
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous CO2 reduction and NADH regeneration using formate and glycerol dehydrogenase enzymes co-immobilized on modified natural zeolite†
Clarissa Cocuzzaa,
Giuseppe Pietricolaa,
Ilaria Zoncaa,
Melodj Dosaa,
Oscar Romero c,
Tonia Tommasia,
Valentina Caudaa,
Debora Finoa,
Carminna Ottone*b and
Marco Piumetti*a
c,
Tonia Tommasia,
Valentina Caudaa,
Debora Finoa,
Carminna Ottone*b and
Marco Piumetti*a
aDepartment of Applied Science and Technology, Politecnico di Torino, Corso Duca degli Abruzzi 24, I-10129 Turin, Italy. E-mail: marco.piumetti@polito.it; Tel: +39 011 0904753
bEscuela de Ingeniería Bioquímica, Pontificia Universidad Católica de Valparaíso, Av. Brasil 2085, Valparaíso, Chile. E-mail: carminna.ottone@pucv.cl; Tel: +56 32 2372018
cBioprocess Engineering and Applied Biocatalysis Group, Departament of Chemical, Biological and Enviromental Engineering, Universitat Autònoma de Barcelona, 08193, Spain
First published on 31st October 2022
Abstract
In this work, the co-immobilization of formate dehydrogenase (FDH) and glycerol dehydrogenase (GlyDH) enzymes is proposed to reduce CO2 into formic acid, an important chemical intermediate. The reduction of carbon dioxide is carried out by FDH to obtain formic acid, simultaneously, the GlyDH regenerated the nicotinamide cofactor in the reduced form (NADH) by the oxidation of glycerol into dihydroxyacetone. Natural zeolite was selected as immobilization support given its good properties and low cost. The natural zeolite was modified with subsequent acid-alkaline attacks to obtain a mesostructurization of the clinoptilolite. The two enzymes were co-immobilized on clinoptilolite, previously hetero-functionalized with amino and glyoxyl groups. The distribution of the enzymes was confirmed by fluorescence microscopy analysis. Furthermore, a great increase in the retained activity for the formate dehydrogenase enzyme was noted, passing from 18% to 89%, when the mesostructured clinoptilolite was used as support. The immobilization yield of formate dehydrogenase and glycerol dehydrogenase is around 100% with all the supports studied. The promising results suggest a possible development of this procedure in enzyme immobilization and biocatalysis. The biocatalysts were characterized to find the optimal pH and temperature. Furthermore, a thermal stability test at 50 °C was carried out on both enzymes, in free and immobilized forms. Finally, it was shown that the biocatalyst is effective in reducing CO2, both by using the cofactor in the reduced form (NADH) or the oxidized form (NAD+), obtaining NADH through the regeneration with glycerol in this latter case.
1 Introduction
In the last two centuries, research in catalysis was focused on pursuing greener and more sustainable technologies for the abatement of atmospheric contaminants and the formation of high-value-added molecules from waste.1,2 Carbon dioxide (CO2) is the predominant greenhouse gas generated both from anthropogenic and natural sources, and its emissions are realistically impossible to nullify. On the other hand, CO2 is involved in several industrial processes, and it could be considered a reagent to produce organic compounds such as urea and salicylic acid.3 As a result, the catalytic reduction of CO2 into high-value-added products or chemical intermediates to decrease its high concentration in the atmosphere has been proposed in recent studies.1,4Mimicking the natural photosynthetic mechanism is a viable route, it is considered an alternative to the more energy-consuming classical heterogeneous catalysis, alongside exploiting biological systems, namely microbial metabolic pathways and enzyme-mediated processes.3,4 Using cellular systems such as bacteria or fungi lead to cheaper processes, decreasing both the operative and the downstream costs. However, they require more stringent operative conditions regarding temperature, pH, aeration, and nutrient supply.2 On the other hand, the production of enzymes is generally more expensive, but since they are single proteins catalyzing single reactions, they provide an incomparable selectivity to the desired product, making the purification process easier and cheaper. Over the years, various enzymes, mainly in the classes of oxidoreductases and lyases, have been investigated to yield carbon-based chemicals with higher value, including methane, methanol, and carbohydrates.5
Obert and Kuwabata developed the first enzymatic cascade reactions to reduce CO2 into methanol.6,7 The process proposed by Obert seems to be very promising and entails three dehydrogenases to work as an assembly line, with the consumption of three moles of reduced nicotinamide adenine dinucleotide cofactor (NADH) for a mole of methanol produced.6 The mechanism mainly consists of the reverse oxidation of methanol: the first enzyme, formate dehydrogenase (FDH, EC 1.2.1.2), carries out the reduction of carbon dioxide into formic acid; then a second oxidoreductase, formaldehyde dehydrogenase (FaldDH, EC 1.2.1.46), converts formate into the corresponding aldehyde; eventually, alcohol dehydrogenase (ADH, EC 1.1.1.1) reduces formaldehyde into methanol.3,6 To obtain better performance, previous studies in the literature suggest using carbonic anhydrase to increase the CO2 solubilization in the reaction medium, before performing the cascade reactions previously described.8 The first step of this cascade reaction is studied in this paper, using the enzyme FDH co-immobilized with glycerol dehydrogenase (GlyDH, EC 1.1.1.6). The second enzyme is necessary to perform a cofactor regeneration during the CO2 reduction reaction.
In fact, in addition to the problems derived from the expensive and burdensome production of the enzymes, and the stable maintenance of their three-dimensional structure, the high cost of the cofactor is another critical issue to scale up this process to the industrial level.5,9 As far as the regeneration of cofactor NADH is concerned, it requires the delivery of electrons to the oxidized form of the molecule. Different approaches have been designed and studied, such as electrochemical, photochemical, and biochemical processes.10–12 The exploitation of solar energy or electricity from renewable sources may be a promising solution for greener applications. However, by using enzymes good efficiency and selectivity have been obtained. Furthermore, it is not necessary to use expensive mediators, such as molecules based on noble metals.5 By using the glycerol dehydrogenase enzyme, it is possible to regenerate the NADH cofactor13 and at the same time, the organic waste from the biodiesel industry can be valorized. This enzyme performs the oxidation of glycerol to dihydroxyacetone using one mole of the oxidized cofactor, returning the reduced form NADH. This reaction has several advantages: the oxidized form of the cofactor (NAD+) is regenerated to NADH and dihydroxyacetone (DHA) is obtained as a co-product, DHA has a market value of about twenty-five times the value of glycerol.14 Finally, there is the possibility to valorize a by-product of the biodiesel industry, which can potentially be almost cost-free.
Furthermore, the immobilization of enzymes will allow the re-using of them, lowering their cost of application. The immobilization process is a critical point because the environment in which the reactions occur significantly affects the global performance of the enzyme: the confinement into materials with specific morphologies and the co-presence of other enzymes in the surroundings are two conditions that can modify the behavior of the enzyme.15 Among all the possible materials, inorganic porous supports, such as silica and zeolites, present good features and are very suitable for enzyme immobilization, although in some cases it is necessary to change the zeolites' morphology due to their reduced pore size.15–17 Natural zeolites such as clinoptilolite can be used as a valid alternative to synthetic ones due to their low cost. Furthermore, they have the advantage of being materials that are widespread in different parts of the world.18,19 Their porous structure can be improved to satisfy the working parameters of a specific process, for example, through a mesostructuring procedure.20 In this way, it is possible to minimize the mass transfer phenomena, caused by too small porosities, and increase enzymatic stability. In particular, zeolites are well-suited for the covalent immobilization technique, which ensures the proper holding of the protein, minimizing the risk of leakage.21 Zeolites can be hetero-functionalized, with glyoxyl and amino groups, to perform a two-step covalent immobilization strategy: the first step at neutral pH enhances the immobilization yield by ionic interactions without damaging the activity of the enzyme; the second step at basic pH allows the multipoint covalent interactions.22 Since the immobilization of enzymes over solid supports involves strong bonds between the support and specific amino acids, this strategy has to be carefully optimized to avoid changes in the conformation of the active site of the protein.23 The multipoint covalent strategy, despite increasing the rigidity of the immobilized enzymes, makes them more stable in the presence of distorting reagents.24,25
In this work, the Candida boidinii FDH enzyme is used along with the geobacillus stearothermophilus GlyDH enzyme to investigate the reduction of CO2 to formic acid, the first step of the cascade reactions proposed by Obert and Kuwabata.6,7 For the first time to our knowledge, both the GlyDH and FDH enzymes were co-immobilized covalently on natural zeolite to perform the CO2 reduction and simultaneously the regeneration of the NADH cofactor. The clinoptilolite was modified with subsequent acid and alkaline attacks to alter its morphology making it more suitable for enzyme immobilization.26 Two different processes of mesostructuring were tested. The first one involved an ion exchange of the original cations of the framework before the mesostructuring process. Instead, the second used the natural zeolite with the native cations originally occupying the cavities of the framework. Subsequently, the zeolites were hetero-functionalized, and finally, FDH and GlyDH were co-immobilized on the support.
The co-immobilization of the two enzymes on the support decreases the delay of the regeneration of the cofactor in its reduced form (NADH) compared to a similar process with FDH and GlyDH immobilized on separate particles, increasing the efficiency of the CO2 reduction. With FDH and GlyDH separately supported, the cofactor would be diluted in a medium and would need more time to migrate from one enzyme to the other. On the other hand, the co-immobilization strategy also has some drawbacks, for example, ones related to the different stability conditions of distinct enzymes. This is another reason why it is important to find operative parameters suitable for both enzymes.27 Another important aspect is the distribution of the FDH and GlyDH on the surface of the support; this can be investigated with fluorescence microscopy. The biocatalyst (enzyme immobilized on support) was also characterized through the retained activity (Ract) and immobilization yield (IY). In addition, the optimum pH and temperature were evaluated as well as the stability factor obtained after the immobilization. Finally, the biocatalyst was tested to carry out the CO2 reduction reaction through FDH. The CO2 reduction was performed using the oxidized form of the nicotinamide cofactor (NAD+) and GlyDH enzyme to regenerate the cofactor, thus confirming the regeneration process of NADH.
2 Materials and methods
2.1 Materials
(3-Glycidyloxypropyl)trimethoxysilane (GPTMS, ≥98%), (3-aminopropyl)triethoxysilane (APTES, ≥98%), sulfuric acid (≥98%), sodium borohydride (≥98%), sodium hydroxide (97%), toluene (99.8%) and hydrochloric acid (37%) were supplied from Sigma-Aldrich. Sodium formate, sodium metaperiodate, and glycerol (86%) were purchased from Merck. Formate dehydrogenase (FDH) from Candida boidinii with a protein concentration of 25 ± 1 mg mL−1 (according to Bradford assay28) was purchased from Megazyme. NAD+ (99.6%) was supplied from PanReach AppliChem. Base Clinoptilolite (B-Clino) was supplied by Zeolado (Greece). Glycerol dehydrogenase (GlyDH) from Geobacillus stearothermophilus was produced and purified as previously reported.292.2 Methods
 | (1) |
 | (2) |
The activity of the immobilized enzyme can be expressed in the form of the retained activity (Ract) shown in eqn (3) as:
 | (3) |
| A = A0 × e−kDt | (4) |
| A = A0[(1 − α)e−kDt + α] | (5) |
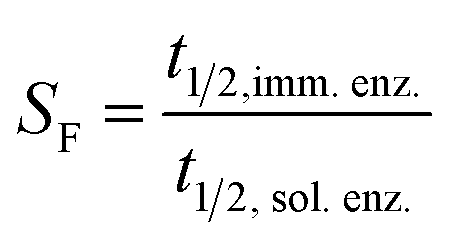 | (6) |
3 Results and discussion
3.1 Support characterization
Table 1 summarizes the results obtained from nitrogen physisorption tests (BET surface area SBET, Langmuir surface area SLang, pore volume Vp, and micropore volume Vmicro). The specific surface area of clinoptilolite (B-Clino) is relatively low, this can be ascribed to the fact that it is a natural zeolite. Due to the mesostructuring process, the SBET of clinoptilolite increased by 427% for M-Clino and 454% for M-Na-Clino while SLang increased by 662% for M-Clino and 697% for M-Na-Clino. This outcome is also observable from the change of shape and position of the isotherms (Fig. 1). The pore size distribution of both mesostructured samples showed multi-modal behavior, with peaks in the meso-macroscopic range between 30 and 200 nm (Fig. S1†). Isotherms of the nitrogen physisorption test, alongside pore size distribution diagrams, were reported to show the entity of Slit-like porosity, typically generated by the flaky morphology of clinoptilolite particles, which was further confirmed by the shape of the hysteresis. Furthermore, the values of the volume of micropores of M-Clino and M-Na-Clino were increased due to acid/alkaline attacks, (total pore volume of the modified clinoptilolites was increased too but to a lesser extent compared to Vmicro values).
| B-Clino | M-Clino | M-Na-Clino | |
|---|---|---|---|
| a Specific surface area obtained through the BET method.b Specific surface area obtained through the Langmuir method.c Total pore volume, calculated with the BJH method.d Volume of micropores calculated applying the t-plot method. | |||
| SBET (m2 g−1)a | 37 | 195 | 216 |
| SLang (m2 g−1)b | 39 | 282 | 311 |
| Vp (cm3 g−1)c | 0.14 | 0.22 | 0.25 |
| Vmicro (cm3 g−1)d | 0.005 | 0.064 | 0.066 |
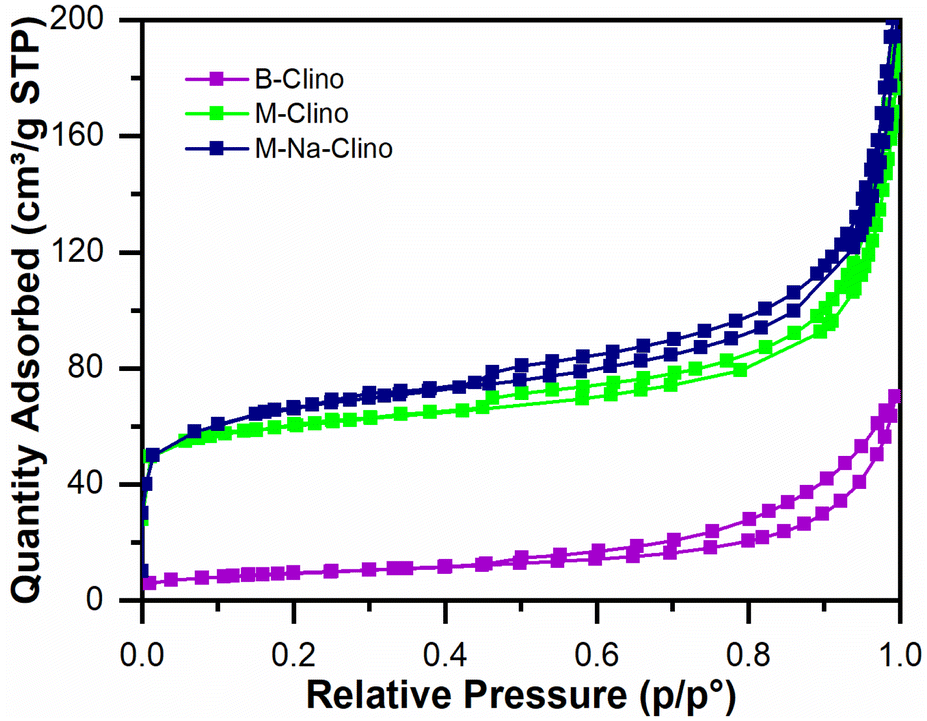 | ||
| Fig. 1 Isotherms from nitrogen physisorption tests for the base (B-Clino) and the mesostructured (M-Clino, M-Na-Clino) clinoptilolite samples. | ||
The mesostructuring process (Fig. S1†). In addition, in our previous work, it was reported that the functionalization procedures with APTES and GPTMS do not affect the mesoporosity of material.16
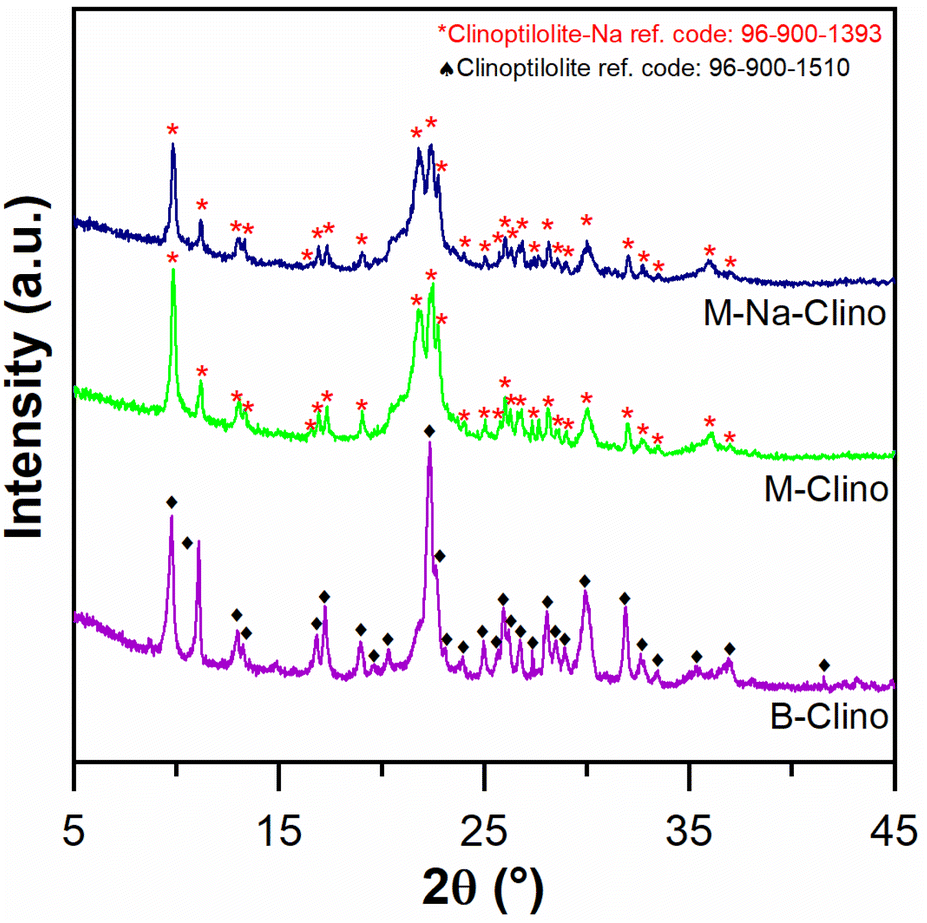 | ||
| Fig. 2 XRD diagram comparison between B-Clino, M-Clino, and M-Na-Clino. | ||
 | ||
| Fig. 3 FE-SEM images for B-Clino (a, b), M-Clino (c, d), and M-Na-Clino (e, f) at magnification 1k× and 10k×. | ||
 | ||
| Fig. 4 HR-TEM images of (a) B-Clino (b) M-Clino and (c) M-Na-Clino samples. | ||
 | ||
| Fig. 5 FT-IR spectra in absorbance of the three functionalized supports: B-Clino, M-Clino, M-Na-Clino. | ||
| Vibrational features | Wavenumber (cm−1) | Reference |
|---|---|---|
| Vibration T–O | 487 | 34 |
Stretching O–![[T with combining circumflex]](https://www.rsc.org/images/entities/char_0054_0302.gif) –O –O |
795 | 34 and 35 |
| Deformation Si–CH2 | 1406 | 36 |
| Bending C–N and bending N–H | 1511 | 37 |
| Water associated with Na and Ca in the zeolite channels | 1637 | 37 and 38 |
| Symmetric vibration CH2 | 2879 | 36 |
| Asymmetric vibration CH2 | 2933 | 35 and 36 |
| Tensile vibration –OH and –NH | 3750–3100 | 26 and 35 |
In addition, it is possible assuming that the M-Clino is the sample for which the process of functionalization has been the most effective: it has the highest ratio between the propylamine vibrational peaks, provided by APTES and GPTMS, and the stretching vibration peak O––O. The FT-IR spectra of the support alone, the functionalized support, and the enzymes individually and co-immobilized, for all the materials used are reported in Fig. S2.†
3.2 Immobilization and co-immobilization of FDH and GlyDH
FDH and GlyDH were individually immobilized and co-immobilized, following a two-step protocol, as previously described, on the three different zeolites. The hetero-functional supports are used to rapidly immobilize enzymes through a multipoint covalent attachment. This technique can be used to immobilize different proteins.25 The first step conducted at pH 7 enables the formation of ionic interactions between the enzymes and the amine groups provided by APTES; while the second step at pH 10 promotes the creation of Schiff's bases between Lysine residues and the glyoxyl groups introduced in the support. The last step of the immobilization involves the use of sodium borohydride to reduce the Schiff's bases and finally obtain covalent bonds. This technique enables the use of hydrophobic support and high ionic strength solutions to be avoided. For example, with epoxy hydrophobic support 1 M buffer phosphate solutions are usually used.25 The immobilization yields (IY) are 100% for all the immobilizations performed. The other immobilization metrics (retained activity, Ract; and specific activity, AIE) of GlyDH and FDH separately immobilized and co-immobilized on the supports are reported in Table 3. Both enzymes immobilized on the mesostructured materials displayed an increased retained activity if compared to the ones on B-Clino. The retained activities obtained immobilizing FDH and GlyDH on the mesostructured zeolites are very similar: the Ract of GlyDH separately immobilized is better on M-Clino, Ract of FDH co-immobilized with GlyDH is slightly higher using M-Clino as support, on the contrary, the Ract of co-immobilized GlyDH is a bit lower on M-Clino. The retained activity obtained on the three supports is comparable with other works where the expressed activity ranged from 5% to 90% with FDH from Candida boidinii immobilized on glyoxyl agarose.39 In our previous works, FDH on glyoxyl silica showed a retained activity of 10–14%, while with FDH on glyoxyl or amino natural zeolite a retained activity from 4 to 40% was obtained.15,16 Approximating the shape of the two enzymes with a sphere,40 a diameter of 5.54 nm and 8.99 nm is obtained, starting with molecular weights of 74 kDa,41 and 31.6 kDa,42 respectively for FDH and GlyDH. Moreover, GlyDH is a multimeric enzyme composed of 8 subunits,42 therefore the loss of activity during immobilization is likely due to the dissociation of its units and their desorption from the support.43 M-Clino support allows to obtain the best Ract of co-immobilized FDH, and although the Ract of co-immobilized GlyDH is slightly lower, it is still sufficiently high to perform the reaction. The yields of immobilization and retained activities of GlyDH are comparable with those obtained by Rocha-Martin and colleagues on glyoxyl agarose,29 with immobilization yields in the 70–100% range and expressed activities in the 5–40% range. The specific activity of the co-immobilized FDH resulted to be approximately 2.5 times the one of GlyDH presents on the same support. For this reason, a further co-immobilization with the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 by weight between FDH and GlyDH was performed, to increase the specific activity of the biocatalyst. Also, the IY of this last co-immobilization resulted to be 100%. The specific activity (AIE) calculated for the co-immobilized FDH (1:4 ratio by weight) is 2.34 UI g−1 while the one of GlyDH is 1.35 UI g−1. Using a 1:4 loading ratio of the two enzymes, the specific activity gap is reduced, in this case, the AIE of FDH resulted to be approximately 1.7 times the one of GlyDH.
:4 by weight between FDH and GlyDH was performed, to increase the specific activity of the biocatalyst. Also, the IY of this last co-immobilization resulted to be 100%. The specific activity (AIE) calculated for the co-immobilized FDH (1:4 ratio by weight) is 2.34 UI g−1 while the one of GlyDH is 1.35 UI g−1. Using a 1:4 loading ratio of the two enzymes, the specific activity gap is reduced, in this case, the AIE of FDH resulted to be approximately 1.7 times the one of GlyDH.
| FDH | FDH co-imm. | GlyDH | GlyDH co-imm. | |||||
|---|---|---|---|---|---|---|---|---|
| AIE (IU g−1) | Ract (%) | AIE (IU g−1) | Ract (%) | AIE (IU g−1) | Ract (%) | AIE (IU g−1) | Ract (%) | |
| B-Clino | 0.64 | 18 | 0.05 | 3 | 0.29 | 16 | 0.09 | 9 |
| M-Na-Clino | 3.22 | 89 | 1.45 | 80 | 0.40 | 22 | 0.77 | 83 |
| M-Clino | 3.17 | 88 | 1.68 | 93 | 0.65 | 35 | 0.69 | 74 |
3.3 Characterization of biocatalysts
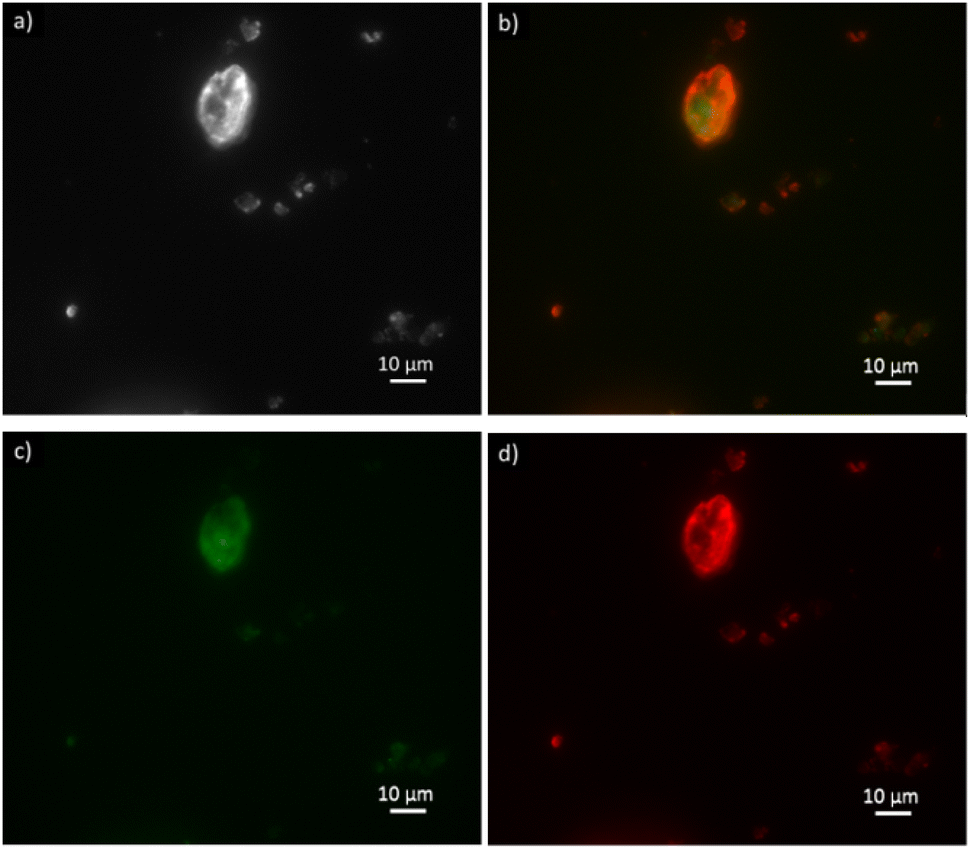 | ||
| Fig. 6 Optical fluorescence microscopy images of clinoptilolite (a) with ATTO-488 labeled FDH enzyme in the green channel (c) and ATTO-550 labeled GlyDH enzyme in the red channel (d) co-immobilized over B-Clino. Image (b) shows the two enzymes in the red and green channels. The image (a) is in brightfield. | ||
FDH is reported in the green emission channel (Fig. 6c), and GlyDH, thus is expected to visualize in the red emission channel (Fig. 6d). Besides, unloaded B-Clino was observed with the same parameters to check that the support itself has no fluorescence emission, as reported in the ESI (Fig. S3†). On the contrary, mesostructured supports (M-Clino and M-Na-Clino) themselves have fluorescence emission preventing the observation of the enzyme, probably due to the reagent used for the mesostructuring and functionalization process. For this reason, the images of FDH and GlyDH immobilized on M-Clino and M-Na-Clino are not reported. From the merged image (Fig. 6b), the two enzymes appear not to be equally distributed over the zeolite particle. In particular, the signal related to ATTO 488 (green), corresponding to FDH, seems to be more uniformly spread, while the one by ATTO 550 (red), labeled GlyDH, is more likely confined on the edges. This can be attributed to the different sizes of the two enzymes, which causes GlyDH to remain at the edges of the particle. This heterogeneous distribution opens opportunities for the development of positional co-immobilization on zeolites.44,45 Furthermore, it raises the question of how the distribution of the enzymes is in the mesostructured zeolites, but unfortunately as mentioned before the autofluorescence of the material did not allow to observe the enzyme distribution. In any case, the fluorescence microscopy also shows the co-presence of the two enzymes in the same sites of the support particles: this is a confirmation of having effectively co-immobilized FDH and GlyDH on Clinoptilolite particles.
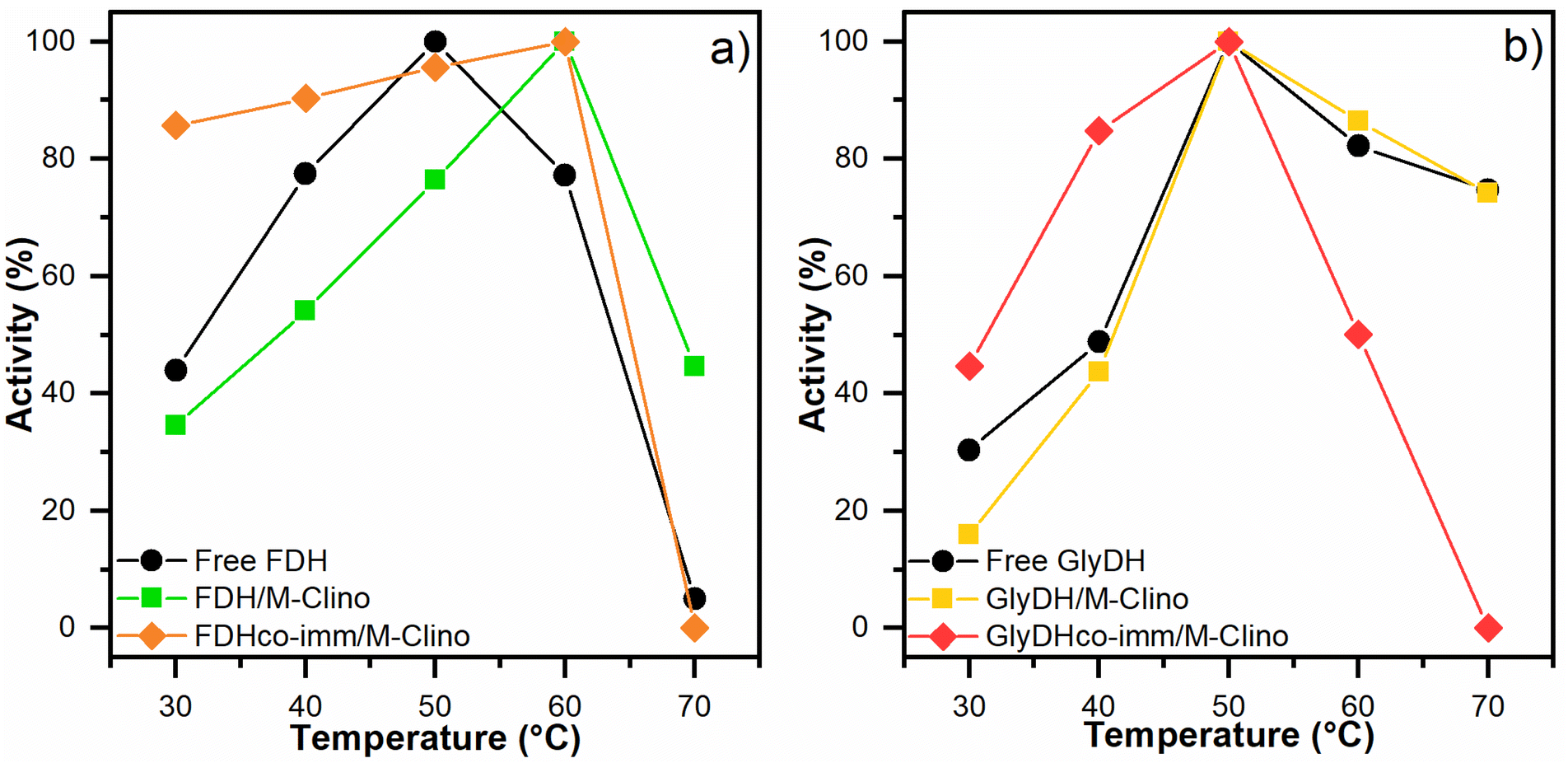 | ||
| Fig. 7 Optimal temperature results for FDH (a) and GlyDH (b). The tests were performed several times. | ||
 | ||
| Fig. 8 Optimal pH results for FDH (a) and GlyDH (b). The tests were performed many times. | ||
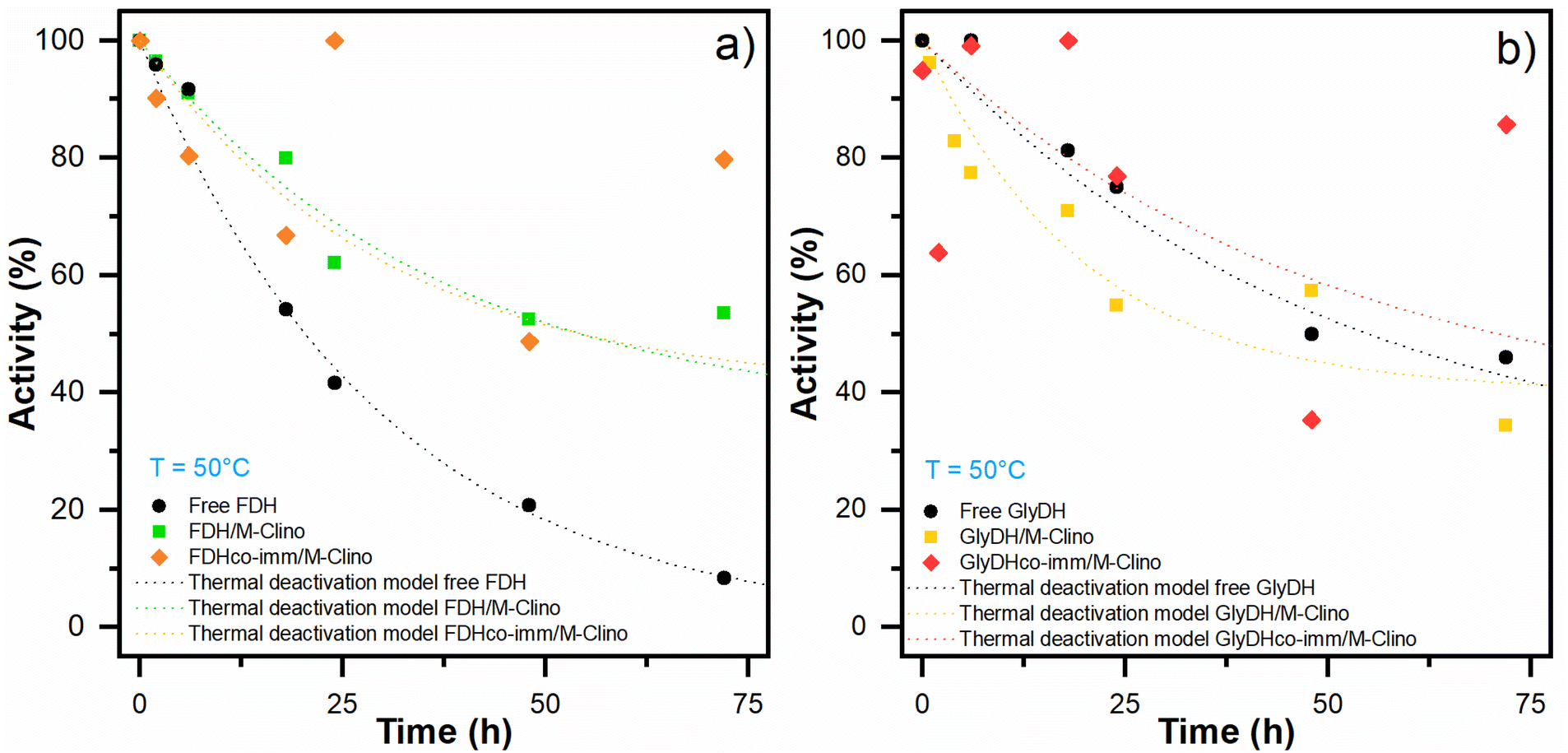 | ||
| Fig. 9 Thermal deactivation of FDH (a) and GlyDH (b): thermal deactivation models for the soluble enzyme (black dot line) and the enzyme immobilized on M-Clino separately (FDH green dot line, GlyDH yellow dot line), and co-immobilized (FDH orange dot line, GlyDH red dot line). The points had been replicated. | ||
| Free FDH | FDH/M-Clino | FDH co-imm/M-Clino | Free GlyDH | GlyDH/M-Clino | GlyDH co-imm/M-Clino | |
|---|---|---|---|---|---|---|
| Decay constant KD (h−1) | 0.034 | 0.027 | 0.037 | 0.020 | 0.050 | 0.020 |
| Residual activity α | 0 | 0.35 | 0.40 | 0.25 | 0.40 | 0.34 |
| Half-life time t1/2 (h) | 20 | 54 | 48 | 55 | 36 | 71 |
With glyoxyl agarose, a stabilization factor of 150 by Bolivar and colleagues was obtained.39 To enhance the biocatalyst stability, the immobilization rate could be optimized, as observed by Mateo and co-workers.47 When FDH is co-immobilized with GlyDH similar results are obtained.
Regarding the stability of GlyDH, no significant differences were observed between its immobilized and soluble forms.
The structural complexity of GlyDH, its large size, and the composition of 8 subunits, make its stabilization very complex. Increased stability of this enzyme has only been reported when its quaternary structure has been stabilized by crosslinking with polymers.29 Considering that both enzymes have similar lifetimes, the use of this type of modification with polymers was discarded.
3.4 CO2 reduction
To carry out the CO2 reduction a 0.1 M buffer phosphate pH 7 was used to avoid the deactivation of the cofactor, which happens at a more basic or acidic pH, during the reaction.48 Furthermore, even if the highest activity was observed at 50 °C, the reaction was carried out at room temperature and atmospheric pressure. Higher temperatures have the drawback of substantially reducing the CO2 solubility in aqueous media and it causes a faster deactivation of cofactor.48,49 The results of the reactions were evaluated by monitoring the production of formic acid (the concentration of DHA in the solution was not measured).Samples of the reacting mixture were collected periodically to evaluate formic acid concentration, for the different reactions. The first reaction is carried out only with FDH supported over M-Clino using NADH 10 mM as a reducing agent, and the results are shown in Fig. S4a.† The second reaction is carried out using FDH and GlyDH co-immobilized on M-Clino (a scheme of the two reactions catalyzed by FDH and GlyDH is reported in Fig. 10a), in this case starting with NAD+ 10 mM, this reaction is conducted in two different cases varying the FDH:GlyDH w/w weight ratio during the immobilization procedure (FDH:GlyDH 1:1 ratio and FDH:GlyDH 1:4 ratio in Fig. S4b and c† respectively). In these tests, the oxidized form of the nicotinamide cofactor was added (NAD+), with glycerol in the solution. If formic acid is observed, the regeneration of the reduced form of the cofactor (NADH) carried out by the GlyDH enzyme is confirmed. Fig. 10b shows the maximum concentration of formic acid obtained in the solution for the three reaction conditions.
 | ||
| Fig. 10 (a) Scheme of the reactions of CO2 conversion into formic acid in the presence of NADH catalyzed by FDH(i) CO2 conversion and oxidation of NAD+ to obtain and regenerate NADH, catalyzed respectively by FDH and GlyDH co-immobilized on M-Clinoptilolite(ii). (b) Maximum concentration of formic produced after 2 hours of reaction due to the reduction of CO2 by FDH (red) FDH:GlyDH w/w ratio 1:1 (blue) FDH:GlyDH w/w ratio 1:4 (yellow). | ||
The reduction of CO2, performed by FDH over M-Clino, proceeds successfully (Fig. S4a†), providing a peak of 5.5 mM of formic acid after 2 hours of reaction. This means that the mass production of formic acid, related to the activity of FDH, is 2.65 mg IUFDH−1. At equilibrium, the conversion settles down at around 50%. A similar result was obtained in our previous work, where a concentration of about 4 mM of formic acid was observed with FDH on glyoxyl and amino mordenite.16
The reaction catalyzed by FDH, with cofactor regenerated by GlyDH, with the same amount in weight of the two enzymes, instead, produced around 100 μM of formic acid (Fig. S4b†). This indicates a mass production of formic acid, related to the activity of FDH, of about 0.05 mg IUFDH−1. These performances may be due to the first reaction of the chain, catalyzed by GlyDH, which is slower than the reaction catalyzed by FDH, producing a “bottleneck” phenomenon. In this case, the formic acid produced is very little, so the concentration of NADH regenerated by GlyDH is not enough to permit a reduction of CO2 catalyzed by FDH. In light of this, to improve the reaction, the loading ratio (in weight) of FDH/GlyDH was raised to 1:4.
By increasing the amount of GlyDH, NADH was produced in a greater amount by oxidizing glycerol to 1,3-dihydroxyacetone, improving the reaction condition, compared to the previous case. Therefore, the regenerated NADH is used to reduce CO2 to formic acid. It is possible to see the reaction mechanism in Fig. 10a-II. After 2 h a maximum of 3 mM of formic acid is reached. This implies that the mass production of formic acid, related to the activity of FDH, is 1.44 mg IUFDH−1. In this way, it is also possible to confirm the regeneration of the NADH cofactor. Subsequently, a part of formic acid is converted back to CO2, in fact after 4 hours the concentration of formic acid in the solution is about 1.5 mM (Fig. S4c†). As the reaction goes ahead, GlyDH is progressively subjected to product inhibition, because DHA is not removed from the reacting medium.50 As a result, the rate of NADH regeneration becomes slower until it reaches zero. This decrease in the regeneration rate leads to an accumulation of NAD+ in the media, which shifts the direction of the reaction to the consumption of formic acid. Finally, after 6 hours a concentration of 2 mM of formic acid was observed, but it does not achieve the maximum reached after 2 hours.
Rocha-Martin and colleagues reported that the GlyDH, as a free enzyme, has a half-maximal inhibitory concentration (IC50) of DHA, produced by the oxidation of glycerol, equal to 0.43 mM.29 Furthermore, its acid sites are the most commonly used to carry out the immobilization on hetero-functionalized support. These sites mainly correspond to the active sites where the non-competitive inhibition by DHA takes place, as demonstrated by Rocha-Martin et al. using a 3D structure,29 increasing the half-maximal inhibitory concentration. To avoid this inhibition and recover the DHA product, alternatives such as in situ product removal have been proposed.51 This could be a strategy to enhance the regeneration of the cofactor and facilitate the purification of the products of the reaction.
The immobilization of FDH and GlyDH on hetero-functionalized support, even if it does not allow an increase in thermal stability, it could be used to carry out CO2 reduction with a biocatalyst that can be recovered and potentially re-used.
4 Conclusions
The conversion of CO2 to formic acid was evaluated using the enzyme FDH from Candida boidinii, covalently immobilized on the hetero-functionalized clinoptilolite. For the first time, the FDH enzyme was co-immobilized on porous support along with the GlyDH enzyme from Geobacillus stearothermophilus, capable of carrying out a regeneration of the NADH cofactor.In particular, the influence of the surface area and the support morphology was investigated. A mesostructuring process was carried out on the support using an acid/base treatment. Through physisorption with N2 at −196 °C, a noticeable increase in surface area and an enlargement of the pores were observed. To carry out a covalent immobilization, a process of hetero-functionalization using APTES and GPTMS was performed. In this way, amino and glyoxyl groups were generated on the supports and the immobilization was carried out on both the pristine support and the mesostructured ones. The co-immobilization and co-localization of both enzymes were confirmed by fluorescence microscopy.
An increase in the activity of the two enzymes was observed when they are immobilized on mesostructured clinoptilolite compared to when untreated clinoptilolite is used as support. Both the free and immobilized enzymes were characterized for optimal pH and T. In addition, a thermal stability test at 50 °C was performed, demonstrating a significant increase in half-life for the FDH enzyme. The GlyDH enzyme, on the other hand, is more stable in free than immobilized form.
Finally, the two enzymes were used to carry out the reduction of CO2 to formic acid. FDH alone produced a concentration of 5 mM, resulting in a conversion of approximately 50% compared to the initially provided NADH cofactor. The reduction reaction was also possible using the two co-immobilized enzymes and starting from the cofactor in an oxidized form. The GlyDH enzyme, using glycerol, generated NADH starting from NAD+ which was then used by the FDH enzyme to carry out the reduction reaction. In this case, at the end of the test, a concentration of about 2 mM of formic acid was obtained. On the other hand, to increase the performance, it would be possible to extract the two enzymes from different microorganisms that are more efficient in carrying out these two reactions.
The strategy of mesostructuring the porous materials and the subsequent immobilization seems very promising and paves the way for the future development of processes applied to a wider array of enzymes.
Author contributions
Conceptualization: M. P., C. O.; methodology: M. P., C. O., T. T.; investigation: G. P., C. C., I. Z., M. D., O. R., V. C.; funding acquisition: C. O., O. R.; resources: M. P., C. O., T. T., V. C., D. F., writing – original draft: I. Z., G. P., C. C.; writing – review and editing: O. R., T. T. C. C.; supervision: M. P., C. O.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Politecnico di Torino for the funding support required to publish this work in gold open access. O. R. acknowledges the support of Generalitat de Catalunya AGAUR for a Beatriu de Pinós H2020 MSCA-Cofund (2019-BP-00180). The funding support provided by ANID-Chile through the project FONDECYT 11180967 is acknowledged.Notes and references
- S. K. Bhatia, R. K. Bhatia, J. M. Jeon, G. Kumar and Y. H. Yang, Renewable Sustainable Energy Rev., 2019, 110, 143–158 CrossRef CAS.
- P. R. Yaashikaa, P. Senthil Kumar, S. J. Varjani and A. Saravanan, J. CO2 Util., 2019, 33, 131–147 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 28, 2975–2992 RSC.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. Dubois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS.
- J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Chem. Soc. Rev., 2015, 44, 5981–6000 RSC.
- R. Obert and B. C. Dave, Ind. Eng. Chem. Res., 1998, 37, 12192–12193 Search PubMed.
- S. Kuwabata, R. Tsuda and H. Yoneyama, Electrochemical Conversion of Carbon Dioxide to Methanol with the Assistance of Formate Dehydrogenase and Methanol Dehydrogenase as Biocatalysts, 1994, vol. 116 Search PubMed.
- C. Mateo, J. M. Bolivar, C. A. Godoy, J. Rocha-Martin, B. C. Pessela, J. A. Curiel, R. Muñoz, J. M. Guisan and G. Fernández-Lorente, Biomacromolecules, 2010, 11, 3112–3117 CrossRef CAS.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., 2017, 129, 789–793 CrossRef.
- F. Marpani, M. Pinelo and A. S. Meyer, Biochem. Eng. J., 2017, 127, 217–228 CrossRef CAS.
- A. Dibenedetto, P. Stufano, W. Macyk, T. Baran, C. Fragale, M. Costa and M. Aresta, ChemSusChem, 2012, 5, 373–378 CrossRef CAS PubMed.
- H. Song, C. Ma, P. Liu, C. You, J. Lin and Z. Zhu, J. CO2 Util., 2019, 34, 568–575 CrossRef CAS.
- R. Cazelles, J. Drone, F. Fajula, O. Ersen, S. Moldovan and A. Galarneau, New J. Chem., 2013, 37, 3721–3730 RSC.
- I. Poljungreed and S. Boonyarattanakalin, J. Chem. Technol. Biotechnol., 2017, 92, 2635–2641 CrossRef CAS.
- G. Pietricola, T. Tommasi, M. Dosa, E. Camelin, E. Berruto, C. Ottone, D. Fino, V. Cauda and M. Piumetti, Int. J. Biol. Macromol., 2021, 177, 261–270 CrossRef CAS PubMed.
- G. Pietricola, C. Ottone, D. Fino and T. Tommasi, J. CO2 Util., 2020, 42, 101343 CrossRef CAS.
- E. Camelin, O. Romero, M. Piumetti, C. Ottone, A. Illanes and D. Fino, in Nanomaterials for biocatalysis, Elsevier, 2021, pp. 105–148 Search PubMed.
- M. Sprynskyy, B. Buszewski, A. P. Terzyk and J. Namieśnik, J. Colloid Interface Sci., 2006, 304, 21–28 CrossRef CAS.
- C. Galletti, M. Dosa, N. Russo and D. Fino, Environ. Sci. Pollut. Res., 2021, 28, 24355–24361 CrossRef CAS.
- S. Mitchell and J. Pérez-Ramírez, Catal. Today, 2011, 168, 28–37 CrossRef CAS.
- M. Hartmann and D. Jung, J. Mater. Chem., 2010, 20, 844–857 RSC.
- N. Ramírez, M. Serey, A. Illanes, M. Piumetti and C. Ottone, J. Photochem. Photobiol., B, 2021, 215, 112113 CrossRef PubMed.
- A. Illanes, Enzyme biocatalysis: Principles and applications, Springer, 2008 Search PubMed.
- R. C. Rodrigues, Á. Berenguer-Murcia, D. Carballares, R. Morellon-Sterling and R. Fernandez-Lafuente, Biotechnol. Adv., 2021, 52, 107821 CrossRef CAS.
- Immobilization of Enzymes and Cells, ed. J. M. Guisan., Humana Totowa, NJ, 3rd edn, 2013 Search PubMed.
- D. Verboekend, T. C. Keller, M. Milina, R. Hauert and J. Pérez-Ramírez, Chem. Mater., 2013, 25, 1947–1959 CrossRef CAS.
- S. Arana-Peña, D. Carballares, R. Morellon-Sterlling, Á. Berenguer-Murcia, A. R. Alcántara, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2021, 51, 107584 CrossRef.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- J. Rocha-Martin, A. Acosta, J. Berenguer, J. M. Guisan and F. Lopez-Gallego, Bioresour. Technol., 2014, 170, 445–453 CrossRef CAS.
- M. Moradi, R. Karimzadeh and E. S. Moosavi, Fuel, 2018, 217, 467–477 CrossRef CAS.
- C. Bernal, L. Sierra and M. Mesa, J. Mol. Catal. B: Enzym., 2012, 84, 166–172 CrossRef CAS.
- J. M. Guisán, Enzyme Microb. Technol., 1988, 10, 375–382 CrossRef.
- J. Rocha-Martín, B. de Las Rivas, R. Muñoz, J. M. Guisán and F. López-Gallego, ChemCatChem, 2012, 4, 1279–1288 CrossRef.
- H. Faghihian and M. Kabiri-Tadi, J. Hazard. Mater., 2010, 178, 66–73 CrossRef CAS.
- M. Babazadeh, H. Abolghasemi, M. Esmaeili and A. Ehsani, Sep. Purif. Technol., 2021, 267, 118601 CrossRef CAS.
- N. Majoul, S. Aouida and B. Bessaïs, Appl. Surf. Sci., 2015, 331, 388–391 CrossRef CAS.
- S. Nezamdoust and D. Seifzadeh, Prog. Org. Coat., 2017, 109, 97–109 CrossRef CAS.
- J. Kabuba and M. Banza, Results Eng., 2020, 8, 100189 CrossRef.
- J. M. Bolivar, L. Wilson, S. A. Ferrarotti, R. Fernandez-Lafuente, J. M. Guisan and C. Mateo, Enzyme Microb. Technol., 2007, 40, 540–546 CrossRef CAS.
- H. P. Erickson, Biol. Proced. Online, 2009, 11, 32–51 CrossRef CAS PubMed.
- H. Schutte, J. Flossdorf, H. Sahm and M.-R. Kula, Eur. J. Biochem., 1976, 62, 151–160 CrossRef.
- S. N. Ruzheinikov, J. Burke, S. Sedelnikova, P. J. Baker, R. Taylor, P. A. Bullough, N. M. Muir, M. G. Gore and D. W. Rice, Structure, 2001, 9, 789–802 CrossRef CAS.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS.
- S. Velasco-Lozano and F. López-Gallego, Biocatal. Biotransform., 2018, 36, 184–194 CrossRef CAS.
- A. H. Orrego, F. López-Gallego, G. Fernandez-Lorente, J. M. Guisan and J. Rocha-Martín, Methods Mol. Biol., 2020, 2100, 297–308 CrossRef CAS.
- B. Binay, D. Alagöz, D. Yildirim, A. Çelik and S. Seyhan Tükel, Beilstein J. Org. Chem., 2016, 12, 271–277 CrossRef CAS.
- C. Mateo, O. Abian, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2000, 26, 509–515 CrossRef CAS.
- O. H. Lowry, J. V. Passonneau and M. K. Rock, J. Biol. Chem., 1961, 236, 2756–2759 CrossRef CAS PubMed.
- R. H. Perry and D. W. Green, Perry's Chemical Engineers' Handbook, 8th edn, McGraw-Hill, 2007 Search PubMed.
- M. Piumetti, Molecular Dynamics and Complexity in Catalysis and Biocatalysis, Nature, First., 2022 Search PubMed.
- Y. Q. Peng, S. Z. Wang, L. Lan, W. Chen and B. S. Fang, Eng. Life Sci., 2013, 13, 479–486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra03459j |
| This journal is © The Royal Society of Chemistry 2022 |