
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanoparticles decorated with ovalbumin-derived epitopes: effect of shape and size on T-cell immune responses†
Elena A. Egorova a,
Gerda E. M. Lamersb,
Fazel Abdolahpur Monikhc,
Aimee L. Boyled,
Bram Slüttere and
Alexander Kros*a
a,
Gerda E. M. Lamersb,
Fazel Abdolahpur Monikhc,
Aimee L. Boyled,
Bram Slüttere and
Alexander Kros*a
aDepartment of Supramolecular & Biomaterials Chemistry, Leiden Institute of Chemistry, Leiden University, The Netherlands. E-mail: a.kros@chem.leidenuniv.nl
bCore Facility Microscopy, Institute of Biology, Leiden University, The Netherlands
cEnvironmental Biology, Institute of Environmental Sciences, Leiden University, The Netherlands
dMacromolecular Biochemistry, Leiden Institute of Chemistry, Leiden University, The Netherlands
eLeiden Academic Centre for Drug Research, Biotherapeutics, Leiden University, The Netherlands
First published on 7th July 2022
Abstract
Gold nanoparticles (GNPs) can be manufactured in various shapes, and their size is programmable, which permits the study of the effects imposed by these parameters on biological processes. However, there is currently no clear evidence that a certain shape or size is beneficial. To address this issue, we have utilised GNPs and gold nanorods (GNRs) functionalised with model epitopes derived from chicken ovalbumin (OVA257–264 and OVA323–339). By using two distinct epitopes, it was possible to draw conclusions regarding the impact of nanoparticle shape and size on different aspects of the immune response. Our findings indicate that the peptide amphiphile-coated GNPs and GNRs are a safe and versatile epitope-presenting system. Smaller GNPs (∼15 nm in diameter) induce significantly less intense T-cell responses. Furthermore, effective antigen presentation via MHC-I was observed for larger spherical particles (∼40 nm in diameter), and to a lesser extent for rod-like particles (40 by 15 nm). At the same time, antigen presentation via MHC-II strongly correlated with the cellular uptake, with smaller GNPs being the least efficient. We believe these findings will have implications for vaccine development, and lead to a better understanding of cellular uptake and antigen egress from lysosomes into the cytosol.
Introduction
The field of nanomedicine focuses on the use of nanoparticles to diagnose, monitor and treat diseases. Within this field, there is a particular focus on using nanoparticles as drug delivery vehicles or as vaccine platforms.1–5 One reason that nanoparticles are suited to this purpose is that their nanometre size range imposes certain interactions between the particles and biological systems, for instance cellular uptake pathways, recognition by immunocompetent cells and clearance from circulation.6–8 However, researchers have reported contradictory data on the effects of nanoparticle size and shape on immune response,2,9,10 or effects of nanoparticle charge on cellular uptake, clearance, immune response,11 and cytotoxicity.12–16 Often, these reports do not provide sufficient information on the antigen loading or particle concentration. Also, they often lack normalisation of the obtained results to a common parameter to aid comparison. Moreover, it is not always possible to find only one determinant parameter attributed to the biological effect of the studied nanoparticles. Parameters including size,17 shape,2,18 surface charge,12,19 rigidity,6 drug or antigen loading,1,2 particle concentration,20,21 total mass of nanoparticles,13,22 and mean surface area available for contact2,20 have all been proposed to affect the biological fate of studied particles.Studying the effect of all these parameters on vaccine efficacy is an enormous undertaking. In order to make a new system suitable for screening the effects of the abovementioned parameters, a new approach is needed. This approach has to allow for thorough control over one parameter while other parameters, such as size, shape, surface charge, and/or surface chemistry are fixed. Gold nanoparticles (GNPs) can be synthesised in a range of sizes and shapes, therefore they are ideal for studying the shape and size dependency of a biological response. Through the use of different sizes and shapes of GNPs, the dependence of cellular uptake,18,21,22 cytotoxicity,13,18 and immune response23 on physico-chemical properties can be studied. In addition, the surface chemistry of GNPs can be easily modified, and as a result, minimised toxicity and modulation of chronic inflammation risk induced by the antigen delivery particles are expected. Another advantage is that antigens are displayed on the GNP surface and cannot be compromised by non-specific interactions with the delivery vehicle itself. For example, peptide-decorated liposomes can show partial encapsulation of the displayed peptides via engulfment into the lipid membrane.24
Here, we studied the effect of nanoparticle shape and size on antigen processing and presentation. For this, gold nanorods (GNRs) of 40 by 15 nm in size and spherical GNPs of 15 and 40 nm in diameter were synthesised. The choice of these sizes was dictated by multiple lines of evidence showing that nanoparticles of 40–50 nm in diameter exhibit more efficient cellular uptake7,21 and immune response compared to other sizes.2,9 GNPs with a diameter of 15 nm are often used in literature and were chosen to serve as a reference for GNRs' width.3,17,19,25 GNRs were introduced to elucidate the effect of shape and their length matched the diameter of the larger GNPs. Incorporation of two spherical particle sizes matching the GNRs dimensions was intended to decouple the effects of shape and size.
A protective coating on the gold surface is required to maintain particle stability in biological media, to prevent aggregation, and to eliminate potential cytotoxicity.12,19,25–28 It was previously shown that, depending on the aggregation state, endocytosis pathways may vary for the same nanoparticle formulation.29 In addition to stabilizing molecules, antigens can be attached to the GNP surface to modulate antigen processing in dendritic cells (DCs).1 Using a common stabilizing molecule for the three selected nanoparticle types results in an identical surface chemistry allowing for a fair comparison of the elicited cellular responses. Previously, we reported the use of thiolated peptide amphiphiles as stabilisers for both GNPs and GNRs.30,31 These coatings insulate the gold surface, prevent particle aggregation under harsh conditions (up to 3 M NaCl, or 1 M competing thiols), and provide GNPs with surface chemistry described as “protein-like”.32 Published data suggest that biologically active moieties displayed on the surface of peptide-capped GNPs preserve their biological function and their availability,32–35 and that GNPs do not impair immune functions of DCs or B cells.19,26,27 Unlike most other metallic nanoparticles, GNPs do not give rise to reactive oxygen species (ROS) in vitro, hence oxidative stress and ROS-related cytotoxicity are less common.25
In this study, 15 nm and 40 nm GNPs and 40 by 15 nm GNRs were coated with a stabilizing peptide amphiphile,30 referred to as the base molecule. Two more peptide amphiphiles were extended with model epitopes derived from the chicken ovalbumin (OVA) protein (OVA257–264 or OVA323–339). The bioactive peptide amphiphile coupled to OVA257–264, the major histocompatibility complex class I (MHC-I) restricting epitope, was abbreviated as 1, and the peptide amphiphile coupled to OVA323–339, the major histocompatibility complex class II (MHC-II) restricting epitope, was denoted as 2.
Immunogenicity of the epitope-decorated GNPs and GNRs was studied using cells derived from the ovalbumin transgenic OT-I and OT-II mice. T cells of these mice express T-cell receptors (TCRs) that are specific for either OVA257–264 (OT-I) or OVA323-339 (OT-II) epitopes and pair with either CD8 or CD4 coreceptors.36 ‘Splitting’ the immune response into two components (OVA257–264 modulates the cytotoxic T-cell response, while OVA323–339 – the helper T-cell response) should provide more clarity as to how the immune system reacts to more complex antigens, depending on their size and shape.
The proposed system can be used as a screening system for evaluation of parameters like epitope sequence, epitope display density, size and shape of the carrier nanoparticle, as well as the charge. With a single mixture of the base and epitope-bearing amphiphiles it is possible to decorate a wide range of GNPs (spheres in the 15–100 nm range and GNRs, according to our previous studies). The procedure takes <2 hours and yields highly stable conjugates in physiological conditions. These GNPs are easily detected, quantified, and accurately dosed based on their optical properties. This system allowed us to thoroughly investigate cytotoxic and T-helper immune responses. Our findings suggest that antigen egress from lysosomes is related to the carrier nanoparticle size or volume. We believe these findings will have implications for vaccine development, and lead to a better understanding of cellular uptake and antigen egress from lysosomes into the cytosol.
Results and discussion
Characterisation of peptide–gold conjugates
In order to maintain identical surface chemistry, GNPs of different shape and sizes were coated with a mixture comprising 9 molar parts of the base peptide amphiphile and 1 molar part of the active peptide amphiphile 1 or 2 (Scheme 1). The base composition was inspired by our previous work and comprised a double thiolated C11 chain conjugated to a KVVVAAAEEEE peptide domain via the N-terminus and the lysine side-chain.30 The thiol group enabled coordination of the ligands to the gold surface via an Au–S bond. The active peptide amphiphiles comprised a single thiolated C11 chain conjugated, via the N-terminus, to a peptide with the sequence V3A3E3, while the epitopes were coupled to the C-terminus of this peptide sequence via a PEG4-spacer. Epitope sequences corresponded to OVA-derived T-cell epitopes (SIINFEKL, OVA257–264, is an MHC-I binding epitope, and ISQAVHAAHAEINEAGR, OVA323–339, is an MHC-II binding epitope). The active peptides 1 and 2 were conjugated to a single alkyl chain instead of a double chain to aid solubility of the resulting compounds. | ||
Scheme 1 Schematic representation of the shell composition (base and epitope-bound peptide amphiphiles 1 and 2) used to stabilise GNPs and GNRs. Base was mixed with either 1 or 2 at a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio to form shells around different GNPs and to provide near-identical surface chemistries. :1 molar ratio to form shells around different GNPs and to provide near-identical surface chemistries. | ||
As-synthesised citrate-stabilised GNPs and CTAB-protected GNRs were characterised with dynamic light scattering (DLS, for GNPs only), ultraviolet-visible spectroscopy (UV-vis), and transmission electron microscopy (TEM) to confirm their size and dispersity (Table 1, Fig. 1 and S1†). Due to their rod-like shape, GNRs were not analysed by DLS, instead only UV-vis spectroscopy and TEM data were collected.
| Sample | 15 nm GNPs | 40 nm GNPs |
|---|---|---|
| a Standard deviation. To determine the core size, citrate-stabilised GNPs were imaged using TEM. The size of peptide amphiphile-coated GNPs was determined using DLS. Both average size and mean hydrodynamic diameter are given in nm. Peptide amphiphile-coated samples were prepared in PBS (pH 7.2). | ||
| Av. size of the core (TEM) | 15.0 ± 1.4a | 43.5 ± 3.3 |
| Citrate | 15.5 ± 0.2 | 40.4 ± 0.5 |
| Base | 26.8 ± 0.3 | 50.8 ± 0.1 |
| 1 | 25.9 ± 0.3 | 50.8 ± 0.8 |
| 2 | 26.6 ± 0.6 | 54.3 ± 0.2 |
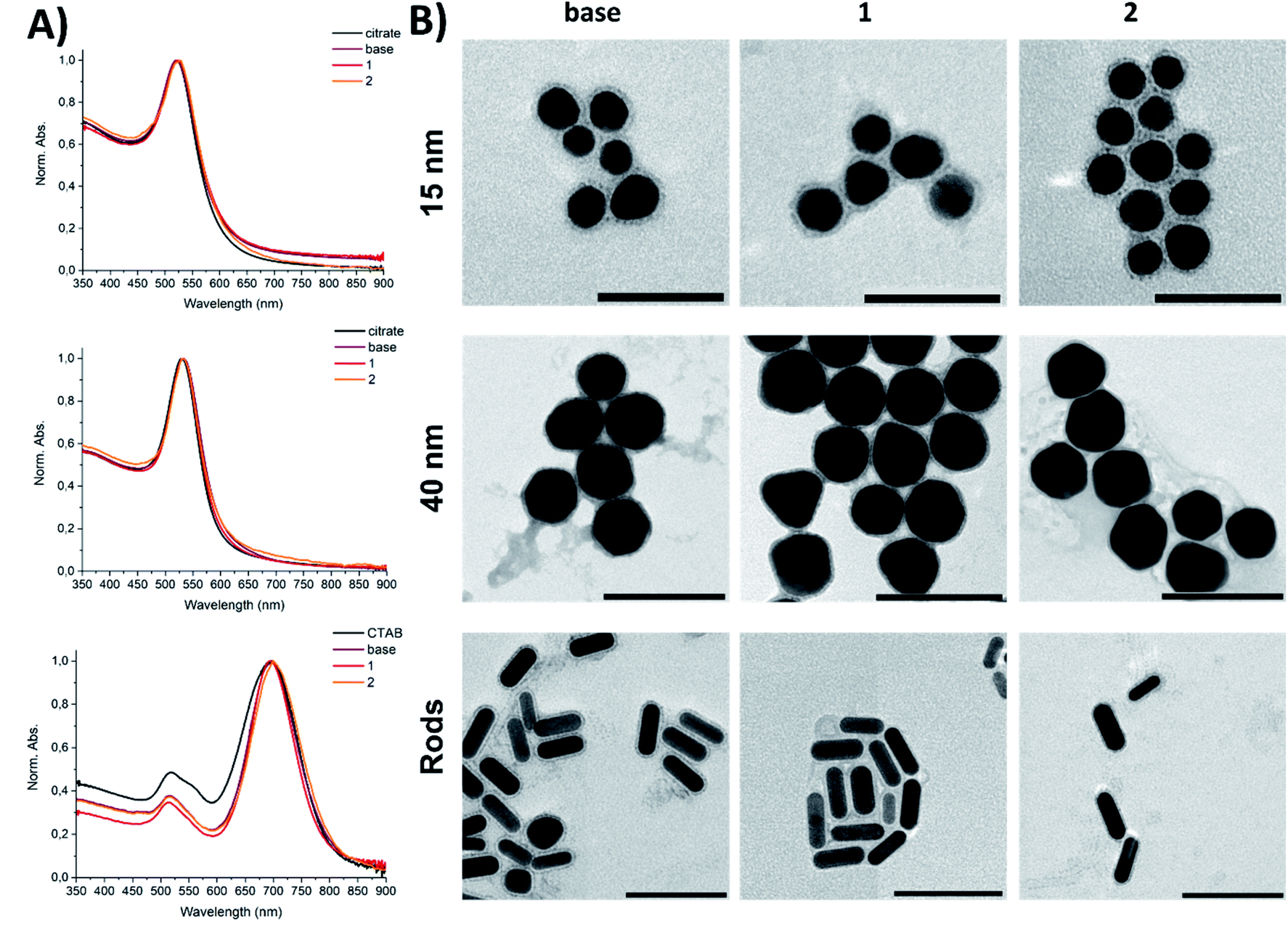 | ||
| Fig. 1 Sample characterisation of the GNPs used in this study: (panel A) UV-vis spectra for coated 15 nm GNPs, 40 nm GNPs, and GNRs; (panel B) TEM images of the corresponding peptide–gold conjugates. Modified samples were prepared in PBS (pH 7.2). Scale bars: 50 nm for 15-base, 15-1, and 15-2; 100 nm for others. Staining – 1% uranyl acetate. The visible shells are the peptide coatings. | ||
Upon modification with the peptide amphiphiles, a slight increase in the hydrodynamic size for all peptide–gold conjugates was observed with DLS. The incorporation of the extended sequences 1 or 2 (10 mol%) into the base peptide shell did not lead to agglomeration of GNPs (Fig. 1; for intensity size distributions see Fig. S1†). The UV-vis spectroscopy supported these findings, since the absence of plasmon band broadening for 15-base, 15-1, and 15-2 relative to 15-citrate indicated that no aggregation occurred after the peptide coatings were applied (Fig. 1A). The same was true for 40 nm GNPs and GNRs. The small (1–7 nm) red-shift of the plasmon band is related to substitution of citrate or CTAB on the gold surface with other molecules, which leads to a change in the dielectric constant.37 Since neither DLS (Fig. S1A and B†) nor TEM imaging (Fig. 1B) indicated aggregation, the peptide-coated GNPs and GNRs were presumed to be monodisperse. TEM imaging revealed particle clustering, which can be explained by so-called drying effects during sample deposition on a TEM grid. This clustering was also facilitated by the presence of salts, since all tested samples were prepared in PBS (pH 7.2), which meant that upon drying the local concentration of salt increased significantly to cause the observed clustering.30 Zeta potential values of as-synthesised citrate-capped GNPs and amphiphile-coated GNPs showed that particles had a negative surface charge (up to −30 mV, Fig. S1C–E†). A slight deviation of surface charge for GNPs coated with 1 and 2 from the samples coated with the base sequence was explained by a slight change in the surface chemistry when the active peptides were incorporated. The GNRs showed a zeta potential change from positive to negative, indicative of CTAB displacement from the GNR surface by the peptide amphiphiles (Fig. S1E†). A negative net charge for peptide amphiphile-coated GNPs and GNRs is advantageous: negatively charged particles cause less cytotoxicity compared to positively charged counterparts, and better cellular uptake than neutral nanoparticles.7
TEM analysis revealed the presence of well-defined peptide shells surrounding the gold core (Fig. 1B). The shell around the GNPs and GNRs ensured that particles were separated from each other with the interparticle distance corresponding to the shell thickness. The thickness of the shells (<5 nm) is in agreement with a peptide-amphiphile monolayer around the particles.30 Notably, in the TEM images there was no obvious difference in the shell thickness for samples with the functional peptides 1 or 2 incorporated, possibly due to a low epitope display density.
According to our previous work, GNPs coated with these amphiphilic molecules are highly stable at elevated salt concentrations and are resistant towards thiols,30 thus the GNPs and GNRs presented in this work were expected to be stable under biologically relevant conditions.
Number concentration and epitope dosage
To study biological effects elicited by nanoparticles of different size and shape, a defined set of shared parameters should be adopted to enable comparison. It can be particle number, particle surface area available for interaction,2,20 or drug/active molecule loading/dosage.21,38 Unfortunately, reports with a normalisation used to truly define the effect of a particle parameter (e.g. of an antigen loading rather than particle mass) on a biological response are scarce.2,13,22 In order to calculate the above-mentioned parameters, inductively coupled plasma mass spectrometry (ICP-MS) was used. This technique has been previously used to determine the thiol-containing ligand coverage on GNPs,39 as well as metal content in cells and tissues.2,13,17,19,20,25,40Peptide amphiphile-coated GNPs and GNRs were digested in aqua regia and analysed with ICP-MS to determine gold mass concentration, which was later translated to particle number concentration (Fig. S2†). The measured concentrations were linked to the optical density (OD) of the GNP or GNR suspension (Table 2). This enables accurate dosing of the GNP and GNR samples according to their OD, acquired with UV-Vis spectroscopy. We determined that a GNP suspension with an OD = 1.0 corresponded to number concentrations within the 1011–1012 particle per mL range regardless of the applied coating, which is in agreement with data for citrate-capped GNPs provided by Sigma Aldrich (https://www.sigmaaldrich.com/NL/en/technical-documents/technical-article/materials-science-and-engineering/biosensors-and-imaging/gold-nanoparticles). Moreover, GNPs with the same gold core but different peptide amphiphile coatings showed similar normalised number concentrations (Table 2), signifying that the employed coating strategy did not result in different optical properties arising from the applied stabiliser. Furthermore, incorporation of 10 mol% of the epitope-displaying functional peptides 1 or 2 did not affect the optical properties either.
| Normalised number concentration,a particles per mL | Epitope concentration, μM | |||||
|---|---|---|---|---|---|---|
| 15 nm GNPs | 40 nm GNPs | GNRs | 15 nm GNPs | 40 nm GNPs | GNRs | |
| a The obtained mass concentrations were divided by the mass of one particle and normalised to ODLSPR = 1.0. Samples were prepared in PBS (pH 7.2). For normalised mass concentrations and areas available for contact, see Fig. S2B,C. n/a – not applicable. | ||||||
| Base | 4.03 × 1012 | 1.49 × 1011 | 3.54 × 1011 | n/a | n/a | n/a |
| 1 | 3.83 × 1012 | 1.25 × 1011 | 4.06 × 1011 | 2.6 | 0.4 | 0.6 |
| 2 | 3.46 × 1012 | 1.46 × 1011 | 3.93 × 1011 | 2.4 | 0.5 | 0.6 |
To quantify the epitope dosage, another peptide amphiphile was synthesised. Mercapto-undecanoyl-V3A3E3G5WG (denoted as W, see Scheme S1†) was composed of a single C11 alkyl chain, the same peptide domain as 1 or 2, and a single Trp (tryptophan, W), conjugated via a Gly5 spacer (glycine, G). GNPs and GNRs were coated with 9 molar parts of the base amphiphile and 1 molar part of W. From the amount of the bound W together with the known relationship between OD and number concentration, the epitope loading was calculated (Table 2).
Due to the high relative surface area and high number concentration, 15 nm GNPs carry a significant epitope load, while carrying a low number of epitopes per particle. Consequently, 40 nm GNPs had a lower number concentration but higher antigen load per particle. Due to their shape, GNRs have a higher relative surface area and number concentration than 40 nm GNPs. In order to study a dose – response effect arising from GNP shape and size for both epitopes, the ≈5–500 nM concentration range in the cell exposure medium equivalent to the epitope content displayed on the gold surface was used for further studies.
Cytotoxicity of coated GNPs and GNRs
An in vitro cytotoxicity test was performed prior to studying immune response, to ensure that the peptide amphiphile-coated GNPs and GNRs were not harmful to cells. BMDCs were used in this assay for two reasons. BMDCs, like other types of dendritic cells (DCs), are antigen-presenting cells most likely to first encounter pathogens. Moreover, the ex vivo assay using transgenic ovalbumin mice (OT-I and OT-II) to probe the immunogenicity requires DCs treatment as the first step.According to an Lactate dehydrogenase (LDH) assay, the observed peptide-gold nanoconjugate cytotoxicity was found to be <10% after 4 hours of exposure and <20% after 24 hours for all tested GNPs and GNRs (negative control = PBS, 0% cytotoxicity; positive control = assay lysis buffer, 100% cytotoxicity, Fig. 2 and S3†). This was true for all particle concentrations tested. Notably, 15-base showed the highest toxicity, most likely attributed to their high number concentration.20,22 On average, cytotoxicity increased after 24 hours compared to the 4 hour exposure for all tested particles. However, there was no statistically significant difference between the cytotoxicity demonstrated by GNPs compared to GNRs after 24 hours.
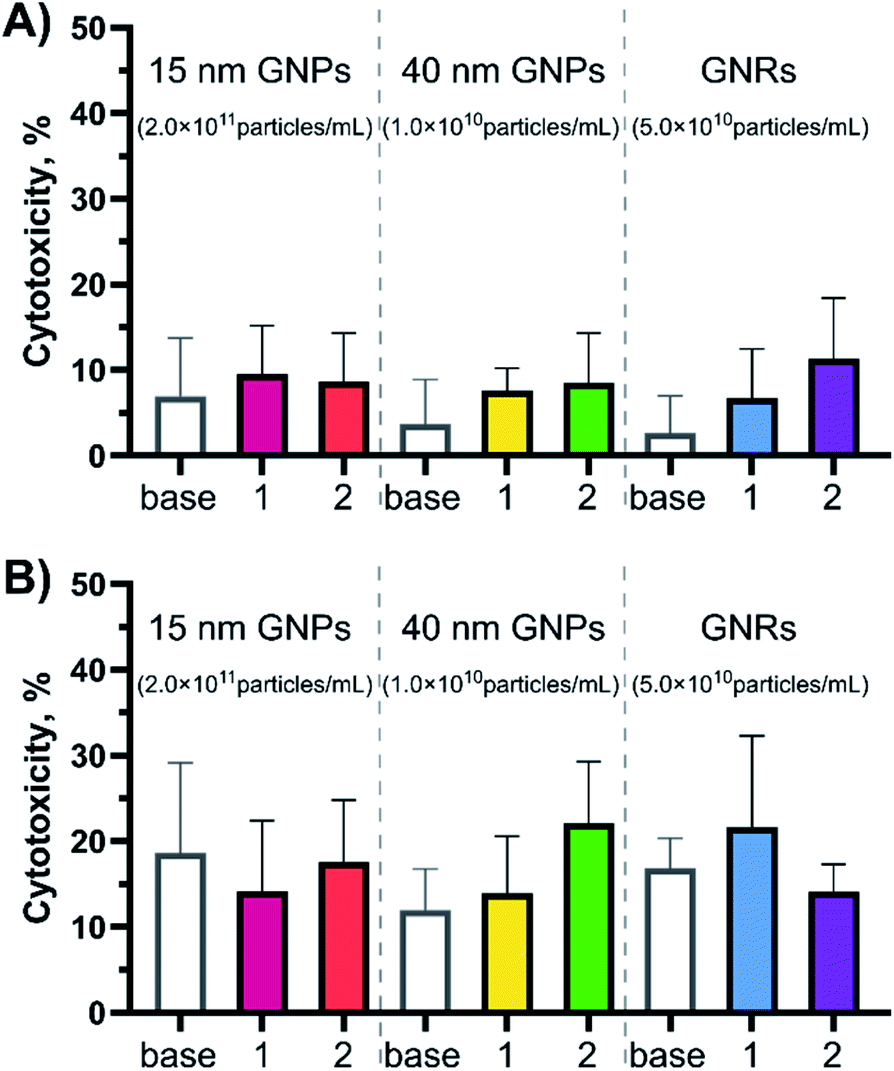 | ||
| Fig. 2 Cytotoxicity profiles for the peptide-coated GNPs and GNRs at different time points (A) 4- and (B) 24 hour exposure times. Cytotoxicity was evaluated in BMDCs (20 × 103 cells per well) with the LDH release assay (N = 2, n = 3) at the highest concentration used for each experimental group (15 nm GNPs – ∼2.0 × 1011 particles per mL; 40 nm GNPs – ∼1.0 × 1010 particles per mL; GNRs – ∼5.0 × 1010 particles per mL in the cell exposure medium). PBS was used as the negative control (0% cytotoxicity) and assay lysis buffer was used as the positive control (100% cytotoxicity). Error bars indicate standard deviations calculated using GraphPad Prism software. ANOWA (one-way) showed there was no significant difference inside the test groups (p ≥ 0.05). For the cytotoxicity profiles for more nanoparticle concentrations see Fig. S3.† | ||
This data is consistent with published results of different GNP samples in different cell types. PEGylated GNPs and GNRs were shown to have a limited effect on LDH release (<20% after 24 hour exposure)14,25 and cell death (<15% after 24 hour exposure).1,17,19,25 According to Fytianos et al. polymer-coated GNPs (15 nm in diameter) did not induce cell death independent of the surface charge,19 while Bhamidipati et al. reported that the surface chemistry contributes towards cytotoxicity, but not the shape when spherical (20 nm in diameter), rod-like (53 by 23 nm), and star-like (63 nm) GNPs were compared.25 Kang et al. reported that neither of 10, 22, or 33 nm OVA protein-coated GNPs induced cytotoxicity.17 It has also been reported that PEGylation of GNRs abolishes cytotoxicity typical for CTAB-stabilised GNRs.25,28 Additionally, changing the surface chemistry by addition of a bioactive peptide did not induce acute cytotoxicity in BMDCs.
Cellular uptake of OVA-decorated GNPs and GNRs by BMDCs
Cellular uptake defines the antigen fate inside the cell and is crucial for establishing a link between the evoked immune reaction and the nanoparticle type. Cellular uptake of peptide-gold nanoconjugates by BMDCs was quantified using ICP-MS and the subcellular localisation of endocytosed GNPs and GNRs was visualised with TEM. For this study, BMDCs were differentiated from bone marrow progenitor cells and exposed to epitope-bearing GNPs and GNRs for 4 hours.GNPs and GNRs were found inside subcellular vesicles but not dispersed in the cytosol or other organelles apart from lysosomes (Fig. 3 and S4†). There was no apparent difference in nanoparticle distribution as a function of peptide amphiphile composition. However, the appearance of these vesicles and number of entrapped particles depended on the nanoparticle core shape and size. For example, 40-1 and 40-2 showed fewer particles per vesicle (<10) than 15-1, 15-2, Rods-1, or Rods-2. The appearance of vesicles entrapping 40-1 and 40-2 may suggest a different endocytosis pathway than in the case of GNRs and 15 nm GNPs. The volume of most of these vesicles was very low and particles appeared to be “encased” in the endosomal membrane. For 15 nm GNPs and GNRs, the vesicles looked similar: larger vesicles with high particle numbers (>10) dominated, although small vesicles (<100 nm) containing fewer particles were also observed.
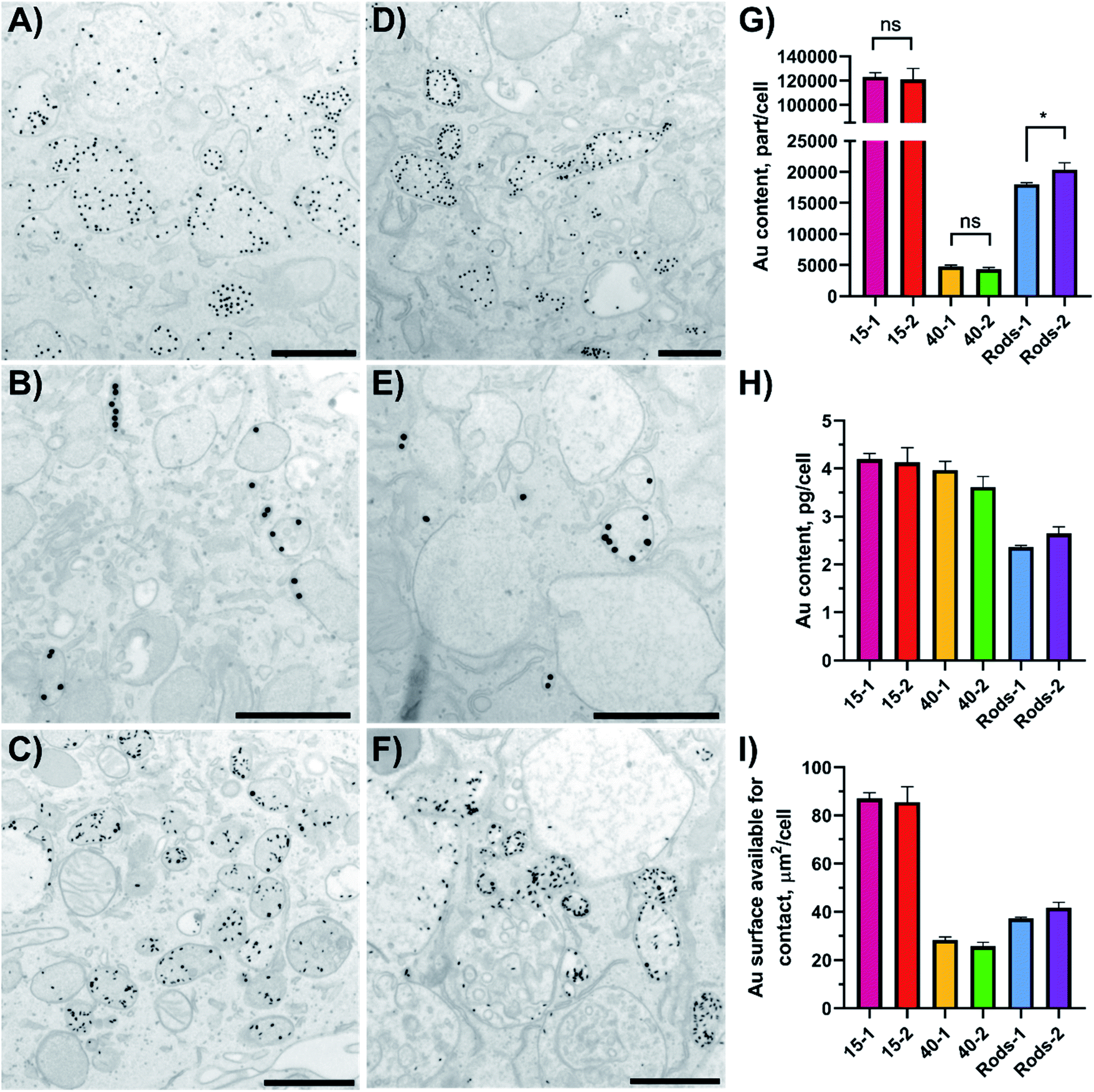 | ||
| Fig. 3 GNPs uptake by BMDCs (A–F) studied using TEM imaging and (G–I) quantified with ICP-MS. Localisation of GNPs and GNRs inside BMDCs sections after a 4 hour incubation was studied using TEM: (A) 15-1; (B) 40-1; (C) Rods-1; (D) 15-2; (E) 40-2; and (F) Rods-2. Scale bars: 500 nm (A,C,D and F) or 1 μm (B and E). A representative image of a DC treated with PBS, low magnification images and their analysis can be found in Fig. S4 and S5.† Error bars in (G–I) indicate standard deviations and were calculated in GraphPad Prism software, as well as the results significance (unpaired t-test; ns = not significant, p ≥ 0.05; * = p within 0.01–0.05). | ||
The type and function of these vesicles could not be determined from TEM analysis, but these vesicles could be divided into several groups based on their size and appearance. Small vesicles with a low number of particles inside are most likely to be endocytic vesicles that are to be fused with endosomes to start the endo-lysosomal pathway.7,8 Reformed endosomes undergo a maturation process and subsequently fuse with lysosomes. Lysosomes can be distinguished by their darker contents in the TEM images.41 Reformed lysosomes perform antigen digestion, which is crucial for antigen presentation through MHC-epitope interactions. Analysis of the TEM images for the vesicles' dimensions and number of entrapped particles showed differences between the different GNPs (Table 3 and Fig. S5† shows analysis for particles decorated with 1, OVA257–264). It should be noted that this analysis was based on a vesicle part restricted to a thin section (70 nm thickness), but not on a vesicle's full volume. There were no vesicles with more than eight 40-1 particles inside and 65.5% of the vesicles contained only one particle (n = 177). Small vesicles (40–120 nm) with low particle numbers (<6) are likely to be endocytic vesicles. On the other hand, 15-1 and R-1 showed a different behaviour. Only 15.3% of vesicles (n = 137) contained one 15-1 particle, while for R-1 this value was 13.0% (n = 200). At the same time, vesicles with ≤10 entrapped particles were moderately present in BMDCs exposed to 15-1 (48.9%), but formed the majority in the case of R-1-exposed cells (74.0%). 15-1 showed a variety of endocytic vesicles: vesicles with one particle inside or ones formed by interactions between the plasma membrane and multiple particles. This suggests that multiple endocytosis pathways are involved for 15-1 uptake. Also, nanoparticles were seen in the cytosol, but these events were rare (<0.5% for all samples).
These findings indicate that different endocytosis pathways might be involved in GNP/GNR uptake. Published data suggests the following pathways for coated GNPs and GNRs: receptor-mediated and macropinocytosis for GNPs within the 15–40 nm size range;21,38 and receptor-independent22 and a combination of clathrin-/caveolae- and lipid raft-mediated pathways for GNRs.18 Ding et al. also reported that endocytosis pathways for GNPs can be influenced through their surface chemistry.22 Solely based on the TEM images it was not possible to distinguish what specific endocytosis pathways peptide amphiphile-coated GNPs and GNRs employ. To address this, more uptake studies focusing on the use of endocytosis inhibitors should be conducted.18,22,42
In order to study the immunological impact of nanoparticles of different size and shape, it was important to determine the gold content per cell. Since quantification of cellular uptake based on the TEM images would not be reflective of the entire cell population, ICP-MS was used to calculate gold content per cell. To make a fair comparison, the number of added nanoparticles was fixed to the gold core size, meaning the same number of 15-1 or 15-2 was added.
After the 4 hour exposure time 15-1 and 15-2 were taken up by BMDCs to the same extent (123 × 103 versus 121 × 103 particle per cell), and 40 nm GNPs and GNRs showed the same trend (Fig. 3G). This suggests that altering the active peptide in the shell composition did not have an effect on GNPs or GNRs uptake by BMDCs. It should be noted that 15 nm and 40 nm GNPs, although taken up in the same mass (in the range of 3.6–4.0 pg per cell), significantly differed in number (∼25× times more for 15 nm GNPs) and delivered total surface area (∼3× times more for 15 nm GNPs, Fig. 3H and I). The surface area available for contact that was delivered into the cells could be correlated to the number of delivered antigens and it decreased in the following order: 15 nm GNPs > GNRs > 40 nm GNPs. In our study epitope-decorated 15 nm GNPs could deliver ∼3× times more antigens than 40 nm GNPs and ∼2× times more than GNRs.
Effect of size and shape on CD4+ and CD8+ T cell proliferation
OT-I and OT-II transgenic mouse lines express T-cell receptors (TCR) specific to OVA-derived epitopes, respectively OVA257–264 and OVA323–339.36 BMDCs obtained from these mice were exposed to the epitope-decorated GNPs and GNRs. BMDCs endocytosed the nanoparticles and antigens were to be cleaved from the gold surface and presented back to the surface of the BMDCs. After 4 hours of exposure, required for antigen processing and presentation to take place, the GNPs were removed, isolated CD8+ (OT-I, samples coated with 1) or CD4+ (OT-II, samples coated with 2) T cells stained with 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE) were added to the activated BMDCs, and the cells proliferated for 72 hours. The degree of T-cell activation was determined with flow cytometry using CD25 gated versus CFSE stain signals (CD25+CFSElow T-cell subset, for gating strategy see Fig. S6†).The assays revealed striking effects of size and shape on the ability of GNPs to induce T-cell mediated responses (Fig. 4).
 | ||
| Fig. 4 Dose-response curves reflecting proliferation rates for (A) OT-II mice derived CD4+ cells and (B) OT-I mice derived CD8+ cells in response to OVA epitope-decorated GNPs (15 and 40 nm in diameter) and GNRs. % of proliferating cells was calculated from the CD25+CFSElow T-cell subset (for gating strategy, see Fig. S6†). Epitope concentration (nM) presented here is the final epitope concentration in the cell exposure medium. The gray line shows the positive controls, free epitopes. Three independent experiments were conducted for each GNP preparation (N = 3, n = 3), and a representative example is shown here. Error bars indicate standard deviations. Data sets were fitted into a dose–response curve using GraphPad Prism software. | ||
Based on the dose response curves for 40 nm GNP and GNRs, the particle shape seems to significantly affect CD4+ T-cell expansion, with GNRs requiring a substantially lower antigen dose to induce OT-II proliferation. Particle size also exhibited a significant impact as 15 nm GNPs were the least effective carrier system. The difference between the behaviour of R-2 and 40-2 can be explained by the MHC-II pathway employed to present processed antigens back to the plasma membrane. MHC-II loads antigens directly inside lysosomes.43 Following this idea, Rods-2 were expected to outperform 40-2, since the former delivers more antigens into cells (Fig. 3I).
Nonetheless, the CD8+ T-cell response was not that trivial to interpret. Similarly to CD4+ T cells, 15 nm GNPs appeared to be the inferior delivery system to induce CD8+ T-cell response, with the 15 nm GNPs showing a clear shift in dose response compared to GNRs. 40-1 was the only formulation that did not show a typical S-shaped dose – response curve: proliferation of CD8+ T cells was suppressed at higher 40-1 concentrations. This suggests that the MHC-I processing pathway is inhibited at a higher concentration of the 40 nm GNPs. MHC-I is supplied with antigens present in the cytosol.43 Egress of antigens from lysosomes into the cytosol is a complex process that is not yet well understood.44
As we already eliminated the possibility of cytotoxicity at higher 40-1 concentrations (Fig. 2), we next assessed whether the effect of 40-1 on the MHC-I processing pathway was indeed dose-dependent. To test this, BMDCs were exposed for 4 h to the various nanoparticle concentrations and the amount of OVA257–264 presented on MHC-I (H2-Kb) was quantified with flow cytometry. In contrast to 15-1 and R-1, with a decreasing concentration of 40-1, an increase in antigen presentation was detected (Fig. S7 and S8†). Moreover, the level of OVA257–264 presentation achieved with 40-1 was higher than that induced by an equivalent amount of whole OVA protein (Fig. S9†). However, the used antibody against the whole MHC-I complex was clearly more sensitive to processing and presentation of free peptide or amphiphile 1 rather than larger constructs, as they showed clear dose-dependent profiles.
‘Splitting’ the T-cell immune response into two components (cytotoxic and T-helper) highlighted several aspects. There is potentially a size threshold below which BMDCs, and hence T cells, only become activated when a very high antigen loading is applied (Fig. 4). Furthermore, CD4+ T-cell activation (T-helper component) is sensitive to the size or shape of a carrier particle and correlates to cellular uptake and the number of delivered antigens. MHC-I processing, and therefore CD8+ T-cell activation (cytotoxic component), does not seem to be directly linked to the amount of material that is taken up, but is influenced by the size and/or shape of carrier nanoparticles. These aspects should be taken into account when a new vaccine platform is being developed and one type of immune response is preferred over the other.
Effect of size and shape on BMDCs activation
In order to obtain additional details on the performance of GNPs and GNRs, activation of BMDCs was studied. Interleukin 12 (IL-12) and interleukin 1 beta (IL-1β) provide information on activation of DCs by particulate formulations.2,3,19,45,46 For this, BMDCs were exposed to epitope-decorated GNPs and GNRs for 24 hours and the cytokine levels were determined with an enzyme-linked immunosorbent assay (ELISA) (Fig. 5 and S10–S12†). | ||
| Fig. 5 Activation of BMDCs 24 h post exposure to epitope-decorated GNPs and GNRs through cytokine release (A) IL-12 and (B) IL-1β or cellular receptor upregulation of (C) CD80 and (D) CD86. The different nanoparticles were used in the following equivalent concentrations: 15 nm GNPs – equivalent to ∼500 nM epitope; 40 nm GNPs – equivalent to ∼125 nM epitope; GNRs – equivalent to ∼300 nM epitope. For comparison, OVA257-264 was added to study cellular receptor upregulation (200 nM in the cell exposure medium). Significance of the datasets mean values was determined through comparison to the negative control (PBS or supplemented DMEM, * – p-value ≤ 0.05, ** – p-value ≤ 0.01, *** – p-value ≤ 0.001, **** – p-value ≤ 0.0001). For more data points, see Fig. S10–S15.† | ||
IL-12 is a cytokine secreted by activated DCs in response to Pathogen Recognition Receptor stimulation and promotes differentiation of naïve T cells into T helper1 cells and therefore production of IFN-γ.46,47 IL-12 also mediates enhancement of CD8+ cytotoxic T-cell activity. Compared to the PBS negative control, only GNRs showed significant levels of IL-12 secretion (Fig. 5A). All tested concentrations of 15-1 and 15-2 failed to induce IL-12 secretion, which indicates that 15 nm GNPs indeed do not activate BMDCs, although they showed potential in delivering high antigen loads (Fig. 3). On the other hand, 40 nm GNPs in high concentrations had almost no impact, while a low number concentration of 40-2 was as effective as high number concentrations of GNRs. GNRs showed a concentration-dependent effect on IL-12 secretion. Additionally, neither of the employed peptide amphiphiles induced IL-12 secretion when administered without the nanoparticles (Fig. S11†). Therefore, our peptide amphiphiles cannot be considered to activate BMDCs, which is the case for other peptide amphiphiles.45 Previously, protein-coated GNPs (40 nm in diameter) and gold nanocubes (40 by 40 nm) were reported to induce IL-12 secretion, but not smaller GNPs (20 nm in diameter) or GNRs.2 Our data therefore supports the assertion that small GNPs do not induce IL-12 secretion, but contradicts the previous report as GNRs are capable of inducing IL-12 secretion in this study.
Additionally, the capacity of epitope-decorated GNPs and GNRs to induce inflammation was studied through IL-1β secretion. None of the tested particles induced significantly elevated levels of IL-1β compared to the negative control (PBS), indicating that the observed activation of BMDCs did not follow an inflammasome-mediated pathway (Fig. 5B and S12†). These findings are in agreement with previous studies using spherical GNPs: irrespective of the GNP size or coating, they did not induce inflammasome formation.2,19 There is no consensus between the results presented here and published data on GNRs. Depending on the coating, GNRs were reported to cause significant levels of IL-1β secretion. For example, this was observed for GNRs coated with the West Nile virus envelope protein, while spherical and cubic GNPs did not induce IL-1β secretion.2 GNRs coated with recombinant Sm29 proteins derived from Schistosoma mansoni also induced high levels of IL-1β, however CTAB-stabilised or cysteamine-stabilised GNRs did not.48 Furthermore, PEGylated GNRs also did not induce IL-1β.28
Next, upregulation in cellular markers of BMDCs under treatment with OVA257–264-decorated particles was studied. Both CD80 and CD86 are necessary for T-cell activation and survival through co-stimulation with CD28 expressed on T cells.47,49 Both receptors are already upregulated on BMDCs due to their handling during the culture procedure, which is reflected in the negative control, DMEM (Fig. S13–S15†). With respect to the negative control, only 40-1 showed an elevated CD80 level (Fig. 5C and S14†). CD86 was more readily activated by 40-1 and less by Rods-1, but not by 15-1 or free OVA257-264 (Fig. 5D and S15†). Enhancement provided by 40-1 was almost double compared to the effect of Rods-1, and showed a dose dependence. This may be explained by the fact that for this assay we used 50 × 103 cells versus previously used 20 × 103 cells. This assay helped to clarify the effect of 40 nm GNPs on BMDCs activation: most likely, suppressed secretion of IL-12 (Fig. 5A) and CD8+ T-cell proliferation (Fig. 4A) at higher particle concentrations could be attributed to BMDCs overstimulation. Moreover, when BMDCs were exposed to the whole OVA protein, no upregulation in CD80 or CD86 was detected (Fig. S17 and S18†). The same was true for peptide amphiphiles 1, 2, and base. This way, the observed effect of 40-1 can be attributed to the presence of GNPs.
The results of CD80 and CD86 activation of BMDCs are consistent with previously reported data: 33 nm GNPs coated with the whole OVA protein were shown to induce greater upregulation in both CD80 and CD86 expression than 10 or 22 nm GNPs coated with OVA.17 Additionally, antigen-coated GNRs were able to induce simultaneous upregulation of CD86, MHC-I, and MHC-II.48
Thus, despite the potential of 15 nm GNPs to deliver higher doses of antigens than 40 nm GNP and GNR counterparts, they failed to activate BMDCs. GNRs were capable of delivering significant antigen loads and caused concentration-dependent activation of BMDCs. 40 nm GNPs were the only particles capable of inducing significant upregulation in BMDCs activation markers. None of the tested particles caused inflammation. These results are in agreement with previously published studies for GNPs, however contradict published data for GNRs.
In summary, our study showed that peptide-gold nanoconjugates comprising a gold core of differing shape and size indeed have an impact on BMDC activation and subsequent T-cell expansion. “Splitting” the two T-cell responses showed two different outcomes for two different pathways: there was no single common shape or size that would be unconditionally beneficial for both T-cell responses.
Conclusions
Peptide-gold nanoconjugates comprising a gold core of differing shape and size (15 and 40 nm in diameter, and rods of 40 by 15 nm) and a peptide shell did not cause cytotoxicity in BMDCs. Cellular uptake of GNPs and GNRs by BMDCs was governed by the gold core parameters, irrespective of the displayed antigen. Core particle parameters such as shape and size indeed have an impact on BMDC activation and subsequent T-cell expansion. Size seemed to play a major role, as the 40 nm GNP showed superior performance in respect to the 15 nm GNPs in every aspect. The shape of GNPs had an effect on the CD8+ T-cell response with the 40 nm GNPs being significantly more effective than the GNRs, despite a lower epitope dose delivered. However, these two samples showed very similar CD4+ T-cell responses. The differences in immune responses were not governed by the total NP uptake but by the intracellular routing of the particles (MHC-I or MHC-II pathway).Experimental
Materials
All chemicals were purchased from Sigma-Aldrich unless stated otherwise. CTAB (≥99%, H6269) and HAuCl4 × 3H2O (49% Au basis, G4022) were purchased from Sigma-Aldrich. Silver nitrate (99.999%) and Oxyma Pure were purchased from Carl Roth GmbH. TFA, piperidine, DMF, DCM, methanol, and acetonitrile were purchased from Biosolve. TEM grids (Formvar/Carbon, 200 mesh, on copper support) were purchased from Electron Microscopy Sciences. FCS, penicillin/streptomycin mixture, L-glutamate, DMEM and RPMI 1640 cell culture media were purchased from Lonza. Mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) was purchased from PeproTech. CD4+ and CD8+ isolation kits (via MACS magnetic separation) were purchased from Miltenyi Biotech. LDH leakage assay (LDH-Cytox™ Assay Kit) and the specific antibody against the whole MHC-I/OVA257–264 complex (PE anti-mouse H-2Kb bound to OVA257–264 antibody, Kb-OVA257–264) were purchased from Biolegend. ELISA standards (IL-12 and IL-1β) and antibodies, TMB substrate, as well as immunostaining antibodies were purchased from eBioscience.Gold nanoparticles synthesis
Spherical particles (GNPs) of 15 and 40 nm in diameter were synthesised via citrate reduction and overgrowth as described elsewhere.30 The average size of the GNPs in suspension was analysed using dynamic light scattering (DLS, Table 1). Size parameters of the obtained GNPs were also determined by transmission electron microscopy (TEM) using ImageJ software (Fiji): mean diameters were 15.0 ± 1.4 nm (n = 185) and 43.5 ± 3.3 nm (n = 220). The mass of one nanoparticle was calculated to be 34.1 and 832.2 ag, respectively; and the surface areas were determined to be 707 and 5945 nm2.20,50Gold nanorods (GNRs) were synthesised via the two-step seed-mediated approach.31 2–3 nm seeds were prepared by mixing CTAB (5 mL, 0.20 M), and HAuCl4 (5 mL, 0.50 mM) with ice-cold NaBH4 (0.60 mL, 10 mM) whilst intensely stirring at room temperature. After 2 min of vigorous stirring, the solution was left undisturbed at room temperature for 2 hours. For the overgrowth, solutions of HAuCl4 (50 mL, 1.0 mM) and AgNO3 (200 μL, 100 mM) were gently mixed with CTAB (50 mL, 0.20 M) at room temperature. After 2 min of stirring, ascorbic acid (550 μL, 100 mM) was added. Next, 120 μL of the seed solution was added under vigorous stirring. After 6 hours the rods were washed with MilliQ to remove the excess CTAB. The average dimensions determined using TEM were 44.0 (±4.0) by 14.8 (±1.9) nm (n = 168). The mass of one nanorod was calculated to be 129.8 ag, and the surface area – 2046 nm2.
Peptide and peptide amphiphile synthesis
Peptides and peptide amphiphiles were synthesised by solid-phase peptide synthesis using standard Fmoc-chemistry protocols. The synthesis was performed on an automated microwave peptide synthesiser, Liberty Blue (CEM), using standard settings. All peptides and amphiphiles were synthesised on a Wang resin preloaded with the corresponding C-terminal residue. 20% piperidine in DMF was employed as the deprotection solution, and N,N′-diisoprorylcarbodiimide (DIC)/OxymaPure (activator/activator base) were used to activate Fmoc-protected α-amino acids. 11-mercaptoundecanoic acid was coupled to the peptide N-terminus using the same protocol as for amino acid coupling but for a longer time (10 min coupling instead of 4 min). The terminal thiol of the acyl chains was blocked with 2,2′-Dithiobis(5-nitropyridine) (DTNP, 1 eq. in DMF, 3 hours). The molecules were cleaved from the resin using a cleavage cocktail comprising 2.5% triisopropylsilane (TIS), 1.5% deionized water, 2.5% phenol in trifluoroacetic acid (TFA, RT, 2 hours). The crude peptide compounds were precipitated into cold diethyl ether, pelleted by centrifugation, redissolved in water, and lyophilised prior to purification. HPLC purification was performed on a Shimadzu system equipped with two LC-20AR pumps, an SPD-20A UV-Vis detector and a Phenomenex Kinetex EVO C18 column (21.2 by 150 mm) with water and acetonitrile as mobile phases. 0.1% TFA was added to the mobile phases, except for purification of the base sequence where 0.1% NH3 was added. Purity of the compounds was confirmed with LC-MS (Fig. S18–S23†). The purified molecules were lyophilised and stored at −20 °C.Preparation and characterisation of peptide amphiphile-coated GNPs and GNRs
Coating of the GNPs was performed via the ligand exchange procedure as described in detail elsewhere.30 Briefly, the desired coating molecules were dissolved in DMSO, mixed at a 9:1 molar ratio (base:1 or base:2), and added to a stirred GNP suspension. The peptide amphiphile molar excess over the GNPs was at least 200000. The final concentration of DMSO in the mixture was 50% (v/v). After a 1 hour incubation the samples were centrifuged to pellet the modified GNPs and the supernatants were discarded and replaced with 25% DMSO (v/v). Samples were incubated overnight and after a final centrifugation cycle, the GNPs were dispersed in 5% DMSO. Size exclusion chromatography (SEC, NAP-25 columns, GE Healthcare) was performed with PBS as eluent to remove free ligands and DMSO.
Particle size distributions, determined by dynamic light scattering (DLS), and zeta-potential before and after modification were obtained using a Zetasizer Nano-7 S (Malvern Instruments). The average of three separate measurements was reported. Transmission electron microscopy (TEM) measurements were performed on a JEM 1400 Plus (JEOL) microscope, with an accelerating voltage of 80 kV and with a CDD camera. Sample preparation was as follows: a 10 μL droplet of the sample was deposited on a carbon-coated copper grid for 20 min. The excess sample was removed by blotting with a fibreless tissue. The grid was washed with MilliQ water once and blotted again. 1% (w/v) uranyl acetate staining was applied, followed by immediate blotting and air drying. UV-vis spectra were recorded in the 900–350 nm range using a Cary 300 UV-vis spectrometer (Agilent). Molar concentrations of GNPs and GNRs were determined by UV-vis using extinction coefficients published by Haiss et al.51 for spherical particles, and by Orendorff et al.52 for rod-like particles.
Determination of number concentrations of GNPs and GNRs
Gold (Au) mass quantification was conducted using inductively coupled plasma mass spectrometry (ICP-MS) on a PerkinElmer NexION 2000 equipped with a quartz nebulizer and a glass cyclonic spray chamber operating in standard mode (1600 W radio frequency power; 18 mL min−1 plasma gas flow; 1.2 L min−1 nebulizer gas flow; and 1.12 L min−1 auxiliary flow). The peptide amphiphile-coated GNPs or GNRs (in suspension, optical density (OD) ∼0.3–0.5) were mixed 1:1 (v/v) with aqua regia (HNO3:HCl = 1:3) and were subjected to digestion at 70 °C for 1 hour. Mixtures were diluted 25× times with MilliQ water and directly measured for 197Au isotope content. Knowing the optical densities of the suspensions before digestion with aqua regia, the mass of digested gold, and the calculated mass of one nanoparticle, it was possible to link the OD of GNP or GNR suspensions with their number concentrations. Deviations in the determined gold concentration between the samples with the same core were not significant (Fig. S2†), and could be attributed to incomplete nanoparticle digestion and analysis error, which can lead to deviations of up to 10%.20
Determination of antigen loading displayed onto GNPs and GNRs
Antigen loading was expressed as shown in eqn (1):| AL = dW × SNP × NNP | (1) |
Coverage density dW was determined using UV-vis spectroscopy. Briefly, base and W peptide amphiphiles were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, 1.0 mg mL−1) and mixed at a molar ratio of 9:1. This mixture was diluted with HFIP to obtain a final concentration of base of 0.50 mg mL−1. Next, it was added to the GNPs and GNRs: for every 0.3 mL of the peptide amphiphile mixture, 0.5 mL of a citrate- or CTAB-protected nanoparticle suspension was taken (OD519 = 4.5 for 15 nm GNPs, OD530 = 2.5 for 40 nm GNPs, and OD756 = 3.2 for GNRs). After a 1 hour incubation with stirring, the nanoparticles were pelleted via centrifugation. The pellets were rehydrated with MilliQ water and lyophilised. The dry residue was redissolved in HFIP and nanoparticles were pelleted again. Both supernatants were analysed to determine concentrations (ε280(W) = 5600 M−1cm−1). Knowing the molar concentrations of GNPs and GNRs and their surface areas, as well as added concentrations of W, the following dW values were calculated: 0.58 peptide per nm2 for 15 nm GNPs; 0.35 peptide per nm2 for 40 nm GNPs; and 0.46 peptide per nm2 for GNRs. Using a simple proportion based on the molecular weight of OVA257–264, and OVA323–339, and particle number concentrations obtained using ICP-MS, the coverage densities were recalculated for epitope loading.
Animals
C57Bl/6, OT-I, and OT-II transgenic mice were purchased from Jackson Laboratory (CA, USA). The animals were bred in-house in agreement with standard laboratory conditions, and provided with water and food ad libitum. The animals were treated in compliance with the Dutch government guidelines and the Directive 2010/63/EU of the European Parliament. Experiments were approved by the Ethics Committee for Animal Experiments of Leiden University.Bone marrow-derived dendritic cells (BMDCs)
Bone marrow was extracted from tibias, femurs, and thigh bones of C57BL/6 or OT-I/OT-II mice. A single-cell suspension of bone marrow cells was prepared using a 70 μm cell trainer (Greiner Bio-One B.V.). The cell culture medium used for the maturation was DMEM supplemented with 2 mM L-glutamine, 5% (v/v) FCS, 100 U mL−1 penicillin/streptomycin. Bone marrow cells were seeded in 95 mm Petri dishes at a density of 8–10 × 106 cells per dish (Greiner Bio-One B.V.) and incubated at 37 °C and 5% CO2 for 10 days. Medium was refreshed four times during the 10 day period, 20 ng mL−1 GM-CSF was added to the medium to induce and maintain the maturation of progenitor cells into BMDCs.LDH leakage assay
LDH leakage assay was performed to determine cytotoxicity of the peptide–gold conjugates in BMDCs. For this assay, BMDCs were cultured as described above. After 10 days of culturing, BMDCs (20 × 103 per well) were plated in 96-well plates (Greiner Bio-One B.V.) and were exposed to GNPs and GNRs (50 μL per well, 200 μL total well volume) for 4 and 24 hours at 37 °C (5% CO2). A negative control (50 μL of PBS) and a positive control (50 μL of the assay lysis buffer) were added to every plate. After the exposure, the plates were centrifuged (1500 rpm, 5 min) and 30 μL of the supernatants were taken for analysis which was performed according to the manufacturer's manual (non-homogenous assay protocol). Cytotoxicity was expressed as the difference between the samples and negative and positive controls (eqn (2)):
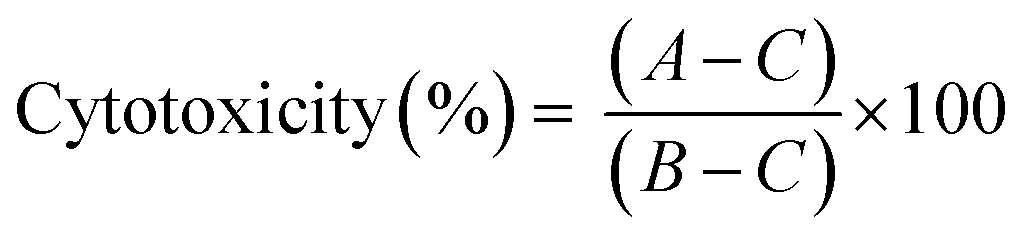 | (2) |
GNPs and GNRs uptake by BMDCs
This step was duplicated for two independent experiments: measuring cellular uptake of GNPs and GNRs via direct detection of the gold content per cell (ICP-MS assisted) and visualisation of subcellular localisation with TEM analysis. Samples analysed included 15-1, 15-2, 40-1, 40-2, Rods-1, and Rods-2. Mature DCs were seeded at a density of 1 × 106 cell per well in 6-well plates (Greiner Bio-One B.V.). 250 μL of sample (15 nm GNPs – equivalent to ≈500 nM epitope; 40 nm GNPs – equivalent to ≈125 nM epitope; GNRs – equivalent to ≈300 nM epitope in the cell exposure medium) were diluted 2× times with DMEM and added to the wells (3 mL total volume). The cells and the samples were incubated for 4 hours at 37 °C (5% CO2). Next, the cells were washed with PBS (pH 7.2) to remove the non-associated nanoparticles.For the cell visualisation experiment, a coverslip (Thermanox, 24 mm in diameter, Nunc) was added to each well. The cells were fixed with 2% (v/v) glutaraldehyde, 2% formaldehyde in sodium cacodylate buffer (0.1 M, pH 7.2, 2 hours at room temperature). After fixation, 1% (w/v) OsO4 with addition of potassium ferrocyanide (0.8%) was applied for 1 hour at room temperature. Next, the cells were dehydrated through ethanol graded series and embedded into Agar 100 resin (Agar Scientific, AGR1043). Thin sections (70 nm) were cut from the resin using a Leica Ultramicrotome equipped with a diamond knife. The images were acquired on a JEM 1400 Plus microscope, fitted with a CCD camera, at an accelerating voltage of 80 kV. The vesicles dimensions were determined using Fiji ImageJ software. Since vesicles are not completely spherical, each vesicle was measured across its longest and shortest axes, and a mean value was reported in Fig. S5.†
ICP-MS was also used to determine the average gold content per cell. After 4 hours of incubation time, the BMDCs were washed with PBS (pH 7.2, 3× times), detached with a cell scraper (Sarstedt) and counted. The cells were lysed with aqua regia (1 mL cell sample volume, 1:1 dilution, 1 hour at 70 °C). Lysates were diluted 25× times with MilliQ water and directly measured in triplicate using the method described above. Since both the cell numbers and total gold content were known, the average gold content per cell could be calculated (Fig. 3G–I).
OT-I and OT-II experiments
BMDCs were cultured as described above for 10 days, and pulsed for 4 hours with GNPs, GNRs or controls (PBS or free epitopes) followed by complete removal of the particles from the cell culture. Spleens were removed from OT-I or OT-II mice and strained through a 70 μm cell strainer to yield a single-cell suspension. Red blood cells (erythrocytes) were lysed with Ammonium-Chloride-Potassium (ACK) lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA; pH 7.3). CD8+ or CD4+ cells were isolated via magnetic separation using a CD8+ or CD4+ T cell isolation kit according to the supplier's protocol. Enriched T-cell fractions were stained with CFSE-FITC and then added to the plates with BMDCs in the ratio of 3:1 = specific T cells:BMDCs in complete RPMI 1640 medium (supplemented with 2 mM glutamine, 5% FCS, 100 U/mL penicillin/streptomycin, and 50 μM β-mercaptoethanol). T cells were left to proliferate for 72 hours in the same plates (37 °C, 5% CO2). Cells were immunostained for Thy1.2, CD4 or CD8, CD25, as well as for viability, and analysed by flow cytometry on a CytoFLEX S (Beckman Coulter). Data analysis was done using the FlowJo software (Treestar). Each experiment was repeated three times (N = 3, n = 3), and a representative example is shown in Fig. 4. The gating strategy can be found in Fig. S6.† The proliferation rate was expressed through the CD25+CFSE-FITClow cell subset. GraphPad Prism version 7.00 for Windows (GraphPad Software) was used to analyse the data and to plot the dose–response curves.
For the analysis of antigen processing and presentation by BMDCs immunostaining for the full MHC-I/OVA257–264 complex, Kb-OVA257–264, was used. Mature BMDCs (50 × 105 cell per well in 96-well plates) were exposed to GNPs and GNRs for 4 hours. Cell culture supernatants were removed and half of the wells were analysed directly, and the other half was supplied with fresh medium and analysed after 24 hours. DMEM was used as a negative control, and free OVA257-264 – as positive one. Flow cytometry was performed on a CytoFLEX S. Data analysis was done using FlowJo software. The gating strategy can be found in Fig. S7.† The data shown is a representative example of two independent experiments (N = 2, n = 3).
Activation of BMDCs
Activation of BMDCs was monitored through cytokine release and upregulation in expression of cellular receptors. Mature BMDCs (20 × 105 cell per well in 96-well plates) were treated with GNPs and GNRs for 24 hours in the same manner as above for the LDH assay. The cell culture supernatants were checked for IL-12 and IL-1β levels. A ‘sandwich’ ELISA was performed according to a standard protocol at room temperature and involved the following steps: (1) coating the 96-well plates (half area, Costar) with capture antibodies (eBioscience, overnight at 4 °C); (2) blocking the wells from non-specific interactions by incubation with BSA (2.5% w/v in PBS, 1 hour); (3) incubation (2 hours) with the supernatant of GNP-exposed cells, PBS-treated samples, IL-12 standard at different concentrations, and blank wells (blocking buffer); (4) addition of detection antibodies and complexation with SAv-HRP (1 hour). The readout was based on the reaction of the HRP conjugate with an TMB substrate (BD Bioscience). The reaction was stopped with 1 N H2SO4 when the blue color in the well became intense. Absorbance at 570 nm was subtracted from absorbance at 450 nm (recorded using a microplate reader Infinite 200 PRO Tecan). Samples from three independent experiments were analysed (N = 3, n = 3). For results see Fig. 5A, B and S10–S12.†Next, mature BMDCs (50 × 105 cell per well in 96-well plates) were exposed to GNPs and GNRs for 4 hours. Cell culture supernatants were removed and half of the wells were analysed directly, and the other half were supplied with fresh medium and analysed after 24 hours. The procedure included immunostaining for CD80, CD86, as well as viability. PBS was used for negative control, and Lipopolysaccharides from Escherichia coli (LPS, Sigma-Aldrich) were used for positive control. Flow cytometry was performed on a CytoFLEX S. Data analysis was done using FlowJo software. The gating strategy can be found in Fig. S13.† The data shown is a representative example out of two independent experiments (N = 2, n = 3).
Author contributions
Conceptualization, E. A. E., B. S., and A. K.; methodology, E. A. E., G. E. M. L., F. A. M., A. L. B., and B. S.; investigation, E. A. E., G. E. M. L., and F. A. M.; formal analysis, E. A. E., F. A. M., and B. S.; writing – original draft, E. A. E., A. L. B., B. S., and A. K.; writing – review and editing, E. A. E., G. E. M. L., F. A. M., A. L. B., B. S., and A. K.; funding acquisition, E. A. E., B. S., A. K.; resources, B. S., A. K.; supervision, A. L. B., B. S., and A. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. A. E. acknowledges the financial support of the Global Education Scholarship Program (Russia). B. S. acknowledges funding by Health-Holland, the Dutch Cooperation of Health Foundations (SGF) and the Dutch Heart Foundation, grant numbers LSHM18056-SGF, ERA-CVD2018T092.Notes and references
- A. Y. Lin, J. Lunsford, A. S. Bear, J. K. Young, P. Eckels, L. Luo, A. E. Foster and R. A. Drezek, Nanoscale Res. Lett., 2013, 8, 72 CrossRef PubMed.
- K. Niikura, T. Matsunaga, T. Suzuki, S. Kobayashi, H. Yamaguchi, Y. Orba, A. Kawaguchi, H. Hasegawa, K. Kajino, T. Ninomiya, K. Ijiro and H. Sawa, ACS Nano, 2013, 7, 3926–3938 CrossRef CAS PubMed.
- N. Climent, I. García, M. Marradi, F. Chiodo, L. Miralles, M. J. Maleno, J. M. Gatell, F. García, S. Penadés and M. Plana, Nanomedicine, 2018, 14, 339–351 CrossRef CAS PubMed.
- C. D. Spicer, C. Jumeaux, B. Gupta and M. M. Stevens, Chem. Soc. Rev., 2018, 47, 3574–3620 RSC.
- P. L. Chariou, O. A. Ortega-Rivera and N. F. Steinmetz, ACS Nano, 2020, 14, 2678–2701 CrossRef CAS PubMed.
- N. Benne, J. van Duijn, J. Kuiper, W. Jiskoot and B. Slütter, J. Controlled Release, 2016, 234, 124–134 CrossRef CAS PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13, 339 CrossRef PubMed.
- B. Rathore, K. Sunwoo, P. Jangili, J. Kim, J. H. Kim, M. Huang, J. Xiong, A. Sharma, Z. Yang, J. Qu and J. S. Kim, Biomaterials, 2019, 211, 25–47 CrossRef CAS PubMed.
- T. Fifis, A. Gamvrellis, B. Crimeen-Irwin, G. A. Pietersz, J. Li, P. L. Mottram, I. F. C. McKenzie and M. Plebanski, J. Immunol., 2004, 173, 3148–3154 CrossRef CAS PubMed.
- L. Xu, X. Wang, W. Wang, M. Sun, W. J. Choi, J.-Y. Kim, C. Hao, S. Li, A. Qu, M. Lu, X. Wu, F. M. Colombari, W. R. Gomes, A. L. Blanco, A. F. de Moura, X. Guo, H. Kuang, N. A. Kotov and C. Xu, Nature, 2022, 601, 366–373 CrossRef CAS PubMed.
- F. Campbell, F. L. Bos, S. Sieber, G. Arias-Alpizar, B. E. Koch, J. Huwyler, A. Kros and J. Bussmann, ACS Nano, 2018, 12, 2138–2150 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- G. Sonavane, K. Tomoda and K. Makino, Colloids Surf., B, 2008, 66, 274–280 CrossRef CAS PubMed.
- Q. Zhang, V. M. Hitchins, A. M. Schrand, S. M. Hussain and P. L. Goering, Nanotoxicology, 2011, 5, 284–295 CrossRef CAS PubMed.
- A. M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS PubMed.
- W. H. de Jong, W. I. Hagens, P. Krystek, M. C. Burger, A. J. A. M. Sips and R. E. Geertsma, Biomaterials, 2008, 29, 1912–1919 CrossRef CAS PubMed.
- S. Kang, S. Ahn, J. Lee, J. Y. Kim, M. Choi, V. Gujrati, H. Kim, J. Kim, E.-C. Shin and S. Jon, J. Controlled Release, 2017, 256, 56–67 CrossRef CAS PubMed.
- X. Xie, J. Liao, X. Shao, Q. Li and Y. Lin, Sci. Rep., 2017, 7, 3827 CrossRef PubMed.
- K. Fytianos, L. Rodriguez-Lorenzo, M. J. D. Clift, F. Blank, D. Vanhecke, C. von Garnier, A. Petri-Fink and B. Rothen-Rutishauser, Nanomedicine, 2015, 11, 633–644 CrossRef CAS PubMed.
- F. Abdolahpur Monikh, B. Fryer, D. Arenas-Lago, M. G. Vijver, G. Krishna Darbha, E. Valsami-Jones and W. J. G. M. Peijnenburg, Environ. Sci. Technol. Lett., 2019, 6, 732–738 CrossRef CAS.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- L. Ding, C. Yao, X. Yin, C. Li, Y. Huang, M. Wu, B. Wang, X. Guo, Y. Wang and M. Wu, Small, 2018, 14, 1801451 CrossRef PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- M. Rabe, C. Aisenbrey, K. Pluhackova, V. de Wert, A. L. Boyle, D. F. Bruggeman, S. A. Kirsch, R. A. Böckmann, A. Kros, J. Raap and B. Bechinger, Biophys. J., 2016, 111, 2162–2175 CrossRef CAS.
- M. Bhamidipati and L. Fabris, Bioconjugate Chem., 2017, 28, 449–460 CrossRef CAS PubMed.
- C. L. Villiers, H. Freitas, R. Couderc, M.-B. Villiers and P. N. Marche, J. Nanopart. Res., 2010, 12, 55–60 CrossRef CAS PubMed.
- S. Hočevar, A. Milošević, L. Rodriguez-Lorenzo, L. Ackermann-Hirschi, I. Mottas, A. Petri-Fink, B. Rothen-Rutishauser, C. Bourquin and M. J. D. Clift, ACS Nano, 2019, 13, 6790–6800 CrossRef PubMed.
- C. Grabinski, N. Schaeublin, A. Wijaya, H. D'Couto, S. H. Baxamusa, K. Hamad-Schifferli and S. M. Hussain, ACS Nano, 2011, 5, 2870–2879 CrossRef CAS PubMed.
- B. Halamoda-Kenzaoui, M. Ceridono, P. Urbán, A. Bogni, J. Ponti, S. Gioria and A. Kinsner-Ovaskainen, J. Nanobiotechnol., 2017, 15, 48 CrossRef PubMed.
- E. A. Egorova, M. M. J. van Rijt, N. Sommerdijk, G. S. Gooris, J. A. Bouwstra, A. L. Boyle and A. Kros, ACS Nano, 2020, 14, 5874–5886 CrossRef CAS.
- E. Egorova, G. Arias Alpizar, R. Vlieg, G. S. Gooris, J. Bouwstra, J. van noort, A. Kros and A. L. Boyle, J. Mater. Chem. B, 2022, 10, 1612–1622 RSC.
- R. Lévy, N. T. K. Thanh, R. C. Doty, I. Hussain, R. J. Nichols, D. J. Schiffrin, M. Brust and D. G. Fernig, J. Am. Chem. Soc., 2004, 126, 10076–10084 CrossRef.
- H. Andresen, M. Mager, M. Grießner, P. Charchar, N. Todorova, N. Bell, G. Theocharidis, S. Bertazzo, I. Yarovsky and M. M. Stevens, Chem. Mater., 2014, 26, 4696–4704 CrossRef CAS.
- Z. Wang, R. Lévy, D. G. Fernig and M. Brust, Bioconjugate Chem., 2005, 16, 497–500 CrossRef CAS PubMed.
- C. Zhao, L. Qiu, P. Lv, A. Han, G. Fang, J. Liu and S. Wang, Analyst, 2019, 144, 1275–1281 RSC.
- K. M. Murphy, A. B. Heimberger and D. Y. Loh, Science, 1990, 250, 1720–1723 CrossRef CAS PubMed.
- P. M. R. Paulo, P. Zijlstra, M. Orrit, E. Garcia-Fernandez, T. C. S. Pace, A. S. Viana and S. M. B. Costa, Langmuir, 2017, 33, 6503–6510 CrossRef CAS PubMed.
- B. D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS PubMed.
- H. Hinterwirth, S. Kappel, T. Waitz, T. Prohaska, W. Lindner and M. Lämmerhofer, ACS Nano, 2013, 7, 1129–1136 CrossRef CAS PubMed.
- S. Huo, H. Ma, K. Huang, J. Liu, T. Wei, S. Jin, J. Zhang, S. He and X.-J. Liang, Cancer Res., 2013, 73, 319–330 CrossRef CAS PubMed.
- H. G. Hagglund, P. A. McSweeney, G. Mathioudakis, B. Bruno, G. E. Georges, M. J. Gass, P. Moore, G. E. Sale, R. Storb and R. A. Nash, Transplantation, 2000, 70, 1437–1442 CrossRef CAS PubMed.
- I. Mäger, K. Langel, T. Lehto, E. Eiríksdóttir and Ü. Langel, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 502–511 CrossRef PubMed.
- E. S. Trombetta and I. Mellman, Annu. Rev. Immunol., 2005, 23, 975–1028 CrossRef CAS.
- L. Cohn and L. Delamarre, Front. Immunol., 2014, 5, 255 Search PubMed.
- I. W. Hamley, Bioconjugate Chem., 2021, 32, 1472–1490 CrossRef CAS PubMed.
- P. Kalinski, C. M. Hilkens, A. Snijders, F. G. Snijdewint and M. L. Kapsenberg, J. Immunol., 1997, 159, 28–35 CAS.
- A. M. Didierlaurent, C. Collignon, P. Bourguignon, S. Wouters, K. Fierens, M. Fochesato, N. Dendouga, C. Langlet, B. Malissen, B. N. Lambrecht, N. Garçon, M. van Mechelen and S. Morel, J. Immunol., 2014, 193, 1920–1930 CrossRef CAS.
- N. R. G. Assis, A. J. Caires, B. C. Figueiredo, S. B. Morais, F. S. Mambelli, F. v. Marinho, L. O. Ladeira and S. C. Oliveira, J. Controlled Release, 2018, 275, 40–52 CrossRef CAS PubMed.
- T. R. Mempel, S. E. Henrickson and U. H. von Andrian, Nature, 2004, 427, 154–159 CrossRef CAS PubMed.
- B. C. Mei, E. Oh, K. Susumu, D. Farrell, T. J. Mountziaris and H. Mattoussi, Langmuir, 2009, 25, 10604–10611 CrossRef CAS PubMed.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990–3994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03027f |
| This journal is © The Royal Society of Chemistry 2022 |