
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInnovative approach to controlled Pt–Rh bimetallic nanoparticle synthesis†
M. Jensen*a,
B. Gonanoa,
W. Kierulf-Vieiraa,
P. J. Kooymanb and
A. O. Sjåstad *a
*a
aCenter for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, P.O. Box 1033 Blindern, N-0315 Oslo, Norway. E-mail: a.o.sjastad@kjemi.uio.no; martin.jensen@kjemi.uio.no
bDepartment of Chemical Engineering, University of Cape Town, Private Bag X3, Rondebosch, 7701, South Africa
First published on 6th July 2022
Abstract
Precise control of the elemental composition and distribution in bimetallic nanoparticles is of great interest for both fundamental studies and applications, e.g. in catalysis. We present a new innovative and facile synthesis strategy for the production of true solid solution Pt1−xRhx nanoparticles. This constitutes a development of the established heat-up method, where undesired shell formation is fully suppressed, despite utilizing metal precursors with different reaction rates. The concept is demonstrated through synthesis of selected Pt1−xRhx solid solution compositions via the polyalcohol reduction approach. In addition, we provide modified procedures, using the same surface stabilizing agent/metal precursors reaction matrix yielding controlled model Rh(core)–Pt(shell) and Pt(core)–Rh(shell) nanoparticles. Tunable bimetallic solid solution and core–shell nanoparticles with the same capping agent are of key importance in systematic fundamental studies, as functional materials properties may be altered by modifying the surface termination.
1 Introduction
Platinum-group metals such as Pt and Rh are used as catalysts in a range of industrial applications, e.g. the Ostwald process for HNO3 production,1 the Andrussow process in the synthesis of HCN,2 and catalytic abatement processes.3 More specifically, Pt, Rh, and Pt–Rh alloys are widely used catalysts in the form of supported nanoparticles, thin films and more robust sub-mm thick wires knitted or woven into nets.4 Reactor design and reaction conditions frequently become governing in deciding optimal metallic architecture, while the chemical reaction and targeted product selectivity dictate the optimal Pt–Rh alloying. For example, in ammonia oxidation to nitric acid for nitrogen-based fertilizer production, robust Pt–Rh nets are used in order to withstand the severe process conditions (temperature > 900 °C; pressure up to 14 atm; 8–12 volume % ammonia in air) and the extreme exothermic nature of the oxidation reaction itself (see e.g. Fjellvåg et al.4 and references therein). In contrast, for NOx abatement processes, ppm levels of NOx are first reduced to nitrogen in a slight excess of ammonia (selective catalytic reduction, SCR). The excess ammonia is subsequently oxidized over a catalyst, which can contain supported Pt–Rh nanoparticles due to the much milder reaction conditions (<400–500 °C and ppm levels of NH3 in air at atmospheric pressure). In this context, a recent combined STM – operando APXPS study demonstrated that product selectivity (nitrogen versus nitrogen oxides) is highly controlled by the nature of the Pt–Rh alloying at the outer atomic layers of the metal.5 Interestingly, although the bulk binary phase diagram of Pt–Rh predicts phase segregation below 760 °C due to the immiscibility dome,6 this has not been verified experimentally.7 In contrast, next to the expected phase-segregated core–shell nanoparticles,8 solid solutions are obtained.9,10 Additionally, for the bimetallic Cu–Ru, Pd–Ru, Au–Ir, Cu–Ir and Au–Ru systems, solid solution nanoparticles have been reported whereas their bulk counterparts cannot be obtained.11–16 These examples represent great achievements with respect to the ability to tune the physicochemical properties of functional materials in general, and to tailor them for applications such as catalysis where e.g. the electronic structure of the nanoparticles is of relevance.14 In this respect, for both applications and fundamental studies, full control of the synthesis of bimetallic nanoparticles is a prerequisite.Achieving control of nanoparticle characteristics such as nanoparticle size, nanoparticle size distribution, and morphology during synthesis is a huge topic in itself.17 The production of bimetallic nanoparticles with predetermined element distributions such as core–shell or solid solution further increases the complexity of the synthesis procedure. In the literature, two main approaches are utilized to produce nanoparticles; the heat-up and the hot-injection methods.18 The advantage of the heat-up over the hot-injection method is that it has no significant operational synthesis parameter which controls the characteristics of the nanoparticle product, e.g. the nanoparticle size distribution.18 Its strength lies in its simplicity, but it is a well-known issue that the obtained solid solution nanoparticles have an additional thin shell of one of the components when the reaction kinetics of the metal precursors are different. This issue can be mitigated by applying the more complex injection-based methods,12–16 or alternatively by searching for a suitable reaction matrix with respect to choice of metal precursors, solvent and surface stabilizing agent(s) that tame the reaction kinetics of the faster metal precursor to match the slower one.11 The first strategy leads to the drawbacks of the hot-injection operation, which requires extreme control of user-dependent synthesis parameters that affect the reproducibility of the product characteristics between batches and operators. The latter strategy is quite an elaborate process, in addition to being very system specific. Consequently, there is a call for the development of the heat-up method, improving it to become a non-system specific synthesis strategy capable of producing full solid solubility while still controlling the chemical composition of the nanoparticles.
To study systematic correlations of the role of elemental distribution within the nanoparticles to a specific functionality (e.g., catalytic performance), it is also of the utmost importance that the series of model materials is produced in similar reaction matrices to avoid the influence of parameters such as the type of surface stabilizing agent.
In this work, we report on three new key aspects of nanoparticle synthesis: (i) development of the established heat-up method, exemplified by production of true solid solution Pt1−xRhx nanoparticles from reactants with different reaction kinetics. A qualitative investigation of the indicative parameters of the relative reaction rates of the two metal precursors suffices to obtain the insight required to quench the reaction before the faster reacting metal precursor is consumed. With this knowhow at hand, the well-known drawback of the heat-up approach is resolved, as shell formation by the slower reacting metal precursor is hindered. (ii) Tuning the gross chemical composition of the solid solution nanoparticles by adding more of the slower-reacting metal precursor to the reaction mixture and quenching the reaction while some of the faster reacting metal precursor is still present. (iii) Successful synthesis of Pt1−xRhx solid solution, Rh(core)–Pt(shell), and Pt(core)–Rh(shell) nanoparticles in the same reaction matrix (precursors and surface stabilizing agent) by manipulation of the synthesis parameters. We anticipate the innovative approach presented in this work will serve as a guideline applicable to other chemical systems.
2 Experimental
Ethylene glycol (EG, ≥99%), 1,4-butanediol (99%), polyvinylpyrrolidone (PVP, average molar mass 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), [Pt(acac)2] (97%), and [Rh(acac)3] (97%) were obtained from Sigma-Aldrich. Acetone (100.0%) and methanol (100.0%) were purchased from VWR. All chemicals were used as received.
000 g mol−1), [Pt(acac)2] (97%), and [Rh(acac)3] (97%) were obtained from Sigma-Aldrich. Acetone (100.0%) and methanol (100.0%) were purchased from VWR. All chemicals were used as received.
Experiments were performed in a three-necked 250 mL round-bottom flask, which was heated in heating blocks mounted on a 825 W Radleys Carousel™ stirring (100–1400 rpm (revolutions per minute)) hotplate. The temperature was monitored using a K-type thermocouple inserted in a glass liner (glass-wall thickness was 1 mm). The response time of the thermocouple when inserted in the glass liner was estimated by measuring the time required to heat it in 20 mL ethylene glycol from 23 °C (room temperature) to 198 °C (boiling point). The average time from four replicates was 20 ± 1 s. A 400 mm Allihn water-cooled condenser was used to avoid evaporation of solvent during the reaction, and an Ar flow was used to ensure inert conditions. To make sure the three-necked round flask and the thermocouple glass liner were properly cleaned between experiments, they were first thoroughly scrubbed and rinsed with plenty of methanol, which is an effective dispersing agent for the nanoparticles we produced, and subsequently rinsed several times with type 2 water. Finally, the round flask and glass liner was sonicated (120 W) with methanol for 10 min to disperse potential residual nanoparticles, followed by rinsing with fresh methanol several times.
2.1 Synthesis of solid solution Pt–Rh nanoparticles and qualitative reaction kinetics experiments
The synthesis of the solid solution Pt–Rh nanoparticles was performed via co-reduction of the precursors. A solution of 2.0 mmol (monomer unit) of PVP in 20 mL of EG and was first dried by heating at 150 °C for 15 minutes under Ar-flow. The solution was cooled to 100–120 °C before the addition of the metal precursors; 0.10 mmol of Rh(acac)3 and 0.10–0.90 mmol of Pt(acac)2 was used. The solution with the highest Pt(acac)2 concentration required a temperature of 120 °C for the precursor to completely dissolve. A condenser was connected before the temperature was increased, then the reduction reaction was initiated by rapid heating to a target temperature of 195 °C. This was achieved in 5–10 min by switching to a higher temperature pre-heated heating block. 15 minutes after reaching 195 °C the reaction mixture was rapidly quenched by transfer to a heating block at room temperature still with stirring. Finally, the nanoparticles were isolated by the standard nanoparticle washing procedure (see below).The precursor reaction kinetics experiments were carried out using 0.10 mmol of Pt(acac)2 or 0.10 mmol of Rh(acac)3, or a mixture of 0.10 mmol Pt(acac)2 and 0.10 mmol Rh(acac)3, by following the procedure for the solid solution nanoparticle synthesis above, but the heating step was extended in time. At equal points in time, aliquots were withdrawn across these experiments to document the color change in the reaction mixture versus time (see Fig. 1).
 | ||
| Fig. 1 Color change as a function of reaction time and temperature as an indicative parameter of the relative reaction rates of 0.10 mmol Pt(acac)2 and 2.0 mmol PVP (a); 0.10 mmol Rh(acac)3 and 2.0 mmol PVP (b); and 0.10 mmol Pt(acac)2 plus 0.10 mmol Rh(acac)3 and 2.0 mmol PVP (c); all in 20 mL ethylene glycol. The aliquots are taken at the same time points in minutes after heating from 100 °C (d) for all three samples. Note that the target reaction temperature of 195 °C was reached after 8 minutes. | ||
2.2 Synthesis of Rh core–Pt shell nanoparticles
To produce the Rh seeds, a solution of 0.50 mmol (monomer unit) of PVP in 20 mL of 1,4-butanediol was first dried by heating at 150 °C for 15 minutes under Ar-flow. The solution was cooled to 100 °C before 0.050 mmol of Rh(acac)3 was dissolved, to ensure the more reactive Rh(acac)3 (compared to Pt(acac)2) would not react immediately. A condenser was connected before the temperature was raised to 220 °C (∼17 °C per minute), followed by 2 hours reaction time and subsequent cooling to room temperature. To form the Pt-shell around the Rh seeds, 0.10 mmol Pt(acac)2 and 0.50 mmol PVP were introduced into the reaction vessel before the temperature was increased to 100 °C under inert conditions to dissolve the Pt(acac)2 precursor. To initiate the mild reduction reaction of Pt, the temperature was raised to 190 °C for 18 hours before cooling to room temperature and isolating the nanoparticles following the standard nanoparticle washing procedure (see below).2.3 Synthesis of Pt core–Rh shell nanoparticles
To produce the Pt seeds, a solution of 0.25 mmol (monomer unit) of PVP and 0.025 mmol Pt(acac)2 in 20 mL of 1,4-butanediol was first dried by heating at 150 °C for 15 minutes under Ar-flow. A condenser was connected before the temperature was raised to 220 °C (∼12 °C per minute) with a reaction time of 2 hours before cooling the reaction mixture to ambient temperature. The Rh-shell was grown on the formed Pt seeds by adding 0.025 mmol Rh(acac)3 to the reaction vessel, followed by heating to 100 °C under Ar-flow to dissolve the Rh(acac)3 precursor and subsequently to 135 °C for 23 hours to expedite the mild reduction reaction of Rh. The synthesis resulted in a bimodal size distribution with larger Pt–Rh core–shell NPs and smaller, undesired, pure Rh NPs. To isolate the core–shell product, the standard washing procedure was followed with the modification that the first centrifugation was performed at low speed (1000 rpm, 5 minutes) to selectively isolate the desired larger core–shell NPs.2.4 Synthesis of Rh nanoparticles for studying stability against oxidation
Samples of pure Rh nanoparticles were synthesized according to the procedure for the Rh seeds in the Rh–Pt core–shell synthesis.2.5 Standard nanoparticle washing procedure
The nanoparticle suspensions were transferred to a glove box (Ar) directly after synthesis and flocculated by the addition of excess acetone. They were sedimented by centrifugation in air-tight sealed centrifuge tubes, first at 7500 rpm (5 minutes), followed by two more repetitions at 4500 rpm (5 minutes). After each centrifugation, the supernatant was discarded, and fresh acetone was added.2.6 Characterization
Scanning transmission electron microscopy (STEM) imaging using a high-angle annular dark field (HAADF) and a bright field (BF) detector, and energy-dispersive X-ray spectroscopy (EDS) maps, were obtained on a FEI Titan G2 60–300 kV equipped with a CEOS DCOR probe-corrector and Super-X-EDS detectors. The FEG electron source was operated at an acceleration voltage of 300 kV. For quasi in situ electron microscopy characterization, a Gatan Double Tilt Vacuum Transfer Holder (Model 648) was used. All EDS analyses were performed using the Velox® software package (version 3). The samples for electron microscopy were prepared by re-dispersing the nanoparticles in methanol after the nanoparticle washing procedure, followed by drop-deposition on microgrid carbon film supported on copper TEM grids (300 square mesh, obtained from Micro to Nano®) and drying under Ar-flow. Powder X-ray diffraction (PXRD) patterns were acquired on a Bruker D8 Discover diffractometer with Bragg–Brentano geometry, CuKα1 radiation (λ = 1.540598 Å; Ge(111)-monochromator) and a LynxEye detector. Samples were prepared by deposition of the nanoparticles onto a single crystal Si flat plate sample holder. A 640b Si-NIST standard (a = 5.430940 ± 0.000035 Å) was mixed with the Pt1−xRhx solid solution nanoparticles to correct for zero point and sample displacement. The samples were scanned twice: first with a short step size (0.020° in 2θ) for increased resolution of the Si reflection positions in the standard, and a larger step size (0.10° in 2θ) optimized for the broader nanoparticle reflections. Unit cell dimensions of the solid solution nanoparticles were obtained from profile refinement using the Topas® software package.19
Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) analyses were performed using A Varian ES 730 ICP-OES to analyze the overall Pt- and Rh content in the samples of solid solution nanoparticles, which were deposited on an alumina support material (0.5–0.6 wt% metal). Microwave-assisted acid digestion in a Mars 6 Microwave Digester was used to dissolve the samples (51.3–51.9 mg) in 10 mL aqua regia at 220 °C. The digested sample was accurately transferred to a volumetric flask and diluted to 50 mL in 2 wt% HNO3. The samples were filtered with a 0.2 μm filter. For the analysis, 1 mL of sample was transferred by pipette to a 100 mL volumetric flask and filled to the mark with 2 wt% HNO3.
3 Results and discussion
3.1 Innovative route toward solid solution Pt1−xRhx nanoparticles
In order to obtain solid solution bi- or multielement nanoparticles, co-precipitation/reduction is a prerequisite. A common strategy to achieve this is by forcing the reaction kinetics of the metal precursors to equalize through an accelerated reaction process via injection-based approaches.12–16 Alternatively, searching for a reaction matrix that provides similar reaction kinetics of all metal components may be both a labor intensive and time consuming excerise.10,11 Therefore, we instead propose to solely manipulate the easily tunable parameters (a) reaction time and (b) molar ratio between the metal components, via a one-pot heat-up method with basis in preliminary qualitative reaction kinetics assessments. Our far more resource-economical heat-up approach yields a true solid solution for any composition Pt1−xRhx. With this simple manipulation, we are successfully preserving the simplicity associated with the heat-up method. We demonstrate the concept via synthesis of Pt1−xRhx nanoparticles using the metal precursors Pt(acac)2 and Rh(acac)3 in ethylene glycol, acting as both reductant and solvent, and PVP as surfactant.:1 mixture of the two metal precursors with a total metal/PVP molar ratio of 0.1. In the first step, at 100 °C, the metal precursors were dissolved and homogenized before the reaction mixture was heated to the targeted reaction temperature of 195 °C. During the heating and the progression of the reaction, small volumes of the colloidal suspensions were withdrawn to visualize the nanoparticle formation in terms of color changes, see Fig. 1.Visual inspection of color change as an indicative parameter is intended as a simple and straightforward tool to qualitatively investigate the relative reaction rates of metal precursors.10,11 The observed color changes from pale yellow to dark brown or black were taken as indications of the formation of nanoparticles; see an experimental validation in ESI (S.I. section S1†), where we carried out a proof-of-concept experiment to document the correlation between the color change and the consumption of metal precursor by high-resolution SEM as deducted from the growth in size of the nanoparticles. In accordance with the relative reaction rates of Pt(acac)2 and Rh(acac)3 in 1,4-butanediol reported by Bundli et al.,10 we also find that the Pt(acac)2 metal precursor transforms to Pt nanoparticles (Fig. 1a) at a much slower rate than the corresponding Rh system (Fig. 1b) in ethylene glycol. Consequently, the reaction window of Pt is longer than that of Rh at the applied conditions.
Notably, for the 1:1 precursor mixture (Fig. 1c), in the initial phase (blue zone) Rh is the main component, followed by precipitation of both components (green zone) while in the third phase Pt is the dominating precipitating component as all the Rh has already reacted (yellow zone). Insight into the start- and end points of the reaction zone (green zone in Fig. 1c) is used as the indicator on when to abort the synthesis to obtain a solid solution. For successful preparation of a solid solution, it is critically important to terminate the reaction before the faster (Rh) precursor is fully consumed, to avoid the slower reducing metal (Pt) forming a shell onto the already grown solid solution. Notably, as the time elapsed between the onset of Rh- and Pt nucleation is very small compared to the end-points of the respective reaction windows, we anticipate the tendency to form a Rh(core) to be negligible in this case.
A few points to consider, using this modified approach as a guide to produce solid solution nanoparticles in other reactant systems, where the difference in precursor reaction rates is more pronounced than in our case, one can imagine a stronger tendency to form a metal core from the faster reacting precursor. Given that the target reaction temperature is sufficient for reaction to occur for both metal precursors, we postulate that this can be counteracted by the use of faster heating. This will decrease the time difference between the onset of metal nucleation from the faster- and slower reacting metal precursors. It is then critical that the reaction rates of the metal precursors allow for overlap of the respective reaction windows (green zone in Fig. 1c). From a practical point of view, higher heating rates can be achieved by placing the reaction vessel in a high-temperature heating block. The maximally attainable heating rate is then limited by factors including the rate of heat transfer from the heating block to the reaction medium, the heat capacity of the system, and the maximum temperature of the heating block.
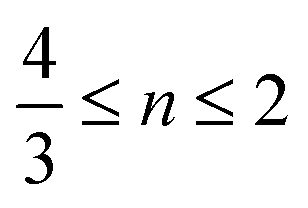 . Since Z(Rh) = 45 and Z(Pt) = 78, areas in the nanoparticle richer in Pt compared to Rh, or of higher thickness, are expected to appear brighter. The HAADF-STEM image of the Pt–Rh solid solution NP (Fig. 2b) shows no trace of a brighter or darker shell around the nanoparticle, and is thus in agreement with the EDS results. The gradual decrease of contrast from the center outwards in Fig. 2b is due to the atomic column length decreasing in the viewing direction.
. Since Z(Rh) = 45 and Z(Pt) = 78, areas in the nanoparticle richer in Pt compared to Rh, or of higher thickness, are expected to appear brighter. The HAADF-STEM image of the Pt–Rh solid solution NP (Fig. 2b) shows no trace of a brighter or darker shell around the nanoparticle, and is thus in agreement with the EDS results. The gradual decrease of contrast from the center outwards in Fig. 2b is due to the atomic column length decreasing in the viewing direction.
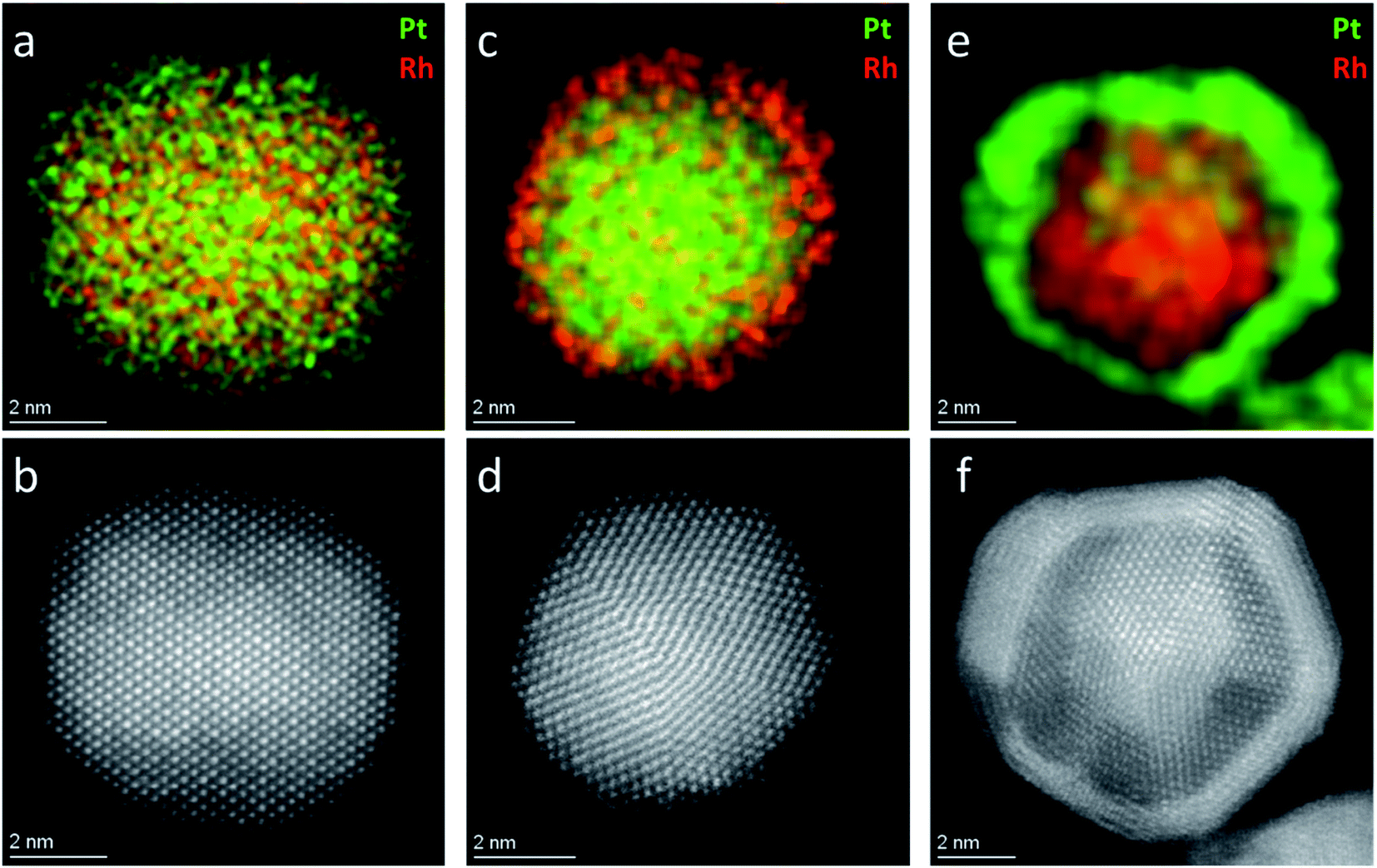 | ||
| Fig. 2 EDS elemental map (a) and HAADF-STEM image (b) of a Pt0.5Rh0.5 solid solution nanoparticle; EDS elemental map (c) and HAADF-STEM image (d) of a Pt(core)–Rh(shell) nanoparticle; EDS elemental map (e) and HAADF-STEM image (f) of a Rh(core)–Pt(shell) nanoparticle. Pt and Rh are mapped in green and red, respectively. | ||
To verify that a Pt-rich shell will form on a solid solution core if the reaction time is prolonged beyond the endpoint of the Rh precursor reaction window (the green zone in Fig. 1c), an additional experiment using an identical starting reaction mixture to the one yielding the Pt0.5Rh0.5 solid solution nanoparticles (Fig. 2a and b) was carried out. The longer reaction time (2 hours) resulted in the formation of a Pt-rich shell around the solid solution nanoparticles, as expected (S.I. section S2†).
As a next step, we developed a strategy to tune the Pt1−xRhx nanoparticle composition by adjusting the relative amounts of Pt(acac)2 and Rh(acac)3 precursors. Three experiments were performed in 20 mL ethylene glycol where the amounts of Rh(acac)3 and PVP were kept constant at 0.10 mmol and 2.0 mmol (monomer unit), respectively. The amount of Pt(acac)2 was varied between 0.10 and 0.90 mmol, corresponding to precursor molar ratios Pt(acac)2:Rh(acac)3 of 1:1, 3:1, and 9:1. All three experiments were quenched 15 minutes after reaching the reaction temperature of 195 °C (Fig. 1d).
The nanoparticle size distributions from TEM for the three samples with Pt(acac)2:Rh(acac)3 molar ratios of 1:1, 3:1, and 9:1 were 8.7 ± 2.5 nm, 6.9 ± 1.4 nm, and 7.0 ± 1.6 nm, respectively (see histograms in Fig. S3†). Quantitative EDS analyses of larger areas containing more than 1000 particles are presented in Fig. 3a, showing the average normalized Pt-content in the samples with precursor ratios of 1:1, 3:1, and 9:1 to be 23 ± 3 at%, 47 ± 8 at%, and 74 ± 14 at%, respectively. Pt-content is normalized with respect to the molar percentage of Pt and Rh. The samples were also analyzed by ICP-OES and the normalized Pt-contents were 26.6 ± 0.1 at%, 52.2 ± 0.5 at%, and 74.6 ± 0.4 at%, respectively, corresponding well with the EDS results. The difference between the molar ratio of Pt and Rh in the precursors and in the nanoparticles is attributed to the difference in precursor reaction rates (Fig. 1).
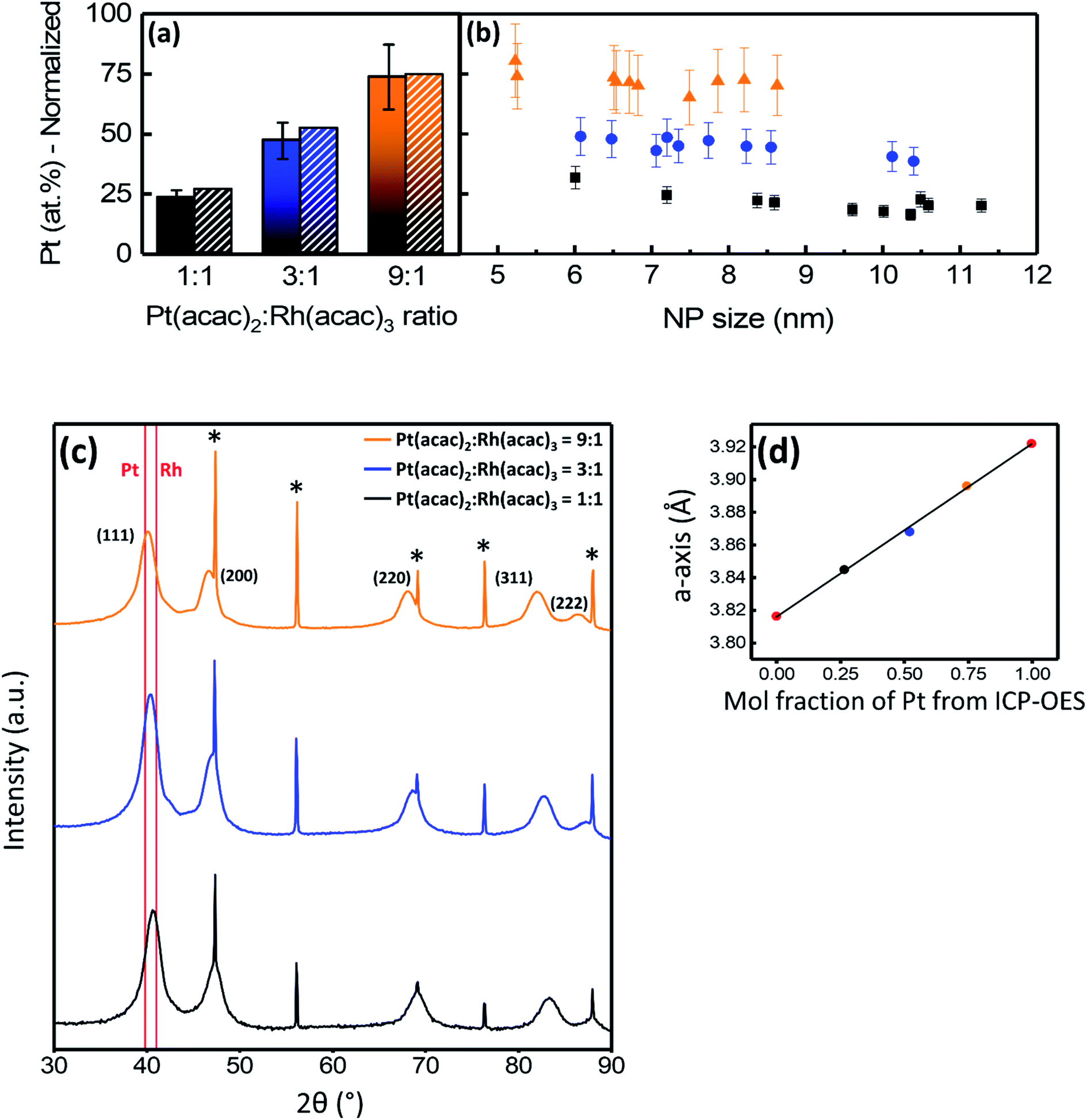 | ||
| Fig. 3 (a) Samples synthesized with Pt(acac)2:Rh(acac)3 ratios = 1:1 (black), 3:1 (blue), and 9:1 (orange). Quantitative EDS results with average normalized Pt content versus precursor ratio used in the synthesis from >1000 nanoparticles (solid bars) and ICP-OES results (shaded bars). (b) Normalized Pt content in individual nanoparticles versus nanoparticle size. Indicated (absolute) error bars represent the error in the EDS-analysis performed with the Velox® software. (c) Powder X-ray diffraction patterns of the solid solution samples with the (111) reflection corresponding to pure Pt and Rh shown in red. (*) corresponds to Bragg reflections from the Si standard. (d) a-axis from refinement of the profiles in (c) plotted versus the mole fraction of Pt (normalized to Pt and Rh) obtained from ICP-OES analysis and analyzed by linear regression (linear equation y = 0.106x + 3.816) with R2 = 0.998. Values for the a-axis dimensions of pure Pt- and Rh nanoparticles are shown in red. | ||
Powder X-ray diffraction (PXRD) was used to evaluate phase content and to determine the unit cell dimension (a-axis) of the Pt1−xRhx solid solution nanoparticles. The diffraction patterns (Fig. 3c) show broad Bragg reflections in accordance with a single ccp phase from the nanoparticles and sharp Bragg reflections corresponding to the Si-standard material used for calibration (see Experimental section for details). The presence of a single ccp phase indicates Pt1−xRhx solid solution nanoparticles have been formed. With increasing Pt(acac)2:Rh(acac)3 precursor ratio, the Bragg reflections shift to lower 2θ values. This indicates more Pt is incorporated into the solid solution structure as Pt has a longer a-axis than Rh (see e.g. ref. 10). As an extended analysis, the a-axes of the three samples were determined from profile refinements of the three nanoparticle scans shown in Fig. 3c. The a-axis of the solid solution ccp phase is plotted versus the atomic composition obtained from the ICP-OES results in Fig. 3d. Values for the unit cell dimensions of pure Pt and Rh nanoparticles, synthesized by a similar approach, are included (red points). A linear relationship between the single a-axis value for each of the samples and the respective chemical composition indicates the solid solution follows Vegard's law. The linear fit of these points in Fig. 3d resulted in an R2 value of 0.998, documenting there is a strong linear correlation between the a-axis value from PXRD and the composition from ICP-OES.
This finding shows that the synthesis strategy successfully allows tuning the average composition of the particles in the batch by simply altering the ratio between the two metal precursors. The lower yield of the slower reacting precursor (Pt) is problematic for industrial applications, but for fundamental studies where atomic composition is directly correlated to materials function, the high level of control, generic nature, and simplicity of the synthesis method far outweigh this matter.
To evaluate the intra- and inter-particle elemental distribution homogeneity, for each of the three metal ratios 10 nanoparticles covering the respective size ranges were selected for quantitative EDS analysis (see Fig. 3b). Overall, Fig. 3b shows the particles in a batch to have the same composition within the regular uncertainty of the method. Additional EDS line scans of the corresponding nanoparticles are reported in Fig. S4† and confirm elemental homogeneity. The normalized Pt content (at%) versus nanoparticle size (Fig. 3b) shows the elemental distribution is homogeneous throughout each sample. The Pt concentration seems to increase slightly with decreasing nanoparticle size. We surmise this trend to originate from the fact that the smallest nanoparticles are most likely formed at a slightly later point in the reaction, where most of the Rh(acac)3 has already reacted and is thus present at lower concentration.
3.2 Core–shell nanoparticles – Pt(core)–Rh(shell) and Rh(core)–Pt(shell)
Frequently, core–shell nanoparticles with “mirrored” nanostructuring are needed to evaluate the role of each of the constituent metals in presence of the second element subsurface. In order to rule out or equalize effects connected to the synthesis reaction matrix, such particles should preferably originate from the same chemical environment during their nucleation- and growth steps. To the best of our knowledge, the successful production of Pt(core)–Rh(shell) and Rh(core)–Pt(shell) nanoparticles in the same reaction matrix has not yet been reported. We here present Pt(core)–Rh(shell) (6.2 ± 1.3 nm) and Rh(core)–Pt(shell) (9.2 ± 1.3 nm) nanoparticles obtained in the reaction matrix 1,4-butanediol, PVP, Pt(acac)2 and Rh(acac)3 (see Fig. S3† for size distribution histograms). In both samples, the shell was formed at milder reducing conditions relative to the core nanoparticles, i.e., at lower temperature and prolonged reaction time, to favor shell overgrowth onto the core nanoparticles rather than self-nucleation.21 In addition, we did an experiment to verify that no oxidation of the core nanoparticles occurred, as this could possibly affect the overgrowth conditions (see S.I. section S5†). For more synthesis details, see the Experimental section. A representative EDS elemental map of a Pt(core)–Rh(shell) nanoparticle is presented in Fig. 2c (see S.I. section S6† for supplementary EDS maps). It clearly shows a Rh-rich shell completely encapsulating a Pt-rich core. From the EDS map, we conclude that the thickness of the Rh shell is less than 1 nm. The HAADF-STEM image (Fig. 2d) alone does not clearly show the presence of a Rh shell, because it is hard to conclude whether the slightly darker outer rim of the nanoparticles is due to Z-dependent contrast by the presence of Rh, or just a thickness contrast effect. Correspondingly, the EDS elemental map of the representative Rh(core)–Pt(shell) nanoparticle sample (Fig. 2e) reveals a 1–2 nm Pt-rich shell surrounding the Rh core of the nanoparticle. This shell is easier to detect with HAADF-STEM imaging, as seen in Fig. 2f, because the Z-dependency of the contrast in the image is not masked by the thickness dependency.In the synthesis of Pt(core)–Rh(shell) nanoparticles, smaller Rh rich nanoparticles were formed next to the core–shell nanoparticles (Fig. 4a, inset). For the reverse system, no additional Pt rich nanoparticles were observed. This may be caused by the relative fast reaction kinetics of the Rh- compared to the Pt precursor, implying the energy barrier for both homogeneous and heterogeneous nucleation is overcome more quickly for Rh, causing simultaneous occurrence of both processes.21 Furthermore, from a kinetical point of view, in addition to the concentration of shell metal precursor, we anticipate successful overgrowth of the shell to depend on the concentration of nanoparticle seeds (the core in the core–shell) onto which overgrowth can take place, which is different in the two systems. Here, it is important to note that the concentration of seeds cannot simply be tuned by the precursor concentration during their synthesis, as changing this concentration will also affect the resulting size distribution in the product. Since we wanted to match the size distributions of the Pt- and Rh seeds, their respective seed concentrations were thus restricted to different levels. It is likely that the lower concentration of seeds in the Pt(core)–Rh(shell) system combined with the higher precursor reactivity contributes to the observed partial self-nucleation of Rh in addition to the shell overgrowth. Therefore, a high precursor reactivity may represent a challenge when the goal is to achieve uniform overgrowth of the shell in seed-mediated core–shell nanoparticle synthesis.
 | ||
| Fig. 4 Representative BF-STEM images of Pt(core)–Rh(shell) NPs centrifuged at 9500 rpm for 5 minutes with EDS elemental map inset showing the distribution of Pt (green) and Rh (red) in the smaller and larger nanoparticles (a), and centrifuged at 1000 rpm for 5 minutes with EDS elemental map inset (b). | ||
To isolate the Pt(core)–Rh(shell) nanoparticles, we used mild centrifugation to selectively cause them to sediment and leave the smaller pure Rh nanoparticles in the suspension. The BF-STEM image in Fig. 4b shows that the smaller Rh nanoparticles are indeed removed when the sample is centrifuged at lower speed (1000 rpm); see S.I. section S6† for supplementary EDS maps and overview images.
4 Conclusions
We synthesized well-defined bimetallic Pt–Rh nanoparticles where the elements are distributed either as a solid solution, a Pt(core)–Rh(shell), or a Rh(core)–Pt(shell) configuration via a modified heat-up method. The key finding of this study is the development of the already established heat-up synthesis strategy now able to produce true solid solution bimetallic Pt1−xRhx nanoparticles, irrespective of relative difference in the precursor reaction kinetics and bulk phase diagram predictions. Success depends on qualitative knowledge of the relative reaction kinetics of the metal precursors and their corresponding reaction windows. By aborting the reaction before the faster reacting metal is consumed completely, the undesired formation of a shell of the slower reacting metal is prevented. The chemical composition is tuned by simply adjusting the ratio between the metal precursors. The same metal precursors and surface stabilizing agent are deliberately used for all three configurations. Having access to nanoparticles with “mirrored” core–shell nanostructuring and solid solutions from the same synthesis environment is of high importance for systematic catalytic studies, where the chemistry of the outermost atomic layers controls the materials functionality. This includes surface termination and potential residues of capping agents. In this perspective, we foresee that sets of nanoparticles analogous to these three Pt–Rh nanoparticle configurations will act as perfect model catalysts for fundamental catalytic studies. Our Pt–Rh nanoparticles will, for example, be important for fundamental studies concerning ammonia oxidation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Research Council of Norway is acknowledged for support through the Norwegian Center for Transmission Electron Microscopy (NORTEM, 197405/F50) and the industrial Catalysis Science and Innovation Centre (iCSI, 237922). The Department of Chemistry, University of Oslo is gratefully acknowledged. The study is also based on the research supported in part by the National Research Foundation of South Africa Grant number 94878. The authors appreciate scientific discussions with and support from Dr Oleksii Ivashenko, and the RECX facility.References
- J. Schäffer, V. A. Kondratenko, N. Steinfeldt, M. Sebek and E. V. Kondratenko, J. Catal., 2013, 301, 210–216 CrossRef.
- V. A. Kondratenko, Appl. Catal., A, 2010, 381, 74–82 CrossRef CAS.
- J. Zheng, O. Ivashenko, H. Fjellvåg, I. M. N. Groot and A. O. Sjåstad, J. Phys. Chem. C, 2018, 122, 26430–26437 CrossRef CAS.
- A. S. Fjellvåg, D. Waller, J. Skjelstad and A. O. Sjåstad, Johnson Matthey Technol. Rev., 2019, 63, 236–246 CrossRef.
- O. Ivashenko, N. Johansson, C. Pettersen, M. Jensen, J. Zheng, J. Schnadt and A. O. Sjåstad, ACS Catal., 2021, 11, 8261–8273 CrossRef CAS.
- H. Okamoto, Platinum-Rhodium binary phase diagram, https://matdata.asminternational.org/apd/index.aspx Search PubMed.
- C. Steiner, B. Schönfeld, M. J. Portmann, M. Kompatscher, G. Kostorz, A. Mazuelas, T. Metzger, J. Kohlbrecher and B. Demé, Phys. Rev. B, 2005, 71, 104204 CrossRef.
- E. W. Harak, K. M. Koczkur, D. W. Harak, P. Patton and S. E. Skrabalak, ChemNanoMat, 2017, 3, 815–821 CrossRef CAS.
- S. Alayoglu and B. Eichhorn, J. Am. Chem. Soc., 2008, 130, 17479–17486 CrossRef CAS PubMed.
- S. Bundli, P. Dhak, M. Jensen, A. E. Gunnæs, P. D. Nguyen, H. Fjellvåg and A. O. Sjåstad, J. Alloys Compd., 2019, 779, 879–885 CrossRef CAS.
- B. Huang, H. Kobayashi, T. Yamamoto, S. Matsumura, Y. Nishida, K. Sato, K. Nagaoka, M. Haneda, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2020, 11, 11413–11418 RSC.
- B. Huang, H. Kobayashi, T. Yamamoto, S. Matsumura, Y. Nishida, K. Sato, K. Nagaoka, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2017, 139, 4643–4646 CrossRef CAS PubMed.
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 1864–1871 CrossRef CAS PubMed.
- K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, W. Xie, M. Koyama, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2019, 10, 652–656 RSC.
- F. Wang, K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, Y. Nanba, M. Koyama and H. Kitagawa, Angew. Chem., Int. Ed., 2018, 57, 4505–4509 CrossRef CAS PubMed.
- Q. Zhang, K. Kusada, D. Wu, N. Ogiwara, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota, T. Honma and H. Kitagawa, Chem. Sci., 2019, 10, 5133–5137 RSC.
- V. De Coster, H. Poelman, J. Dendooven, C. Detavernier and V. V. Galvita, Molecules, 2020, 25(16), 3735 CrossRef CAS PubMed.
- S. G. Kwon and T. Hyeon, Small, 2011, 7, 2685–2702 CrossRef CAS PubMed.
- TOPAS V6: General profile and structure analysis software for powder diffraction data, Bruker-AXS, Karlsruhe, Germany, 2017 Search PubMed.
- M. M. J. Treacy, Microsc. Microanal., 2011, 17, 847–858 CrossRef CAS PubMed.
- Y. Sun, Natl. Sci. Rev., 2015, 2, 329–348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03373a |
| This journal is © The Royal Society of Chemistry 2022 |