
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, electropolymerization and functionalization via click chemistry of N-alkynylated dithieno[3,2-b:2′,3′-d]pyrrole†
Yuriy Banderaab,
Haley W. Jones ab,
Benjamin Grantab,
Sarah Mellab and
Stephen H. Foulger*abc
ab,
Benjamin Grantab,
Sarah Mellab and
Stephen H. Foulger*abc
aCenter for Optical Materials Science and Engineering Technologies (COMSET), Clemson University, Anderson, SC 29625, USA
bDepartment of Materials Science and Engineering, Clemson University, Clemson, SC 29634, USA
cDepartment of Bioengineering, Clemson University, Clemson, SC 29634, USA. E-mail: foulger@clemson.edu
First published on 13th October 2022
Abstract
A new N-alkynylated dithieno[3,2-b:2′,3′-d]pyrrole (DTP) monomer was synthesized using a Buchwald–Hartwig amination of 3,3′-dibromo-2,2′-bithiophene with pent-4-yn-1-amine. The obtained monomer was investigated for the possibility of a pre-polymerization modification via Huisgen 1,3-dipolar cycloaddition (“click”) reaction with azide-containing organic compounds. The synthesized N-alkynylated DTP monomer is soluble in a number of organic solvents and reacts with organic azides via “click” reactions in mild conditions, achieving high yields. The N-alkynylated DTP monomer and its “click”-modified derivative can be electropolymerized to form polymeric films. Herein, the synthesis and characterization of a “click” modified DTP monomer, its pre-modified derivative, and their corresponding polymers are described. The developed method is a facile route to synthesize a new generation of various N-functionalized DTP homopolymers.
Introduction
Dithieno[3,2-b:2′,3′-d]pyrrole (DTP) and its N-substituted derivatives are popular building blocks to prepare a wide array of conjugated organic polymers.1,2 Due to the tunable material properties achieved by changing the structure of the monomeric unit, DTP-based polymers are employed in various applications. The optical and electronic properties of various polyDTPs have been investigated and applied in organic electronics, such as organic light-emitting diodes (OLEDs),3,4 organic field effect transistors (OFETs),5–9 electrochromic devices,6 organic photovoltaic devices8,10–14 and organic batteries.15 Additionally, N-alkyl substituted DTP homopolymers have been used as highly fluorescent materials16 and biosensors.17,18The majority of DTP monomers and their polymeric systems are based on dithienopyrroles with traditional alkyl and aryl side chains and are termed first generation DTPs.2 So-called second generation DTPs are N-acylated DTPs, which show more stable HOMO and LUMO energy levels than first generation DTPs.19 Recently, the synthesis of the first N-functionalized DTP monomer with an ethynyl group was described.20 The presence of a terminal triple bond allows for the pre- or post-polymerization modification of the corresponding monomer or polymer via 1,3-dipolar cycloaddition “click” reactions21,22 or Sonogashira cross-coupling reactions.22 This method can be prospective for an easy and efficient route to synthesize new bio-conjugated DTP homopolymers containing various pendant groups attached to the conjugated polymer backbone via “click” reactions with corresponding organic azides. However, this technique would be easily applicable in practice only if the new N-alkynylated DTP monomer is a compact molecule, well soluble in organic solvents, and has a relatively simple synthetic preparation. To the best of our knowledge, the first alkynylated DTP monomer described in literature20 is the only known N-functionalized DTP with a terminal triple bond. Its preparation required two steps, beginning with a Buchwald–Hartwig cross-coupling reaction of 3,3′-dibromo-2,2′-bithiophene and 4-[(trimethylsilyl)ethynyl]-aniline, followed by the deprotection of the alkynyl group. In this product, the terminal triple bond was attached to the DTP ring by the phenyl group, which resulted in a bulky DTP molecule due to the additional aromatic ring in its structure.
Herein, a one-step synthesis of a new N-alkynylated DTP monomer via a Buchwald-Hartwig cross-coupling reaction23 with pent-4-yn-1-amine and commercially available 3,3′-dibromo-2,2′-bithiophene is described. Additionally, a facile method to functionalize the new DTP monomer prior to polymerization via a copper-catalyzed azide/alkyne cycloaddition reaction is demonstrated. This methodology provides a rapid approach to fabricate different functionalized polyDTPs.
Materials and methods
Reagents and solvents
All chemicals were purchased from Alfa Aesar or TCI America and used without further purification. All solvents used for reactions were distilled under nitrogen after drying over an appropriate drying reagent. All manipulations involving air- and/or moisture-sensitive compounds were carried out with the standard Schlenk technique under nitrogen.Characterization
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on JEOL ECX-300 spectrometer. Chemical shifts for protons are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CDCl3: δ 7.26 ppm, D2O: δ 4.79 ppm). Chemical shifts for carbons are reported in ppm downfield from tetramethylsilane and are referenced to the carbon resonances of the solvent (CDCl3: δ 77.16 ppm). Melting points were determined on an EZ-Melt automated melting point apparatus. Analytical thin-layer chromatography was performed on glass plates coated with 0.25 mm 230–400 mesh silica gel containing a fluorescent indicator. Column chromatography was performed using silica gel (spherical neutral, particle size 63–210 μm). Electrospray (ESI) mass spectra were obtained using an Agilent 6230 Time-of-Flight (TOF) spectrometer. Fourier transform infrared (FTIR) spectra were obtained with a Thermo Scientific Nicolet 6700 FTIR spectrometer and all spectra were measured using an attenuated total reflection (ATR) attachment with a diamond head. Absorption spectra were obtained using a PerkinElmer Lambda 900 UV-vis/NIR spectrophotometer. A BASi 100A Electrochemical Analyzer and a C3 Cell Stand were used to perform cyclic voltammetry (CV) measurements. A KLA Alpha-HQ Profilometer was used to measure film thickness.Synthesis
The same reaction as previously described was also performed with pent-4-yn-1-amine base (3) (3 mol excess) and powdered sodium methoxide (3 mol excess) in boiling toluene for 40 hours. This reaction resulted in AlkyneDTP with a 48% yield.
Electropolymerization
Cyclovoltammetry (CV) was performed with a BASi 100A Electrochemical Analyzer coupled to a C3 Cell Stand. p-AlkyneDTP was formed by adding 2 mL of a 0.01 M solution of AlkyneDTP in acetonitrile to 6 mL of 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) solution in acetonitrile in a C3 Cell Stand. The cell was purged with nitrogen for 20 minutes, followed by the insertion of a 3-electrode configuration with a silver/silver chloride (Ag/AgCl) reference, platinum (Pt) counter, and either a templated indium tin oxide (ITO) or Pt button working electrode. To electropolymerize the monomer into a film, the electrodes were cycled over 50 times at a scan rate of 100 mV s−1 from −0.5 V to +0.9 V (vs. Ag/AgCl). To ensure the films were fully polymerized, the films were oxidized in pure electrolyte at +1 V for 120 seconds. AlkyneDTP resulted in p-AlkyneDTP films that were blue colored when in the oxidized form (+0.9 V) and yellow colored when in the reduced form (−0.5 V). The p-AlkyneDTP film thickness was measured to be 255.5 nm.p-DTPNap was formed by adding 1 mL of a 0.01 M solution of DTPNap in dichloromethane to 6 mL of 0.1 M TBAPF6 solution in dichloromethane in a C3 Cell Stand. The cell was purged with nitrogen for 20 minutes, followed by the insertion of a 3-electrode configuration with a Ag/AgCl reference, Pt counter, and a templated ITO working electrode. To electropolymerize the monomer into a film, the electrodes were cycled over 50 times at a scan rate of 100 mV s−1 from −0.5 V to +0.9 V (vs. Ag/AgCl). To ensure the films were fully polymerized, the films were oxidized in pure electrolyte at +1 V for 120 seconds. DTPNap resulted in p-DTPNap films that were blue colored when in the oxidized form (+0.9 V) and red colored when in the reduced form (−0.5 V). The p-AlkyneDTP film thickness was measured to be 528.3 nm. Data presented in the IUPAC redox voltage and current convention (positive potential to the right of the graph; positive/oxidizing current at the top of the graph).
Results and discussion
Synthesis
The unfunctionalized, parent dithieno[3,2-b:2′,3′-d]pyrrole (DTP) molecule was first synthesized in 1983.26 Nearly a decade later, N-alkylated DTPs were prepared by the alkylation of the parent DTP.27 Shortly thereafter, an improved method of N-alkyl DTP preparation was developed using Buchwald–Hartwig amination of 3-bromothiophene.28 The current preferred synthetic method to prepare N-alkyl DTPs (cf. Scheme 1) is based on a Pd-catalyzed Buchwald-Hartwig coupling reaction of the primary amine with 3,3′-dibromo-2,2′-bithiophene.29,30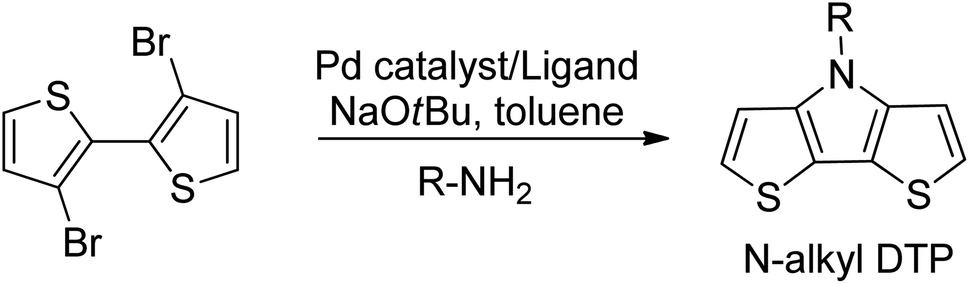 | ||
| Scheme 1 Synthesis of N-alkyl DTPs. | ||
This reaction achieves the highest yields with primary alkylamines by using a tris(dibenzylideneacetone)dipalladium(0) (Pd2(DBA)3) catalyst and a 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) ligand in the presence of sodium tert-butoxide (NaOtBu). However, there is no literature regarding the preparation of alkynylated dithieno[3,2-b:2′,3′-d]pyrroles using Buchwald–Hartwig amination with primary alkylamines containing terminal triple bonds. This approach does not work with primary alkylamines, such as propargylamine or its higher homologs. When the reaction is performed using a Pd2(DBA)3 catalyst, BINAP ligand and a NaOtBu base, only the starting compounds are detected at the end of the reaction with no final product. In the efforts to determine a synthetic route to an alkyne-terminated DTP monomer, pent-4-yn-1-amine hydrochloride (2), it's free base (3), and commercially available propargylamine were used as starting alkylamines for the Buchwald–Hartwig amination of 3,3′-dibromo-2,2′-bithiophene.
Pent-4-yn-1-amine hydrochloride (2) was synthesized starting from commercially available 5-chloropent-1-yne (cf. Scheme 2). In brief, 5-chloropent-1-yne was converted into its azide derivative 5-azidopent-1-yne (1). Compound (1) was then reduced by a Staudinger reaction to the hydrochloric salt of pent-4-yn-1-amine (2). Pent-4-yn-1-amine free base (3) was isolated from its hydrochloride salt (2) by the extraction of a saturated, basified aqueous solution of compound (2) with diethyl ether. However, it was preferable to work with hydrochloride (2) because the free base (3) is volatile and well soluble in water, which makes it difficult to separate at high yields.
 | ||
| Scheme 2 Synthesis of 5-azidopent-1-yne (1) (yield 97%), pent-4-yn-1-amine hydrochloride (2) (yield 71%), and pent-4-yn-1-amine (3) (yield 50%). | ||
Having all amines in hand, a Buchwald–Hartwig coupling amination with 3,3′-dibromo-2,2′-bithiophene was investigated. It is important to note that when the common base for this type of coupling reaction, NaOtBu, was used, only starting compounds were observed in the reaction media after 24 hours in boiling toluene. NaOtBu base was substituted with NaOMe, and it was found that pent-4-yn-1-amine hydrochloride (2) or its free base (3) reacted with 3,3′-dibromo-2,2′-bithiophene in a refluxed toluene solution in the presence of a Pd2(DBA)3 catalyst, BINAP ligand, and excess MeONa to form N-alkynylated DTP AlkyneDTP (cf. Scheme 3). The same coupling reaction with propargylamine, a lower homolog of pent-4-yn-1-amine (3), was performed in a closed flask and no product was detected by NMR analysis after several hours at 120 °C in a toluene solution. Propargylamine is less nucleophilic than its higher homologs and does not react with 3,3′-dibromo-2,2′-bithiophene under the reaction conditions. The greatest AlkyneDTP yield (48%) was obtained when 3 moles (excess) of the amine base (3) or 2 moles of its hydrochloride salt (2) were used. The fastest reaction occurred when using the free amine base (3) and was complete after 40 hours in boiling toluene under a nitrogen atmosphere with 3 moles MeONa. When using the hydrochloride salt (2), the reaction required 5 moles of MeONa and 48 hours in boiling toluene under a nitrogen atmosphere and ended with lower AlkyneDTP yields (37%). The reaction time could be twice reduced by raising the temperature to 130 °C and using a closed, heavy wall pressure vessel.
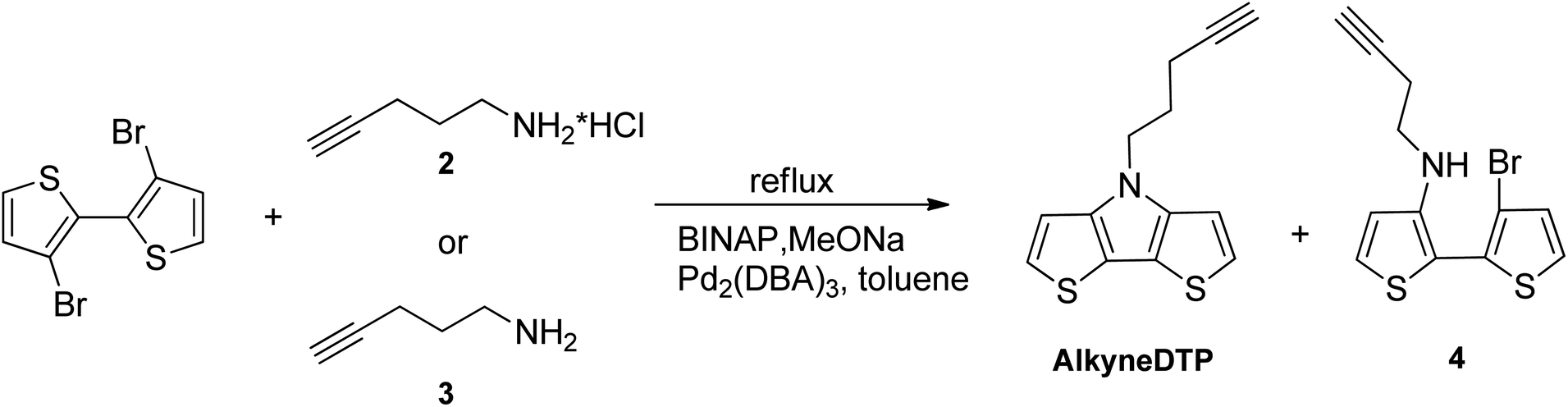 | ||
| Scheme 3 Synthesis of AlkyneDTP (yield 37%, 48%) and mono-aminated intermediate (4). | ||
The amination of 3,3′-dibromo-2,2′-bithiophene with pent-4-yn-1-amine began with the formation of a mono-aminated intermediate (4). The secondary amino group in intermediate (4) finally reacted with a second bromine atom and formed the corresponding dithienopyrrole AlkyneDTP. The formation of the mono-aminated intermediate (4) occurred relatively quickly under the reaction conditions, usually within the first 6 hours, and could be easily tracked by NMR or TLC analyses. In the NMR spectra, the methylene group (–NH–CH2–) in intermediate (4) has a distinct triplet at 3.34 ppm in deuterated chloroform, while the final AlkyneDTP product has this signal shifted to 4.37 ppm. The ring closure and the formation of AlkyneDTP was a slower process and took more time than the formation of the mono-aminated intermediate (4). Usually after 24 hours at 130◦C, the reaction with pent-4-yn-1-amine hydrochloride (2) produced a mix of the final product AlkyneDTP and the mono-aminated intermediate (4) with a ratio 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 correspondingly. Increasing the reaction time to fully complete the process did not improve the yield because of the formation of side products. However, intermediate (4) could be easily separated from AlkyneDTP by column chromatography and converted to the final AlkyneDTP with a high yield (90%) by a repeated coupling reaction.
:3 correspondingly. Increasing the reaction time to fully complete the process did not improve the yield because of the formation of side products. However, intermediate (4) could be easily separated from AlkyneDTP by column chromatography and converted to the final AlkyneDTP with a high yield (90%) by a repeated coupling reaction.
To investigate the pre-polymerization functionalization of AlkyneDTP via copper-catalyzed 1,3-dipolar cycloaddition, an azide-modified naphthalimide dye (AzNap)25 was used (cf. Scheme 4). The AzNap dye has a maximum absorption at 400 nm that can be easily tracked by UV-vis analysis when attached to the DTP monomer (DTPNap) and corresponding polymer (p-DTPNap).
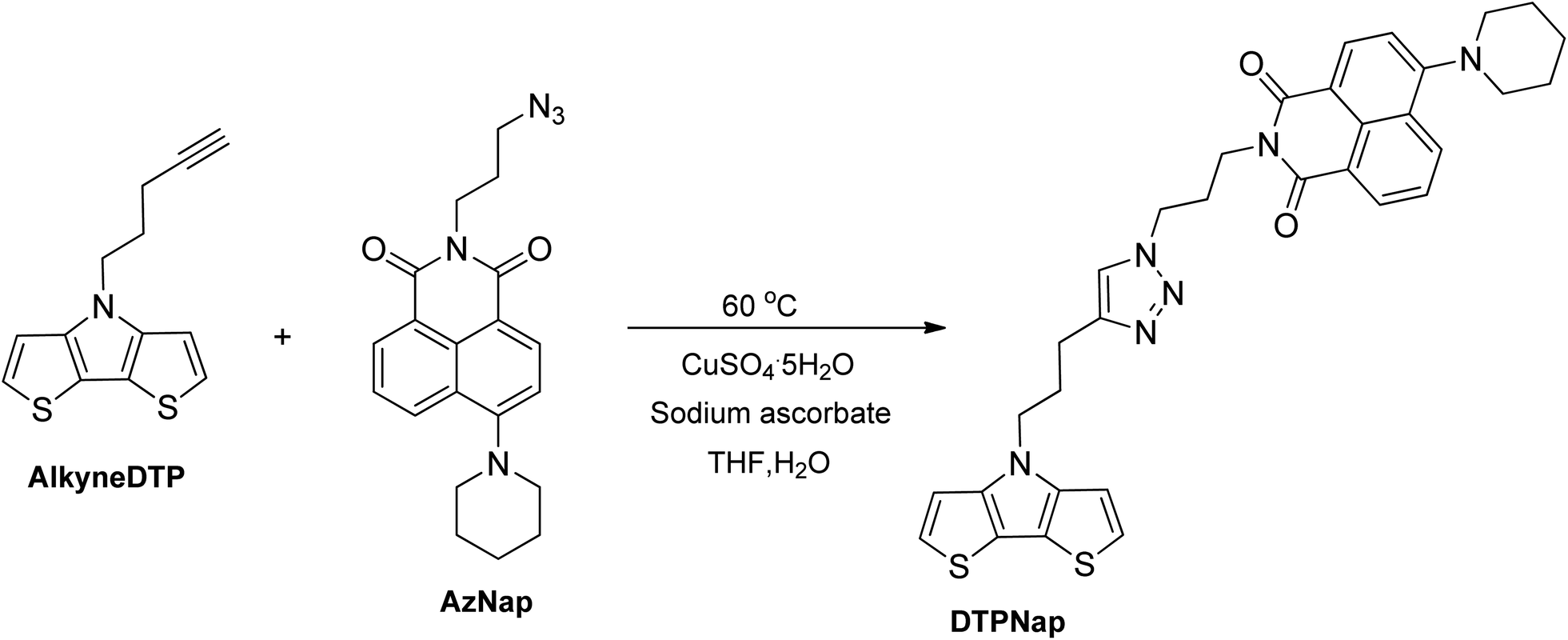 | ||
| Scheme 4 Synthesis of DTPNap (yield 81%) via 1,3-dipolar cycloaddition. | ||
A cycloaddition reaction of AlkyneDTP and AzNap was performed in a tetrahydrofuran/water solution and catalyzed with aqueous solutions of copper(II) sulfate pentahydrate and sodium ascorbate to generate Cu(I) in situ.31 This reaction did not occur at room temperature and required heating at 60 °C for completion. The reaction is regioselective21,32 and, as expected, a single regioisomer of DTPNap monomer was isolated with an 81% yield.
Electropolymerization
Monomers comprising of the dithieno[3,2-b:2,3-d]pyrrole (DTP) backbone can be electrochemically and chemically polymerized.16,27–29The two synthesized monomers, AlkyneDTP and DTPNap, were electrochemically polymerized to form films of p-AlkyneDTP and p-DTPNap (cf. Scheme 5), respectively, on Pt button electrodes. Fig. 1a presents the 1st and 50th cyclic voltammogram of the AlkyneDTP monomer in an acetonitrile/0.1 M TBAPF6 electrolyte. In the 1st cycle, the irreversible peak oxidation of the monomer has an onset at 770 mV.
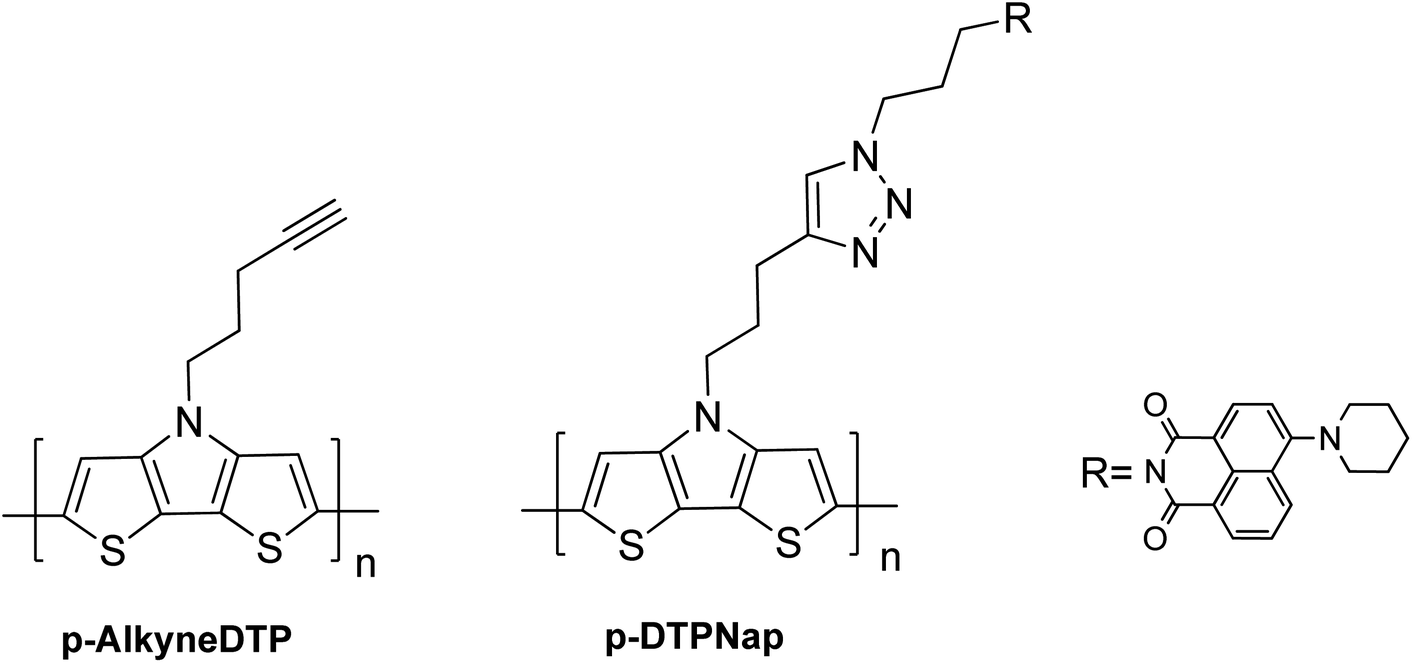 | ||
| Scheme 5 Polymers p-AlkyneDTP and p-DTPNap. | ||
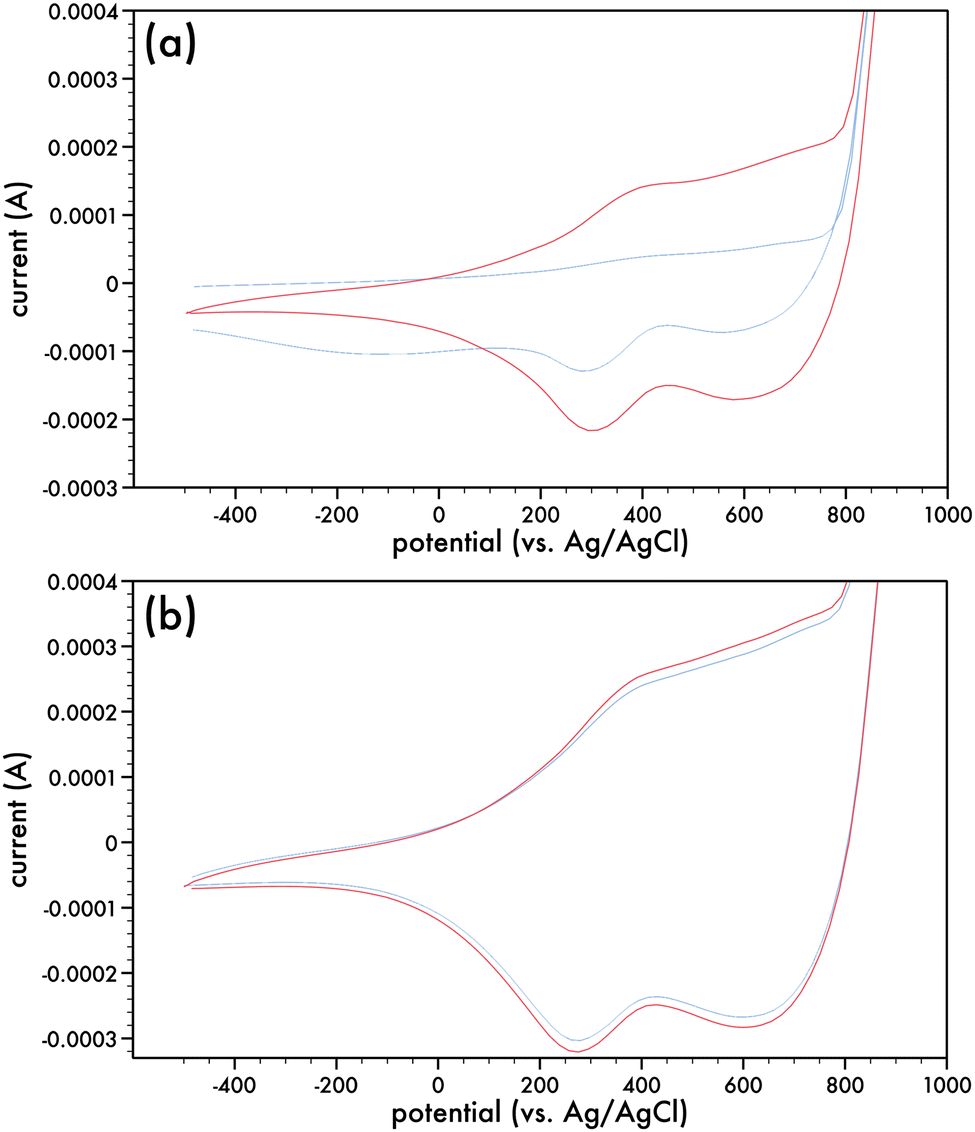 | ||
| Fig. 1 Cyclic voltammograms for the oxidation of (a) AlkyneDTP monomer (c = 2.5 mM) in acetonitrile/0.1 M TBAPF6; 1st cycle (blue) and 50th cycle (red). (b) p-AlkyneDTP; 25th cycle (blue) and 50th cycle (red). Electrolyte was acetonitrile/0.1 M TBAPF6 and scan rate was 100 mV s−1. | ||
Two very small reduction peaks at ca. 695 mV and 277 mV (which are much more evident in the 50th cycle) on the back-sweep are indicative of the reduction of the initially deposited polymer. Films of polymerized AlkyneDTP (p-AlkyneDTP) were formed by substituting the Pt button working electrode with ITO-coated glass slides or Pt strips. Electropolymerizing the monomer at +1000 mV (vs. Ag/AgCl) for 150 seconds resulted in thick adherent dark-blue films that were insoluble in water or organic solvents. This insolubility has been previously reported with electropolymerized DTP monomers.16 These films were conditioned prior to running their cyclic voltammograms by repeatedly cycling them in a monomer-free solution of acetonitrile/0.1 M TBAPF6. Cyclic voltammograms were run from −500 mV to +900 mV (100 mV s−1) (cf. Fig. 1b) and indicate the reversibility of the oxidation process of the p-AlkyneDTP films by the re-tracings of the cyclic runs. This suggests that no electrochemical degradation has taken place and the onset of the oxidation potential can be related to the ionization potential (IP). The onset potential (E0) was estimated from the intersection of two tangent lines drawn at the rising oxidation current and background current to give a value of 60 mV. Referring the potential value to the standard hydrogen electrode (SHE) and in turn to the vacuum level reference, the IP (estimated through IP (eV) = e(E0 + 4.4)) for p-AlkyneDTP is 4.46 eV.33,34 Readily oxidizable conjugated polymers are characterized by IP values under 5 eV, such as poly(pyrrole) and poly(1,3-ethylenedioxythiophene), which are more stable in their poly(cationic) (doped) state than in their neutral state at ambient conditions.35 For comparison, the electrochemical properties of poly(4-(6-hexyl)-4H-dithieno[3,2-b:2′,3′-d]pyrrole) (p6DTP) have been recently presented and the IP of p6DTP is 4.4 eV, a value similar to reported energy levels in some N-substituted alkyl and alkyl ether polyDTPs.36–38
Fig. 2a presents the 1st cyclic voltammogram of the DTPNap monomer in a dichloro-methane/0.1 M TBAPF6 electrolyte and is relatively featureless in comparison to the initial cyclic voltammogram of the AlkyneDTP monomer (cf. Fig. 1a). Nonetheless, there is an irreversible oxidation peak of the monomer, which has an onset at 200 mV. Films of p-DTPNap were formed with ITO-coated glass slides or Pt strips as the working electrode. Electropolymerizing the monomer at +1000 mV (vs. Ag/AgCl) for 150 seconds resulted in thick adherent dark-blue films that were conditioned in monomer-free electrolyte prior to running cyclic voltammograms. Fig. 2b presents a number of the cyclic voltammograms of the p-DTPNap films indicating the reversibility of the oxidation process when cycled from −500 mV to +900 mV. With an E0 of 160 mV, the IP is estimated to be 4.56 eV for p-AlkyneDTP.
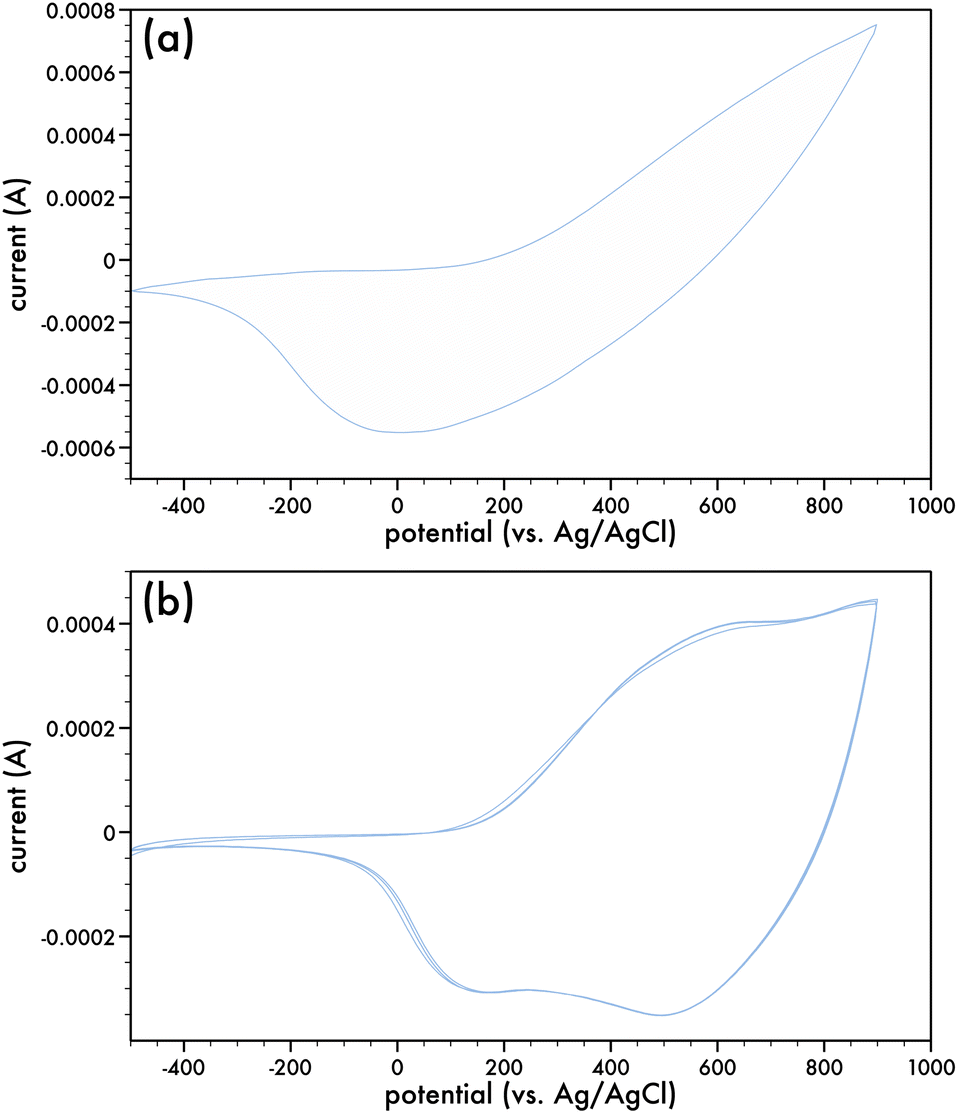 | ||
| Fig. 2 Cyclic voltammograms for the oxidation of (a) DTPNap monomer (c = 2.5 mM) in dichloromethane/0.1 M TBAPF6; 1st cycle (blue). (b) p-DTPNap; multiple cycles. Electrolyte was dichloromethane/0.1 M TBAPF6 and scan rate was 100 mV s−1. | ||
FTIR spectroscopy
Fourier transform infrared (FTIR) spectra of the monomers were performed and compared to that of the corresponding polymers (cf. Fig. 3). As expected, the AlkyneDTP monomer exhibits the characteristic –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– stretch vibration as a weak narrow peak at 2116 cm−1 and a terminal alkyne C–H bond stretch appears as a strong narrow band at 3266 cm−1. The FTIR spectra of the electropolymerized polymer p-AlkyneDTP does not show any of the characteristic alkyne bond adsorption signals. However, the FTIR spectra of p-AlkyneDTP does show strong bands in the 3000–2850 cm−1 region, confirming the presence of aliphatic –CH2– groups. These results indicate that the terminal alkyne group in AlkyneDTP was not stable during electropolymerization. Because the synthesized polymer p-AlkyneDTP no longer contained alkyne functional groups, it was not a suitable material for post-polymerization functionalization via “click” reactions with organic azides.
C– stretch vibration as a weak narrow peak at 2116 cm−1 and a terminal alkyne C–H bond stretch appears as a strong narrow band at 3266 cm−1. The FTIR spectra of the electropolymerized polymer p-AlkyneDTP does not show any of the characteristic alkyne bond adsorption signals. However, the FTIR spectra of p-AlkyneDTP does show strong bands in the 3000–2850 cm−1 region, confirming the presence of aliphatic –CH2– groups. These results indicate that the terminal alkyne group in AlkyneDTP was not stable during electropolymerization. Because the synthesized polymer p-AlkyneDTP no longer contained alkyne functional groups, it was not a suitable material for post-polymerization functionalization via “click” reactions with organic azides.
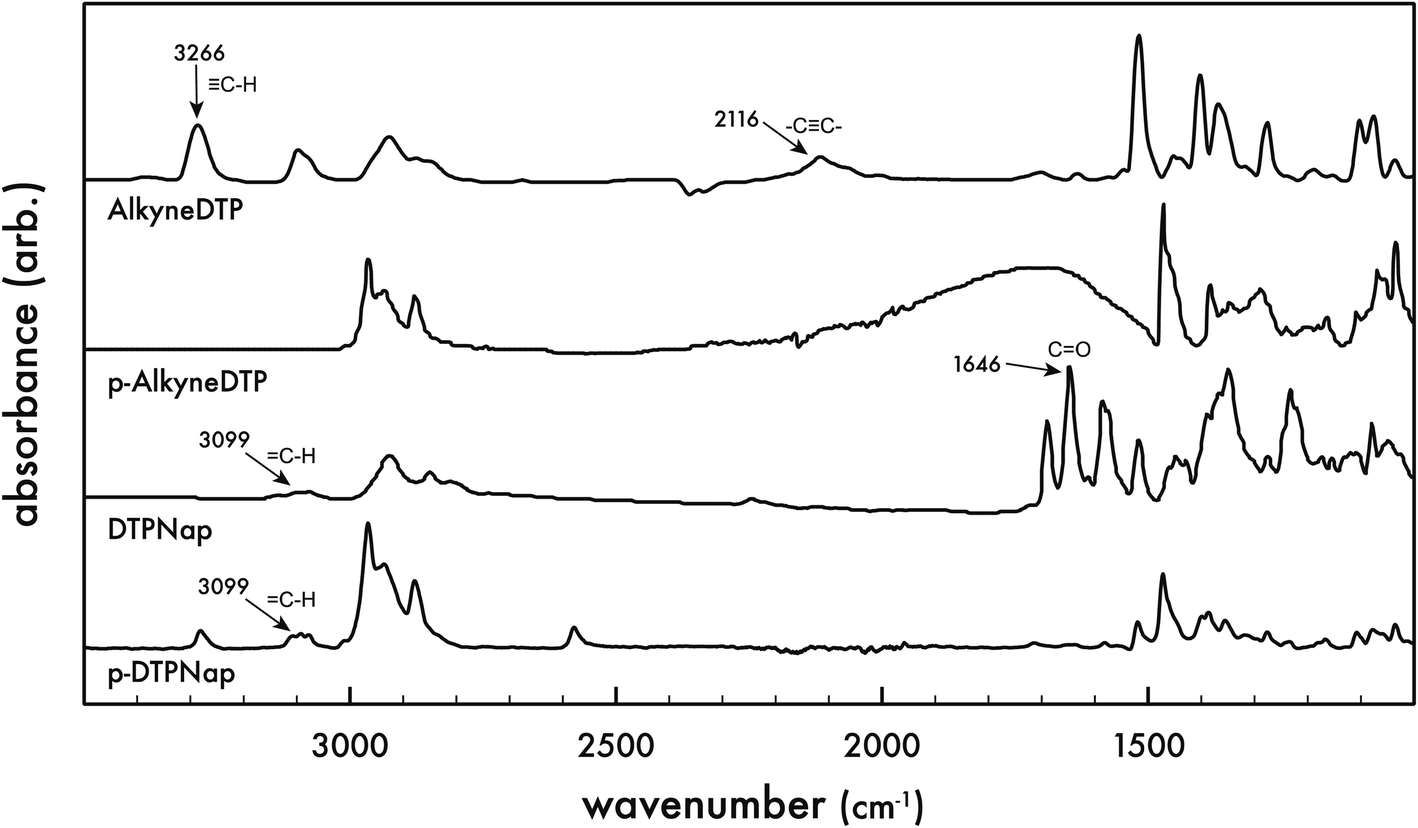 | ||
| Fig. 3 Fourier transform infrared (FTIR) spectra of monomers, AlkyneDTP and DTPNap, and corresponding polymers, p-AlkyneDTP and p-DTPNap. | ||
FTIR spectra of the DTPNap monomer contains a few bands at 3099 cm−1, which are stretching vibrations of the triazole ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H bond and aromatic C–H groups. Similar to the AlkyneDTP monomer, there are aliphatic –CH2– bands observed at 2925–2849 cm−1. The CO carbonyl groups in the DTPNap monomer have a strong band at 1646 cm−1 but this signal as well as other bands around 1640 cm−1 disappear after electropolymerization, which indicates the absence of carbonyl groups in p-DTPNap polymer due to their partial or complete reduction or another electrochemical reaction during electropolymerization. Similar to the DTPNap monomer, the FTIR spectra of p-DTPNap polymer has bands ranging from 3100–2800 cm−1 that correspond to aliphatic, aromatic C–H and triazole = C–H groups. These results indicate that the pre-polymerization functionalization of AlkyneDTP via a 1,3-dipolar cycloaddition reaction with an azide-modified molecule is a promising route to synthesize new DTP-based monomers and electropolymerization polymers.
C–H bond and aromatic C–H groups. Similar to the AlkyneDTP monomer, there are aliphatic –CH2– bands observed at 2925–2849 cm−1. The CO carbonyl groups in the DTPNap monomer have a strong band at 1646 cm−1 but this signal as well as other bands around 1640 cm−1 disappear after electropolymerization, which indicates the absence of carbonyl groups in p-DTPNap polymer due to their partial or complete reduction or another electrochemical reaction during electropolymerization. Similar to the DTPNap monomer, the FTIR spectra of p-DTPNap polymer has bands ranging from 3100–2800 cm−1 that correspond to aliphatic, aromatic C–H and triazole = C–H groups. These results indicate that the pre-polymerization functionalization of AlkyneDTP via a 1,3-dipolar cycloaddition reaction with an azide-modified molecule is a promising route to synthesize new DTP-based monomers and electropolymerization polymers.
UV-vis spectroscopy
The optical absorbance of monomers AlkyneDTP and DTPNap in dichloromethane are presented in Fig. 4. The monomers exhibit similar absorption peaks with maximums at 227, 298, and 310 nm, as well as a shoulder at ca. 288 nm. Additionally, DTPNap exhibits a peak with a maximum at 413 nm, which is attributed to the naphthalimide dye (AzNap) attached to the monomer via a “click” reaction.39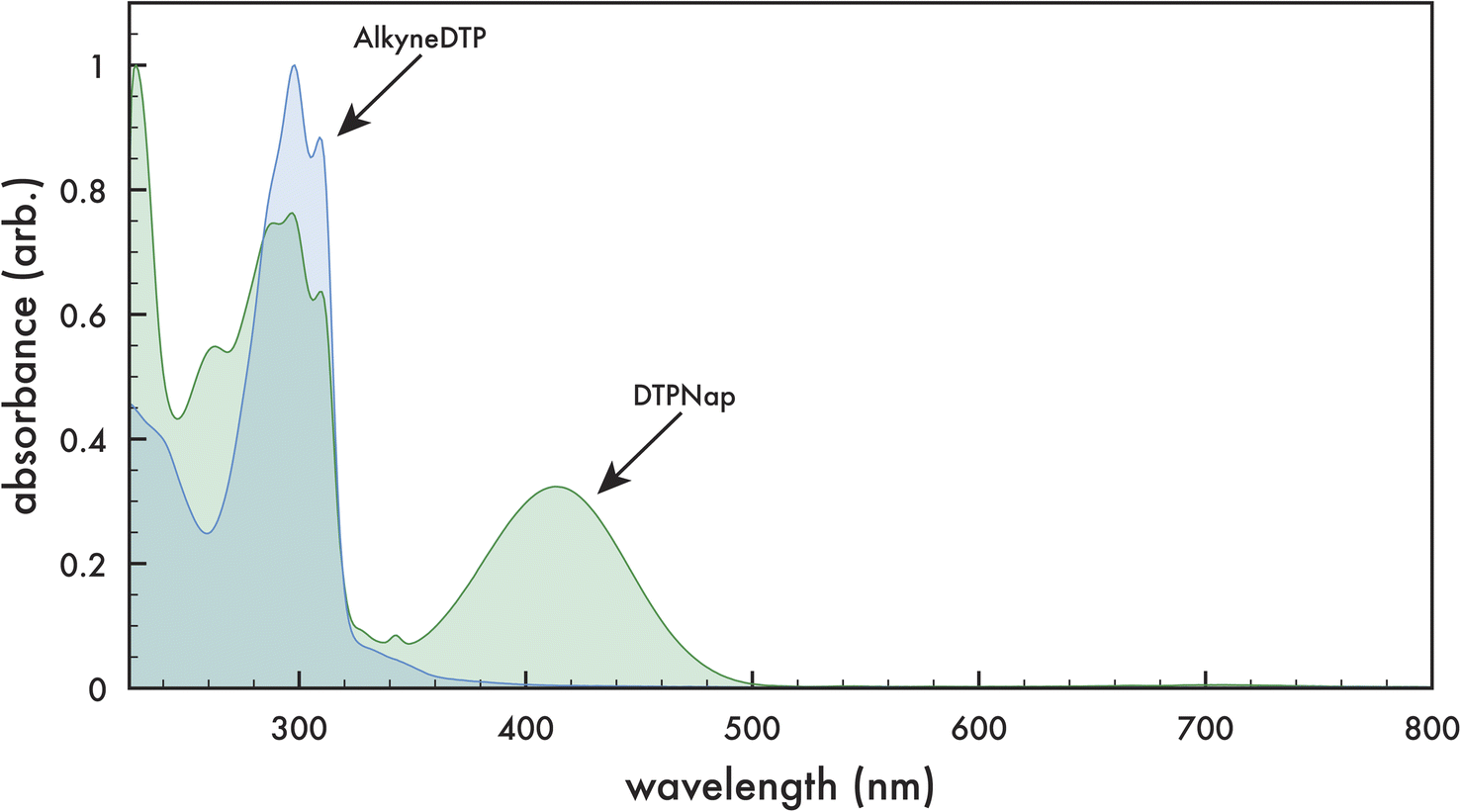 | ||
| Fig. 4 Optical absorption spectra of AlkyneDTP (blue) and DTPNap (green) monomers in dichloromethane. | ||
Spectroelectrochemical absorption properties were obtained on the corresponding polymers p-AlkyneDTP (cf. Fig. 5a) and p-DTPNap (cf. Fig. 5b) in the solid state on ITO-coated glass slides. To measure the spectroelectrochemical properties, a 3-terminal electrochemical cell with a Pt wire counter electrode, Ag/AgCl reference electrode, and the electropolymerized films on ITO-coated glass as working electrodes were tested in a dichloromethane/0.1 M TBAPF6 electrolyte solution. Each film was probed at a specified voltage (+900 mV, +500 mV, −200 mV, and −500 mV (vs. Ag/AgCl)) for 120 seconds prior to measuring the optical properties by UV-vis spectroscopy. Both polymers demonstrate typical spectra for conjugated polymers undergoing redox reactions. The reduced form of p-AlkyneDTP (−500 mV (vs. Ag/AgCl)) exhibits a maximum absorption peak at 545 nm. By increasing the potential across the p-AlkyneDTP film up to +900 mV (vs. Ag/AgCl), maximum absorption peaks at 388 nm and 760 nm appear as the reduced peak at 545 nm diminishes. Additionally, as voltage to the working electrode increased, a featureless absorption tail appeared extending from 800 nm to beyond 1350 nm. The reduced form of p-DTPNap exhibits a maximum absorption peak at 431 nm when charged at −500 mV (vs. Ag/AgCl). This maximum peak is attributed to the naphthalimide dye (AzNap) attached to the polymer via “click” reaction. By increasing the potential across the p-DTPNap film up to +900 mV (vs. Ag/AgCl), the maximum peak blue-shifts to 409 nm and a new local maximum absorption peak appears at 737 nm. Similar to p-AlkyneDTP, a featureless absorption tail appears extending from 820 nm to beyond 1200 nm.
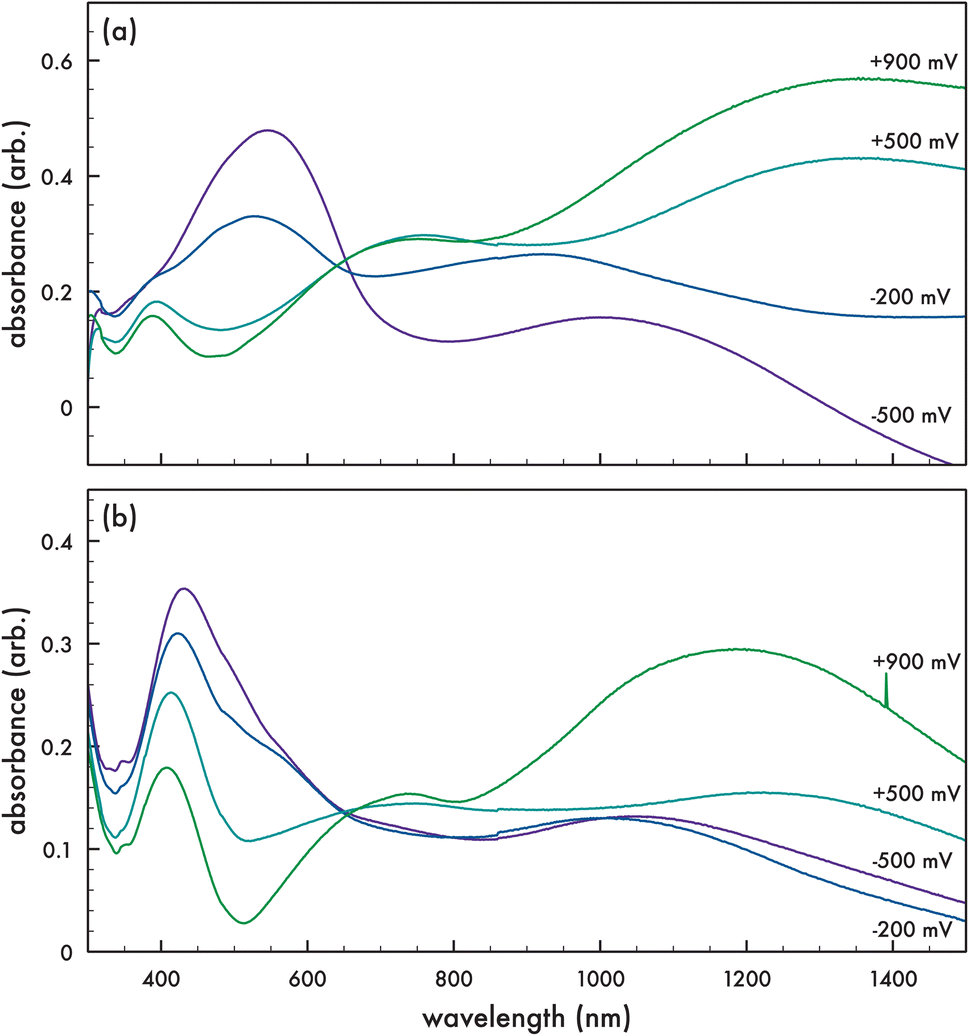 | ||
| Fig. 5 Optical absorption spectra of (a) p-AlkyneDTP and (b) p-DTPNap in the solid state. | ||
The electronic bandgap (ΔEuv) for these chemistries could be calculated from the optical absorption band edge that is observed at the absorbance onset of the neutral form of the polymer. The onset of the absorption peak for p-AlkyneDTP and p-DTPNap is observed at 703 nm and 680 nm, respectively, giving a ΔEuv of 1.76 eV for p-AlkyneDTP and 1.82 eV for p-DTPNap (cf. Table 1). By coupling the ΔEuv obtained by UV-vis with the cyclic voltammetry derived IP (i.e., HOMO) of the electropolymerized films, the LUMO was estimated for each polymer (cf. Table 1). The p-AlkyneDTP and p-DTPNap polymers exhibit similar HOMO/LUMO energies to that of previously reported polyDTPs.36–38
| UV-vis | Cyclic voltammetry | ||
|---|---|---|---|
| ΔEuv | HOMO | LUMO | |
| p-AlkyneDTP | 1.76 | −4.46 | −2.70 |
| p-DTPNap | 1.82 | −4.56 | −2.74 |
Conclusion
In summary, a new N-alkylated dithieno[3,2-b:2′,3′-d]pyrrole (DTP) monomer with a terminal triple bond (AlkyneDTP) was synthesized and electropolymerized (p-AlkyneDTP). The AlkyneDTP monomer was successfully functionalized before polymerization via a 1,3- dipolar cycloaddition with an azide-modified dye (AzNap) to yield a DTPNap monomer. In addition to the electropolymerization of AlkyneDTP, the DTPNap monomer was also electropolymerized (p-DTPNap). The monomers and subsequent polymers were analyzed by Fourier transform infrared (FTIR) and UV-vis spectroscopy. It was found that the alkyne functional group in the AlkyneDTP monomer was not stable during electropolymerization and was not detected in the FTIR spectra of p-AlkyneDTP. Thus, eliminating p-AlkyneDTP's candidacy for post-polymerization functionalization via 1,3-dipolar cycloaddition. However, p-DTPNap retained its pre-polymerization functionalization throughout the electropolymerization. These results afford the possibility to prepare various N-functionalized DTP monomers and corresponding polymers using 1,3-dipolar cycloaddition reactions with organic azides. The developed method allows for the addition of different functional groups to the polymeric DTP-backbone at high yields.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Gregg-Graniteville Foundation and the National Science Foundation (OIA-1632881) for financial support. The authors thank the National Institute of Health (S10 OD021758-01A1) for funding the electrospray (ESI) mass spectra unit.References
- I. F. Perepichka and D. F. Perepichka, Handbook of Thiophene Based Materials, Hoboken, NJ, John Wiley & Sons, 2009 Search PubMed.
- S. C. Rasmussen and S. J. Evenson, Dithieno[3,2-b:2′,3′-d]pyrrole-based materials: Synthesis and application to organic electronics, Prog. Polym. Sci., 2013, 38(12), 1773–1804 CrossRef CAS.
- M. Nakamura, C. Yang, E. Zhou, K. Tajima and K. Hashimoto, Polymer Bulk Heterojunction Photovoltaic Devices with Multilayer Structures Prepared by Thermal Lamination, ACS Appl. Mater. Interfaces, 2009, 1(12), 2703–2706 CrossRef CAS PubMed.
- S. J. Evenson, M. J. Mumm, K. I. Pokhodnya and S. C. Rasmussen, Highly Fluorescent Dithieno[3,2-b:2′,3′-d]pyrrole-Based Materials: Synthesis, Characterization, and OLED Device Applications, Macromolecules, 2011, 44(4), 835–841 CrossRef CAS.
- T. L. Nelson, T. M. Young, J. Liu, S. P. Mishra, J. A. Belot and C. L. Balliet, et al. Transistor Paint: High Mobilities in Small Bandgap Polymer Semiconductor Based on the Strong Acceptor, Diketopyrrolopyrrole and Strong Donor, Dithienopyrrole, Adv. Mater., 2010, 22(41), 4617–4621 CrossRef CAS PubMed.
- X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage and S. P. Tiwari, et al. Dithienopyrrole-based donor–acceptor copolymers: low band-gap materials for charge transport, photovoltaics and electrochromism, J. Mater. Chem., 2010, 20(1), 123–134 RSC.
- J. Liu, R. Zhang, G. Sauvé, T. Kowalewski and R. D. McCullough, Highly Disordered Polymer Field Effect Transistors: N-Alkyl Dithieno[3,2-b:2′,3′-d]pyrrole-Based Copolymers with Surprisingly High Charge Carrier Mobilities, J. Am. Chem. Soc., 2008, 130(39), 13167–13176 CrossRef CAS PubMed.
- X. Zhang, J. W. Shim, S. P. Tiwari, Q. Zhang, J. E. Norton and P.-T. Wu, et al. Dithienopyrrole–quinoxaline/pyridopyrazine donor–acceptor polymers: synthesis and electrochemical, optical, charge-transport, and photovoltaic properties, J. Mater. Chem., 2011, 21(13), 4971–4982 RSC.
- J. Liu, R. Zhang, I. Osaka, S. Mishra, A. E. Javier and D.-M. Smilgies, et al. Transistor Paint: Environmentally Stable N-alkyldithienopyrrole and Bithiazole-Based Copolymer Thin-Film Transistors Show Reproducible High Mobilities without Annealing, Adv. Funct. Mater., 2009, 19(21), 3427–3434 CrossRef CAS.
- W. Belcher, S. Rasmussen and P. Dastoor, Characterization of N-Functionalized Poly(dithieno[3,2-b:2′,3′-d]pyrrole)s in Ternary Blend Bulk Heterojunction Structured Organic Electronic Devices, Polym. Prepr., 2007, 48, 11–12 CAS.
- R. S. Ashraf, J. Gilot and R. A. J. Janssen, Fused ring thiophene-based poly(heteroarylene ethynylene)s for organic solar cells, Sol. Energy Mater. Sol. Cells, 2010, 94(10), 1759–1766 CrossRef CAS.
- S. C. Price, A. C. Stuart and W. You, Polycyclic Aromatics with Flanking Thiophenes: Tuning Energy Level and Band Gap of Conjugated Polymers for Bulk Heterojunction Photovoltaics, Macromolecules, 2009, 43(2), 797–804 CrossRef.
- D. Sahu, H. Padhy, D. Patra, J.-F. Yin, Y.-C. Hsu and J.-T. S. Lin, et al. Synthesis and applications of novel acceptor–donor–acceptor organic dyes with dithienopyrrole- and fluorene-cores for dye-sensitized solar cells, Tetrahedron, 2011, 67(2), 303–311 CrossRef CAS.
- A. Yassin, T. Rousseau, P. Leriche, A. Cravino and J. Roncali, Evaluation of bis-dicyanovinyl short-chain conjugated systems as donor materials for organic solar cells, Sol. Energy Mater. Sol. Cells, 2011, 95(2), 462–468 CrossRef CAS.
- P.-O. Schwartz, S. Förtsch, E. Mena-Osteritz, D. Weirather-Köstner, M. Wachtler and P. Bäuerle, Ferrocene-functionalized polyheteroacenes for the use as cathode active material in rechargeable batteries, RSC Adv., 2018, 8(26), 14193–14200 RSC.
- K. Ogawa and S. C. Rasmussen, N-Functionalized Poly(dithieno[3,2-b:2′,3′-d]pyrrole)s: Highly Fluorescent Materials with Reduced Band Gaps, Macromolecules, 2006, 39(5), 1771–1778 CrossRef CAS.
- H. Azak, H. B. Yildiz and B. Bezgin Carbas, Synthesis and characterization of a new poly(dithieno (3,2-b:2′,3′-d) pyrrole) derivative conjugated polymer: Its electrochromic and biosensing applications, Polymer, 2018, 134, 44–52 CrossRef CAS.
- B. S. Dakshayini, K. R. Reddy, A. Mishra, N. P. Shetti, S. J. Malode and S. Basu, et al. Role of conducting polymer and metal oxide-based hybrids for applications in ampereometric sensors and biosensors, Microchem. J., 2019, 147, 7–24 CrossRef CAS.
- S. J. Evenson and S. C. Rasmussen, N-Acyldithieno[3,2-b:2′,3′-d]pyrroles: Second Generation Dithieno[3,2-b:2′,3′-d]pyrrole Building Blocks with Stabilized Energy Levels, Org. Lett., 2010, 12(18), 4054–4057 CrossRef CAS PubMed.
- S. Schmid, J. Gačanin, Y. Wu, T. Weil and P. Bäuerle, Synthesis and bioconjugation of first alkynylated poly(dithieno[3,2-b:2′,3′-d]pyrrole)s, Polym. Chem., 2017, 8(46), 7113–7118 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41(14), 2596–2599 CrossRef CAS PubMed.
- X. Huang, L. Yang, R. Emanuelsson, J. Bergquist, M. Stromme and M. Sjodin, et al. A versatile route to polythiophenes with functional pendant groups using alkyne chemistry, Beilstein J. Org. Chem., 2016, 12, 2682–2688 CrossRef CAS PubMed.
- J. P. Wolfe, S. Wagaw and S. L. Buchwald, An Improved Catalyst System for Aromatic Carbon–Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates, J. Am. Chem. Soc., 1996, 118(30), 7215–7216 CrossRef CAS.
- Y. Saito, K. Matsumoto, S. S. Bag, S. Ogasawara, K. Fujimoto and K. Hanawa, et al. C8-alkynyl- and alkylamino substituted 2′-deoxyguanosines: a universal linker for nucleic acids modification, Tetrahedron, 2008, 64(16), 3578–3588 CrossRef CAS.
- M. K. Burdette, H. W. Jones, Y. Bandera and S. H. Foulger, X-ray radioluminescent hydrogel stabilized crystalline colloidal arrays, Opt. Mater. Express, 2019, 9(3), 1416–1429 CrossRef CAS.
- P. Zanirato, P. Spagnolo and G. Zanardi, Thermal decomposition of o-azidobithienyls, J. Chem. Soc., Perkin Trans. 1, 1983, 1, 2551–2554 RSC.
- A. Berlin, G. Pagani, G. Zotti and G. Schiavon, Electrochemical polymerization of 1H,7H-pyrrolo[2′,3′:4,5]-thieno[3,2-b]pyrrole and 4H-dithieno[3,2-b;2′,3′-d]pyrrole, Makromol. Chem., 1992, 193(2), 399–409 CrossRef CAS.
- K. Ogawa and S. C. Rasmussen, A Simple and Efficient Route to N-Functionalized Dithieno[3,2-b:2′,3′-d]pyrroles: Fused-Ring Building Blocks for New Conjugated Polymeric Systems, J. Org. Chem., 2003, 68(7), 2921–2928 CrossRef CAS PubMed.
- G. Koeckelberghs, L. De Cremer, W. Vanormelingen, W. Dehaen, T. Verbiest and A. Persoons, et al. Improved synthesis of N-alkyl substituted dithieno[3,2-b:2′,3′-d]pyrroles, Tetrahedron, 2005, 61(3), 687–691 CrossRef CAS.
- G. Koeckelberghs, L. De Cremer, A. Persoons and T. Verbiest, Influence of the Substituent and Polymerization Methodology on the Properties of Chiral Poly(dithieno[3,2-b:2′,3′-d]pyrrole)s, Macromolecules, 2007, 40(12), 4173–4181 CrossRef CAS.
- R. Berg and B. F. Straub, Advancements in the mechanistic understanding of the copper-catalyzed azide-alkyne cycloaddition, Beilstein J. Org. Chem., 2013, 9, 2715–2750 CrossRef PubMed.
- F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman and K. B. Sharpless, et al. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates, J. Am. Chem. Soc., 2005, 127(1), 210–216 CrossRef CAS PubMed.
- L. Micaroni, F. Nart and I. Hümmelgen, Considerations about the electrochemical estimation of the ionization potential of conducting polymers, J. Solid State Electrochem., 2002, 7(1), 55–59 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications, Adv. Mater., 2011, 23(20), 2367–2371 CrossRef CAS PubMed.
- P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Polymers for electronics and spintronics, Chem. Soc. Rev., 2013, 42(23), 8895–8999 RSC.
- J. F. Mike, L. Shao, J.-W. Jeon and J. L. Lutkenhaus, Charge Storage in Decyl- and 3,6,9-Trioxadecyl-Substituted Poly(dithieno[3,2-b:2,3-d]pyrrole) Electrodes, Macromolecules, 2014, 47(1), 79–88 CrossRef CAS.
- S. Förtsch and P. Bäuerle, Synthesis and characterization of two isomeric dithienopyrrole series and the corresponding electropolymers, Polym. Chem., 2017, 8(23), 3586–3595 RSC.
- S. H. Foulger, Y. Bandera, B. Grant, J. Vilčáková and P. Sáha, Exploiting multiple percolation in two-terminal memristor to achieve a multitude of resistive states, J. Mater. Chem. C, 2021, 9(28), 8975–8986 RSC.
- M. K. Burdette, Y. P. Bandera, G. M. Gray and S. H. Foulger, Dynamic Emission Tuning of X-ray Radioluminescent Crystalline Colloidal Arrays: Coupling the Optical Stop Band with Sequential Förster Resonance Energy Transfers, Adv. Opt. Mater., 2019, 7(2), 1801142 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra03265a |
| This journal is © The Royal Society of Chemistry 2022 |