
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting the singlet oxygen production from H2O2 activation with highly dispersed Co–N-graphene for pollutant removal†
Yang-Yang Yuac,
Wen-Zhu Quana,
Yuanyuan Caod,
Qijian Niue,
Yilin Luc,
Xiang Xiao c and
Liang Cheng*ab
c and
Liang Cheng*ab
aSchool of the Environment and Safety Engineering, Jiangsu University, 301 Xuefu Road, Zhenjiang 212013, China. E-mail: clcheng@ujs.edu.cn
bInstitute of Environmental Health and Ecological Security, School of the Environment and Safety Engineering, Jiangsu University, 301 Xuefu Road, Zhenjiang 212013, China
cInformation Materials and Intelligent Sensing Laboratory of Anhui Province, Institutes of Physical Science and Information Technology, Anhui University, Hefei, 230601, China
dInstitute of Medicine & Chemical Engineering, Zhenjiang College, Zhenjiang 212000, China
eKey Laboratory of Modern Agricultural Equipment and Technology, Ministry of Education, School of Agricultural Equipment Engineering, Institute of Agricultural Engineering, Jiangsu University, Zhenjiang, Jiangsu 212013, China
First published on 16th June 2022
Abstract
Singlet oxygen (1O2) is a promising reactive species for the selective degradation of organic pollutants. However, it is difficult to generate 1O2 from H2O2 activation with high efficiency and selectivity. In this work, a graphene-supported highly dispersed cobalt catalyst with abundant Co–Nx active sites (Co–N-graphene) was synthesized for activating H2O2. The Co–N-graphene catalyzed H2O2 reaction system selectively catalyzed 1O2 production associated with the superoxide radical (O2˙−) as the critical intermediate, as proven by scavenger experiments, electron spin resonance (ESR) spin trapping and a kinetic solvent isotope effect study. This resulted in excellent degradation efficiency towards the model organic pollutant methylene blue (MB), with an outstanding pseudo-first-order kinetic rate constant of 0.432 min−1 (g Lcatalyst−1)−1 under optimal reaction conditions (CH2O2 = 400 mM, initial pH = 9). Furthermore, this Co–N-graphene catalyst enabled strong synergy with HCO3− in accelerating MB degradation, whereas the scavenger experiment implied that the synergy herein differed significantly from the current Co2+–HCO3− reaction system, in which contribution of O2˙− was only validated with a Co–N-graphene catalyst. Therefore, this work developed a novel catalyst for boosting 1O2 production from H2O2 activation and will extend the inventory of catalysts for advanced oxidation processes.
1. Introduction
H2O2 is a mild and commonly used oxidant due to its environmental friendliness. H2O2 can be activated by heat, illumination, ultrasonication, and versatile homogeneous and heterogenous catalysts, to produce derivative reactive oxygen species (ROS) including the hydroxyl radical (˙OH), superoxide radical (O2˙−), and singlet oxygen (1O2) with extremely high reactivity.1–3 Among them, the hydroxyl radical can be easily produced in Fenton or Fenton-like processes, exhibiting impressive application potential in refractory wastewater treatment via the indiscriminate oxidation of organic pollutants. In contrast, singlet oxygen shows selective reactivity towards organic compounds with electron-rich moieties, attracting vast interest in pollutant remediation as well as fine chemical synthesis and photodynamic therapy.4,5 Besides, 1O2 oxidation can proceed over a wide pH range from 3 to 11 and is rarely influenced by the anions in water, showing unique advantages over ˙OH oxidation.6,7 Hence, 1O2 oxidation is attracting vast research attention, especially in pollutant remediation.8 Generally, 1O2 can be produced in a photocatalytic process9 or through the disproportionation of H2O2 catalyzed by high-valent transition-metal ions, such as MoO4.10 However, 1O2 evolution from H2O2 activation is usually accompanied by the production of undesirable ˙OH following the Haber–Weiss reaction.11 Therefore, exploring new catalysts that specifically boost 1O2 generation from H2O2 activation is of great significance.Due to the positive redox potential of the Co3+/Co2+ half reaction, Co2+ does not efficiently activate H2O2 (Xu et al., 2011). However, coordinating Co2+ to form a Co(II) complex significantly reduces the redox potential and promotes H2O2 activation. For example, Co(II) complexes of phthalocyanine derivatives, monoethanolamine and polyampholyte were found to effectively activate H2O2, degrading diverse organic pollutants such as C.I. Acid Red, Orange II and methyl orange.12–15 Besides, Co(II) complexes of inorganic ligands, especially bicarbonate, greatly accelerated H2O2 activation and pollutant degradation.16–18 Moreover, cobalt-containing mixed-metal nanomaterials, such as spinel CoCr2−xFexO4, inverse spinel Ni2CoS4, Fe–Co nanosheets, and CoxMn-layered double hydroxide, were synthesized and exhibited excellent H2O2 activation performance.19–22 Impressively, even though all these cobalt-containing catalysts could efficiently activate H2O2, the activation mechanism and major ROS were found to be significantly different. For instance, ˙OH was accounted the dominant ROS in most H2O2 activation processes by cobalt-containing mixed-metal nanomaterials, probably due to the easy regeneration of metal ions for O–O bond dissection.19–21 Moreover, ˙OH was also dominant when H2O2 was activated by soluble coordinated Co(II) complexes of monoethanolamine and bicarbonate.14,16 Surprisingly, immobilizing the coordinated Co(II) complex into a solid matrix, such as a hydrogel, mesoporous carbon or diatomite, to form a heterogenous cobalt catalyst inhibited the generation of ˙OH but promoted O2˙− and/or 1O2.13,15,17 Since O2˙− is an important intermediate in 1O2 evolution, highly selective 1O2 oxidation might be achieved in heterogenous catalysts with coordinated Co(II) complexes as active sites.
Carbon-based nitrogen coordinated atomically dispersed metal catalysts (M–N–C) are the most active new frontier in heterogeneous catalysis.23 M–N–C catalysts including Fe, Mn and Cu have already proved their value in activating Fenton-like reactions.24–26 Co–N–C was also applied for activating persulfate in pollutant remediation,27,28 whereas their role in H2O2 activation has not been fully addressed yet. In this work, a highly dispersed Co–N-graphene catalyst was prepared in a two-step process by the incorporation of Co and N into the graphene matrix. The activation of H2O2 and catalytic oxidation were investigated with a model pollutant, methylene blue (MB). The decisive 1O2 mediated oxidation was confirmed with an ROS scavenger experiment, electron spin resonance (ESR) spin trapping and a kinetic solvent isotope effect study with D2O as an alternative solvent. The selective 1O2 generation mechanism is discussed based on a comparative study with the current soluble Co2+–HCO3− reaction system.
2. Materials and methods
2.1. Chemicals
Commercial graphene oxide (GO) powder (99.0% purity, average 10 μm in size) was purchased from Carbon Thin Technologies Co., Ltd. (China). L-Tryptophan (C11H12N2O2, AR) and cobaltosic oxide (Co3O4, AR) were purchased from Shanghai Macklin Biochemical Co., Ltd. (China). Deuterium oxide (D2O, 99.9%) was purchased from Manalab (Guangdong, China). All the other chemicals were in AR and purchased from Sinopharm Group Co. Ltd. (China), and used directly unless otherwise indicated.2.2. Catalyst fabrication
The Co–N-graphene catalyst was prepared in a two-step process.29 Typically, 0.5 g of GO powder was dispersed in 500 mL of DI water by sonification for 30 min. After that, 169 mg of Co(Ac)2˙4H2O (8 wt% of cobalt to GO) and 338 mg of dicyandiamide were successively added and dispersed in the GO solution. After magnetic stirring for 30 min, the mixture was dried in an oven overnight (60 °C). The obtained solid mixture was ground into fine powder, and placed in a tube furnace for carbonization in an N2 atmosphere. The carbonization followed a two-step temperature programming (550 °C for 2 h and 700 °C for 1 h) with heating rates of 5 °C min−1. After the calcination processes, the oven was cooled down to room temperature and a black powder was obtained. Catalysts with different cobalt contents were fabricated using the same procedure by varying the Co(Ac)2˙4H2O dosage but fixing the mass ratio of dicyandiamide to Co(Ac)2˙4H2O at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The cobalt-free catalyst (N-graphene) was synthesized similarly, but only 338 mg of dicyandiamide was added. Acid treatment of Co–N-graphene proceeded as in previous work, by refluxing the product in 1 M H2SO4 for 4 h 3 times.27
:1. The cobalt-free catalyst (N-graphene) was synthesized similarly, but only 338 mg of dicyandiamide was added. Acid treatment of Co–N-graphene proceeded as in previous work, by refluxing the product in 1 M H2SO4 for 4 h 3 times.27
2.3. Batch experiment for MB degradation
The methylene blue (MB) degradation was carried out in a 100 mL conical flask at room temperature with magnetic stirring (200 rpm). Typically, the catalyst powder was dispersed in DI water under sonification to prepare a stock solution of the catalyst (1 g L−1). After that, 1.5 mL of stock solution of the catalyst was mixed with 28.5 mL of MB solution (finally, Ccat = 50 mg L−1, CMB = 50 μM). The pH of the solution was adjusted with 1 M HCl or 1 M NaOH, and hydrogen peroxide was added to initiate the MB degradation. 1 mL of solution was sampled each time, centrifuged (7500 rpm, 1 min, TG18G, Hunan Kaida Sci. Inst. Co. LTD, China) to obtain the supernatant and the absorption at 660 nm was measured with UV-vis spectroscopy (DU-800, Beckman Coulter, USA). All the concentrations of MB were reported as C/C0, where C is the absorption at the time of sampling and C0 is the absorption before H2O2 addition. In the scavenger experiments, the scavengers of ˙OH (1 mL of tert-butanol or TBA), O2˙− (1 mM of benzoquinone or BQ) or 1O2 (10 mM of tryptophan or TRP) were applied before the addition of H2O2. In the kinetic solvent isotope effect (KSIE) study, D2O/H2O mixed solvent (1:1 molar ratio) was used, and the reaction solution was stabilized for an extra 0.5 h before adding H2O2, which improved the fitting of the degradation kinetics to the pseudo-first-order kinetic equation (data not shown). The reusability of the catalysts was tested by five successive batch cycles, and the catalyst in the sample and reaction solution was harvested by centrifugation and washed with DI water and reused for the next batch experiment.
2.4. Characterization
The morphology of the catalyst was observed with field emission scanning electron microscopy (FESEM, JSM-7800F, Jeol, Japan), transmission electron microscopy (TEM, HT7800, Hitachi, Japan) and spherical-aberration-corrected transmission electron microscopy (STEM, JEM-ARM200F, Jeol, Japan) with a JEM-ARM200F (URP) ED for energy-dispersive X-ray spectroscopy (EDS) analyses. The BET specific surface area was characterized with a TriStarII3020 (Micromeritics Instr. Corp., USA). The chemical status and crystalline structure of the catalysts were analyzed by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi, Thermo, UK) and X-ray diffractometry (XRD, D8 ADVANCE, Germany), respectively. The cobalt content of the catalyst was calculated from the XPS spectrum and also measured with inductively coupled plasma mass spectrometry (ICP-MS, iCAP QC, USA). The chemical oxygen demand (COD) of the sample was quantified with a COD meter (Model 6B-200, Shengaohua Environ. Protect. Technol. Co., Ltd, China). Electron spin resonance (ESR) spin trapping investigation was conducted with an A300 spectrometer (Bruker, USA), with a center field at 3510 G and a sweep width of 100 G at room temperature. DMPO (100 mM) was used as the trapping agent for ˙OH (water as solvent) and O2˙− (methanol as solvent), and TEMP (50 mM) as the 1O2 trapping agent.303. Results and discussion
3.1. Synthesis and characterization of highly dispersed Co–N-graphene
The Co–N-graphene catalyst was synthesized in a two-step process, in which water-dispersed GO was reacted with Co(Ac)2˙4H2O and dicyandiamide of different dosages, and then dried and carbonized in an N2 atmosphere. For the catalyst prepared with 8% cobalt (wt% to GO), SEM imaging indicated the reservation of a graphene-like structure on the surface (Fig. S1†), and the BET specific surface area was determined to be 97.5 m2 g−1 (Fig. S2†). Meanwhile, the spherical-aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images disclosed a large quantity of bright spots that probably corresponded to single cobalt atoms (Fig. 1a). In addition, the HAADF-STEM and EDS (Fig. 1b–e) images revealed that C, N and Co elements were homogeneously distributed over the entire catalyst, indicating the successful incorporation of Co and N into the graphene matrix. Moreover, the high dispersity of cobalt was also supported by XRD analysis, in which only the reflections belonging to GO (12.9° and 26.7°) can be discriminated,31,32 whereas the typical reflection of cobalt species cannot be observed (Fig. S3†).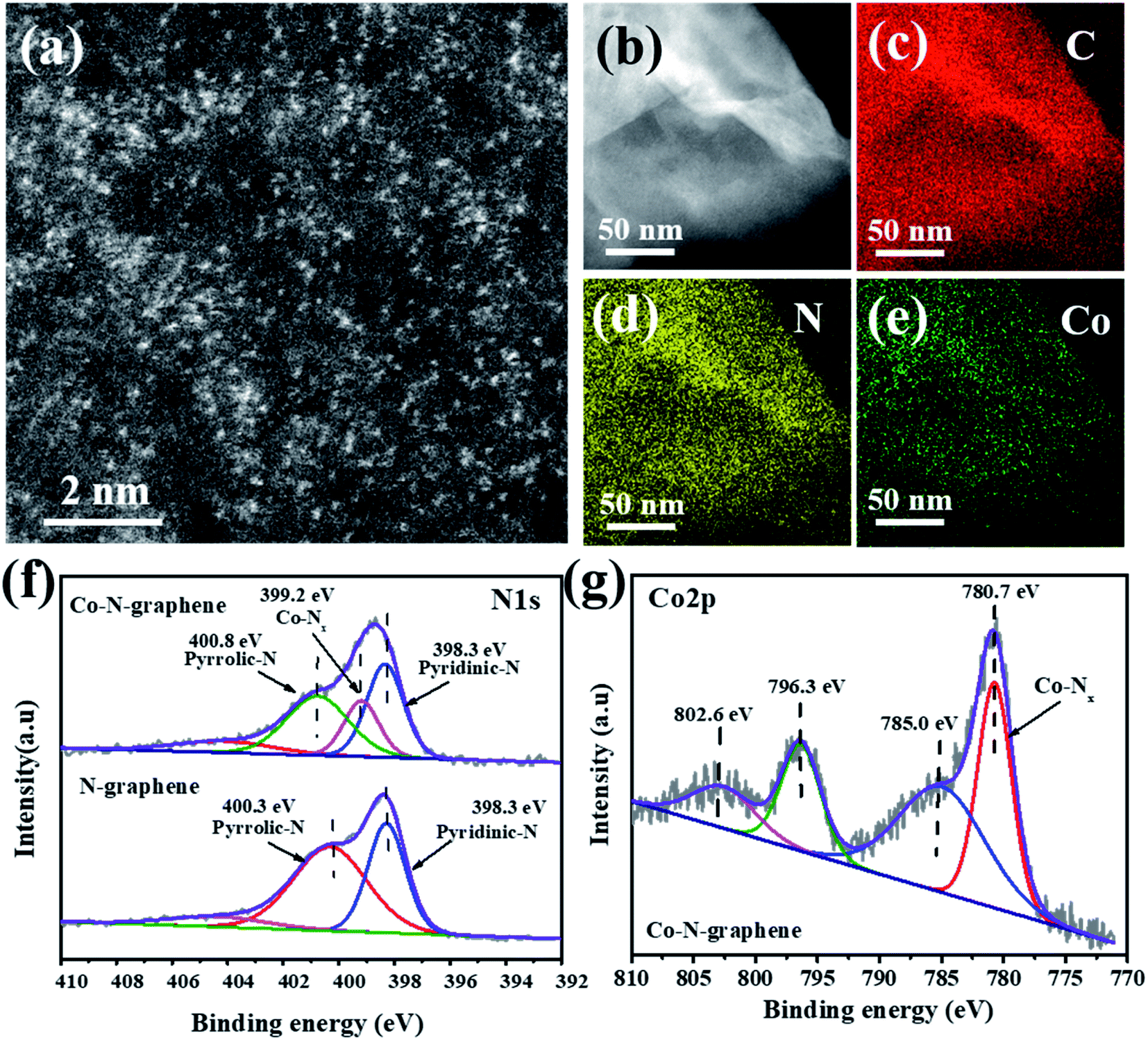 | ||
| Fig. 1 (a and b) HAADF-STEM images; (c–e) EDS mapping of C, N or Co elements of Co–N-graphene; (f) N1s and (g) Co2p XPS spectra of N-graphene and Co–N-graphene. | ||
Next, XPS analysis was used to investigate the chemical status of the catalyst. The emergence of adsorptions around 790 eV also confirmed the presence of cobalt in the catalyst (Fig. S4†), and the surface elemental contents were determined to be C 79.77%, O 11.87%, Co 1.2%, and N 7.17%, equaling a cobalt content of 5.36 wt%. Moreover, a similar cobalt content (5.78 wt%) was suggested by ICP-MS analysis. Furthermore, the high-solution N1s peaks revealed subpeaks of pyridinic (398.3 eV) and pyrrolic (400.3 eV) nitrogen species when dicyandiamide reacted only with GO. With the presence of cobalt, an additional subpeak at 399.2 eV was discriminated (Fig. 1f), and the peak location was consistent with reported Co–Nx.18 In the high-solution Co2p peaks, the subpeaks of Co2p3/2 (780.7 eV) and Co2p1/2 (796.3 eV) and satellite peak of Co2p3/2 (785 eV) were observed (Fig. 1g), and the peak at 780.7 eV was consistent with previously observed Co–Nx.33 Therefore, all these characterizations indicated the formation of highly dispersed Co–N-graphene.
3.2. Catalytic performance
The catalytic performance was investigated in a magnetically stirred flask containing catalyst, H2O2 and a model pollutant, methylene blue (MB). The study was initiated with an assay containing 50 mg L−1 of catalyst, 300 mM of H2O2 and 50 μM of MB. Decolorization of MB was observed in all assays, but the highest decolorization ratio of 86.7 ± 3.9% was only achieved with Co–N-graphene as the catalyst after treatment for 180 min, whereas the value was only 27.1 ± 0.1% for the assay with N-graphene as the catalyst and 19.3 ± 0.8% in the absence of a catalyst (only H2O2). Moreover, a decolorization ratio of 22.2 ± 0.1% was also confirmed when only Co–N-graphene was adopted (without H2O2) (Fig. 2a). Hence, although there was a certain adsorption of MB by the catalyst involved, the decolorization of MB should be attributed to catalytic removal by the Co–N-graphene activated H2O2. Consistently, UV-vis adsorption belonging to the chromophoric group of MB (N–S conjugated system on the central aromatic heterocycle)34 simultaneously disappeared (Fig. S5†) and 53.1% COD was removed after treatment (from 52.7 to 24.7 mg L−1), confirming that MB was decolorized via oxidative degradation rather than physical adsorption or reduction. | ||
| Fig. 2 (a) Removal of MB in the presence or absence of different catalysts or H2O2. Influence of (b) cobalt wt% to GO, (c) different initial pH and (d) H2O2 dosage on the removal of MB. Basic reaction conditions: Ccat: 50 mg L−1, CH2O2: 300 mM, CMB: 50 μM, pH: 7. | ||
Moreover, nanoparticles belonging to the aggregated cobalt also formed on GO during the fabrication of the catalyst (Fig. S6a†). In order to exclude the potential contribution of these aggregated nanoparticles, Co–N-graphene was acid treated via refluxing in 1 M H2SO4 for 4 h 3 times.27 The SEM image confirmed the successful removal of nanoparticles (Fig. S6b†); meanwhile the XRD and BET patterns were almost identical to those of untreated Co–N-graphene. However, the N1s XPS spectrum showed slight differences, with a much more pronounced Co–Nx subpeak observed in the N1s XPS spectrum (Fig. S7†). Moreover, the catalytic performance of acid-treated Co–N-graphene was also tested. Interestingly, the MB degradation efficiency by acid-treated Co–N-graphene was almost the same as that without acid treatment (Fig. 2a), suggesting the reaction was simply catalyzed by the dispersed Co(II), even though almost 40% of the cobalt was removed after acid treatment (3.63 wt% cobalt remaining after acid treatment, based on ICP-MS analyses). The above postulation is reasonable since cobalt-containing nanoparticles alone are not effective for H2O2 activation.18 Therefore, the influence of aggregated nanoparticles was neglected and only Co–N-graphene was used for the rest of the study.
To further discover the catalytic capacity of Co–N-graphene, the reaction conditions, including the dosage of cobalt precursor, pH and H2O2 concentration of the reaction system were optimized. Firstly, catalysts with different dosages of cobalt precursor were synthesized (1 wt%, 2 wt%, 4 wt%, 8 wt%, 12 wt% and 20 wt% of cobalt to GO) with a fixed mass ratio of Co(Ac)2˙4H2O to dicyandiamide (1:2). Unsurprisingly, the cobalt dosage significantly influenced MB degradation (Fig. 2b). With the lowest cobalt dosage (1 wt%), 46.7 ± 0.6% MB was degraded, which was obviously higher than with N-graphene (27.1 ± 0.1%, Fig. 2a). Increasing the cobalt dosage synchronously promoted the MB removal efficiency and a highest removal ratio of 98.3 ± 0.1% was achieved with the 8 wt% catalysts. However, further increasing the cobalt dosage inhibited MB degradation, and a removal ratio that was almost identical to that of N-graphene (27.1 ± 0.1%) was recorded for 20 wt% cobalt dosage (28.4 ± 0.7%). These results imply that even a small amount of Co–N active sites can efficiently activate H2O2; increasing the cobalt dosage probably increased the amount of Co–N active sites and promoted MB degradation. However, with the formation of non-reactive nanoparticles on 8% catalysts (Fig. S6a†), an even higher cobalt dosage may result in more severe aggregation, reducing the availability of dispersed active sites. Therefore, 8% was determined as the optimal cobalt dosage and the corresponding Co–N-graphene was employed in the rest of the study.
The influence of initial pH was examined over a wide pH range covering 3, 5, 7, 8, 9, 10, 11. As expected, pH was critical and the highest degradation efficiency was achieved at pH = 9, in which 98.6 ± 0.1% MB was degraded within 100 min. At pH as high as 10 and 11, slightly lower degradation efficiencies of 89.4 ± 0.1% and 86.4 ± 0.2% were obtained within 100 min, respectively. In contrast, acidic pH (3 and 5) severely inhibited MB degradation, and degradation efficiencies as low as 28.1 ± 1.1% and 52.3 ± 1.8% were recorded after 100 min, respectively. Moreover, the role of H2O2 concentration was also investigated, and the highest degradation efficiency was observed with 400 mM H2O2. A low H2O2 dosage cannot generate sufficient ROS to promote effective MB degradation due to the substrate limitation, whereas radical scavenging by H2O2 became dominant once the optimal dosage was exceeded and the MB degradation was constrained as well. Notably, the observed optimal H2O2 concentration (400 mM) was higher than in previous work with ˙OH as the main ROS (Table S1†), implying that this reaction system may be not as sensitive to scavenging ˙OH. Hence, pH = 9 and 400 mM were set as the optimal pH and H2O2 dosage, respectively.
Since MB is a well-known photosensitizer which produces ROS including 1O2 with excellent quantum yield,35 the potential role of self-produced ROS in MB degradation was investigated by comparing the performance in the presence or absence of environmental light. The results showed that enveloping the conical reaction flask with aluminum foil only resulted in a slight suppression of MB degradation (Fig. S8†). Hence, MB was degraded mainly by the ROS produced from Co–N-graphene catalyzed H2O2 activation, and the influence of environmental light was neglected. Moreover, this catalyst showed excellent reusability under the optimal reaction conditions, and only a slight reduction in the performance was observed in five consecutive MB degradation cycles (Fig. S9†). Such performance reduction might be caused by the demetallation of Co–N-graphene in the presence of H2O2.36 Besides, H2O2 may be activated by the released Co2+. To exclude this, the Co2+ ions in the reaction solution after MB degradation were quantified and the same dosage of the measured soluble Co2+ (0.066 mg L−1) was added to N-graphene for MB degradation. The results showed that such a small content of Co2+ rarely influenced MB degradation (Fig. S10†). Therefore, the Co2+ released from Co–N-graphene demetallation did not contribute to MB degradation.
3.3. 1O2 mediated MB degradation
Next, the main ROS contributing to MB degradation was identified. Firstly, an ESR spin trapping investigation was conducted to disclose the ROS species generated from H2O2 activation. Since the presence of ˙OH, O2˙− and 1O2 was observed in the diverse cobalt-containing catalyst activated H2O2,16–18 DMPO (100 mM, for ˙OH with water as solvent and O2˙− with methanol as solvent) and TEMP (50 mM, for 1O2) were used as trapping agents. The results showed very weak peaks belonging to the DMPO–˙OH adduct but revealed peaks corresponding to O2˙− and 1O2 (Fig. 3a) (Luo et al., 2019). Therefore, O2˙− and 1O2 were the dominant ROS species when H2O2 was activated by as-prepared Co–N-graphene. | ||
| Fig. 3 (a) ESR spectra of Co–N-graphene activated H2O2, with DMPO and TEMP as trapping agents. (b) Effect of ROS scavengers on the removal of MB. TBA: 1 mL of tert-butanal (for ˙OH), BQ: 1 mM of benzoquinone (for O2˙−), TRP: 10 mM of tryptophan (for 1O2). Effect of D2O on (c) the removal of MB and (d) fitting with pseudo-first-order kinetic model. Ccat: 50 mg L−1, CH2O2: 400 mM, CMB: 50 μM, pH: 9. | ||
After that, the scavenger experiments were processed to investigate the contribution of these ROS species to MB degradation. tert-Butanol (TBA) shows good reactivity with ˙OH (k˙OH = 6.0 × 108 M−1 s−1), and was selected as the ˙OH scavenger.37 Meanwhile, benzoquinone (BQ, 1 mM) and tryptophan (TRP, 10 mM) were used due to their good reactivity and selectivity towards O2˙− (1.1 × 108 M−1 s−1) and 1O2 (108 to 109 M−1 s−1), respectively.38,39 Impressively, TBA exhibited a negligible effect on MB degradation (Fig. 3b, blue and red), implying that ˙OH hardly contributed, which was consistent with the weak peaks of the DPMO–˙OH adduct observed in ESR. In contrast, BQ and TRP substantially inhibited MB degradation (Fig. 3b, green and pink). In particular, only 8.4 ± 0.8% MB was removed when 10 mM TRP was added, which was similar to H2O2 alone (10.0 ± 0.2%) (Fig. S11†). Hence, scavenging 1O2 alone almost completely inhibited MB degradation. These results were remarkably different from previous observations, in which more than one ROS species usually contributed to substrate degradation in the cobalt-containing catalyst activated H2O2 system, and certain degree of catalytic degradation could still be obtained when even combined ROS scavengers such as TBA and NaN3 (another 1O2 scavenger) were applied.17
To further confirm the dominant role of 1O2, a kinetic solvent isotope effect (KSIE) study was carried out by applying deuterium oxide (D2O) as an alternative solvent. Because quenching of 1O2 is solvent-dependent, it was reported that the rate constant is 16 times slower in D2O (kD2O, 1.6 × 104 s−1) than in H2O (kH2O, 2.5 × 105 s−1).7 Since the fast quenching of 1O2 was decisive for the overall reaction,40 the reaction rate constant of MB with 1O2 in a D2O and H2O mixed solvent (kobs, mix) follows the equation below :41
 | (1) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group of MB first,42,43 broke the C–S bond and then decomposed the macromolecular intermediates to form multiple benzene derivatives.44 Finally, the benzene ring was destroyed by an attack by 1O2 to form smaller organisms.45
C group of MB first,42,43 broke the C–S bond and then decomposed the macromolecular intermediates to form multiple benzene derivatives.44 Finally, the benzene ring was destroyed by an attack by 1O2 to form smaller organisms.45
Table S1† summarizes representative recent achievements of using diverse heterogeneous nanocatalysts to activate H2O2 and degrade MB. For a reasonable comparison, the observed pseudo-first-order kinetic rate constants (kobs) were also normalized to the catalyst dosage (k1) and metal amount in the catalyst (k2). The k1 herein (0.432 min−1 (g L−1)−1) was obviously larger than most nanocatalysts in directly activating H2O2 to degrade MB, except those anchoring Fe2O3 nanoparticles inside a nanoporous carbon matrix to promote catalytic efficiency via a nanoconfinement effect.3,46 Strikingly, the direct catalytic efficiency was also comparable to most heterogeneous photo-Fenton catalysts. Moreover, since H2O2 was activated by metal species, the small atomic fraction of cobalt in Co–N-graphene (1.25%) indicated an impressively large k2 compared with aggregated nanocatalysts, demonstrating excellent atomic efficiency. Besides this, 1O2 was the ROS solely contributing to MB degradation when H2O2 was activated by this dispersed Co–N-graphene catalyst.
In these previous approaches, the decisive contribution of 1O2 was observed only with the nanoconfined Fe2O3@FCNT-H, which was attributed to the nanoconfinement provided by carbon nanotubes enabling pulse-like water transmission with concerted and rapid motion along the tube axis, promoting 1O2 generation by accelerating the one-electron reductive generation of intermediate HO2˙/O2˙− due to the strong electronic interaction between CNT and nanoconfined Fe2O3 nanoparticles.3 However, the dispersed Co–N-graphene catalyst produced herein features 1O2 dominated oxidation from H2O2 activation in an “open” aqueous environment, and better catalytic efficiency than most aggregated heterogeneous nanocatalysts.
The “dark” production of 1O2 from a peroxide such as H2O2 is significant due to its independence of illumination.10 This process involves one-step two-electron or two-step one-electron oxidation of H2O2.47 In this work, MB was almost completely degraded by 1O2, whereas O2˙− was also detected by ESR (Fig. 3a) and scavenging of O2˙− significantly inhibited MB degradation (Fig. 3b). Therefore, O2˙− was the key intermediate here, and 1O2 was likely to have been generated in the two-step one-electron processes following the Haber–Weiss reaction.3 In particular, Co(III) was generated when coordinated Co(II) was initially oxidized by H2O2, and then carried on the one-electron oxidation of H2O2 to form O2˙−. Another one-electron oxidation proceeded to generate 1O2 thereafter. The second step one-electron oxidation might be carried out by ˙OH, H2O2 or cobalt species. The involvement of ˙OH in 1O2 formation was observed in a diatomite-supported cobalt powder catalyst activated H2O2 system.17 However, since the scavenging of ˙OH rarely influenced MB degradation, the oxidation by ˙OH is probably marginal in this study. In contrast, O2˙− is more likely to be oxidized by H2O2, making the high optimal concentration of H2O2 (400 mM) reasonable since the reaction rate constant of O2˙− with H2O2 is several orders lower than with ˙OH.48 Also, the oxidation of O2˙− by cobalt species cannot be excluded in this study due to recent reports showing the critical role of high-valent cobalt–oxo complexes in persulfate oxidation.49–51 In brief, O2˙− from the one-electron oxidation of H2O2 was the critical intermediate here and 1O2 was most likely generated via the oxidation of O2˙− by H2O2 (Fig. 4).
 | ||
| Fig. 4 Proposed H2O2 activation and 1O2 evolution mechanism catalyzed by Co–N-graphene. | ||
3.4. The influence of anions in the wastewater
Anions in the wastewater can react with radicals generated, consuming the ROS available to the organic pollutants and produce secondary radicals, having a contradictory influence on the degradation efficiency (Luo et al., 2019). Therefore, the effect of anions was studied. Among the common anions in the wastewater, HCO3− is specific for a cobalt–H2O2 catalytic system because it might coordinate with Co2+ to form a Co2+–HCO3− complex that is extremely effective for Fenton-like reactions and also to generate secondary carbonate radicals for pollutant degradation.16–18 Consistent with previous reports, Co2+ (50 mg L−1) alone marginally catalyzed MB degradation (Fig. S11†), but supplying HCO3− (10 mM) remarkably enhanced the degradation efficiency, with 96.6 ± 0.1% MB removed within 20 min (Fig. 5a). Impressively, HCO3− significantly promoted MB degradation in Co–N-graphene catalyzed reactions as well, recording an extremely high catalytic efficiency (88.6 ± 0.1% MB removed within 5 min and 99.6 ± 0.2% within 10 min) (Fig. 5b). Moreover, the scavenging experiments disclosed a different inhibition pattern between Co2+–HCO3− and Co–N-graphene–HCO3− reaction systems. As shown in Fig. 5, MB degradation was significantly inhibited when scavenging 1O2 in the Co2+–HCO3− reaction system, but not O2˙− and ˙OH. However, in the Co–N-graphene–HCO3− system, scavenging 1O2 and O2˙− both exhibited a remarkable inhibition effect, which was the same as for Co–N-graphene alone. Therefore, 1O2 was the dominant reactive radical contributing to these systems, but it may be generated in quite a different way. In a diatomite-supported cobalt powder and HCO3− co-catalyzed H2O2 reaction system, 1O2 and ˙OH contributed to MB degradation but O2˙− did not participate. It was postulated that 1O2 was generated in a process involving ˙OH, but O2˙− did not contribute due to the fast radical–radical recombination.17 The negligible role of O2˙− was similar to the Co2+–HCO3− reaction system here, but differed significantly from the Co–N-graphene–HCO3− reaction system. Therefore, the observed O2˙− intermediated 1O2 evolution regardless of the presence or absence of HCO3− with dispersed Co–N-graphene catalyst seems unique compared with those previous works, which might be attributed to the Co–N-graphene catalyst being very efficient for catalyzing the one-electron oxidation of H2O2 to form O2˙−. On the other hand, although HCO3− effectively promoted MB degradation, its role in coordinating cobalt to form a metal complex as well as the formation of a derivative carbonate radical is not evident here. Taking all these results together, Co–N-graphene here possessed distinguished characteristics to generate O2˙− for 1O2 evolution, and the synergy with HCO3− probably derived from a currently unaddressed pathway, in which the generation or sustaining of 1O2 might be enhanced. | ||
Fig. 5 Removal of MB in the (a) Co2+–HCO3− and (b) Co–N-graphene–HCO3− reaction system in the absence or presence of ROS scavengers. Ccat: 50 mg L−1,  , CH2O2: 400 mM, CMB: 50 μM, pH: 9. , CH2O2: 400 mM, CMB: 50 μM, pH: 9. | ||
In addition, the influence of other anions in wastewater including H2PO4−, Cl−, NO3−, and SO42− was also studied. The result showed that the anions tested rarely influenced MB degradation even at a high concentration of 500 mM (Fig. 6). A similar phenomenon was observed in the 1O2 dominated peroxymonosulfate (PMS) activation processes, except that 500 mM Cl− remarkably enhanced the pollutant degradation due to the formation of HOCl via the direct reaction between Cl− and PMS (Luo et al., 2019). Hence, this dispersed Co–N-graphene catalyzed H2O2 reaction system enables excellent resistance to side reactions caused by the anions in the wastewater and is advantageous in practical applications.
 | ||
| Fig. 6 Effect of common anions in the wastewater on the removal of MB. Ccat: 50 mg L−1, CH2O2: 400 mM, CMB: 50 μM, pH: 9. | ||
4. Conclusion
In summary, a highly dispersed Co–N-graphene catalyst was synthesized in a two-step process by the incorporation of Co and N into the graphene matrix. The prepared catalyst showed excellent performance in activating H2O2 for methylene blue degradation. After optimizing the dosage of cobalt precursor (8 wt% of cobalt to GO), reaction pH (initial pH = 9) and content of H2O2 in the reaction (400 mM), the catalyst dosage normalized pseudo-first-order kinetic rate constant was determined to be 0.432 min−1 (g L−1)−1, outperforming most current aggregated nanocatalysts in the heterogeneous Fenton or Fenton-like reaction for MB degradation. Moreover, the ESR, scavenger experiment and kinetic solvent isotope effect study suggested that MB was completely degraded by 1O2, which was produced in an O2˙− intermediated two-step one-electron oxidation of H2O2. Furthermore, this highly dispersed Co–N-graphene catalyst enabled strong synergy with bicarbonate anions, but the underlying mechanism of this synergistic interaction differed remarkably from the current Co2+–HCO3− reaction system, suggesting the unrecognized role of bicarbonate in the generation or sustaining of 1O2 in this reaction system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20211600), the Jiangsu Agricultural Science and Technology Innovation Fund (CX(21)3056), and the Science and Technology Planning Social Development Project of Zhenjiang City (SH2021017). The authors are also thankful for the support from the program of Jiangsu Distinguished Professor and the Innovation/Entrepreneurship Program of Jiangsu Province.References
- Y. Zhang and M. Zhou, J. Hazard. Mater., 2019, 362, 436–450 CrossRef CAS PubMed.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- Z. Yang, J. Qian, A. Yu and B. Pan, Natl. Acad. Sci., 2019, 116, 6659–6664 CrossRef CAS PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- W. Wu, X. Shao, J. Zhao and M. Wu, Adv. Sci., 2017, 4, 1700113 CrossRef.
- S. Liu, Z. Zhang, F. Huang, Y. Liu, L. Feng, J. Jiang, L. Zhang, F. Qi and C. Liu, Appl. Catal. B Environ., 2021, 286, 119921 CrossRef CAS.
- T. Liu, D. Zhang, K. Yin, C. Yang, S. Luo and J. C. Crittenden, Chem. Eng. J., 2020, 388, 124264 CrossRef CAS.
- J. Hynek, M. K. Chahal, D. T. Payne, J. Labuta and J. P. Hill, Coord. Chem. Rev., 2020, 425, 213541 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2007, 129, 6916–6926 CrossRef CAS PubMed.
- L. S. Zhang, X. H. Jiang, Z. A. Zhong, L. Tian, Q. Sun, Y. T. Cui, X. Lu, J. P. Zou and S. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 21751–21755 CrossRef CAS.
- C. Shen, S. Song, L. Zang, X. Kang, Y. Wen, W. Liu and L. Fu, J. Hazard. Mater., 2010, 177, 560–566 CrossRef CAS PubMed.
- N. Li, W. Lu, K. Pei, Y. Yao and W. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 5869–5876 CrossRef CAS PubMed.
- M. Chen, K. Peng, H. Wang, Z. Yang, Q. Zeng and A. Xu, Chem. Eng. J., 2012, 197, 110–115 CrossRef CAS.
- L. V. Lombardo Lupano, J. M. Lázaro Martínez, L. L. Piehl, E. Rubin de Celis and V. Campo Dall' Orto, Appl. Catal. Gen., 2013, 467, 342–354 CrossRef CAS.
- A. Xu, X. Li, S. Ye, G. Yin and Q. Zeng, Appl. Catal. B Environ., 2011, 102, 37–43 CrossRef CAS.
- L. Zhou, W. Song, Z. Chen and G. Yin, Environ. Sci. Technol., 2013, 47, 3833–3839 CrossRef CAS PubMed.
- H. Dong, X. Feng, Y. Guo, Z. Jia, X. Zhang, A. Xu and X. Li, Colloids Surf. A Physicochem. Eng. Asp., 2021, 630, 127645 CrossRef CAS.
- T. Tatarchuk, A. Shyichuk, I. Trawczyńska, I. Yaremiy, A. T. Pędziwiatr, P. Kurzydło, B. F. Bogacz and R. Gargula, Ceram. Int., 2020, 46, 27517–27530 CrossRef CAS.
- M. Kim, S. H. Kim, J. H. Lee and J. Kim, J. Hazard. Mater., 2020, 392, 122347 CrossRef CAS.
- M. Nie, Y. Li, L. Li, J. He, P. Hong, K. Zhang, X. Cai, L. Kong and J. Liu, Appl. Surf. Sci., 2021, 535, 147655 CrossRef CAS.
- H. Farhat, C. Taviot-Gueho, G. Monier, V. Briois, C. Forano and C. Mousty, J. Phys. Chem. C, 2020, 124, 15585–15599 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang, Y. Li, D. Vuckovic, Y. Li and S. Dai, Nat. Sustain., 2021, 4, 233–241 CrossRef PubMed.
- M. Liu, Z. Feng, X. Luan, W. Chu, H. Zhao and G. Zhao, Environ. Sci. Technol., 2021, 55, 6042–6051 CrossRef CAS PubMed.
- Z. Guo, Y. Xie, J. Xiao, Z. J. Zhao, Y. Wang, Z. Xu, Y. Zhang, L. Yin, H. Cao and J. Gong, J. Am. Chem. Soc., 2019, 141, 12005–12010 CrossRef CAS PubMed.
- H. Xu, N. Jiang, D. Wang, L. Wang, Y. Song, Z. Chen, J. Ma and T. Zhang, Appl. Catal. B Environ., 2020, 263, 118350 CrossRef CAS.
- Y. Qi, J. Li, Y. Zhang, Q. Cao, Y. Si, Z. Wu, M. Akram and X. Xu, Appl. Catal. B Environ., 2021, 286, 119910 CrossRef CAS.
- F. Chen, X.-L. Wu, L. Yang, C. Chen, H. Lin and J. Chen, Chem. Eng. J., 2020, 394, 124904 CrossRef CAS.
- M. P. Rayaroth, K. P. Prasanthkumar, Y. G. Kang, C. S. Lee and Y. S. Chang, Chem. Eng. J., 2020, 382, 122828 CrossRef CAS.
- J. Sun, S. Bai, Y. Tian, Y. Zhao, N. Han, R. Luo, D. Li and A. Chen, Sens. Actuators, B, 2018, 257, 29–36 CrossRef CAS.
- Y. Ping, J.-M. Yan, Z. L. Wang, H. L. Wang and Q. Jiang, J. Mater. Chem. A, 2013, 1, 12188–12191 RSC.
- C. Tang, B. Wang, H. F. Wang and Q. Zhang, Adv. Mater., 2017, 29, 1703185 CrossRef PubMed.
- C. Yang, W. Dong, G. Cui, Y. Zhao, X. Shi, X. Xia, B. Tang and W. Wang, RSC Adv., 2017, 7, 23699–23708 RSC.
- F. Ronzani, A. Trivella, E. Arzoumanian, S. Blanc, M. Sarakha, C. Richard, E. Oliveros and S. Lacombe, Photochem. Photobiol. Sci., 2013, 12, 2160–2169 CrossRef CAS PubMed.
- X. Xie, C. He, B. Li, Y. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. Nie, J. Liu, D. J. Myers, P. Zelenay, G. Wang, G. Wu, V. Ramani and Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- R. Luo, M. Li, C. Wang, M. Zhang, M. A. N. Khan, X. Sun, J. Shen, W. Han, L. Wang and J. Li, Water Res., 2019, 148, 416–424 CrossRef CAS PubMed.
- M. Zhu, J. Lu, Y. Hu, Y. Liu, S. Hu and C. Zhu, Environ. Sci. Pollut. Res., 2020, 27, 31289–31299 CrossRef CAS PubMed.
- R. L. Jensen, J. Arnbjerg and P. R. Ogilby, J. Am. Chem. Soc., 2012, 134, 9820–9826 CrossRef CAS PubMed.
- X. Lu, W. Qiu, J. Ma, H. Xu, D. Wang, H. Cheng, W. Zhang and X. He, Chem. Eng. J., 2020, 401, 126128 CrossRef CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Refer. Data, 1995, 24, 663–677 CrossRef CAS.
- G. Zhu, J. Zhu, X. Fu, Q. Liu, F. Cao, Y.-n. Li, Q. Qin and M. Jiao, PCCP, 2020, 22, 15340–15353 RSC.
- G. Zhu, J. Zhu, Q. Liu, X. Fu, Z. Chen, K. Li, F. Cao, Q. Qin and M. Jiao, Phys. Chem. Chem. Phys., 2021, 23, 5283–5297 RSC.
- S. Su, Y. Liu, X. Liu, W. Jin and Y. Zhao, Chemosphere, 2019, 218, 83–92 CrossRef CAS PubMed.
- S. Tang, Z. Wang, D. Yuan, C. Zhang, Y. Rao, Z. Wang and K. Yin, J. Clean. Prod., 2020, 268, 122253 CrossRef CAS.
- S. Zhang, M. Sun, T. Hedtke, A. Deshmukh, X. Zhou, S. Weon, M. Elimelech and J. H. Kim, Environ. Sci. Technol., 2020, 54, 10868–10875 CrossRef CAS PubMed.
- D. F. Evans and M. W. Upton, J. Chem. Soc., Dalton Trans., 1985, 124, 2525–2529 RSC.
- J. Weinstein and B. H. Bielski, J. Am. Chem. Soc., 1979, 101, 58–62 CrossRef CAS.
- Y. Zong, X. Guan, J. Xu, Y. Feng, Y. Mao, L. Xu, H. Chu and D. Wu, Environ. Sci. Technol., 2020, 54, 16231–16239 CrossRef CAS PubMed.
- B. Liu, W. Guo, H. Wang, S. Zheng, Q. Si, Q. Zhao, H. Luo and N. Ren, J. Hazard. Mater., 2021, 416, 125679 CrossRef CAS PubMed.
- B. Liu, W. Guo, W. Jia, H. Wang, S. Zheng, Q. Si, Q. Zhao, H. Luo, J. Jiang and N. Ren, Water Res., 2021, 117313 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02491h |
| This journal is © The Royal Society of Chemistry 2022 |