
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of hybrid compounds as novel drugs and medicines
Abdulaziz H. Alkhzem
,
Timothy J. Woodman
and
Ian S. Blagbrough
 *
*
Department of Pharmacy and Pharmacology, University of Bath, Bath BA2 7AY, UK. E-mail: prsisb@bath.ac.uk
First published on 6th July 2022
Abstract
The development of highly effective conjugate chemistry approaches is a way to improve the quality of drugs and of medicines. The aim of this paper is to highlight and review such hybrid compounds and the strategies underpinning their design. A variety of unique hybrid compounds provide an excellent toolkit for novel biological activity, e.g. anticancer and non-viral gene therapy (NVGT), and as templates for killing bacteria and preventing antibiotic drug resistance. First we discuss the anticancer potential of hybrid compounds, containing daunorubicin, benzyl- or tetrahydroisoquinoline-coumarin, and cytotoxic NSAID-pyrrolizidine/indolizine hybrids, then NVGT cationic lipid-based delivery agents, where steroids or long chain fatty acids as the lipid moiety are bound to polyamines as the cationic moiety. These polyamines can be linear as in spermidine or spermine, or on a polycyclic sugar template, aminoglycosides kanamycin and neomycin B, the latter substituted with six amino groups. They are highly efficient for the delivery of both fluorescent DNA and siRNA. Molecular precedents can be found for the design of hybrid compounds in the natural world, e.g., squalamine, the first representative of a previously unknown class of natural antibiotics of animal origin. These polyamine-bile acid (e.g. cholic acid type) conjugates display many exciting biological activities with the bile acids acting as a lipidic region and spermidine as the polycationic region. Analogues of squalamine can act as vectors in NVGT. Their natural role is as antibiotics. Novel antibacterial materials are urgently needed as recalcitrant bacterial infection is a worldwide problem for human health. Ribosome inhibitors founded upon dimers of tobramycin or neomycin, bound as ethers by a 1,6-hexyl linker or a more complex diether-disulfide linker, improved upon the antibiotic activity of aminoglycoside monomers by 20- to 1200-fold. Other hybrids, linked by click chemistry, conjugated ciprofloxacin to neomycin, trimethoprim, or tedizolid, which is now in clinical trials.
 Abdulaziz H. Alkhzem | Abdulaziz H. Alkhzem was born in August 1991, Makkah, Saudi Arabia. He graduated with a BSc in Pharmacy in 2015 from Umm Al-Qura University in Makkah, Saudi Arabia. He then received an MPhil from the University of Bath under the supervision of Dr I S Blagbrough and Dr T J Woodman on the analysis of clinically important aminoglycoside antibiotics, in particular measuring their individual pKa values by 1H, 13C and 15N NMR spectroscopy. He has recently submitted his PhD Thesis on the topic of this review with a particular focus on polyamine medicinal chemistry. |
 Timothy J. Woodman | Dr Timothy J. Woodman graduated with a BSc in Chemistry from the University of Warwick in 1994, then completed a PhD at the same institution in 1998. Following a Royal Society Fellowship to Auckland, NZ under the supervision of Professor Warren Roper, he joined the research group of Professor Manfred Bochmann as a Postdoctoral Research Fellow, first at the University of Leeds and then at the University of East Anglia. He was appointed as NMR Spectroscopist at the University of Bath in 2005, and became a Senior NMR Spectroscopist in 2011. His current research interests combine NMR and the synthesis and biological activity of capsaicinoids, molecules related to the biologically active components of chillies. |
 Ian S. Blagbrough | Dr Ian S. Blagbrough is a chemist with a keen interest in many aspects of natural products and pharmaceutical analysis. He graduated in chemistry in 1980 and 1983 from the University of Nottingham and his PhD supervisor was Prof Gerry Pattenden FRS. This was followed by an NIH Post-Doctoral Research Fellowship at Texas A&M University with Prof A. Ian Scott FRS and Prof Neil E. MacKenzie (1983–85). At the University of Bath, he has maintained an active research group of 6 PhD students for over 20 years where his research interests are on Delphinium and Aconitum norditerpenoid alkaloids, on spider and wasp polyamine amides, on spermine and spermidine conjugates for cation channel block, as anticancer cytotoxics, for non-viral gene therapy, and as vectors for siRNA delivery. His three children having all graduated, he spends more time with his four grandchildren. |
Introduction
In recent decades, there has been a general decrease in the registration of new medications for treating pathologies. Some therapeutic areas have suffered as a result of this decrease, including the treatment of infectious diseases, neurodegenerative diseases, and cancers that have a huge economic impact. This contrasts with the growth of technologies and science aimed to improve human quality of life.1 Some of the reappearing diseases linked to resistant microorganisms such as malaria, novel viruses (as now need no introduction), tropical diseases, and autoimmune diseases still present significant challenges to the pharmaceutical sector.1 These challenging conditions call for further ongoing effort in the development of new treatment strategies that are more selective, efficient, and economically acceptable. The potential of novel hybrid compounds over more traditional combinations of medicines is discussed.Combination therapy
Combination therapy has been used against bacteria for at least 40 years. It is common for clinicians to prescribe two or more antibiotics at the same time with the aim of ensuring that bacterial pathogens and potential routes for resistance developing are covered.2 For instance, Augmentin is a clinically useful broad spectrum antibiotic combination of amoxicillin augmented by clavulanic acid.3 Clavulanic acid is a β-lactamase inhibitor that acts together with the β-lactam amoxicillin to stop the proliferation of bacteria. β-Lactamase inhibitors, including clavulanic acid, prevent such β-lactam-hydrolysing enzymes from functioning.4 Antibiotic-adjuvant combination is the name given to this combination. Antibiotic–antibiotic therapy is another type of combination of drugs. Here the aim is to achieve synergies between the different drug components in a manner that boosts treatment efficacy and affects several pathogens, especially working together to overcome the development of bacterial resistance in some strains to some antibiotics.5,6Even though some successes have been scored recently with antibiotic–antibiotic combinations, the strategy still has its weaknesses; a number of pharmacological questions remain unanswered. For example, there is clinical evidence supporting the idea that antibiotic–antibiotic combinations suppress antibiotic resistance in tuberculosis (TB).5,6 Multidrug-resistant (MDR)-TB is present in ∼4% of new TB patients. Moreover, ∼20% of TB patients have a history of previous treatment. Indeed, it is estimated that ∼10% of MDR-TB cases have extensively drug-resistant (XDR)-TB.7 Isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA), as a combination therapy, are first-line anti-TB medications. The initial phase of such combination treatment is vital in order to prevent the emergence of drug resistance due to the differences in the mechanism of action, which are inhibiting the mycolic acid synthesis that interferes with cell wall synthesis, for INH, inhibiting DNA-dependent RNA polymerase, leading to a suppression of RNA synthesis, for RIF, inhibiting the arabinosyltransferase that interferes with cell wall synthesis, for EMB, and inhibiting the synthesis of coenzyme A that play an important role in the synthesis of fatty acids, according to the current hypothesis, for PZA.8 Added to this, a lack of pharmacokinetic (PK) information between various medications could also lead to inconsistencies between clinical observations and in vitro data,9 where each drug may be absorbed or distributed in the human body to varying levels.5 It is therefore sensible to come up with a new approach that is able to solve these challenges, for example, hybrid compounds, rather than combination therapies.
Critical review of hybrid compounds
A hybrid compound is an artificial assembly of two or more pharmacophores that can be categorised under a recognised agent acknowledged for triggering the desired activity.10–12 Such artificial scaffolds could produce antibiotics with the ability to overcome drug resistance, intensify activity, and increase binding affinity. Scientific creativity has resulted in the establishment of molecular hybrids, see Fig. 1. Such hybrids were created by covalently binding various biologically active agents with the aim of retaining the pharmacological actions of each counterpart. A molecular linker is usually employed to attach the constituent agents through a covalent bond. The method of covalent binding can also be made cleavable or non-cleavable (see Fig. 1). It is expected that a cleavable linker will be bio-transformed as soon as the hybrid gets to the site of action. On the other hand, a non-cleavable linker maintains its structure right through the period it remains in the body. The cleavable linker is a pro-drug strategy. The non-cleavable linker is a hybrid drug approach.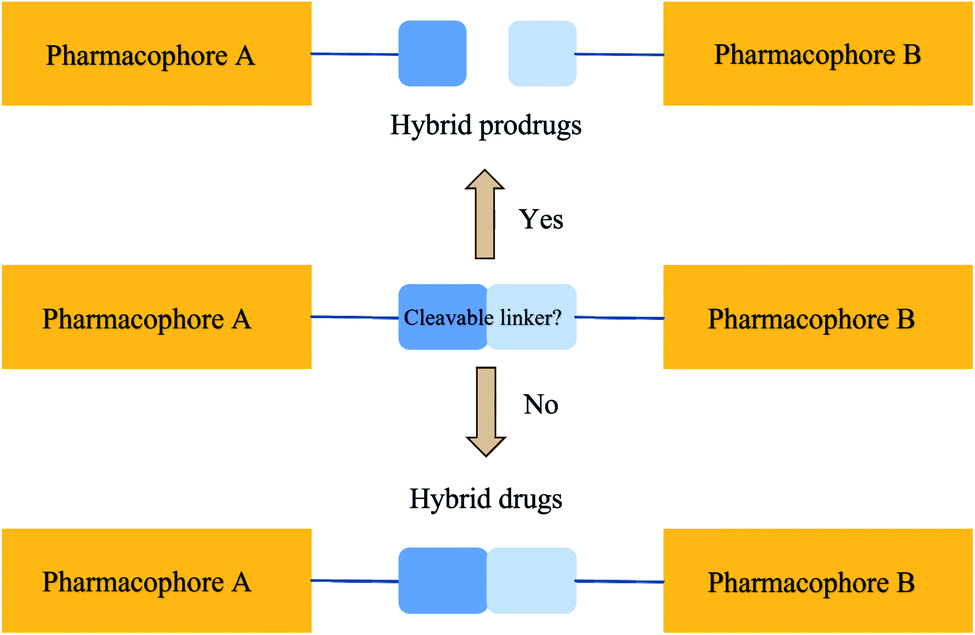 | ||
| Fig. 1 A linker that can be degraded enzymatically (by bacterium-specific enzymes with regard to a hybrid antibacterial drug) results in two useful drugs in a hybrid pro-drug approach. A linker that cannot be degraded enzymatically and holds two the same or different pharmacophores bound together represents the hybrid drug strategy. | ||
The hybrid compound hypothesis assimilates the working idea of decreasing drug resistance, escalating the binding affinity and biological activity in combination therapy into monotherapy, in the process offering a single molecular agent with one PK profile. Even though not predictable, it is possible that antibacterial effectiveness can be retained in fighting pathogens that retain resistance to the drug components. This is because the hybridisation process may also introduce supplementary physicochemical properties with the potential to change the pharmacological spectrum of the hybrid. For example, hybridisation of two therapeutic agents could improve the effectiveness or even convey a novel antibacterial action to the resultant hybrid agent.
Challenges of hybrid compounds
For hybrid agents with a molecular mass higher than 600 Da, inadequate cellular penetration through the protecting outer and inner layers of Gram-negative bacteria is the main initial concern. At present, no infallible permeation guidelines exist to help medicinal chemists design therapeutic agents with the ability to pass through bacterial lipid bilayers. If the molecular mass of antibacterial agents is high, the agents might not permeate nonselective porin channels, hampering cellular entry by passive diffusion. It is possible that a hybrid drug can be designed to maintain the uptake mechanism of the parent drugs and deal with this permeability challenge. For example, aminoglycosides are known to penetrate Gram-negative bacteria's outer membrane by a self-promoted uptake mechanism followed by inner membrane uptake that is via two energy dependant phases as it follows a path to the cytosol to trigger the antibacterial properties.13 Therefore, an aminoglycoside containing hybrid can be developed potentially maintaining the aminoglycoside's intrinsic uptake mode. Undoubtedly, the impairments linked to permeability as a result of the high molecular mass (>600 Da) of hybrid drugs are a leading reason the majority of such agents have restricted activity against dual membrane Gram-negative bacteria.5,14 Emerging reports, however, project a good prognosis for this strategy, as several hybrid drugs that are capable of eradicating MDR Gram-negative bacteria and presumably are able to delay the onset of drug resistance are in preclinical/clinical evaluation (https://www.pewtrusts.org/en/multimedia/data-visualizations/2014/antibiotics-currently-in-clinical-development).15The first ligand in such a dimeric molecule (where the two ligands might be the same or different) on binding to its target, e.g. RNA for aminoglycosides, will bring the second covalently linked ligand closer to its binding site.16–18 This effect has experimental support with a synthetic dimer of tobramycin, Tob–Tob, 1 μM, which achieved more than was achieved with 2 μM of (free) Tob.18 Perhaps the mechanism is partial dissociation of one ligand, followed by rapid re-association due to the dissociated moiety being held near to its binding site by the linked molecule remaining bound at one binding site, see Fig. 2.18 Another challenge can be found within the major concept of covalently binding two pharmacological agents. The general activity of the hybrid relies on the choice of physicochemical properties of the selected linker and the point of attachment. A pharmacophore is composed of “the collective steric and electronic properties of a molecule that are essential for interaction with the biological target and to elicit response”.19 Preferably, the covalent bonding of the two ligand molecules should not be in the pharmacophoric region in order to maintain the integrity of the biological activity. Any such group bound in the area of the pharmacophores of the drugs attached may well adversely affect the biological activity of the created hybrid.
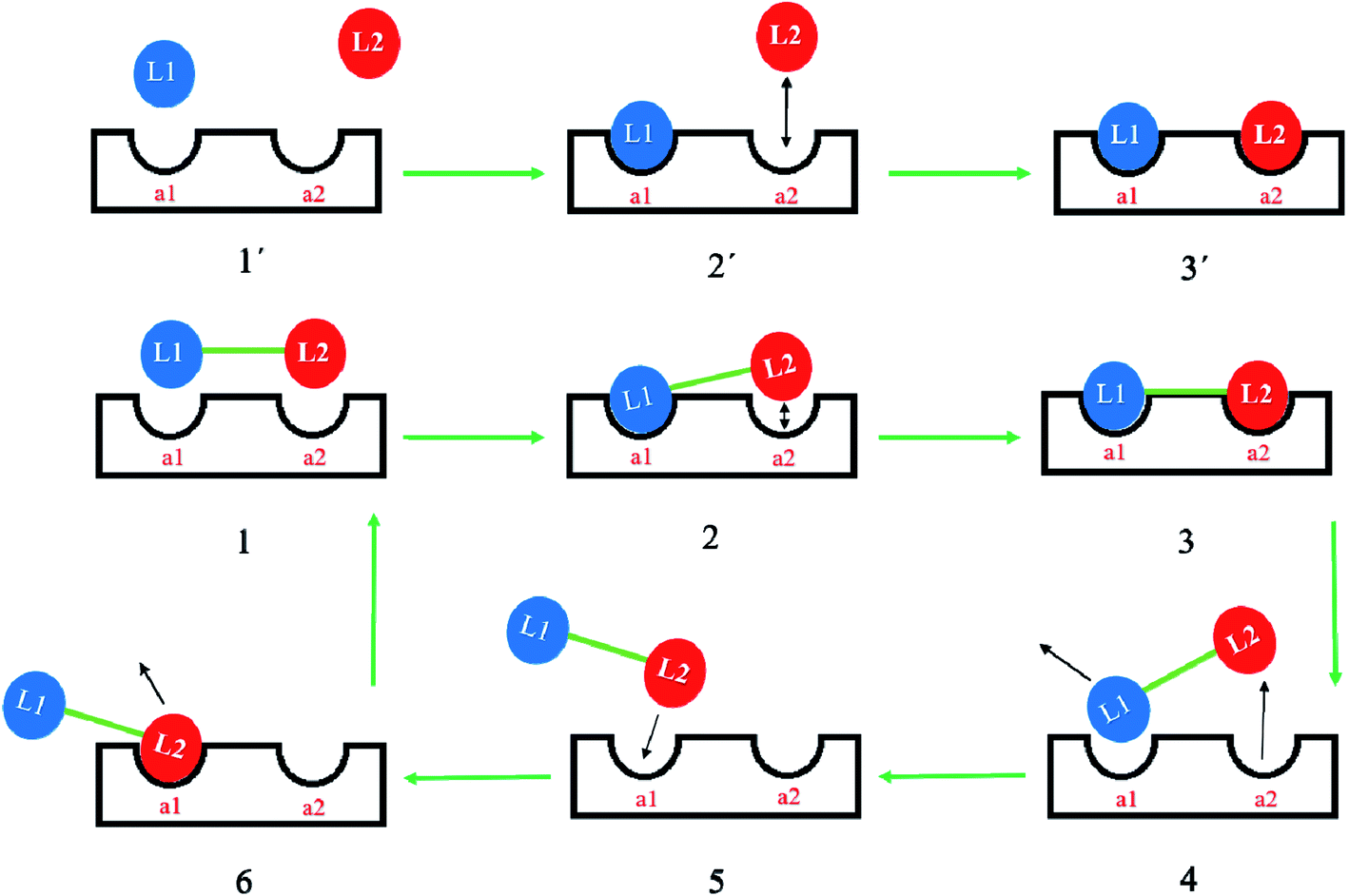 | ||
| Fig. 2 Dimeric ligands possess potential advantages over monomers. The hypothetical model for monomeric ligand binding shown in 1′, 2′, and 3′ show that monomeric ligands, L1 and L2 (which may possess different biological activity, e.g., aminoglycoside antibiotics, DNA intercalators, polyamines), may dissociate rapidly after binding to their binding sites (a1 and a2) in an independent manner. The hypothetical model for dimeric ligands, shown in panels 1–6, will allow for changed kinetics. Thus in 1–3, L2 may be brought into close proximity to a2 by the binding of L1 to a1, and may be held close to the site even after dissociation, and thus will have an increased chance of binding again. In addition, should both moieties dissociate there is an increased chance that one of the moieties will interact with the other binding site, as shown in panels 4–6. This is therefore an advantage of incorporating the linker (shown in green) that might then allow the additional association of L2 to a1. | ||
Examples of hybrid compounds
Anticancer
Combination therapy has been used against cancer for approximately 60 years since acute lymphocytic leukemia (ALL) was treated with the combination of methotrexate, 6-mercaptopurine, vincristine and prednisone (the POMP regimen) which was successful in reducing tumour burden and prolonging remission. Anti-cancer research then became more focused on investigating combination therapies that target different pathways to create a synergistic or additive effect. Sabutoclax, a pan-Bcl-2 inhibitor, in combination with minocycline, an antibiotic that has previously displayed anti-cancer effects acted synergistically on the intrinsic apoptotic pathway. This combination displayed selective toxicity and a reduction in tumour growth in vitro and in vivo on pancreatic ductal adenocarcinoma. There is therefore the potential now to develop anticancer hybrid compounds and investigate if they do deliver an improvement. A challenge is the choice of linker. The synthesis of compounds containing two pharmacophores linked by a simple spacer functional group or a longer linker is a promising approach to not only minimize the drawbacks of using medications in combination therapy, but also to improve their affinity and potency.Amino groups of two daunorubicin molecules were linked to form a bis-daunorubicin, leading to potent bis-intercalating compounds.20–22 The linker was chosen for its suitable length (∼6 Å) and its ability to lie in the DNA minor groove without steric effects based on the high-resolution crystal structure of daunorubicin intercalating to its target DNA. In the crystal structure, a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the compound intercalating the DNA was observed, with the sugar moieties of the daunorubicin molecules directed towards each other, see Fig. 3. This arrangement brought the –NH2 groups of the two ligands to within 6 Å. Crucially, daunorubicin linked-dimers exhibited more cytotoxic activity than daunorubicin itself. Over-long linkers might not fit well into the DNA minor groove. For this reason, a p-xylene (WP631) and a (click chemistry product) triazole were selected and synthesised as linkers. In tests on the MDR breast carcinoma MCF-7/VP-16 cell line, WP631 exhibited more cytotoxicity than free doxorubicin, although the opposite effect was noted for the daunorubicin dimer linked via a triazole against the leukaemia cell line K562, than the parent daunorubicin (Fig. 3).20–22
:1 stoichiometry of the compound intercalating the DNA was observed, with the sugar moieties of the daunorubicin molecules directed towards each other, see Fig. 3. This arrangement brought the –NH2 groups of the two ligands to within 6 Å. Crucially, daunorubicin linked-dimers exhibited more cytotoxic activity than daunorubicin itself. Over-long linkers might not fit well into the DNA minor groove. For this reason, a p-xylene (WP631) and a (click chemistry product) triazole were selected and synthesised as linkers. In tests on the MDR breast carcinoma MCF-7/VP-16 cell line, WP631 exhibited more cytotoxicity than free doxorubicin, although the opposite effect was noted for the daunorubicin dimer linked via a triazole against the leukaemia cell line K562, than the parent daunorubicin (Fig. 3).20–22
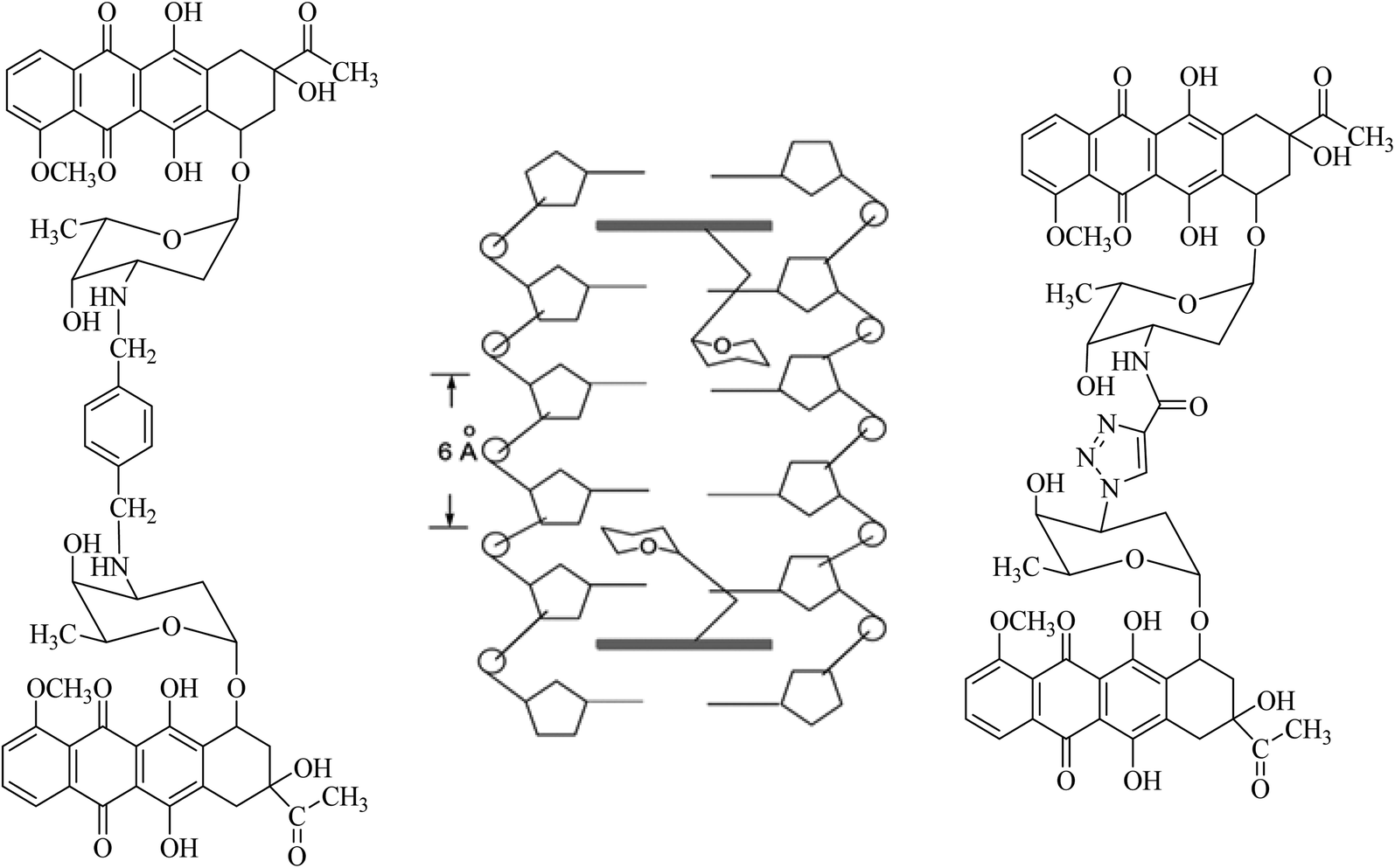 | ||
| Fig. 3 Daunorubicin dimer linked by a p-xylene (WP631) (left), monomeric daunorubicin bound to DNA, the NH2 groups are within 6 Å of one another (middle), daunorubicin dimer linked by a triazole via click-chemistry (right). | ||
There has been extensive research with polyamine conjugates as anti-cancer agents. Polyamines and their biochemistry and metabolism as targets, polyamine transporters for the cellular uptake of anti-cancer cargoes, and using polyamines for their high affinity electrostatic binding to DNA or (si)RNA. With their wide variety of cellular effects, polyamine containing hybrid compounds may yet be useful agents in the chemoprevention of cancer.23
Brel and co-workerrs24 showed that covalently linking cancerostatic daunorubicin and a natural antiproliferative agent, piperonal and its dimethoxy derivative by a simple amine spacer makes a designed conjugate with higher anticancer activity compared to an amide spacer synthesised from piperonylic acid (Fig. 4). These conjugates were tested in four types of cancer cells: lung carcinoma (A549), rhabdomyosarcoma (RD), large intestine carcinoma (HCT116), and breast adenocarcinoma (MCF7). The analogues linked with an amide spacer were shown to be 100-times less toxic to normal healthy cells and had lower anticancer activity compared to analogues linked with an amine spacer, which showed higher toxicity both to healthy and to the four cancer cell lines.24
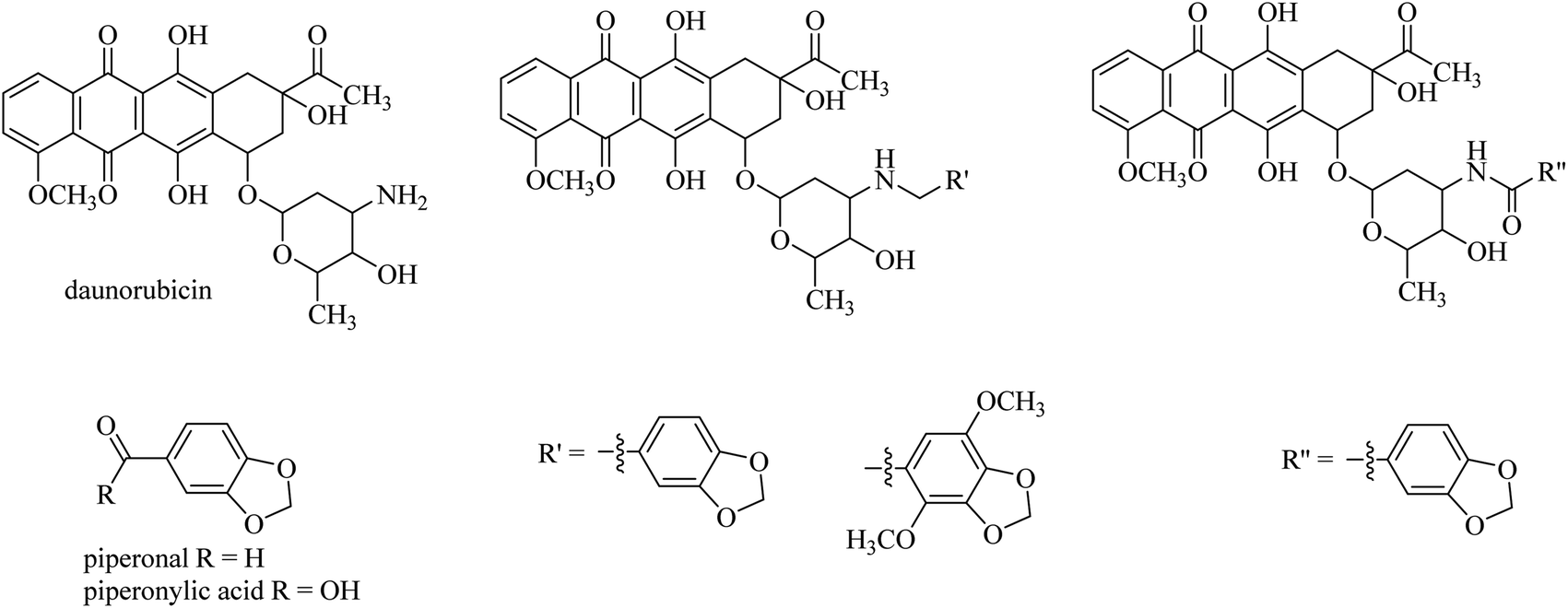 | ||
| Fig. 4 Daunorubicin secondary amine conjugates (centre) prepared from the aldehyde piperonal (left) and its dimethoxy derivative (centre) and daunorubicin amide from piperonylic acid (right). | ||
Benzylcoumarin has good anticancer activity by inhibiting several targets, e.g., 17b-HSD3, MEK1, and NQO1.25–27 Wang et al. reported the synthesis of a series of coumarin-benzimidazole analogues.28 However, unsubstituted A, mono- and di-methyl, B and C, respectively, substituted benzimidazole-coumarin hybrids showed no activity against five human cancer cell lines, HL-60, SMMC-7721, A549, MCF-7, and SW480 (Fig. 5). On the other hand, naphthylation at position N-1 on the benzimidazole D was found to be active against those five cancer cell lines (Fig. 5).28
 | ||
| Fig. 5 Coumarin-benzimidazole hybrids. | ||
Rullo et al. synthesised a conjugate consisting of 1,2,3,4-tetrahydroisoquinoline (THIQ) moiety linked to a coumarin moiety via a penta-methylene linker.29 The hybrid (Fig. 6) was tested in Madin–Darby Canine Kidney (MDCK) cells overexpressing P-glycoprotein (P-gp) and MRP1. The THIQ-coumarin analogue showed a nanomolar P-gp inhibition potency.29 In some cases, amidation or esterification of the carboxylic acid functional group in ibuprofen showed an improvement in the anti-inflammatory activity30,31 as well as antiproliferative activity compared to the parent drug.32 Encouraged by the above findings, Abourehab et al. designed and synthesised a new series of hybrids by linking the pyrrolizine/indolizine derivatives with ibuprofen or ketoprofen NSAIDs.33 These two scaffolds were selected based on their promising cytotoxic activity. The antiproliferative activities of these compounds/hybrids were investigated and showed 4–71% inhibition of the growth of three cancer cell lines, MCF-7, A549, and HT-29. Compounds A, B, and C, see Fig. 7, the most active compounds, revealed IC50 values of 7.6, 1.1, and 3.2 μM, respectively, against MCF-7 cells.33
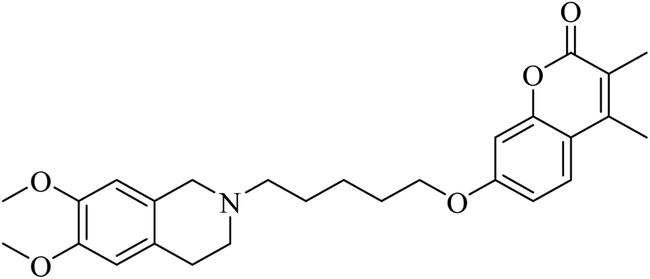 | ||
| Fig. 6 THIQ-coumarin hybrid. | ||
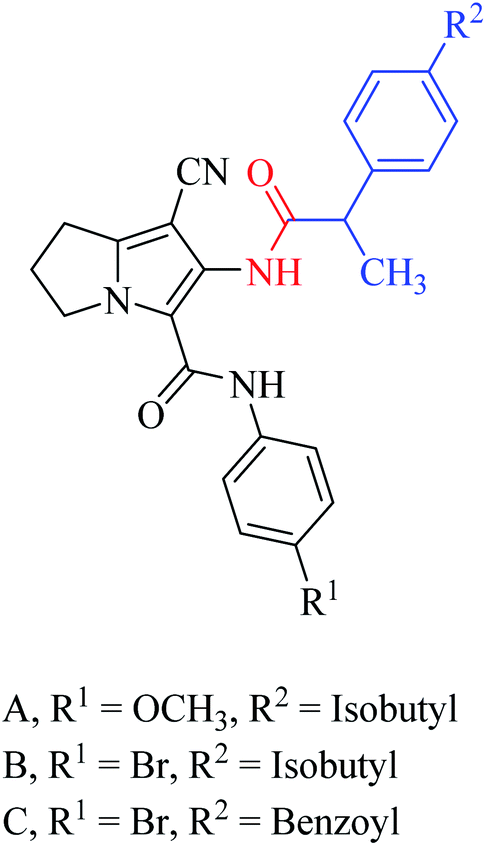 | ||
| Fig. 7 Ibuprofen or ketoprofen-pyrrolizine hybrids. | ||
Non-viral gene therapy (NVGT)
The application of using two pharmacophores attached by a linker is not limited to improvements in the binding affinity. This approach can also be used to design a compound that might help in gene delivery where therapeutic strategies are based on supplementing genes where faulty ones exist, introducing genes with new functions, or interrupting (silencing) gene expression. These strategies are growing in terms of scope and diversity in their use to treat diseases resulting from gene malfunction. Gene therapy is a technique based on introducing genetic materials into cells to assist with producing therapeutic proteins or obstructing the production of harmful (signalling) proteins. The production of therapeutic proteins involves amending genetic defects in target cells. The strategy is used chiefly when treating diseases with single-gene disorders (such as cystic fibrosis, severe combined immunodeficiency syndromes, muscular dystrophy, haemophilia, β-thalassemia, or sickle cell anaemia) and malignant tumours, including ovarian carcinoma. The strategy involving the obstruction of harmful proteins' production involves destroying target cells using a cytotoxic pathway. It is mainly used in treating malignant tumours, e.g., breast, ovarian, and endometrial carcinoma.34,35 The last decade has witnessed a remarkable improvement in gene therapy technologies, as shown by many clinical trials reported.Systems for gene delivery can be divided into two categories: viral vectors and non-viral vectors. Research in gene therapy is divided into two broad fields. One relates to developing a therapeutic gene, which involves developing an effective therapeutic gene for specific diseases or a regulated gene expression system. The second area of research is focused on the development of a safe and efficient delivery system. If gene therapy is to be developed into a successful therapeutic method, optimising delivery vectors is an area that will need to be given (still more) serious attention. Developing effective and safe delivery methods is one of the most significant challenges preventing the use of gene therapy and DNA vaccines. The non-viral delivery systems include physical methods (naked DNA) and chemical techniques, which include those with cationic polymers (polyplexes) or synthetic cationic lipids (lipoplexes). Notwithstanding the reality that lipopolyamines are not as efficient as viral vectors, however, they are less toxic when compared to viral vectors. This has spurred efforts to develop novel lipopolyamines with enhanced transfection efficiency.34,35
Selecting a delivery system is guided by the characteristics of the disease being treated and the period of the gene expression in question. Even though clinical trials are primarily focused on viral systems, numerous clinical trials focus on cationic lipids when used to treat cancer, fibrosis, and recently, cardiovascular diseases (an exhaustive list of clinical trials can be found at: http://www.wiley.co.uk/genmed/clinical/). Even though some encouraging results have been reported, it is an essential requirement if NVGT is to succeed that the vectors must be more efficient. There are several benefits linked to non-viral vectors, even though the most efficient way of delivering genes to cells is viral vectors. The main differences between viral and non-viral vectors are that the latter is cheaper and has low immunogenicity and cytotoxicity. Several gene carriers have been developed to meet the needs of gene delivery to humans. Such delivery mechanisms protect and condense plasmid DNA from the DNase enzyme, resulting in improved efficiency in gene delivery. However, some issues still need to be resolved, such as overall transfection efficiency and biocompatibility.
The majority of cationic lipid non-viral gene carriers possess positive charges on amine functional groups. These positive charges interact with the phosphate groups' negative charges in the plasmid DNA, leading to condensation of the plasmid DNA or siRNA. There is the interaction between the negatively charged cell membranes and the positively charged complexes, a process that facilitates lipoplex/plasmid DNA complexes' cellular uptake through endocytosis. When nucleases degrade plasmid DNA, the result is a loss of gene expression. This is why plasmid DNA needs to be protected from the nucleases by the cells. When plasmid DNA is condensed by the carriers, the nucleases are prevented from accessing the plasmid DNA, a process that boosts plasmid DNA resistance against enzymatic degradation. In the non-viral delivery system, cationic lipids are seen as the main gene carriers. They assist in delivering to the nucleus by their ability to condense DNA into particles that can freely be endocytosed by cultured cells and then escape. In the cellular transfection achieved and DNA condensation, the covalent adding of lipid moiety, usually a single or two alkenyl chains or alkyl, or a steroid introduces more efficiency. Therefore, efficient RNA or DNA condensation and the resulting drug delivery is possible through the use of novel polyamine conjugates.36 Usually, the charged head group is linked to between one and three hydrocarbon chains by a spacer. The type of linkage between the cationic and lipophilic moieties determines how stable the cationic lipids in biological systems are. It was concluded that the ester bond was easily hydrolysed by endogenous esterase in the cell. Carbamate bonds, were, however, observed to have better stability when compared to ester bonds. Thus, carbamate bonds tend to be used more often.37–39 The covalent addition of a lipid moiety usually involves a single or two alkenyl chains, alkyl, or a steroid. Usually, the hydrocarbon chains consist of 14 or more carbon atoms (Fig. 8).
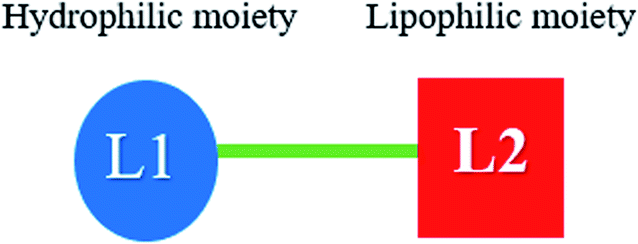 | ||
| Fig. 8 The three parts of a cationic lipid: the hydrophilic moiety (L1), e.g., a polyamine, the linker (shown here as a green line), e.g., a carbamate or amide functional group, and a lipophilic moiety (L2). | ||
Geall and Blagbrough synthesised novel lipopolyamines by coupling selectively protected triBoc-spermine with fatty acids. They found that the binding affinity of these lipophilic polyamines to DNA is fully dependent on the lipid part attached by a linker to positively charged polyamines, see Fig. 9.40
 | ||
| Fig. 9 Coupling spermine with long chain fatty acids. | ||
Five different long-chain fatty acids with different chain length and oxidation state (C22:1, C18:2, C18:1, C18:0, C12:0), were linked to the naturally occurring polyamine, spermine (Fig. 10). Compounds 1–5 were tested for gene, siRNA delivery, and knock-down. The results showed that the unsaturated fatty acid amides of spermine are non-toxic and efficient for gene and siRNA delivery.41
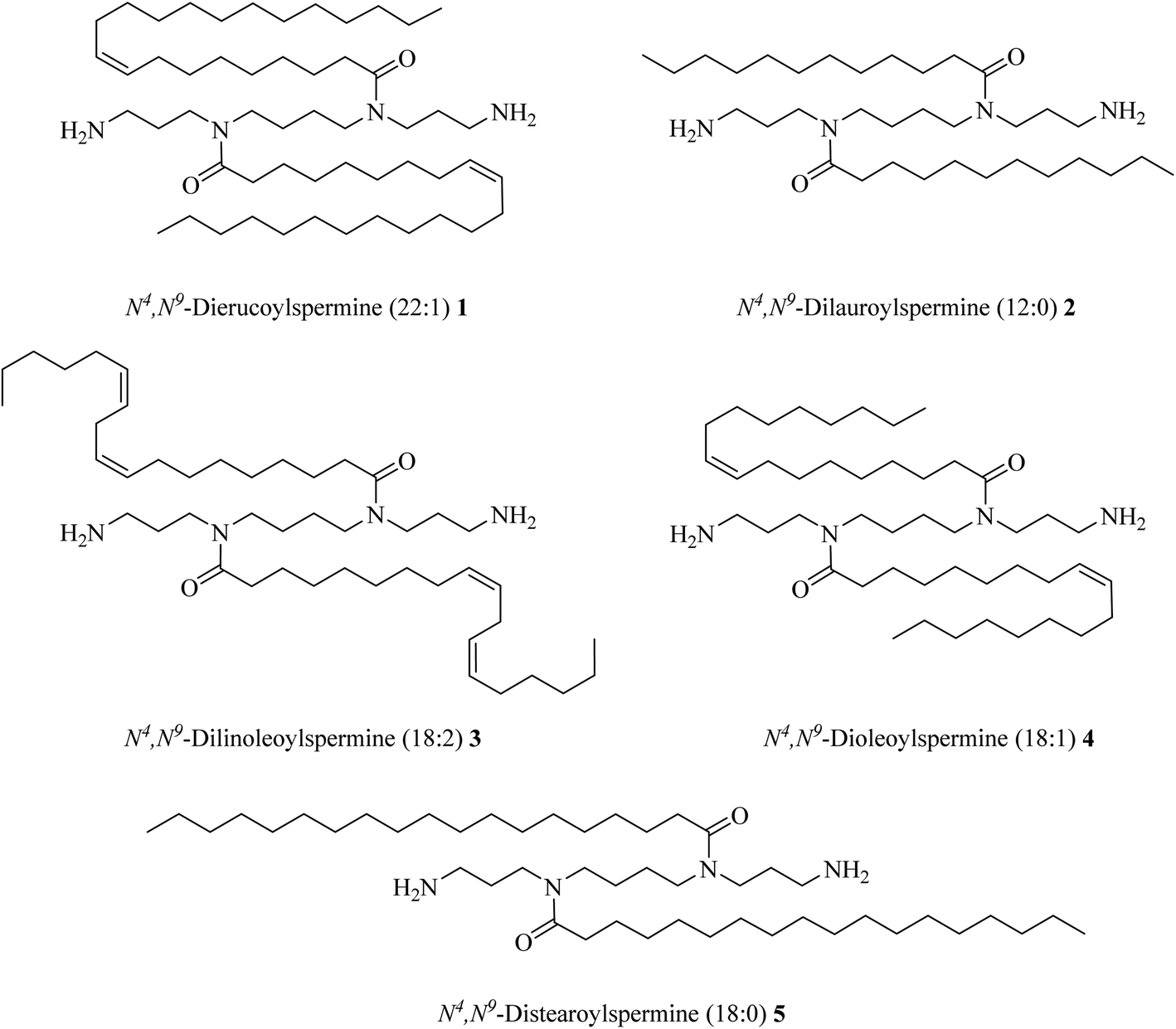 | ||
| Fig. 10 Di-fatty acid amides of spermine. | ||
Another type of cationic cholesterol conjugate with an aminoglycoside head-group has also been synthesised. Kanamycin with a cholesterol lipid moiety is effective in NVGT (Fig. 11).42 Moreover, we demonstrated that a synthetic cholesteryl conjugate with neomycin B (substituted with six amino groups) is highly efficient for the delivery of fluorescent siRNA (85–90%) and DNA (65–80%) (Fig. 11).43 Such findings agree with the hypothesis that for an efficient nucleic acid delivery system, the positively charged polyamines and the lipophilic moiety both play significant roles. Thus, such rationally designed and easily prepared novel conjugates can find a crucial role in NVGT.
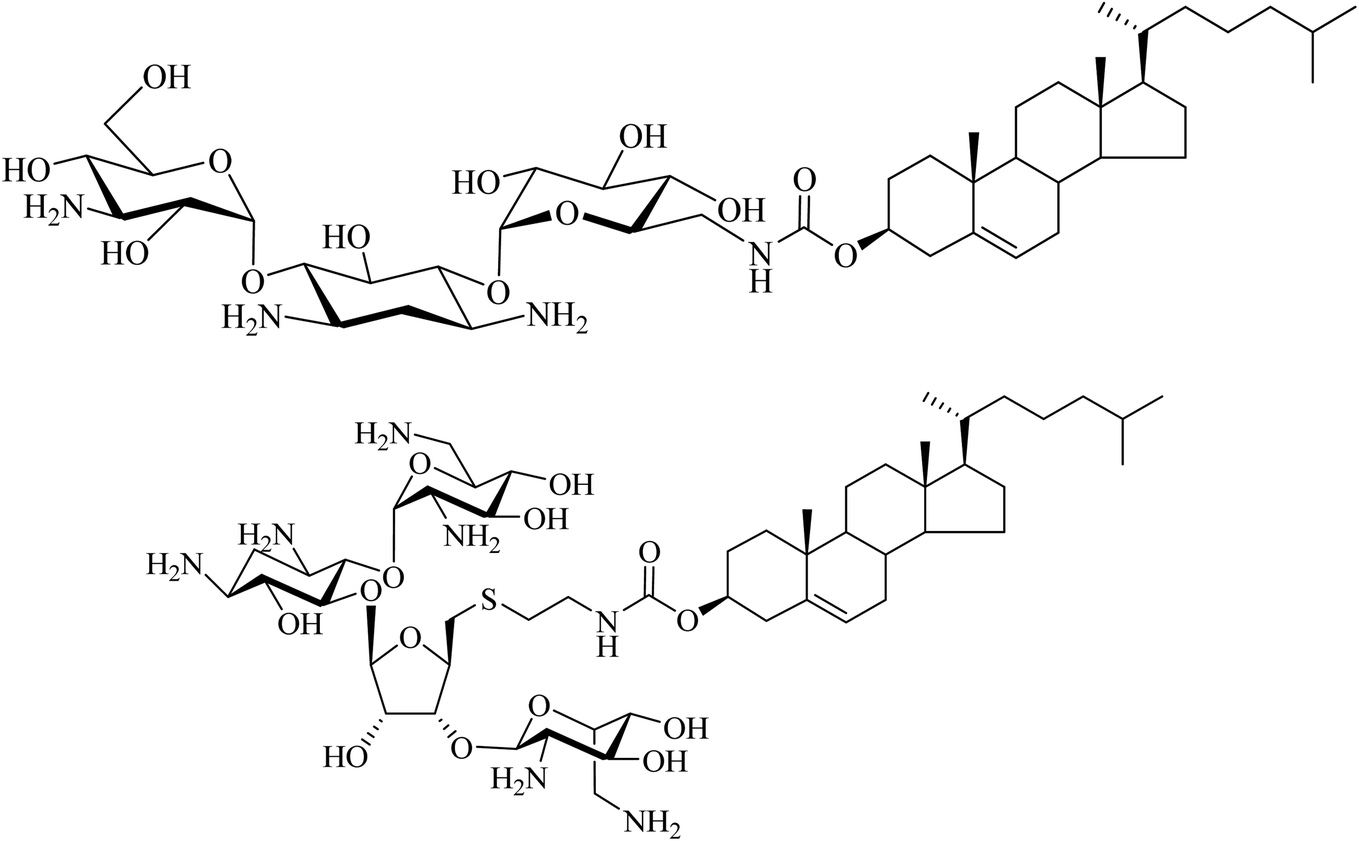 | ||
| Fig. 11 Coupling kanamycin (upper) or neomycin (lower) to a cholesteryl moiety. | ||
Molecular precedents can be found for the design of hybrid compounds in the natural world. Such conjugates and their derivatives can be used as natural product leads for drugs or even as medicines for their formulation in NVGT. In terms of sharks healing their wounds and certainly possessing antibacterial agents, the 1993 report of squalamine was a novel discovery.44 Squalamine (Fig. 12) is the first representative of a previously unknown class of natural antibiotics of animal origin. These polyamine-bile acid (e.g. cholic acid type) conjugates display many exciting biological activities.45 Squalamine analogues also demonstrate the possibility to form supramolecular networks, opening up many possibilities in their use in drug delivery systems in serum or other body fluids. The research area of steroid polyamines, e.g., squalamine, trodusquemine, ceragenins, claramine, and their diverse analogues and derivatives, has been comprehensively reviewed.45 As well as an antibacterial, such compounds display antiviral, neuroprotective, antiangiogenic, antitumor, antiobesity and weight-loss activity, antiatherogenic, regenerative, and anxiolytic properties. We and others have shown that cholic acid and other bile acids act as a lipidic region,46–48 so that after the many phosphate negative charges along the DNA or siRNA drug have been neutralised by the polycationic polyamine region, the nucleic acid thereby being condensed, the formed nanoparticle is coated in the steroid lipid.49–51 Taken together, these hybrid compounds act as efficient delivery devices in NVGT.52,53
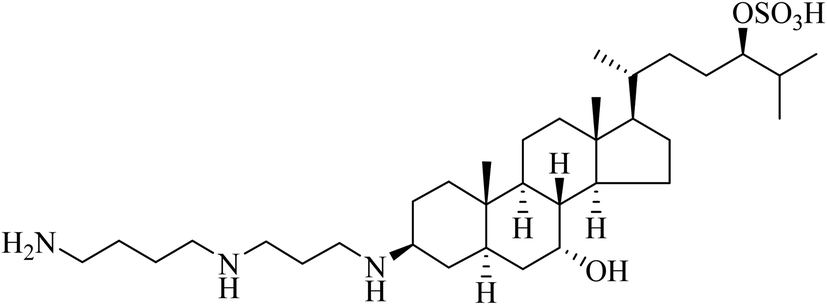 | ||
| Fig. 12 Squalamine, a naturally occurring spermidine bile acid conjugate found in sharks. | ||
Antimicrobial
The fast proliferation of Gram-positive and Gram-negative bacterial pathogens resistant to prevailing antimicrobial therapies, whether in community or hospital settings across the world, is an indication of the advent of potentially grave global health crises. Out of all such pathogens, the ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) bacteria are responsible for most of the global nosocomial infections, with an annual escalation of drug resistance.54The prevalence of clinical isolates that can be grouped under the ESKAPE category that shows either pandrug resistance (PDR), extensively drug resistant (XDR), or multidrug resistant (MDR) has reached levels that some scholars have started to call alarming.55–57 MDR denotes a situation of nonsusceptibility to a minimum of one agent in three antibiotic classes that are chemically different. XDR involves nonsusceptibility to a minimum of one agent in all except two antibiotic classes that are chemically different. PDT denotes a situation where nonsusceptibility is in all agents in every antibiotic class.58,59 There have been few novel antibacterial drug entities containing new modes of action approved for use in a clinical setting in over half a century, except novel combinations of drugs.54
Numerous organisations involved in healthcare provision have voiced concern about the pressing need to develop novel antibiotics, particularly for dealing with drug-resistant Gram-negative ESKAPE bacilli. The World Health Organisation (WHO), for instance, has been referring to the idea of the post-antibiotic era, characterised by rising mortality and morbidity rates even for minor injuries and common infections.60 The inadequate accessibility of antibiotics for treating MDR Gram-negative bacterial infections continues to be a grave challenge.
Another strategy for some kinds of bacteria to develop antimicrobial resistance is by forming biofilms. More than 70% of human bacterial infections are related to biofilm formation.61 The existence of microbial cells within the biofilm affords the protection of bacteria cells from both antibacterial compounds and the human immune system.62,63 A biofilm grows in three stages: attachment, micro colonies formation, and maturation.64–67 Initially, the free cells attach to a surface which can be biotic or abiotic. These free bacterial cells can either attach on their own or form aggregated colonies.61,65 After irreversible attachment which allows the bacteria to stick to a surface without being washed off, biofilm maturation begins.67 Several factors play a role in developing the irreversible attachment to the surface such as hydrophobicity, quorum sensing (QS) and surface roughness.68–70
One of the effective strategies for dealing with the challenge of antibiotic resistance lies in the development of agents that are not bactericidal but still indirectly stop the bacterial communication.71,72 For instance, obstructing bacterial quorum sensing is a practical approach. The concept of quorum sensing involves the creation, and discharge of molecules by bacteria as a way through which adjacent bacteria communicate.68–70 Environmental factors, such as chemical or physical, in microbial communities activate this network of communication. The detection of these signalling molecules activates metabolic and physiological changes in the gene expression of bacteria. Consequently, the bacteria produce bio-molecules that are required for biofilm formation. It can therefore be predicted that preventing QS could lead to the pathogen being unable to harm the host.70 There are several compounds that have been suggested and evaluated for their antibiofilm ability, such as QS modifying molecules and polyamines.71,72
Tor and co-workers led the way utilising aminoglycoside–RNA interactions for the design and synthesis of hybrid compounds where dimerized aminoglycosides achieved enhanced RNA binding by electrostatic interactions.73 Following experimental results, 2-deoxystreptamine (DOS) fragment I and fragment II, that are present in neomycin, paromomycin, kanamycin, tobramycin, amikacin, and gentamicin aminoglycosides,74,75 are important for electrostatic binding with the rRNA of prokaryotic (blue part, Fig. 13).16 The primary amines at positions 1 and 3 in the 2-DOS ring provide extra electrostatic interactions with the rRNA binding sites.16,18,73 On the other hand, other studies have shown that the OH groups at position 6′′′ in fragment III of kanamycin and tobramycin and the OH group at position 5′′ in fragment III of neomycin and paromomycin are not essential for rRNA binding (highlighted hydroxy groups in red, Fig. 13).18,73
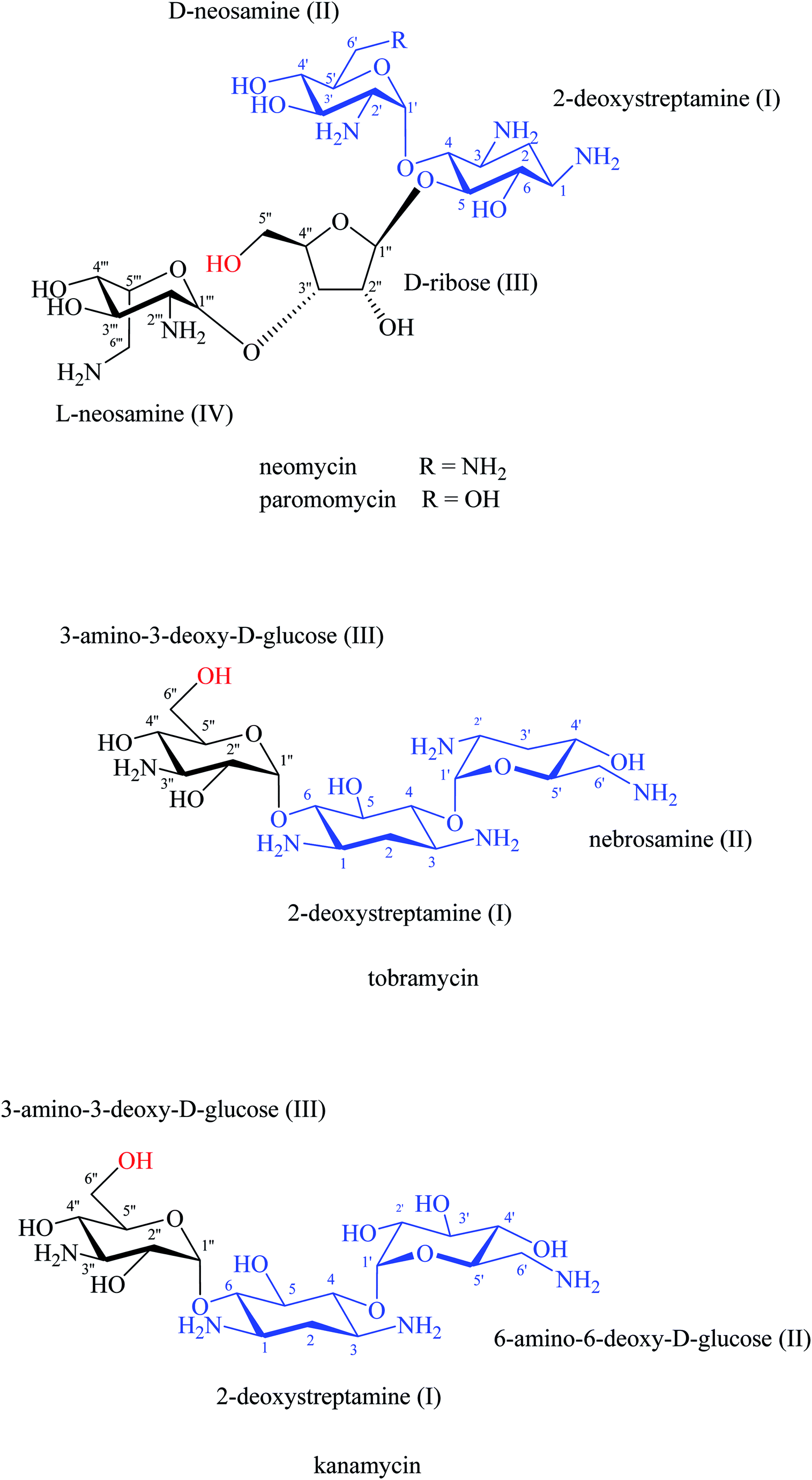 | ||
| Fig. 13 Aminoglycoside antibiotics. | ||
Tor and co-workers reported that they synthesised a dimeric neomycin linked by a disulfide linker containing 12 other atoms (Fig. 14).18,73 This was designed, linking via an ether of the not essential OH group at position 5′′ in fragment III of neomycin, in order to allow the aminoglycosides to find or scan for the second RNA binding site. The activity as a ribosome inhibitor of this derivative improved from 20- to 1200-fold compared to the monomeric counterpart. This enhancement in its activity might be due to the interaction with two sites in bacterial RNA.73 This has experimental support with a dimer of tobramycin, Tob–Tob (1 μM) covalently bound by a chain linker containing a disulfide which decreased the substrate cleavage rate more than was achieved with 2 μM of (free) Tob.18 Another related method was reported by Santana et al. who synthesised, in 75% yield, Tob dimers using 1,6-dibromohexane as a source of the linker through DOS ethers (Fig. 15). They argued that the increase in cationic charge might reinforce the overall binding of aminoglycosides to RNA.76
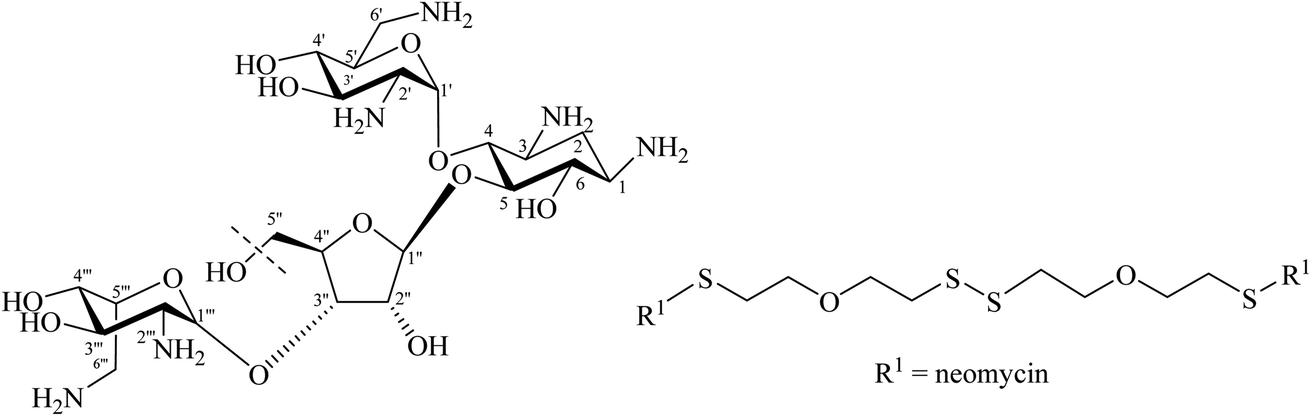 | ||
| Fig. 14 Dimeric neomycin bound by a disulfide containing chain linker. | ||
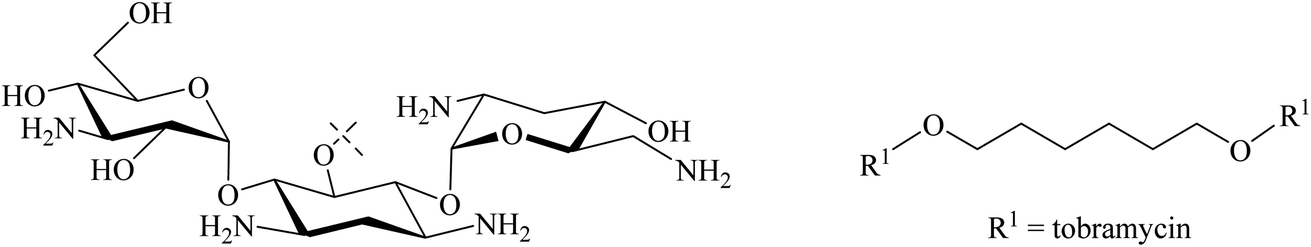 | ||
| Fig. 15 Dimeric tobramycin bound by a 1,6-hexyl diether linker. | ||
Idowu et al. reported that combining a homo dimeric Tob (Fig. 16) with ceftolozane potentiates the antimicrobial activity of ceftolozane against MDR/XDR in P. aeruginosa in vitro. In contrast, the combination of tobramycin monomer and ceftolozane does not potentiate the antimicrobial activity of ceftolozane against MDR/XDR in P. aeruginosa under the same conditions.77
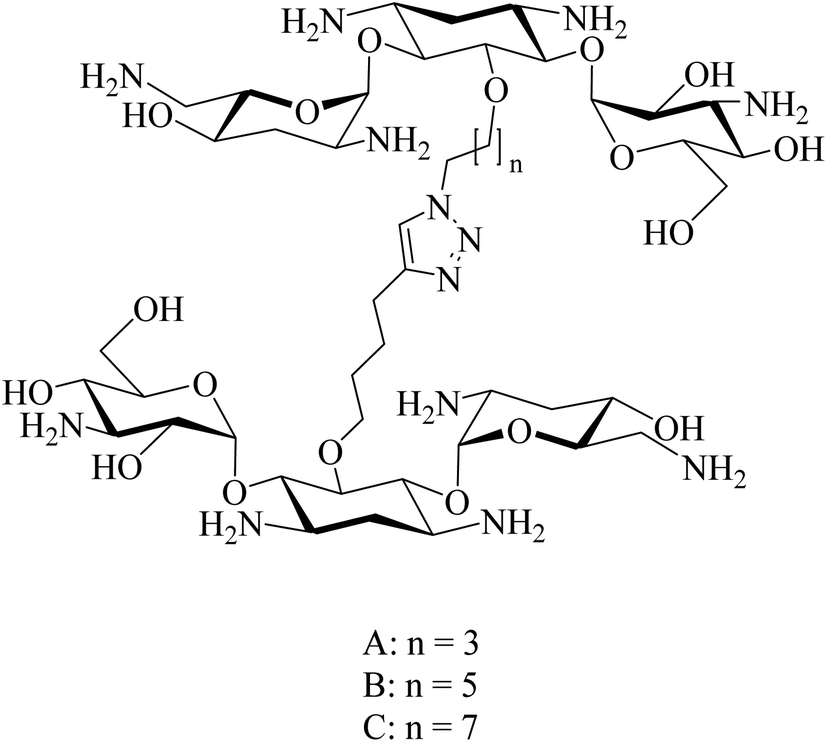 | ||
| Fig. 16 A homo-dimeric tobramycin linked triazole synthesized via click-chemistry. | ||
Remarkably, the majority of the reported antimicrobial hybrid compounds possess a chemical stable fluoroquinolone moiety, such as ciprofloxacin. These hybrids showed antibacterial activity against Gram-negative bacteria. Moreover, the structure-activity relationship (SAR) and the broad-spectrum of activity of fluoroquinolone antibiotics are well-known.78,79 Pokrovskaya et al. reported that neomycin B-ciprofloxacin hybrid compounds were covalently bound via different linkers in order to achieve the optimum linker length and physicochemical properties required for the best activity.80 Neomycin B-ciprofloxacin hybrids, with an aromatic triazole linker and aliphatic triazole linker (Fig. 17), exhibited more antibacterial activity than (free) neomycin B, but not ciprofloxacin. In a different experiment, selected neomycin B-ciprofloxacin hybrid compounds showed up to 32-fold more potent inhibition of TopoIV and DNA gyrase than ciprofloxacin. The presence of six amino groups (positively charged) in neomycin B might increase the binding affinity of ciprofloxacin to its target site (DNA). Moreover, a significant delay in any resistance formation was noticed in both Gram-positive and negative bacteria that were treated with neomycin B-ciprofloxacin hybrid compounds compared to neomycin B and ciprofloxacin separately or their 1:1 equivalent mixture.80
 | ||
| Fig. 17 Neomycin B-ciprofloxacin hybrids, with aromatic and aliphatic bound triazole linkers. | ||
Pyrimidine-fluoroquinolone hybrids have been reported in a patent.81 Trimethoprim was linked to ciprofloxacin and other fluoroquinolone moieties through an aryl ether bond (Fig. 18). Ciprofloxacin and trimethoprim MICs are 32 and 4 μg mL−1, respectively, and the MIC of the 1:1 mixture is 8 μg mL−1. However, these hybrids displayed a better antibacterial activity than trimethoprim against S. aureus and S. epidermidis (Gram-positive) and Escherichia coli (Gram-negative). Indeed, they have potent activity against ciprofloxacin-resistant S. aureus strain NRS19, as low as MIC = 1 μg mL−1.81
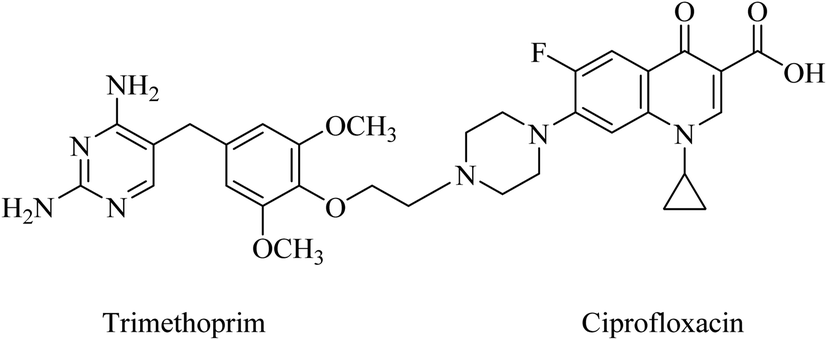 | ||
| Fig. 18 Trimethoprim-ciprofloxacin hybrid. | ||
Cadazolid, a hybrid consisting of two pharmacophores derived from tedizolid and ciprofloxacin, see Fig. 19, has successfully completed phase III clinical trials for the treatment of Clostridium difficile Associated Diarrhoea (CDAD) in comparison to vancomycin (https://clinicaltrials.gov/show/NCT01987895).82 Two polyamine-based analogues, A and B, with two hydrophobic moieties were linked to linear polyamines via thioamides, see Fig. 20. The analogue B inhibits biofilm formation of P. aeruginosa at 64 μg mL−1 by targeting the bacterial membrane.83 Long lipophilic chains were linked to norspermidine-di-amino acid analogues (compounds C and D, see Fig. 20). These analogues displayed antibacterial activity against several Gram-positive and Gram-negative bacteria, e.g., S. aureus, E. coli, and K. pneumoniae. Moreover, compound D showed antibiofilm activity at 60 μg mL−1 against S. aureus MTCC 737.84
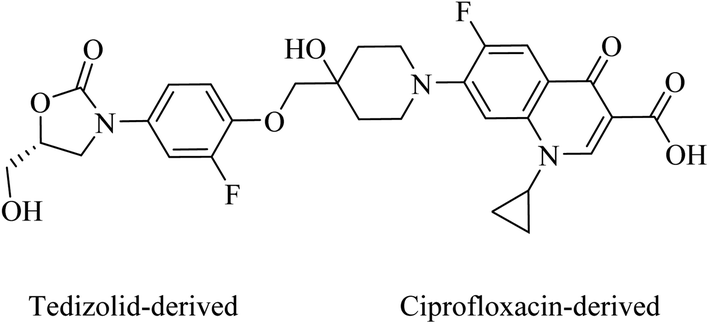 | ||
| Fig. 19 Cadazolid was synthesised by linking tedizolid and ciprofloxacin. | ||
 | ||
| Fig. 20 Dithiourea-diamine (A and B) and lipidic polyamine (C and D) hybrid molecules with antibacterial and antibiofilm activity. | ||
Conclusions
There is a need to develop new drugs/medications that can treat infectious diseases, especially those caused by MDR bacteria and the many different cancers. We have highlighted several approaches to develop new antibiotics and anticancer agents. Considering their potential advantages, the concept of hybrid compounds is attractive. However, molecular complexity, inflexible/indirect chemical synthesis, and the effort needed to find the mechanism of action over conventional medications need to be considered. Despite these related challenges, employing the hybrid technique is a practical approach to enlarge the chemotherapeutic space of our current medications. The application of using two pharmacophores attached by a covalent linker is not limited to improvements in the binding affinity or reducing the side effects of antibiotics or anticancer drugs and medicines, this approach can also be used to design compounds that might be applied to treat other diseases or in gene or siRNA delivery.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Government of the Kingdom of Saudi Arabia for the Studentship to A. H. A.References
- C. Viegas-Junior, A. Danuello, V. da Silva Bolzani, E. J. Barreiro and C. A. M. Fraga, Molecular hybridization: A useful tool in the design of new drug prototypes, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef CAS PubMed.
- M. Hilf, V. L. Yu, J. Sharp, J. J. Zuravleff, J. A. Korvick and R. R. Muder, Antibiotic therapy for Pseudomonas aeruginosa bacteremia: Outcome correlations in a prospective study of 200 patients, Am. J. Med., 1989, 87, 540–546 CrossRef CAS PubMed.
- A. R. White, Augmentin(R) (amoxicillin/clavulanate) in the treatment of community-acquired respiratory tract infection: a review of the continuing development of an innovative antimicrobial agent, J. Antimicrob. Chemother., 2004, 53, 3–20 CrossRef PubMed.
- S. M. Drawz and R. A. Bonomo, Three decades of β-lactamase inhibitors, Clin. Microbiol. Rev., 2010, 23, 160–201 CrossRef CAS PubMed.
- J. G. Kmeid, M. M. Youssef, Z. A. Kanafani and S. S. Kanj, Combination therapy for Gram-negative bacteria: what is the evidence?, Expert Rev. Anti-Infect. Ther., 2013, 11, 1355–1362 CrossRef CAS PubMed.
- R. Domalaon, T. Idowu, G. G. Zhanel and F. Schweizer, Antibiotic hybrids: The next generation of agents and adjuvants against Gram-negative pathogens?, Clin. Microbiol. Rev., 2018, 31, 1–45 CrossRef PubMed.
- K. J. Seung, S. Keshavjee and M. L. Rich, Multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis, Cold Spring Harbor Perspect. Med., 2015, 5, 163–178 Search PubMed.
- J. A. Caminero, G. Sotgiu, A. Zumla and G. B. Migliori, Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis, Lancet Infect. Dis., 2010, 10, 621–629 CrossRef CAS PubMed.
- P. D. Tamma, S. E. Cosgrove and L. L. Maragakis, Combination therapy for treatment of infections with Gram-negative bacteria, Clin. Microbiol. Rev., 2012, 25, 450–470 CrossRef CAS PubMed.
- A. Basak and R. Pal, Synthesis of β-lactam nucleoside chimera via Kinugasa reaction and evaluation of their antibacterial activity, Bioorg. Med. Chem. Lett., 2005, 15, 2015–2018 CrossRef CAS PubMed.
- D. D. Long, J. B. Aggen, B. G. Christensen, J. K. Judice, S. S. Hegde, K. Kaniga, K. M. Krause, M. S. Linsell, E. J. Moran and J. L. Pace, A multivalent approach to drug discovery for novel antibiotics, J. Antibiot., 2008, 61, 595–602 CrossRef CAS PubMed.
- T. Karoli, S. K. Mamidyala, J. Zuegg, S. R. Fry, E. H. L. Tee, T. A. Bradford, P. K. Madala, J. X. Huang, S. Ramu, M. S. Butler and M. A. Cooper, Structure aided design of chimeric antibiotics, Bioorg. Med. Chem. Lett., 2012, 22, 2428–2433 CrossRef CAS PubMed.
- B. Becker and M. A. Cooper, Aminoglycoside antibiotics in the 21st century, ACS Chem. Biol., 2013, 105–115 CrossRef CAS PubMed.
- H. Brotzoesterhelt and N. Brunner, How many modes of action should an antibiotic have?, Curr. Opin. Pharmacol., 2008, 8, 564–573 CrossRef PubMed.
- https://www.pewtrusts.org/en/multimedia/data-visualizations/2014/antibiotics-currently-in-clinical-development.
- M. Hendrix, E. S. Priestley, G. F. Joyce and C. H. Wong, Direct observation of aminoglycoside−RNA interactions by surface plasmon resonance, J. Am. Chem. Soc., 1997, 119, 3641–3648 CrossRef CAS PubMed.
- D. Williams and M. Westwell, Aspects of weak interactions, Chem. Soc. Rev., 1998, 27, 57–64 RSC.
- K. Michael, H. Wang and Y. Tor, Enhanced RNA binding of dimerized aminoglycosides, Bioorg. Med. Chem., 1999, 7, 1361–1371 CrossRef CAS PubMed.
- C. G. Wermuth, C. R. Ganellin, P. Lindberg and L. A. Mitscher, Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998), Pure Appl. Chem., 1998, 70, 1129–1143 CrossRef CAS.
- J. B. Chaires, F. Leng, T. Przewloka, I. Fokt, Y. H. Ling, R. Perez-Soler and W. Priebe, Structure-based design of a new bisintercalating anthracycline antibiotic, J. Med. Chem., 1997, 40, 261–266 CrossRef CAS PubMed.
- G. G. Hu, X. Shui, F. Leng, W. Priebe, J. B. Chaires and L. D. Williams, Structure of a DNA-bisdaunomycin complex, Biochemistry, 1997, 36, 5940–5946 CrossRef CAS PubMed.
- G. Zhang, L. Fang, L. Zhu, D. Sun and P. G. Wang, Syntheses and biological activity of bisdaunorubicins, Bioorg. Med. Chem., 2006, 14, 426–434 CrossRef CAS PubMed.
- I. S. Blagbrough and M. K. Soltan, Chapter 13: Polyamine conjugates as anti-cancer agents, in Biological aspects of biogenic amines, polyamines and conjugates, ed G. Dandrifosse, Research Signpost, India, 2009, pp. 343–370 Search PubMed.
- A. A. Moiseeva, O. I. Artyushin, L. V. Anikina and V. K. Brel, Synthesis and antitumor activity of daunorubicin conjugates with of 3,4-methylendioxybenzaldehyde, Bioorg. Med. Chem. Lett., 2019, 29, 126617 CrossRef CAS PubMed.
- S. Han, V. Zhou, S. Pan, Y. Liu, M. Hornsby, D. McMullan, H. E. Klock, J. Haugen, S. A. Lesley, N. Gray, J. Caldwell and X. Gu, Identification of coumarin derivatives as a novel class of allosteric MEK1 inhibitors, Bioorg. Med. Chem. Lett., 2005, 15, 5467–5473 CrossRef CAS PubMed.
- K. A. Nolan, J. R. Doncaster, M. S. Dunstan, K. A. Scott, A. D. Frenkel, D. Siegel, D. Ross, J. Barnes, C. Levy, D. Leys, R. C. Whitehead, I. J. Stratford and R. A. Bryce, Synthesis and biological evaluation of coumarin-based inhibitors of NAD(P)H: quinone oxidoreductase-1 (NQO1), J. Med. Chem., 2009, 52, 7142–7156 CrossRef CAS PubMed.
- K. Harada, H. Kubo, Y. Tomigahara, K. Nishioka, J. Takahashi, M. Momose, S. Inoue and A. Kojima, Coumarins as novel 17β-hydroxysteroid dehydrogenase type 3 inhibitors for potential treatment of prostate cancer, Bioorg. Med. Chem. Lett., 2010, 20, 272–275 CrossRef CAS PubMed.
- X.-Q. Wang, X.-B. Chen, P.-T. Ye, Z.-X. Yang, M.-J. Bai, S.-Y. Duan, Y. Li and X.-D. Yang, Synthesis and biological evaluation of novel 3-benzylcoumarin-imidazolium salts, Bioorg. Med. Chem. Lett., 2020, 30, 126896 CrossRef CAS PubMed.
- M. Rullo, M. Niso, L. Pisani, A. Carrieri, N. A. Colabufo, S. Cellamare and C. D. Altomare, 1,2,3,4-Tetrahydroisoquinoline/2H-chromen-2-one conjugates as nanomolar P-glycoprotein inhibitors: Molecular determinants for affinity and selectivity over multidrug resistance associated protein 1, Eur. J. Med. Chem., 2019, 161, 433–444 CrossRef CAS PubMed.
- V. R. Shanbhag, A. M. Crider, R. Gokhale, A. Harpalani and R. M. Dick, Ester and amide prodrugs of ibuprofen and naproxen: Synthesis, anti-inflammatory activity, and gastrointestinal toxicity, J. Pharm. Sci., 1992, 81, 149–154 CrossRef CAS PubMed.
- V. D. Dhakane, H. V. Chavan, V. N. Thakare, L. K. Adsul, S. N. Shringare and B. P. Bandgar, Novel ibuprofen prodrugs with improved pharmacokinetics and non-ulcerogenic potential, Med. Chem. Res., 2014, 23, 503–517 CrossRef CAS.
- A. M. Gouda, E. A. Beshr, F. A. Almalki, H. H. Halawah, B. F. Taj, A. F. Alnafaei, R. S. Alharazi, W. M. Kazi and M. M. AlMatrafi, Arylpropionic acid-derived NSAIDs: New insights on derivatization, anticancer activity and potential mechanism of action, Bioorg. Chem., 2019, 92, 103224–103230 CrossRef CAS PubMed.
- M. A. S. Abourehab, A. M. Alqahtani, F. A. Almalki, D. M. Zaher, A. N. Abdalla, A. M. Gouda and E. A. M. Beshr, Pyrrolizine/indolizine-NSAID hybrids: design, synthesis, biological evaluation, and molecular docking studies, Molecules, 2021, 26, 6582 CrossRef CAS PubMed.
- B. Martin, M. Sainlos, A. Aissaoui, N. Oudrhiri, M. Hauchecorne, J.-P. Vigneron, J.-M. Lehn and P. Lehn, The design of cationic lipids for gene delivery, Curr. Pharm. Des., 2005, 11, 375–394 CrossRef CAS PubMed.
- I. S. Blagbrough and A. A. Metwally, Chapter 7: siRNA and gene formulation for efficient gene therapy, in Gene Therapy – Tools and Potential Applications, ed. F. Martin, InTech, Open Access, 2013, pp. 135–173 doi DOI:10.5772/55518.
- I. S. Blagbrough and H. M. Ghonaim, Chapter 4: Polyamines and their conjugates for gene and siRNA delivery. in Biological aspects of biogenic amines, polyamines and conjugates, ed. G. Dandrifosse, Research Signpost: India, 2009, pp. 81–112 Search PubMed.
- J. P. Vigneron, N. Oudrhiri, M. Fauquet, L. Vergely, J. C. Bradley, M. Basseville, P. Lehn and J. M. Lehn, Guanidinium-cholesterol cationic lipids: efficient vectors for the transfection of eukaryotic cells, Proc. Natl. Acad. Sci. U.S.A., 1996, 93, 9682–9686 CrossRef CAS PubMed.
- Y. K. Ghosh, S. S. Visweswariah and S. Bhattacharya, Nature of linkage between the cationic headgroup and cholesteryl skeleton controls gene transfection efficiency, FEBS Lett., 2000, 473, 341–344 CrossRef CAS PubMed.
- A. A. Metwally and I. S. Blagbrough, Self-assembled lipoplexes of short interfering RNA (siRNA) using spermine-based fatty acid amide guanidines: Effect on gene silencing efficiency, Pharmaceutics, 2011, 3, 406–424, DOI:10.3390/pharmaceutics3030406.
- A. J. Geall and I. S. Blagbrough, Homologation of polyamines in the rapid synthesis of lipospermine conjugates and related lipoplexes, Tetrahedron, 2000, 56, 2449–2460 CrossRef CAS.
- A. A. Metwally, C. Pourzand and I. S. Blagbrough, Efficient gene silencing by self-assembled complexes of siRNA and symmetrical fatty acid amides of spermine, Pharmaceutics, 2011, 3, 125–140 CrossRef CAS PubMed.
- P. Belmont, A. Aissaoui, M. Hauchecorne, N. Oudrhiri, L. Petit, J.-P. Vigneron, J.-M. Lehn and P. Lehn, Aminoglycoside-derived cationic lipids as efficient vectors for gene transfection in vitro and in vivo, J. Gene Med., 2002, 4, 517–526 CrossRef CAS PubMed.
- H. M. Ghonaim and I. S. Blagbrough, Synthesis and formulations of lipid aminoglycoside conjugates: Nanoparticles for efficient gene and siRNA delivery, Int. J. Pharmacol. Pharm. Sci., 2014, 6, 153–157 Search PubMed.
- K. S. Moore, S. Wehrli, H. Roder, M. Rogers, J. N. Forrest Jr, D. McCrimmon and M. S. Zasloff, An aminosterol antibiotic from the shark, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1354–1358 CrossRef CAS PubMed.
- O. Kazakova, G. Giniyatullina, D. Babkov and Z. Wimmer, From marine metabolites to the drugs of the future: Squalamine, trodusquemine, their steroid and triterpene analogues, Int. J. Mol. Sci., 2022, 23, 1075, DOI:10.3390/ijms23031075.
- A. J. Geall, D. Al-Hadithi and I. S. Blagbrough, Spermine and thermine conjugates of cholic acid condense DNA, but lithocholic acid polyamine conjugates do so more efficiently, Chem. Commun., 1998, 2035–2036 RSC.
- I. S. Blagbrough, D. Al-Hadithi and A. J. Geall, Cheno-, urso-, and deoxycholic acid spermine conjugates: relative binding affinities for calf thymus DNA, Tetrahedron, 2000, 56, 3439–3447 CrossRef CAS.
- A. J. Geall, D. Al-Hadithi and I. S. Blagbrough, Efficient calf thymus DNA condensation upon binding with novel bile acid polyamine amides, Bioconjugate Chem., 2002, 13, 481–490 CrossRef CAS PubMed.
- A. J. Geall, M. A. W. Eaton, T. Baker, C. Catterall and I. S. Blagbrough, The regiochemical distribution of positive charges along cholesterol polyamine carbamates plays significant roles in modulating DNA binding affinity and lipofection, FEBS Lett., 1999, 459, 337–342 CrossRef CAS PubMed.
- A. J. Geall, R. J. Taylor, M. E. Earll, M. A. W. Eaton and I. S. Blagbrough, Synthesis of cholesteryl polyamine carbamates: pKa studies and condensation of calf thymus DNA, Bioconjugate Chem., 2000, 11, 314–326 CrossRef CAS PubMed.
- O. A. A. Ahmed, C. Pourzand and I. S. Blagbrough, Varying the unsaturation in N-4,N-9-dioctadecanoyl spermines: Nonviral lipopolyamine vectors for more efficient plasmid DNA formulation, Pharm. Res., 2006, 23, 31–40 CrossRef CAS PubMed.
- I. S. Blagbrough, A. J. Geall and A. P. Neal, Polyamines and novel polyamine conjugates interact with DNA in ways that can be exploited in non-viral gene therapy, Biochem. Soc. Trans., 2003, 31, 397–406 CrossRef CAS PubMed.
- I. S. Blagbrough, A. A. Metwally and O. A. A. Ahmed, Chapter 9: Polyamine-based agents for gene and siRNA transfer, in Polyamine Drug Discovery, ed. P. M. Woster and R. A. Casero Jr., RSC, Cambridge, 2012, pp. 205–237 Search PubMed.
- (a) H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America, Clin. Infect. Dis., 2009, 48, 1–12 CrossRef PubMed; (b) L. B. Rice, Progress and challenges in implementing the research on ESKAPE pathogens, Infect. Control Hosp. Epidemiol., 2010, 31, S7–S10 CrossRef PubMed.
- T. Cardoso, O. Ribeiro, I. C. Aragão, A. Costa-Pereira and A. E. Sarmento, Additional risk factors for infection by multidrug-resistant pathogens in healthcare-associated infection: a large cohort study, BMC Infect. Dis., 2012, 12, 375 CrossRef PubMed.
- J. A. Karlowsky, D. J. Hoban, M. A. Hackel, S. H. Lob and D. F. Sahm, Antimicrobial susceptibility of Gram-negative ESKAPE pathogens isolated from hospitalized patients with intra-abdominal and urinary tract infections in Asia–Pacific countries: SMART 2013–2015, J. Med. Microbiol., 2017, 66, 61–69 CrossRef PubMed.
- J. A. Karlowsky, D. J. Hoban, M. A. Hackel, S. H. Lob and D. F. Sahm, Resistance among Gram-negative ESKAPE pathogens isolated from hospitalized patients with intra-abdominal and urinary tract infections in Latin American countries: SMART 2013–2015, Braz. J. Infect. Dis., 2017, 21, 343–348 CrossRef PubMed.
- A.-P. Magiorakos, A. Srinivasan, R. B. Carey, Y. Carmeli, M. E. Falagas, C. G. Giske, S. Harbarth, J. F. Hindler, G. Kahlmeter, B. Olsson-Liljequist, D. L. Paterson, L. B. Rice, J. Stelling, M. J. Struelens, A. Vatopoulos, J. T. Weber and D. L. Monnet, Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance, Clin. Microbiol. Infect., 2012, 18, 268–281 CrossRef CAS PubMed.
- J. Ho, P. A. Tambyah and D. L. Paterson, Multiresistant Gram-negative infections: A global perspective, Curr. Opin. Infect. Dis., 2010, 23, 546–553 CrossRef PubMed.
- WHO. Antimicrobial resistance. Global report on surveillance. World Health Organ., 2014 Search PubMed.
- J. W. Costerton, Bacterial biofilms: A common cause of persistent infections, Sci. Am., 1999, 284, 1318–1322 CAS.
- R. J. Worthington, J. J. Richards and C. Melander, Small molecule control of bacterial biofilms, Org. Biomol. Chem., 2012, 10, 7457–7474 RSC.
- A. E. Barsoumian, K. Mende, C. J. Sanchez, M. L. Beckius, J. C. Wenke, C. K. Murray and K. S. Akers, Clinical infectious outcomes associated with biofilm-related bacterial infections: a retrospective chart review, BMC Infect. Dis., 2015, 15, 223 CrossRef PubMed.
- R. M. Donlan, Biofilms: microbial life on surfaces, Emerging Infect. Dis., 2002, 8, 881–890 CrossRef PubMed.
- L. Tan and C. Darby, A movable surface: Formation of Yersinia sp. biofilms on motile Caenorhabditis elegans, J. Bacteriol., 2004, 186, 5087–5092 CrossRef CAS PubMed.
- B. R. Boles and A. R. Horswill, Staphylococcal biofilm disassembly, Trends Microbiol., 2011, 19, 449–455 CrossRef CAS PubMed.
- Y. Abouelhassan, A. Basak, H. Yousaf and R. W. Huigens, Identification of N-arylated NH125 analogues as rapid eradicating agents against MRSA persister cells and potent biofilm killers of Gram-positive pathogens, ChemBioChem, 2017, 18, 352–357 CrossRef CAS PubMed.
- J. Lv, Y. Wang, C. Zhong, Y. Li, W. Hao and J. Zhu, The microbial attachment potential and quorum sensing measurement of aerobic granular activated sludge and flocculent activated sludge, Bioresour. Technol., 2014, 151, 291–296 CrossRef CAS PubMed.
- D. Mack, H. Rohde, L. G. Harris, A. P. Davies, M. A. Horstkotte and J. K.-M. Knobloch, Biofilm formation in medical device-related infection, Int. J. Artif. Organs, 2006, 29, 343–359 CrossRef CAS PubMed.
- K. Papenfort and B. L. Bassler, Quorum sensing signal–response systems in Gram-negative bacteria, Nat. Rev. Microbiol., 2016, 14, 576–588 CrossRef CAS PubMed.
- C. de la Fuente-Núñez, F. Reffuveille, E. F. Haney, S. K. Straus and R. E. W. Hancock, Broad-spectrum anti-biofilm peptide that targets a cellular stress response, PLoS Pathog., 2014, 10, e1004152 CrossRef PubMed.
- C. de la Fuente-Núñez, F. Reffuveille, S. C. Mansour, S. L. Reckseidler-Zenteno, D. Hernández, G. Brackman, T. Coenye and R. E. W. Hancock, D-Enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections, Chem. Biol., 2015, 22, 196–205 CrossRef PubMed.
- H. Wang and Y. Tor, Electrostatic interactions in RNA aminoglycosides binding, J. Am. Chem. Soc., 1997, 119, 8734–8735 CrossRef CAS.
- A. H. Alkhzem, T. J. Woodman and I. S. Blagbrough, Individual pKa values of tobramycin, kanamycin B, amikacin, sisomicin, and netilmicin determined by multinuclear NMR spectroscopy, ACS Omega, 2020, 5, 21094–21103, DOI:10.1021/acsomega.0c02744.
- A. H. Alkhzem, T. J. Woodman and I. S. Blagbrough, Multinuclear nuclear magnetic resonance spectroscopy is used to determine rapidly and accurately the individual pKa values of 2-deoxystreptamine, neamine, neomycin, paromomycin, and streptomycin, ACS Omega, 2021, 6, 2824–2835, DOI:10.1021/acsomega.0c05138.
- A. G. Santana, Á. Bastida, T. M. Del Campo, J. L. Asensio and J. Revuelta, An efficient and general route to the synthesis of novel aminoglycosides for RNA binding, Synlett, 2011, 2011, 219–222 CrossRef.
- T. Idowu, G. G. Zhanel and F. A. Schweizer, Dimer, but not monomer, of tobramycin potentiates ceftolozane against multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa and delays resistance development, Antimicrob. Agents Chemother., 2020, 64 Search PubMed.
- G. G. Zhanel, S. Fontaine, H. Adam, K. Schurek, M. Mayer, A. M. Noreddin, A. S. Gin, E. Rubinstein and D. J. Hoban, A review of new fluoroquinolones, Treat. Respir. Med., 2006, 5, 437–465 CrossRef CAS PubMed.
- J. Sousa, G. Alves, A. Fortuna and A. Falcao, Third and fourth generation fluoroquinolone antibacterials: A systematic review of safety and toxicity profiles, Curr. Drug Saf., 2014, 9, 89–105 CrossRef CAS PubMed.
- V. Pokrovskaya, V. Belakhov, M. Hainrichson, S. Yaron and T. Baasov, Design, synthesis, and evaluation of novel fluoroquinolone - Aminoglycoside hybrid antibiotics, J. Med. Chem., 2009, 52, 2243–2254 CrossRef CAS PubMed.
- H. Labischinski; J. Cherian; C. Cleofe and R. S. BoyceHybrid antimicrobial compounds and their use, WO2010025906, 2010.
- D. Baldoni, M. Gutierrez, W. Timmer and J. Dingemanse, Cadazolid, a novel antibiotic with potent activity against Clostridium difficile: safety, tolerability and pharmacokinetics in healthy subjects following single and multiple oral doses, J. Antimicrob. Chemother., 2014, 69, 706–714 CrossRef CAS PubMed.
- B. Wang, B. Pachaiyappan, J. D. Gruber, M. G. Schmidt, Y.-M. Zhang and P. M. Woster, Antibacterial diamines targeting bacterial membranes, J. Med. Chem., 2016, 59, 3140–3151 CrossRef CAS PubMed.
- M. M. Konai, C. Ghosh, V. Yarlagadda, S. Samaddar and J. Haldar, Membrane active phenylalanine conjugated lipophilic norspermidine derivatives with selective antibacterial activity, J. Med. Chem., 2014, 57, 9409–9423 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |