli, ACS Earth Space Chem., 2020, 4, 488–490 CrossRef CAS.
- K.-J. Chuang, G. Fedoseev, D. Qasim, S. Ioppolo, E. van Dishoeck and H. Linnartz, Mon. Not. R. Astron. Soc., 2017, 467, 2552–2565 CAS.
- A. Ciaravella, A. Jiménez-Escobar, G. Cosentino, C. Cecchi-Pestellini, G. Peres, R. Candia, A. Collura, M. Barbera, G. D. Cicca, S. Varisco and A. M. Venezia, Astrophys. J., 2018, 858, 35 CrossRef.
- K.-J. Chuang, G. Fedoseev, D. Qasim, S. Ioppolo, C. Jäger, Th. Henning, M. E. Palumbo, E. F. van Dishoeck and H. Linnartz, Astron. Astrophys., 2020, 635, A199 CrossRef CAS.
- C. A. Gottlieb, Detection of Acetaldehyde in Sagittarius. Molecules in the Galactic Environment, 1973, p. 181 Search PubMed.
- N. Fourikis, M. W. Sinclair, B. J. Robinson, P. D. Godfrey and R. D. Brown, Aust. J. Phys., 1974, 27(3), 425–430 CrossRef CAS.
- A. Bacmann, V. Taquet, A. Faure, C. Kahane and C. Ceccarelli, Astron. Astrophys., 2012, 541, L12 CrossRef.
- J. Cernicharo, N. Marcelino, E. Roueff, M. Gerin, A. Jiménez-Escobar and G. M. M. Caro, Astrophys. J., 2012, 759, L43 CrossRef.
- V. Taquet, E. S. Wirström, S. B. Charnley, A. Faure, A. López-Sepulcre and C. M. Persson, Astron. Astrophys., 2017, 607, A20 CrossRef.
- B. E. Turner, R. Terzieva and E. Herbst, Astrophys. J., 1999, 518, 699–732 CrossRef CAS.
- A. Bacmann, A. Faure and J. Berteaud, ACS Earth Space Chem., 2019, 3, 1000–1013 CrossRef CAS.
- J. Crovisier, D. Bockelée-Morvan, N. Biver, P. Colom, D. Despois and D. C. Lis, Astron. Astrophys., 2004, 418, L35–L38 CrossRef CAS.
- R. T. Garrod and E. Herbst, Astron. Astrophys., 2006, 457, 927–936 CrossRef CAS.
- G. Jungclaus, G. Yuen, C. Moore and J. Lawless, Meteoritics, 1976, 11, 231–237 CrossRef CAS.
- P. Clarke, D. Smith, A. Steer and N. Bia, Chem. Commun., 2017, 53, 10362–10365 RSC , Ⓒ 2017, The Royal Society of Chemistry. This is an author-produced version of the published paper. Uploaded in accordance with the publisher's self-archiving policy. Further copying may not be permitted; contact the publisher for details..
- H. J. Cleaves II, Monatsh. Chem., 2003, 134, 585–593 CrossRef.
- A. Hjalmarson, P. Bergman and A. Nummelin, 1st European Workshop on Exo-/astro-biology, 2001, p. 263 Search PubMed.
- S. Charnley, A. Tielens and T. Millar, Astrophys. J., 1992, 399, L71 CrossRef CAS.
- S. Charnley, Adv. Space Res., 2004, 33, 23–30 CrossRef CAS.
- J. Enrique-Romero, A. Rimola, C. Ceccarelli and N. Balucani, Mon. Not. Roy. Astron. Soc. Lett., 2016, 459, L6–L10 CrossRef CAS.
- J. Enrique-Romero, A. Rimola, C. Ceccarelli, P. Ugliengo, N. Balucani and D. Skouteris, ACS Earth Space Chem., 2019, 3, 2158–2170 CrossRef CAS.
- J. Enrique-Romero, S. Alvarez-Barcia, F. J. Kolb, A. Rimola, C. Ceccarelli, N. Balucani, J. Meisner, P. Ugliengo, T. Lamberts and J. Kastner, Mon. Not. R. Astron. Soc., 2020, 493, 2523–2527 CrossRef CAS.
- S. Maity, R. I. Kaiser and B. M. Jones, Phys. Chem. Chem. Phys., 2015, 17, 3081–3114 RSC.
- M. J. Abplanalp, S. Gozem, A. I. Krylov, C. N. Shingledecker, E. Herbst and R. I. Kaiser, Mon. Not. Roy. Astron. Soc. Lett., 2016, 113, 7727–7732 CAS.
- T. Lamberts, M. N. Markmeyer, F. J. Kolb and J. Kastner, ACS Earth Space Chem., 2019, 3, 958–963 CrossRef CAS.
- V. Wakelam, et al., Astrophys. J. Suppl., 2012, 199, 21 CrossRef.
- D. McElroy, C. Walsh, A. J. Markwick, M. A. Cordiner, K. Smith and T. J. Millar, Astron. Astrophys., 2013, 550, A36 CrossRef.
- M. J. Abplanalp and R. I. Kaiser, Phys. Chem. Chem. Phys., 2019, 21, 16949–16980 RSC.
- K. I. Öberg, R. T. Garrod, E. F. van Dishoeck and H. Linnartz, Astron. Astrophys., 2009, 504, 891–913 CrossRef.
- R. Martín-Doménech, G. M. Muñoz Caro and G. A. Cruz-Díaz, Astron. Astrophys., 2016, 589, A107 CrossRef.
- N. F. Kleimeier, A. M. Turner, R. C. Fortenberry and R. I. Kaiser, ChemPhysChem, 2020, 21, 1531–1540 CrossRef CAS.
- R. L. Hudson and M. H. Moore, Astrophys. J., 2018, 857, 89 CrossRef.
- K. K. Singh, P. Tandon and A. Misra, Formation of Acetaldehyde in the Interstellar Medium from the Reaction of Methanol and Atomic Carbon in Interstellar Water Ice, Advances in Spectroscopy: Molecules to Materials, Singapore, 2019, pp. 415–422 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- M. J. Frisch et al., Gaussian 16 Revision C.01, Gaussian Inc, Wallingford CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. M. L. Martin, J. P. François and R. Gijbels, J. Comput. Chem., 1989, 10, 875–886 CrossRef CAS.
- D. K. Singh, S. Cha, D. Nam, H. Cheong, S.-W. Joo and D. Kim, ChemPhysChem, 2016, 17, 3040–3046 CrossRef CAS PubMed.
- D. K. Singh, B. Rathke, J. Kiefer and A. Materny, J. Phys. Chem., 2016, 120, 6274–6286 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Theory Comput., 2005, 1, 61–69 CrossRef CAS.
- J. Zheng et al., Polyrate-version 2017-C, University of Minnesota, Minneapolis, 2017 Search PubMed.
- D. G. Truhlar and B. C. Garrett, Acc. Chem. Res., 1980, 13, 440–448 CrossRef CAS.
- Y. P. Liu, G. C. Lynch, T. N. Truong, D. H. Lu, D. G. Truhlar and B. C. Garrett, J. Am. Chem. Soc., 1993, 115, 2408–2415 CrossRef CAS.
- R. T. Skodje, D. G. Truhlar and B. C. Garrett, J. Phys. Chem., 1981, 85, 3019–3023 CrossRef CAS.
- A. G. Vandeputte, M. K. Sabbe, M.-F. Reyniers, V. Van Speybroeck, M. Waroquier and G. B. Marin, J. Phys. Chem., 2007, 111, 11771–11786 CrossRef CAS.
- M. A. Ali, Sci. Rep., 2020, 10, 10995–11008 CrossRef CAS.
- M. Akbar Ali, M. Balaganesh and K. C. Lin, Phys. Chem. Chem. Phys., 2018, 20, 4297–4307 RSC.
- M. A. Ali, M. Balaganesh and S. Jang, Atmos. Environ., 2019, 207, 82–92 CrossRef CAS.
- A. Parandaman, C. B. Tangtartharakul, M. Kumar, J. S. Francisco and A. Sinha, J. Phys. Chem., 2017, 121, 8465–8473 CrossRef CAS PubMed.
- I. Oueslati, B. Kerkeni, A. Spielfiedel, W.-L. Tchang-Brillet and N. Feautrier, J. Phys. Chem., 2014, 118, 791–802 CrossRef CAS PubMed.
- N. Henriksen and F. Hansen, Theories of molecular reaction dynamics: the microscopic foundation of chemical kinetics, Oxford University Press, United States, 1st edn, Oxford graduate texts, 2008 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The following files are available free of charge, COORD: ASCII files for all XYZ coordinates pertaining to I1, I2, P1, P2, P3, TS1, TS2, TS3 and TS4 for possible spin states S = 0, 1. This material is available free of charge via the Internet at https://pubs.acs.org/. See https://doi.org/10.1039/d2ra03555c |
| This journal is © The Royal Society of Chemistry 2022 |