
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrograming a cyanide-free transformation of aldehydes to nitriles and one-pot synthesis of amides through tandem chemo-enzymatic cascades†
Haoteng Zhenga,
Qinjie Xiaoa,
Feiying Maoa,
Anming Wang *a,
Mu Lib,
Qiuyan Wangc,
Pengfei Zhanga and
Xiaolin Pei*a
*a,
Mu Lib,
Qiuyan Wangc,
Pengfei Zhanga and
Xiaolin Pei*a
aCollege of Material, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou, 311121, PR China. E-mail: pxl@hznu.edu.cn; waming@hznu.edu.cn
bCollege of Food Science and Technology, Huazhong Agricultural University, Wuhan, 430070, PR China
cSchool of Basic Medical Sciences, Hangzhou Normal University, Hangzhou, 311121, PR China
First published on 16th June 2022
Abstract
Nitriles are broadly applied to synthesize pharmaceuticals, agrochemicals, and materials because of their versatile transformation. Although various methods have been developed for introducing a nitrile group into organic molecules, most of them entail the use of highly toxic chemicals, transition metals, or harsh conditions. In this work, we reported a greener chemo-enzymatic cascade to synthesize alky and aryl nitriles from readily accessible aldehydes, that were further transformed into corresponding amides via an artificial enzyme cascade. A biphasic reaction system was designed to bridge chemical synthesis and enzymatic catalysis through simple phase separation. The biphasic system mainly perfectly avoided the inactivation of hydroxylamine on aldoxime dehydratase from Pseudomonas putida (OxdF1) and nitrile hydratase from Aurantimonas manganoxydans ATCC BAA-1229 (NHase1229). For the synthesis of various nitriles, moderate isolation yields of approximately 60% were obtained by the chemo-enzymatic cascade. Interestingly, two seemingly conflicting reactions of dehydration and hydration were sequentially proceeded to synthesize amides by the synergistic catalysis of OxdF1 and NHase1229 in E. coli cells. An isolation yield of approximately 62% was achieved for benzamide at the one-liter scale. In addition, the shuttle transport of substrates and products between two phases is convenient for the product separation and n-hexane recycling. Thus, the chemo-enzymatic cascade shows a potential application in the cyanide-free and large-scale synthesis of nitriles and amides.
1. Introduction
Nitriles and amides are widely present as important structural motifs in pharmaceuticals, agrochemicals, materials, and natural products.1–3 Moreover, nitriles can serve as ubiquitous building precursors in organic synthesis, which can be readily converted into versatile functional groups, such as amides, amines, carboxylic acids, and heterocycles.4,5 So far, successive efforts have been devoted to introducing a nitrile group into organic molecules. The most general method is the nucleophilic substitution reaction of alkyl and aryl halides with stoichiometric hydrogen cyanide (HCN) or metal cyanides, such as Kolbe nitrile synthesis,6 Sandmeyer cyanation,7 and Rosenmund-von Braun reaction.8 HCN and metal cyanides belong to highly toxic chemicals, and their large-scale use has been severely restricted. Therefore, the cyanide-free synthesis of nitrile has attracted great attention from chemical scientists.9 A variety of strategies have been recently developed using less toxic cyanide sources from different substrates in the last decade, including alkenes,10,11 amines,12,13 carboxylic acids,14,15 primary alcohols,16,17 and aldehydes.18–20 However, most of them depended on transition metals (e.g., Cu, Fe, Ru, Pd, etc.) and harsh conditions (e.g., industrial ammoxidation process, >300 °C in ammonia gas).21 Considering the ready availability of aldehydes, nitriles were prepared from aldehydes via aldoxime as intermediate by chemical dehydrating reagents, such as POCl3,22 H-zeolites,23 NTSI,24 BOP reagent,25 PhSe(O)OH,26 NH4SCN,27 etc. Aldoximes are important metabolic nitrogen-containing metabolites in all kingdoms of life and are also easily accessible by spontaneous condensation of aldehydes with hydroxylamine hydrochloride (NH2OH·HCl) at room temperature.28,29 The dehydration of aldoximes provides a green method for synthesizing nitriles with H2O as the only by-product. However, some shortcomings still limit the widespread application of the chemical dehydration of aldoximes to nitriles, including the harsh reaction conditions, waster production, and special chemicals.30 Therefore, developing greener alternative catalytic technology for preparing nitriles has attracted extensive attention from both academia and industry.Biocatalysis has become an increasingly attractive alternative to conventional chemical synthesis for producing bulk and fine chemicals.31–33 Aldoxime dehydratase (Oxd, EC 4.99.1.5) catalyses the dehydration of aldoximes to corresponding nitriles in the water medium, which were firstly characterized in Bacillus and Rhodococcus strains by Asano group.34,35 Oxds, as a special class of heme-binding protein, are different from other heme-containing monooxygenases (e.g., cytochrome P450) catalysing the oxidation of non-activated hydrocarbons by transferring an oxygen atom from H2O2 or O2.36 Instead, most Oxds appear to lose catalytic activity under aerobic conditions, which is suspected to be associated with the oxidation state of heme group (Fe2+/Fe3+).37 So far, about eight Oxd genes have been discovered from diverse bacteria including Bacillus, Pseudomonas, Rhodococcus, etc.34,38,39 Moreover, some Oxds have been applied for the synthesis of aliphatic nitriles,40–42 and chiral β-hydroxy nitriles.43,44 Prof. Gröger group designed a chemo-enzymatic cascade to synthesize n-octanenitrile from 1-octene.45 To avoid the inhibition effect of hydroxylamine on the activity of Oxds, an in situ thermal decompositions (16 h, 100 °C) strategy has been applied to remove residual NH2OH·HCl in the reaction solutions from chemical aldoxime formation. Furthermore, Prof. Gröger group combined TEMPO oxidation, aldoxime formation, and Oxd dehydration to synthesize aliphatic nitriles from alcohols without isolation of the aldehyde and aldoxime intermediates.46 The product aliphatic nitriles were used as the organic solvent to connect three reaction steps in biphasic reaction systems. Recently, Prof. Winkler group also constructed a novel chemo-enzymatic cascade to synthesize nitriles from carboxylic acids.47 These cascade reactions can bridge the advantages of chemo-catalysis and bio-catalysis to avoid the isolation of intermediates. Thus, designing more efficient cascades have been becoming a concerned topic for the large-scale preparation of high-valued nitriles under moderate reaction condition.
In the present work, we represented a chemo-enzymatic cascade for cyanide-freely synthesizing nitriles and amides from aldehydes by a temporal–spatial compartmentalization strategy. The starting material aldehydes were converted to the corresponding aldoximes with NH2OH·HCl in a biphasic reaction system. Then, the organic phase containing aldoximes was separated and directly mixed with another aqueous solution to synthesize nitriles by an Oxd or to synthesize amides by a multi-enzymes system including OxdF1 and nitrile hydratase (OxdF1–NHase1229). We investigated the possibility of an organic solvent to couple chemo- and enzymatic catalysis for the large-scale synthesis of nitriles and amides (Fig. 1). The biphasic reaction system would completely remove residual NH2OH·HCl in the reaction mixture by a convenient phase separation step, and be efficient to prepare nitriles and amides without the isolation of intermediates.
 | ||
| Fig. 1 Chemo-enzymatic synthesis of nitriles and amides in a biphasic system. | ||
2. Experimental
2.1. Chemicals and bacterial strains
All chemicals used in this study were obtained from Sinopharm Chemical Reagent (Shanghai, China) unless otherwise specified. DNA polymerase (PrimeSTAR), T4 DNA ligase, and restriction endonucleases (Nde I and EcoR V) were purchased from Takara Biomedical Technology (Beijing) Co., Ltd. Isopropyl-β-D-thiogalactopyranoside (IPTG) was purchased from Sigma-Aldrich (Merck KGaA, Germany). Escherichia coli BL21 (DE3) (Dingguo Biotechnology, China) were used for protein expression.2.2. Recombinant plasmids construction and protein expression
Plasmid pRSFDuet-1 (Novagen™, Merck, Germany) was employed to construct a recombinant vector for expressing recombinant aldoxime dehydratase from P. putida F1 (OxdF1)48 and nitrile hydratase from A. manganoxydans ATCC BAA-1229 (NHase1229),49 which DNA sequences and the deduced amino acid sequences were shown in ESI.† The pairs of primers in this study are described in Table S1.† Recombinant plasmid pROxdF1 had been constructed to express OxdF1 in E. coli cells (Fig. S1†). The NHase1229 gene was amplified with plasmid pET-NHase1229 as a template using primers NHase1229_Up and NHase1229_Down. PCRs were conducted with PrimeSTAR™ Max DNA polymerase [Takara (Dalian) Biotech, China]. PCRs were carried out as follows: a preliminary denaturation was conducted at 95 °C for 2 min, followed by 30 cycles of 10 s denaturation at 95 °C, 15 s annealing at 58 °C, and 15 s extensions at 72 °C. A final extension was conducted at 72 °C for 5 min. The amplified PCR fragment of approximately 1700 bp was inserted into the second MCS of plasmid pROxdF1 to obtain recombinant plasmid pROxdF1-NHase (Fig. S2 and S3†). Recombinant plasmids pROxdF1 and pROxdF1-NHase were respectively transformed into E. coli BL21 (DE3), and the positive clones were screened on LB agar medium containing 50 μg mL−1 kanamycin and further confirmed by gene sequencing (Qingke Biological Technology, Hangzhou, China).2.3. Chemical synthesis of aldoximes in a biphasic reaction system
Aldoximes were prepared by the reaction of aldehydes with NH2OH·HCl in a biphasic reaction system with benzaldehyde as a model substrate. Benzaldehyde (1a, 200 mM) was dissolved in 2 mL various types of organic solvents, including n-hexane, ethyl acetate, ether, amyl acetate, toluene, and dichloromethane. Hydroxylamine hydrochloride (NH2OH·HCl, 1.5 eq., 300 mM) and sodium carbonate (Na2CO3, 0.75 eq., 150 mM) were dissolved in 2 mL pure water. The biphasic reaction was magnetically stirred at 150 rpm and maintained at room temperature until all aldehydes were completely converted to benzaldoxime (BAOx, 1b). The product in the organic phase was obtained by a simple phase separation. A control reaction was performed in pure water as the reaction medium containing 1.5 eq. NH2OH·HCl and 0.75 eq. Na2CO3. Benzaldehyde was dissolved in 0.4 mL ethanol (10%, v/v) as a co-solvent was added into the above aqueous solution. The reaction was stirred at room temperature. The reaction product was extracted three times with ethyl acetate. To avoid the deterioration of NH2OH·HCl on the biocatalyst OxdF1, the organic phase was further washed three times using water. To analyse BAOx, the organic solution was dried with anhydrous MgSO4, filtered, and then rotary evaporated to obtain the target product.2.4. Biocatalysis of aldoxime dehydratase in a biphasic reaction system
The enzymatic catalysis was performed in a biphasic reaction system using BAOx as a model substrate to synthesize benzonitrile (BN, 1c). 200 mM BAOx was dissolved in 2 mL organic solvents including n-hexane, ethyl acetate, ether, amyl acetate, toluene, and dichloromethane. Recombinant E. coli OxdF1 whole cells [30 mg mL−1, dry cell weight, (dcw)] were suspended in 2 mL 50 mM potassium phosphate buffer (PPB, pH 7.0). The two phases were mixed and magnetically stirred at 150 rpm and maintained at room temperature for 2 h. A control reaction was performed in PPB solution (50 mM, pH 7.0), and was magnetically stirred at room temperature for 2 h. The reaction mixture was centrifuged to separate two phases and remove E. coli cells (8000×g, 5 min). For the control reaction mixture, the product was obtained by ethyl acetate extraction. The BN concentration was determined by HPLC method to calculate the conversion rate and yield.2.5. Biocatalysis of nitrile hydratase in a biphasic reaction system
The enzymatic catalysis was performed in a biphasic reaction system with BN as a model substrate. 200 mM BN was dissolved in 2 mL of different organic solvents including n-hexane, amyl acetate, and toluene. Recombinant E. coli OxdF1-NHase whole cells (30 mg dcw mL−1) were suspended in 2 mL 50 mM potassium phosphate buffer (PPB, pH 7.0). The two phases were mixed and magnetically stirred for 2 h at room temperature. A control reaction was performed using PPB (50 mM, pH 7.0) as the reaction medium. The reaction was stirred at room temperature for 2 h in a round-bottomed flask. The reaction mixture was centrifuged at 8000×g for 5 min to separate two phases and remove E. coli cells. The concentration of benzamide (BAM, 1d) was determined by HPLC to calculate the conversion rate and the corresponding yield.2.6. Two-step chemo-enzymatic cascade to nitriles without isolation of intermediates
The chemo-enzymatic cascade was performed in a biphasic reaction system. Aldehyde (200 mM) was dissolved in 2 mL n-hexane. NH2OH·HCl (1.5 eq., 300 mM) and Na2CO3 (0.75 eq., 150 mM) were dissolved in 2 mL pure water. The reaction was started by mixing the organic phase and the aqueous phase and was magnetically stirred at room temperature until aldehydes were completely converted to corresponding aldoximes (confirmed by thin-layer chromatography, TLC). The organic phase was separated and immediately mixed with another aqueous phase (2 mL 50 mM PPB, pH 7.0) containing 30 mg mL−1 E. coli OxdF1 whole cells in a round bottom flask and further magnetically stirred for 2 h at room temperature. The reaction mixture was centrifuged to separate two phases and remove E. coli cells (8000×g, 5 min). The concentration of nitrile was determined by HPLC or GC method to calculate the conversion rate and yield. For 1H-NMR analysis of nitriles, the organic phase was evaporated, the target products were purified by column chromatography, and then dissolved in deuterated reagent (CDCl3) for scanning.2.7. Three-step chemo-enzymatic cascade to amides without isolation of intermediates
The chemo-enzymatic cascade was performed in a biphasic reaction system. Aldehydes (200 mM) were dissolved in 2 mL n-hexane. NH2OH·HCl (1.5 eq., 300 mM) and Na2CO3 (0.75 eq., 150 mM) were dissolved in 2 mL pure water. The reaction was started by mixing the organic phase and the aqueous phase and was stirred at room temperature until aldehydes were completely converted to aldoximes (confirmed by TLC). The organic phase was separated and immediately mixed with another aqueous phase (2 mL 50 mM PPB, pH 7.0) containing 30 mg mL−1 E. coli OxdF1–NHase1229 whole cells in a round bottom flask and stirred for 2 h at room temperature. The reaction mixture was centrifuged to separate two phases and remove E. coli cells (8000×g, 5 min). The concentration of amide was determined by HPLC or GC method to calculate the conversion rate and yield. For 1H-NMR analysis of amides, the products were obtained by dichloromethane extraction, the organic phase was evaporated and further dissolved in deuterated reagent (DMSO) for scanning.2.8. The scale-up preparation of nitrile and amide from aldehyde
Chemical synthesis of BAOx was carried out in a biphasic system. Benzaldehyde (1 M) was dissolved in 450 mL n-hexane, NH2OH·HCl (1.5 eq., 1.5 M) and sodium carbonate (0.55 eq., 0.75 M) were dissolved in 450 mL water. Two solutions were mixed and stirred at room temperature until all benzaldehyde was completely conversed to BAOx. The organic phase was separated, in which the concentration of BAOx was identified by HPLC. The organic was directly mixed with E. coli OxdF1 suspension (30 g dcw L−1 cell in 50 mM PPB, pH 7.0) resulting in a BAOx concentration of 500 mM. The biphasic reaction system was stirred at 150 rpm and room temperature until BAOx was completely conversed to BN. Then, another batch of 200 mM BAOx was added into the reaction mixture, which was further stirred to synthesize BN. The process for preparing BAM was similar to the above method. E. coli OxdF1–NHase1229 cells were used as catalyst. The BAOx concentration in the upper organic phase was 100 mM. When BAOx was completely conversed into BAM, other three batches of 100 mM BAOx were separately added into the reaction mixture for producing BAM.2.9. Analysis method
The concentrations of aromatic reactants and products were determined by high-pressure liquid chromatography (HPLC, Agilent 1100) with a C18 reverse-phase column (5 μm, 4.6 mm × 250 mm). The elution solvent consisted of 0.5% trifluoroacetic acid (TFA) ddH20 and acetonitrile (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60, v/v) at a flow rate of 0.5 mL min−1. The absorbance was measured at 230 nm. For aliphatic substrate and products, the concentration was determined by gas chromatography (GC, Agilent Technologies Inc., USA) with a flame ionization detector and an HP-5 capillary column (0.1 μm, 30 m × 0.25 mm). The initial column oven temperature was regulated at 35 °C and held for 2 min, then heated to 200 °C at 5 °C min−1, and further heated to 280 °C at 20 °C min−1. Nitrogen gas was set at 2.0 mL min−1 as the carrier.
:60, v/v) at a flow rate of 0.5 mL min−1. The absorbance was measured at 230 nm. For aliphatic substrate and products, the concentration was determined by gas chromatography (GC, Agilent Technologies Inc., USA) with a flame ionization detector and an HP-5 capillary column (0.1 μm, 30 m × 0.25 mm). The initial column oven temperature was regulated at 35 °C and held for 2 min, then heated to 200 °C at 5 °C min−1, and further heated to 280 °C at 20 °C min−1. Nitrogen gas was set at 2.0 mL min−1 as the carrier.
3. Results and discussion
3.1. Chemical synthesis of benzaldoxime from benzaldehyde in biphasic systems
The first step is to prepare aldoximes from aldehydes and NH2OH·HCl. The aldoximation of aldehydes is a basic addition–elimination condensation reaction with high chemoselectivity.50 Previous studies found that the nitrogen donor NH2OH·HCl seriously deteriorated the activities of biocatalyst Oxds, even at a very low concentration of 0.5–1.0 mM.45 To solve the problem, some necessary operations were adopted to remove the residual NH2OH·HCl, such as water washing after organic extraction or heating decomposition at 100 °C for 16 h.45,46 In the present work, we investigated a biphasic reaction system to connect the chemical aldoximation and enzymatic dehydration based on the insolubility of NH2OH·HCl in a suitable solvent. Different biphasic reaction systems were adopted to investigate the effect of organic solvents on the aldoximation of benzaldehyde (Table 1). Encouragingly, 100 mM benzaldehyde was completely converted into benzaldoxime (BAOx) in all biphasic reaction systems (>99%). However, the moderate isolation yields were usually obtained in the range of 74–90%, which is mainly because of the partition coefficient of benzaldehyde oxime in different organic solvents.
|
|||
|---|---|---|---|
| Solvent | Conversiona [%] | Isolated yieldb [%] | |
| a The conversion of benzaldehyde to BAOx was determined by HPLC analysis.b After phase separation or extraction with ethyl acetate (EtOAc), BAOx was obtained by rotary evaporation to determine the isolated yield.c The reaction was carried out in pure water at room temperature. After the reaction was completed, an equal volume of EtOAc was used to extract the target product BAOx. | |||
| 1 | n-Hexane | >99% | 83.4% |
| 2 | Amyl acetate | >99% | 88.2% |
| 3 | Ether | >99% | 74.3% |
| 4 | Ethyl acetate | >99% | 85.6% |
| 5 | Toluene | >99% | 80.3% |
| 6 | Dichloromethane | >99% | 90.1% |
| 7 | Nonec | >99% | 85.3% |

3.2. Enzymatic synthesis of benzonitrile from benzaldoxime in biphasic systems
Although enzyme catalysis is usually carried out in an aqueous medium, the use of organic solvents to construct aqueous/organic biphasic system has been proven to be beneficial for expanding the adaptability of biocatalysis, in particular for poorly water-soluble organic substrates.51 In the second step, an enzymatic process was performed to synthesize nitrile from aldoxime by the recombinant OxdF1 expressed in E. coli BL21 whole cells as the catalyst in different biphasic aqueous/organic solvent systems [1:1 (v/v) solvent/PPB buffer, 50 mM, pH 7.0, total 4 mL]. The expression of recombinant OxdF1 in E. coli was shown in Fig. S4.† The whole cells were suspended in the aqueous PPB buffer and then mixed with 100 mM BAOx in different organic solvents. After the reactions were completed, the organic and aqueous phases were separated and analysed to determine the conversion rate of BAOx to benzonitrile (Table 2). In the case of n-hexane, amyl acetate, and toluene, above 99% of BAOx were converted to benzonitrile, moreover, and almost all of benzonitrile existed in the organic phase. However, the reaction hardly took place in ether and dichloromethane, and ethyl acetate gave a very low conversion of approximately 3%.
|
|||
|---|---|---|---|
| Solvent | Substrate [mM] | Conversiona [%] | |
| a The conversion of BAOx to benzonitrile was determined by HPLC analysis.b The biotransformation was performed in the pure organic solvent.c Ethanol (10%, v/v) was used as co-solvent, and the conversion was determined after extraction of the reaction mixture with EtOAc. | |||
| 1 | n-Hexane | 100 | >99 |
| 2 | n-Hexaneb | 100 | >99 |
| 3 | Amyl acetate | 100 | 84.1 |
| 4 | Amyl acetateb | 100 | >99 |
| 5 | Toluene | 100 | >99 |
| 6 | Tolueneb | 100 | >99 |
| 7 | Ether | 100 | <1 |
| 8 | Ethyl acetate | 100 | 3.2 |
| 9 | Dichloromethane | 100 | <1 |
| 10 | Nonec | 100 | >99 |

In addition, we investigated the conversion in pure organic solvents using the whole E. coli cells as the catalyst, and 100 mM BAOx was completely converted to BN in pure toluene and n-hexane. Prof. Gröger group had investigated the dehydration of n-octanaloxime to n-octanenitrile by OxdB (aldoxime dehydratase from Bacillus sp. OxdB-1) in pure organic solvents, however, all solvents directly led to the inactivation of OxdB.52 Organic solvents commonly inactivate natural biocatalysts, but also can regulate the concentration of toxic substrate and product to relieve the inhibition on the activity of enzymes.53,54 Therefore, the good biocompatibility and partitioning behaviour of substrate and product are restrictive criteria for selecting a suitable organic solvent. In this work, we found a clear correlation between the partition coefficient (logP) and the OxdF1 activity. The high logP values (>2) give the excellent catalytic property (Fig. 2). n-Hexane and toluene showed better biocompatibility for the enzymatic hydration by OxdF1.
 | ||
| Fig. 2 Effect of organic solvents in biphasic reaction system on the dehydration of BAOx to BN by OxdF1. logP values are plotted to correlate enzyme activity with the hydrophobicity of the organic solvents. | ||
Suitable solvents can combine chemical and enzymatic catalysis by a temporal compartmentalization strategy, which can avoid the coexistence of incompatible components in a one-pot cascade reaction.55 Furthermore, we investigated the catalytic performance of the whole E. coli cells with OxdF1 in a biphasic reaction system with the organic phase directly extracted from the first chemical step (Table S2†). After the chemical synthesis of BAOx was completed in the PPB buffer, recombinant E. coli cells with OxdF1 were added into the aqueous medium to catalyse the synthesis of BN. It is disappointing that only approximately 5.4% of BAOx was converted to BN. If BAOx was extracted from the aqueous mixture by ethyl acetate, water washing, and rotary evaporation, 100 mM BAOx was completely converted to BN in the aqueous medium using 10% ethanol as co-solvent. For the chemical synthesis of BAOx in the biphasic system with n-hexane as the organic solvent, the organic phase was obtained by simple phase separation, and directly mixed with the E. coli cell suspension. After 2 h of reaction, 100 mM BAOx was completely converted to BN. However, this is unsatisfactory for amyl acetate that approximately 76% of BAOx was converted to BN. These results suggest that n-hexane is an ideal solvent in the chemo-enzymatic cascade reaction for the synthesis of BN from benzaldehyde, which can effectively avoid the inhibition effect of NH2OH·HCl on the recombinant OxdF1.
3.3. Enzymatic synthesis of benzamide from benzonitrile in biphasic systems
In the third step, we investigated an enzymatic process for catalysing BN to BAM using E. coli BL21 whole cells containing NHase1229 as the catalyst in a biphasic reaction system. The NHase1229 from A. manganoxydans ATCC BAA-1229 had been functionally expressed in E. coli, and can efficiently hydrate various aryl and alkyl nitriles to corresponding amides with high thermal stability.49 The expression of recombinant NHase1229 in E. coli was shown in Fig. S4 and S5.† Because of the hydrophobicity of most substrates, we investigated the catalytic efficiency of NHase1229 in different biphasic systems using BN as the model substrate. After 2 h of reaction, the organic and aqueous phases were analysed to determine the conversion of BN to BAM (Table 3). The use of n-hexane, amyl acetate, and toluene gave very high conversions exceeding 99%, which is similar to the control group using pure PPB as the reaction medium. Different biphasic systems resulted in different partition fraction of BAM in organic and aqueous phases. The n-hexane/PPB system led to the complete distribution of BAM in the aqueous phase, while BAM completely existed in the organic phase in the amyl acetate/PPB system. When utilizing toluene as an organic solvent in the biphasic system, BAM existed in all aqueous and organic phases, and the proportion was approximately 3.3:1. After comprehensive considerations of various factors, n-hexane exhibited high suitability in the biphasic reaction medium. Meanwhile, n-hexane was easily obtained by the phase separation after the reaction was completed, which was reused in the first step of chemical BAM to realize the organic solvent recycling.
|
||||
|---|---|---|---|---|
| Solvent | Conversiona [%] | Distribution of amide [%] | ||
| Organic phase | Aqueous phase | |||
| a The conversion of BN to BAM was determined by HPLC analysis.b No product was detected.c The control group was carried out in PPB (pH 7.0).d The catalysis was performed in a pure aqueous solution, so the analysis was not conducted. | ||||
| 1 | n-Hexane | >99 | n.d.b | >99 |
| 2 | Amyl acetate | >99 | >99 | n.d. |
| 3 | Toluene | >99 | 23.3 | 76.7 |
| 4 | Nonec | >99 | n.d.d | >99 |

3.4. Synthesis of nitriles by two-step chemo-enzymatic cascade without isolation of intermediates
Then, our first goal was to develop a chemo-enzymatic cascade to synthesize nitriles from aldehydes without isolation of the intermediate aldoximes. The major challenge is the inhibition of NH2OH·HCl on the Oxds-catalysed dehydration reaction. Aliphatic and aromatic aldoximes were efficiently synthesized in the n-hexane/water reaction system at room temperature. After simple phase separation, the organic phase containing oximes was directly mixed with the E. coli OxdF1 whole cells suspension, and were converted to the corresponding nitriles. The reaction process is monitored by TLC analysis, and the final conversion rate and isolated yield are determined by HPLC of GC analysis (Fig. S6–S19†). The Nuclear Magnetic Resonance (NMR) spectra and GC-MS spectra were shown in Fig. S20–S47.† For all substrates, high conversion efficiencies were achieved to convert aldehydes into corresponding nitriles (Fig. 3). Meanwhile, the moderate isolated yields were achieved in all cases, approximately within 40–70%. Because of their hydrophobicity, the product nitriles almost entirely existed in n-hexane of the biphasic phase reaction system, which will be beneficial to release the inhibition of products on the activity of enzyme OxdF1. The organic phase was easily separated and further evaporated to achieve the target product nitriles with high purity. The resulted n-hexane could be recycled in the first step of chemical catalysis. Prof. Gröger recently developed a chemo-enzymatic cascade to synthesize aliphatic nitriles from primary alcohols using the product nitriles as a solvent.46 Although the “solvent-free” cascade opened the possibility for efficiently preparing aliphatic nitriles, the main challenge is to broaden its universality, in particular aromatic and heterocyclic nitriles. In this work, we developed a chemo-enzymatic cascade reaction to synthesize aromatic and aliphatic nitriles from aldehydes in an aqueous/organic biphasic system. n-Hexane was used to bridge chemical and enzymatic catalysis, which successfully avoided the coexistence of inhibitor NH2OH·HCl and biocatalyst OxdF1.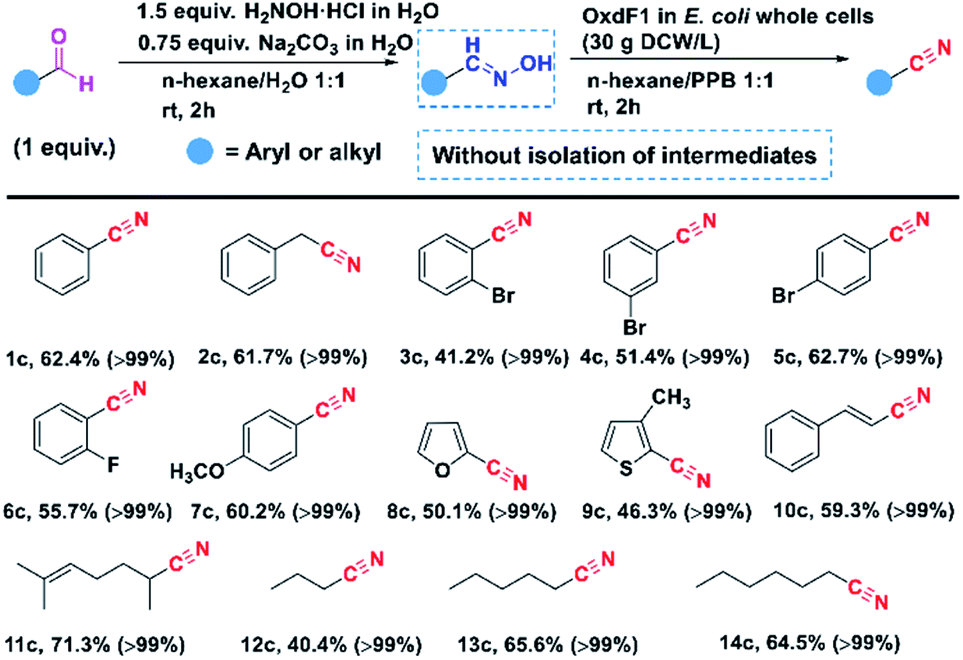 | ||
| Fig. 3 Chemo-enzymatic cascade reaction to synthesize nitriles from aldehydes. The data inside parentheses are the total conversion of two steps, while the values outside parentheses are the overall isolated yields of nitriles from aldehydes by column chromatography. | ||
3.5. Synthesis of amides by three-step chemo-enzymatic cascade without isolation of intermediates
Furthermore, another chemo-enzymatic cascade was designed for the one-pot synthesis of amides from aldehydes without isolation of the intermediates. A Co-type nitrile hydratase from A. manganoxydans (NHase1229) was cloned and co-expressed with OxdF1 in E. coli cells (Fig. S5†). The two seemingly conflicting reactions of dehydration and hydration were sequentially proceeded by the synergistic catalysis of OxdF1 and NHase1229 in E. coli cells. Because of the compartmentalized substrate processing chamber in microbial cells, various amides were efficiently synthesized from aldehydes in the biphasic reaction system. Different aldehydes and f NH2OH·HCl were condensed to obtain oximes in the n-hexane/water biphasic system. After the complete reaction, the organic phase was obtained by simple phase separation, and mixed with an equal volume of PPB solution containing 30 mg mL−1 recombinant E. coli OxdF1–NHase1229 whole cells. As shown in Fig. 4, most of aldehydes were efficiently converted into corresponding amides with 99% conversion every step. The chemo-enzymatic cascade reaction is suitable for aryl- and alkyl aldehydes with a satisfactory separation yield of 40–70%. However, in the case of 2-bromobenzaldehyde (3a) toward 2-bromobenzamide (3d), a relatively low conversion was obtained, only approximately 30.7%. Correspondingly, the 2-fluorobenzaldehyde (6a) at the same substitution position can be completely converted, and the conversion rate of 3-bromobenzaldehyde (4a) and 4-bromobenzaldehyde (5a) with the same substituent group is also >99%. These phenomena together prove that the steric hindrance of the substituents plays a vital role in the NHase1229 catalysis. In addition, the conversion rate of citronellal (11a) to the corresponding amide is only 25%, which also may be because of this reason. As for p-methoxybenzaldehyde (7a), the accumulation of the intermediate aldoxime results in a conversion rate of only 44% to p-methoxybenzamide (7d) by the cascade method. In the three-step chemo-enzymatic cascade, the resulting amides completely existed in the aqueous phase. Consequently, the target products were easily obtained after removing E. coli cells by centrifugation, and n-hexane was enough pure to be recycled. These results suggest that the chemo-enzymatic cascade using a biphasic system can realize the efficient synthesis of amides from aldehydes with aldoxime and nitrile as the intermediate. | ||
| Fig. 4 Chemo-enzymatic cascade reaction to synthesize amides from aldehydes. The data inside parentheses are the total conversion of two steps, while the values outside parentheses are the overall isolated yields of amides from aldehydes by column chromatography. | ||
3.6. The scale-up preparation of nitrile and amide from aldehyde
The chemical synthesis of BAOx was scaled up to 1 L in an n-hexane/H2O biphasic reaction system. The reaction mixture was magnetically stirred at room temperature, and 1 M benzaldehyde was completed converted to BAOx after 2 h. The upper organic phase containing BAOx was separated and directly mixed with recombinant E. coli whole cells suspension, which further convert to benzonitrile and benzamide (Fig. S48†). OxdF1 and NHase1229 were expressed in E. coli as the whole-cell catalysts, which were prepared in a 5 L fermenter with an initial 3 L of modified Terrific Broth (TB) medium. The time course of recombinant E. coli cultivation and the expression of target proteins are shown in Fig. S49–S52.† Firstly, the biotransformation reaction to synthesize nitrile was performed on a 1 L scale in a screwcap bottle with a magnetic stirrer (Fig. S53†). The initial batch reaction was done at 0.5 M benzaldehyde oxime concentration, and the substrate was fully converted to benzonitrile within 3 h (Fig. 5A). Further 200 mM substrate was added into the reaction mixture, and approximately 90% of substrate was converted to the corresponding nitrile after a further 4 h reaction time. The final concentration of benzonitrile reached 0.62 M (63.9 g L−1). Another biotransformation reaction to synthesize benzamide was carried out on 1 L scale (Fig. 5B). The initial concentration of benzaldoxime was 100 mM, which was completely converted to benzamide within 1 h. The other batches of 100 mM substrate were added into the reaction mixture at different times. The final concentration of benzamide was 0.33 M, and the productivity was calculated to be approximately 10 g h−1 L−1. These results indicate the chemo-enzymatic process can efficiently convert aldehyde to corresponding nitriles and amides without the isolation of intermediates.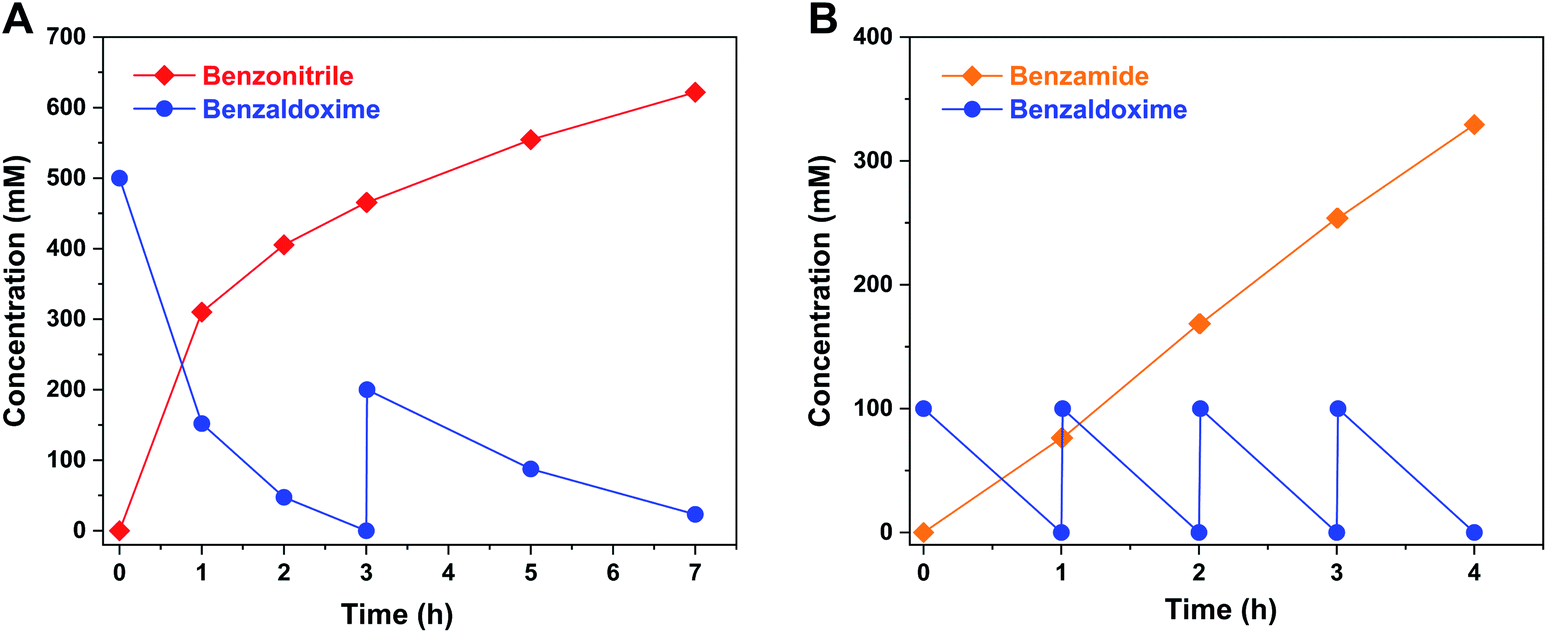 | ||
| Fig. 5 Time course of fed-batch synthesis of BN (A) and BAM (B) at the one-liter scale. Chemical synthesis of BAOx was carried out in a biphasic system. The organic was directly mixed with E. coli OxdF1 (or OxdF1–NHase1229) suspension (30 g dcw L−1 cell in 50 mM PPB, pH 7.0) resulting in a BAOx concentration of 500 mM (or 100 mM). The biphasic reaction system was stirred at 150 rpm and room temperature until BAOx was completely conversed to BN or BAM. | ||
4. Conclusions
In summary, we presented an efficient chemo-enzymatic cascade from aldehydes to corresponding nitriles and amides in a biphasic catalysis system using n-hexane as the organic solvent. The cascade reaction starts from readily accessible aldehydes that condensed with hydroxylamine to corresponding aldoximes. The upper organic phase containing aldoximes was separated and directly mixed with recombinant E. coli whole cells suspension to undergo enzymatic dehydration to the desired nitriles, or successive dehydration and hydration to amides. The biphasic system avoided the inactivation of hydroxylamine on enzymes, and bridges two worlds of chemical and enzymatic catalysis for efficiently preparing alkyl and aryl nitriles or amides by one phase separation step, and approximately 60% yields were obtained without isolation of the aldoximes intermediates. In addition, the shuttle transport of substrates and products between two phases is convenient for the products separation and the solvents recycling. Thus, the chemo-enzymatic cascade provided an alternative selection to environmentally synthesize nitriles and amides, which has great potential applications in the industry.Author contributions
Haoteng Zheng: data curation, writing – original draft; Qinjie Xiao: investigation; Feiying Mao: validation; Anming Wang: funding acquisition; Mu Li: resources; Qiuyan Wang: visualization; Pengfei Zhang: supervision; Xiaolin Pei: conceptualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Natural Science Foundation of Zhejiang Province (No. LY22B060003), the National Natural Science Foundation of China (No. 22078079), Ten Thousand People Plan of Zhejiang Province (No. 2019R51012), and the College Students' Science and Technology Innovation Project of Zhejiang Province: New Talent Plan (No. 2021R426040).References
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
- F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597–606 RSC.
- Y. Wang, Y. Du and N. Huang, Future Med. Chem., 2018, 10, 2713–2728 CrossRef CAS PubMed.
- M. X. Wang, Acc. Chem. Res., 2015, 48, 602–611 CrossRef CAS PubMed.
- Y. Nakao, Chem. Rev., 2021, 121, 327–344 CrossRef CAS PubMed.
- Z. Wang, in Comprehensive Organic Name Reactions and Reagents, ed. Z. Wang, John Wiley & Sons, 2010, pp. 1661–1663, DOI:10.1002/9780470638859.conrr370.
- I. P. Beletskaya, A. S. Sigeev, A. S. Peregudov and P. V. Petrovskii, J. Organomet. Chem., 2004, 689, 3810–3812 CrossRef CAS.
- A. Pradal and G. Evano, Chem. Commun., 2014, 50, 11907–11910 RSC.
- X. Fang, P. Yu and B. Morandi, Science, 2016, 351, 832–836 CrossRef CAS PubMed.
- D. Yang, H. Huang, M. H. Li, X. J. Si, H. Zhang, J. L. Niu and M. P. Song, Org. Lett., 2020, 22, 4333–4338 CrossRef CAS PubMed.
- X. Li, C. You, J. Yang, S. Li, D. Zhang, H. Lv and X. Zhang, Angew. Chem., Int. Ed., 2019, 58, 10928–10931 CrossRef CAS PubMed.
- I. Satyanarayana, K. B. Manjappa and D.-Y. Yang, Green Chem., 2020, 22, 8316–8322 RSC.
- R. D. Patil and M. K. Gupta, Adv. Synth. Catal., 2020, 362, 3987–4009 CrossRef CAS.
- L. Vanoye, A. Hammoud, H. Gérard, A. Barnes, R. Philippe, P. Fongarland, C. de Bellefon and A. Favre-Réguillon, ACS Catal., 2019, 9, 9705–9714 CrossRef CAS.
- A.-P. Xing, Z. Shen, Z. Zhao, X. Tian and Y.-L. Ren, Catal. Commun., 2021, 149, 106175 CrossRef CAS.
- K.-k. Sun, J.-l. Sun, G.-P. Lu and C. Cai, Green Chem., 2019, 21, 4334–4340 RSC.
- R. V. Jagadeesh, H. Junge and M. Beller, Nat. Commun., 2014, 5, 4123 CrossRef CAS PubMed.
- B. Chatterjee, S. Jena, V. Chugh, T. Weyhermüller and C. Werlé, ACS Catal., 2021, 11, 7176–7185 CrossRef CAS.
- J. Liu, C. Zhang, Z. Zhang, X. Wen, X. Dou, J. Wei, X. Qiu, S. Song and N. Jiao, Science, 2020, 367, 281–285 CrossRef CAS PubMed.
- K. Murugesan, T. Senthamarai, M. Sohail, M. Sharif, N. V. Kalevaru and R. V. Jagadeesh, Green Chem., 2018, 20, 266–273 RSC.
- A. Martin and V. N. Kalevaru, ChemCatChem, 2010, 2, 1504–1522 CrossRef CAS.
- G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068–4105 CrossRef CAS.
- B. Thomas, S. Prathapan and S. Sugunan, Chem. Eng. J., 2007, 133, 59–68 CrossRef CAS.
- W. Zhang, J. H. Lin, P. Zhang and J. C. Xiao, Chem. Commun., 2020, 56, 6221–6224 RSC.
- M. K. Singh and M. K. Lakshman, J. Org. Chem., 2009, 74, 3079–3084 CrossRef CAS PubMed.
- X. Zhang, J. Sun, Y. Ding and L. Yu, Org. Lett., 2015, 17, 5840–5842 CrossRef CAS PubMed.
- Y. L. Ban, J. L. Dai, X. L. Jin, Q. B. Zhang and Q. Liu, Chem. Commun., 2019, 55, 9701–9704 RSC.
- J.-J. Guo, T.-S. Jin, S.-L. Zhang and T.-S. Li, Green Chem., 2001, 3, 193–195 RSC.
- M. Sorensen, E. H. J. Neilson and B. L. Moller, Mol. Plant, 2018, 11, 95–117 CrossRef PubMed.
- N. Uludag, Russian J. Org. Chem., 2020, 56, 1640–1645 CrossRef CAS.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- J. B. Pyser, S. Chakrabarty, E. O. Romero and A. R. H. Narayan, ACS Cent. Sci., 2021, 7, 1105–1116 CrossRef CAS PubMed.
- Y. Asano and Y. Kato, FEMS Microbiol. Lett., 1998, 158, 185–190 CrossRef CAS.
- Y. Kato, R. Ooi and Y. Asano, J. Mol. Catal. B: Enzym., 1999, 6, 249–256 CrossRef CAS.
- K. Konishi, T. Ohta, K. Oinuma, Y. Hashimoto, T. Kitagawa and M. Kobayashi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 564–568 CrossRef CAS PubMed.
- K. Kobayashi, S. Yoshioka, Y. Kato, Y. Asano and S. Aono, J. Biol. Chem., 2005, 280, 5486–5490 CrossRef CAS PubMed.
- K. Oinuma, Y. Hashimoto, K. Konishi, M. Goda, T. Noguchi, H. Higashibata and M. Kobayashi, J. Biol. Chem., 2003, 278, 29600–29608 CrossRef CAS PubMed.
- S. X. Xie, Y. Kato, H. Komeda, S. Yoshida and Y. Asano, Biochemistry, 2003, 42, 12056–12066 CrossRef CAS PubMed.
- T. Betke, M. Maier, H. Gruber-Wolfler and H. Gröger, Nat. Commun., 2018, 9, 5112 CrossRef PubMed.
- A. Hinzmann, S. Glinski, M. Worm and H. Gröger, J. Org. Chem., 2019, 84, 4867–4872 CrossRef CAS PubMed.
- T. Betke, P. Rommelmann, K. Oike, Y. Asano and H. Gröger, Angew. Chem., Int. Ed., 2017, 56, 12361–12366 CrossRef CAS PubMed.
- D. Zheng and Y. Asano, Green Chem., 2020, 22, 4930–4936 RSC.
- D. Zheng and Y. Asano, ChemCatChem, 2021, 13, 4237–4242 CrossRef CAS.
- C. Plass, A. Hinzmann, M. Terhorst, W. Brauer, K. Oike, H. Yavuzer, Y. Asano, A. J. Vorholt, T. Betke and H. Gröger, ACS Catal., 2019, 9, 5198–5203 CrossRef CAS.
- A. Hinzmann, M. Stricker and H. Gröger, ACS Sustain. Chem. Eng., 2020, 8, 17088–17096 CrossRef CAS.
- M. Horvat, V. Weilch, R. Rädisch, S. Hecko, A. Schiefer, F. Rudroff, B. Wilding, N. Klempier, M. Pátek, L. Martínková and M. Winkler, Catal. Sci. Technol., 2022, 12, 62–66 RSC.
- Z. Chen, F. Mao, H. Zheng, Q. Xiao, Z. Ding, A. Wang and X. Pei, Enzyme Microb. Technol., 2021, 150, 109883 CrossRef CAS PubMed.
- X. Pei, H. Zhang, L. Meng, G. Xu, L. Yang and J. Wu, Process Biochem., 2013, 48, 1921–1927 CrossRef CAS.
- X. Jiang, X. Xu, Y. Lin, Y. Yan, P. Li, R. Bai and Y. Xie, Tetrahedron, 2018, 74, 5879–5885 CrossRef CAS.
- Y. Ni and J. H. Xu, Biotechnol. Adv., 2012, 30, 1279–1288 CrossRef CAS PubMed.
- A. Hinzmann, N. Adebar, T. Betke, M. Leppin and H. Gröger, Eur. J. Org. Chem., 2019, 2019, 6911–6916 CrossRef CAS.
- T. Harada, Y. Kubota, T. Kamitanaka, K. Nakamura and T. Matsuda, Tetrahedron Lett., 2009, 50, 4934–4936 CrossRef CAS.
- Z. Cheng, L. Peplowski, W. Cui, Y. Xia, Z. Liu, J. Zhang, M. Kobayashi and Z. Zhou, Biotechnol. Bioeng., 2018, 115, 524–535 CrossRef CAS PubMed.
- Y. Liu, P. Liu, S. Gao, Z. Wang, P. Luan, J. González-Sabín and Y. Jiang, Chem. Eng. J., 2021, 420, 127659 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra03256b |
| This journal is © The Royal Society of Chemistry 2022 |