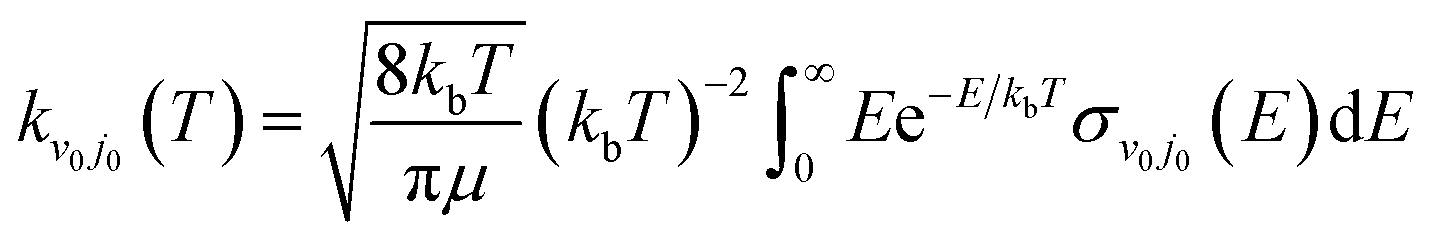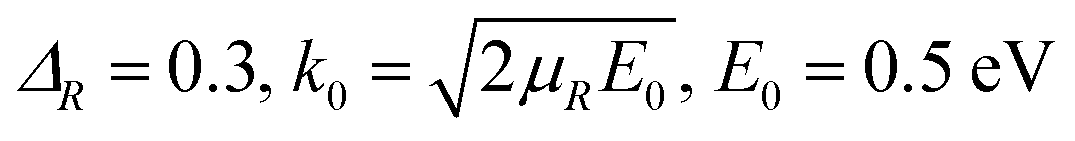DOI:
10.1039/D2RA03028D
(Paper)
RSC Adv., 2022,
12, 19751-19762
Nonadiabatic dynamics studies of the H(2S) + RbH(X1Σ+) reaction: based on new diabatic potential energy surfaces
Received
13th May 2022
, Accepted 14th June 2022
First published on 7th July 2022
Abstract
The global diabatic potential energy surfaces (PESs) that correspond to the ground (12A′) and first excited states (22A′) of the RbH2 system PES are constructed based on 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 786 ab initio points. The neural network method is used to fit the PESs and the topographic features of the new diabatic PESs are discussed in detail. Based on the newly constructed diabatic PESs, the dynamics calculations of the H(2S) + RbH(X1Σ+) → Rb(52S) + H2(X1Σg+)/Rb(52P) + H2(X1Σg+) reactions are performed using the time-dependent wave packet method. The dynamics properties of these two channels such as the reaction probabilities, integral cross sections, and differential cross sections (DCSs) are calculated at state-to-state level of theory. The nonadiabatic effects are discussed in detail, and the results indicate that the adiabatic results are overestimated from the dynamics values. The DCSs of these two channels are forward biased, which indicates that the abstraction mechanism plays a dominant role in the reaction.
786 ab initio points. The neural network method is used to fit the PESs and the topographic features of the new diabatic PESs are discussed in detail. Based on the newly constructed diabatic PESs, the dynamics calculations of the H(2S) + RbH(X1Σ+) → Rb(52S) + H2(X1Σg+)/Rb(52P) + H2(X1Σg+) reactions are performed using the time-dependent wave packet method. The dynamics properties of these two channels such as the reaction probabilities, integral cross sections, and differential cross sections (DCSs) are calculated at state-to-state level of theory. The nonadiabatic effects are discussed in detail, and the results indicate that the adiabatic results are overestimated from the dynamics values. The DCSs of these two channels are forward biased, which indicates that the abstraction mechanism plays a dominant role in the reaction.
1. Introduction
The Born–Oppenheimer (BO) approximation separates the motions of the electrons and nucleus of an atom and is the foundation of modern theoretical chemistry. However, the BO approximation fails at describing electronic state crossings or degeneracies (nonadiabatic effects), and these phenomena are ubiquitous in molecular reaction dynamics. Thus, to further our understanding of reaction mechanisms, accurate diabatic potential energy surfaces (PESs) are necessary. Solving for the mixing angle between different electronic states is a key step during the construction, and is often challenging. A rigorous approach to obtaining the mixing angle is to calculate the integral of the nonadiabatic coupling matrix elements (NACMEs) between different electronic states. However, some drawbacks are found when the NACME approach is applied to polyatomic systems. First of all, the NACMEs are not equal to zero at positions far from the conical intersection area. Secondly, singularities in the NACMEs and abrupt changes within certain ranges of configuration space result in discontinuities in the derivatives near the conical intersection area. Thirdly, the integration path through NACME is not unique for systems containing more than two atoms. As a result, some approximation methods have been developed to build diabatic PESs, such as the CI coefficients,1–3 molecular properties,4–6 and diabatization by Ansatz7 methods.
Reactions of X + H2 (X = Li, Na, K, Rb, Cs) are highly endothermic, and nonadiabatic coupling occurs along the reaction path. Thus, X + H2 reactions and their reverse reactions are ideal models for the study of nonadiabatic processes. In present work, the H + RbH reaction was chosen. Because Rb is highly endergonic, it is always excited from ground state to an excited state in experiments using a laser. For example, using a crossed beam apparatus, Cuvellier and co-worker8 studied the electronic-to-vibrational (E–V) energy transfer processes of the Rb(7S) + H2(j = 1) → Rb(5D) + H2(j = 3) reaction in 1986. The results indicated that the near-resonant process has a larger cross section than the non-resonant process at low collision velocities. In 1997, the E-V energy transfer processes between Rb(52P1/2,3/2) atom and H2 molecule were investigated by Chen et al.9 using coherent anti-stokes Raman scattering spectroscopy. To best of our knowledge, only Fan et al.10 studied the reaction between Rb(52D, 72S) atom and H2 in 1999. The relative reactivity of different excited states of Rb with H2 followed Rb(72S1/2) > Rb(52D3/2) > Rb(52D5/2) and was determined by comparing the spectral intensities of RbH action spectra and Rb atomic fluorescence excitation spectra. In addition, the results also indicated that the available energy was mainly transformed into translational energy (80%).
Theoretical studies of the H + RbH reaction were mainly carried out by the group of Pascale11,12 in 1987. The non-reaction collisions processes between Rb(52P) and H2, D2 molecules were studied using close-coupling quantum mechanical methods based on a diabatic PES the authors constructed. The fine-structure transition cross sections of between Rb(52P1/2) and Rb(52P3/2) were calculated and compared with available experimental data.
As far as we know, no theoretical studies of the Rb + H2 reaction and its reverse reactions until now. The main reason is that few PESs are available for this system. Thus, the aims of present paper are to present new global nonadiabatic PESs for the RbH2 system and to study the nonadiabatic H + RbH (v0 = 0, j0 = 0) reaction process. This paper is organized as follows: the theoretical methods used in the ab initio calculations as well as the PES fitting and dynamics calculations are shown in Section 2; the properties of the diabatic PESs and the dynamical results are presented in Section 3; and the conclusions are presented in Section 4.
2. Theoretical methods
2.1 Construction of the diabatic PES
2.1.1 Ab initio calculations. The ab initio energy points used in the PES fitting were calculated using the internally contracted multi-reference configuration interaction (MRCI) method13,14 with the aug-cc-pVQZ15,16 basis set for the H atoms and pseudopotential basis set Def2-QZVPP17 for the Rb atom. The ab initio calculations were first performed using the Hartree–Fock calculation, and then state-averaged complete active space self-consistent field (SA-CASSCF)18,19 calculations over three lowest electronic states (12A′, 22A′ and 12A′′) with equal weight were performed. The active space of the RbH2 system was composed of 12 active orbitals (10a′ + 2a′′). In detail, 3a′ and 1a′′ are closed and related to the 4s and 4p orbitals of Rb, and the 7a′ and 1a′′ active orbitals correspond to the 1s, 2s orbitals of H and the 5s, 5p and 4d orbitals of Rb. Subsequently, the internally contracted MRCI calculation was performed based on the reference orbits determined in the SA-CASSCF calculation. The Davidson correction was applied to the final energies to compensate for the termination errors of high-order terms. Jacobi coordinates (RQ, RHH, θ) were used to cover the entire configuration space, and 17786 ab initio energy points were calculated on a non-uniform grid. In detail, RQ is the distance between Rb atom and the midpoint of H2 molecule, and varied between 0.4 ≤ RQ/a0 ≤ 25; RHH is the bond length of H2 molecule, and varied between 0.6 ≤ RHH/a0 ≤ 25; θ is the angle between RQ and RHH varied from 1° to 89°. The geometrical structures used in the ab initio calculations are presented in Table 1. In addition, all ab initio calculations were performed using the MOLPRO20 program and using Cs symmetry.
Table 1 The geometrical structures used in the ab initio calculation
| RQ (bohr) |
0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.2, 2.4, 2.6, 2.8, 3.0, 3.3, 3.6, 3.9, 4.2, 4.5, 4.9, 5.3, 5.7, 6.1, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5, 10.0, 11.0, 12.0, 13.0, 14.0, 15.0, 16.0, 18.0, 20.0, 22.0, 25.0 |
| RHH (bohr) |
0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.2, 2.4, 2.6, 2.8, 3.0, 3.3, 3.6, 3.9, 4.2, 4.5, 4.9, 5.3, 5.7, 6.1, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, 9.5, 10.0, 11.0, 12.0, 13.0, 14.0, 15.0, 16.0, 18.0, 20.0, 22.0, 25.0 |
| θ (°) |
1°, 15°, 30°, 45°, 60°, 75°, 89° |
2.1.2 Diabatization scheme. As discussed in the introduction, because it is very difficult to determine the mixing angle between the coupled electronic states from the integral of NACMEs, the molecule property method was used in the present calculations. After thorough testing, the molecule property method accurately reflected the transition characteristics between coupled states in the RbH2 system. The diabatization scheme has been widely used in previous theoretical studies.4–6 Herein, a brief introduction is given, and more details can be found in previous literature.21–23 In Cs symmetry, the 12A′ and 22A′ states in the RbH2 system are correlated. As the symmetry changes to C2v, the 12A′ and 22A′ states change to the 12A1 and 12B2 states, respectively.As the diabatic coupling is considered between 12A′ and 22A′ states, the diabatic wave function ϕi was obtained from the adiabatic wave function ψi using a unitary transformation
| |
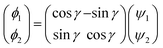 | (1) |
where
γ is the mixing angle. Then the diabatic energies
Vii were obtained from adiabatic energies following
| | |
V11 = cos2γE1 + sin2γE2
| (2) |
| | |
V22 = cos2γE2 + sin2γE1
| (3) |
| | |
V12 = (E2 − E1)cosγ × sinγ
| (4) |
where
V11 and
V22 are diabatic energies corresponding to the diabatic states, and
V12 and
V21 are the coupling potential energies between 1
2A′ and 2
2A′ states. In present calculation, the dipole operator transition moments are used to obtain the mixing angle. To obtain the mixing angle, the
ψ3 wave function for the 1
2A′′ state was taken and was not coupled with the 1
2A′ and 2
2A′ states. Thus, according to
eqn (1), the matrix elements 〈
ψ3|
![[P with combining circumflex]](https://www.rsc.org/images/entities/i_char_0050_0302.gif)
|
ψ1〉 and 〈
ψ3|
|
ψ2〉 can be presented as
| | |
〈ψ3||ψ1〉 = cosγ〈ψ3||ϕ1〉 + sinγ〈ψ3||ϕ2〉
| (6) |
| | |
〈ψ3||ψ2〉 = cosγ〈ψ3||ϕ2〉 − sinγ〈ψ3||ϕ1〉
| (7) |
The matrix elements 〈ψ3||ϕ1〉 = 0 and 〈ψ3||ϕ2〉 = 1 at high symmetry conditions. In the present work, the approximation that 〈ψ3||ϕ1〉 = 0 and 〈ψ3||ϕ2〉 = 1 was used for all geometries, not just for the high symmetry geometries. Then, the mixing angle was obtained by the following formula
| |
 | (8) |
2.1.3 PES fitting. Compared with traditional fitting methods, the neural network (NN) method has several numerical advantages and is more accurate. As such, the NN method has been widely used in the construction of PESs for the F + H2,24 O+ + H2,25 Br + H2 (ref. 26) reactions among others. In present work, the permutation invariant polynomial neural network (PIP-NN)27,28 method was used to fit the PES. Herein, we only make a brief introduction to the method. For the calculations, three bond lengths in RbH2 were transformed into PRbHa = exp(−0.2 × rRbHa), PRbHb = exp(−0.2 × rRbHb) and PHH = exp(−0.2 × rHH). To avoid the discontinuities in the derivatives, the permutation invariant polynomial method was introduced.| |
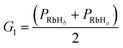 | (9) |
To ensure a random sample, the data for {G1, G2, G3} were subjected to a normalization treatment
| |
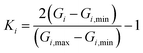 | (12) |
| |
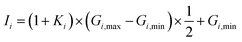 | (13) |
where
Gi,min and
Gi,max are the minimum and maximum values of
Gi, and
Ii is the input data. In this work, two hidden layers were used and 15 neurons were included in each layer. The Levenberg–Marquardt method
29 was used to train the neural network and the expansion of neural network can be written as
| |
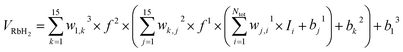 | (14) |
where
Ntot is the number of energy points,
wj,il are connection weights of the
ith neuron in (
l − 1)th layer and the
jth neuron in
lth layer, and
bjl are biases of the
jth neuron in
lth layer,
f1 and
f2 are training function and it can be presented as
| |
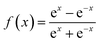 | (15) |
Over-fitting is a well-known problem in neural network calculations. To avoid over-fitting, the input data were randomly sampling into three portions, namely the training part (90 percent), testing part (5 percent) and validation part (5 percent).
2.2 Time-dependent wave packet method
The time-dependent wave packet (TDWP) method is a powerful theoretical tool for reaction scattering calculations, and it is widely used to calculate atom-diatom collision scattering30–32 as well as the reactions involving polyatomic system.33,34 Herein, we only make a brief introduction to the TDWP method used in the atom-diatom reaction scattering. In body-fixed frame, the Hamiltonian of the H(2S) + RbH(X1Σ+) reaction for a given total angular momentum J can be presented as| |
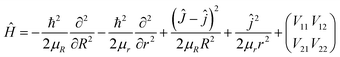 | (16) |
where R is the distance between the H atom and the center of mass of RbH molecule, and r is the vibrational coordinate of RbH. Ĵ and ĵ are angular momentum operators for the RbH2 system and RbH molecule, respectively, and μR and μr are the reduced masses. The 2 × 2 matrix is the potential energy obtained from the diabatic PES of RbH2 system.
Before the propagation, an initial wave packet needs to be defined. Here, at t = 0, the initial wave packet can be presented as
| |
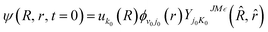 | (17) |
where
uk0(
R) is a Gaussian function, and
ϕv0j0(
r) is the ro-vibrational eigenfunction of RbH.
In the calculation, the split operator scheme35 was used to propagate the initial wave packet. Finally, the reactant coordinates-based method36,37 was used to extract the state-to-state information. Using the state-to-state S-matrix, the state-to-state reaction probability was obtained as
| |
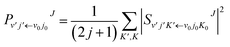 | (18) |
the state-to-state integral cross section (ICS) was obtained as
| |
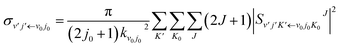 | (19) |
and the state-to-state differential cross section (DCS) was obtained as
| |
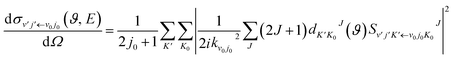 | (20) |
where
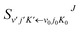
is the state-to-state
S-matrix,
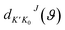
is the reduced rotation matrix element, and
ϑ is the scattering angle.
The rate constant of the H(2S) + RbH(X1Σ+) reaction was obtained by thermally averaging the translational energy of the corresponding ICS, and the formula is presented as
| |
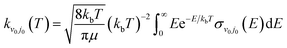 | (21) |
where
kb is Boltzmann's constant.
3. Results and discussion
3.1 The features of the diabatic PES
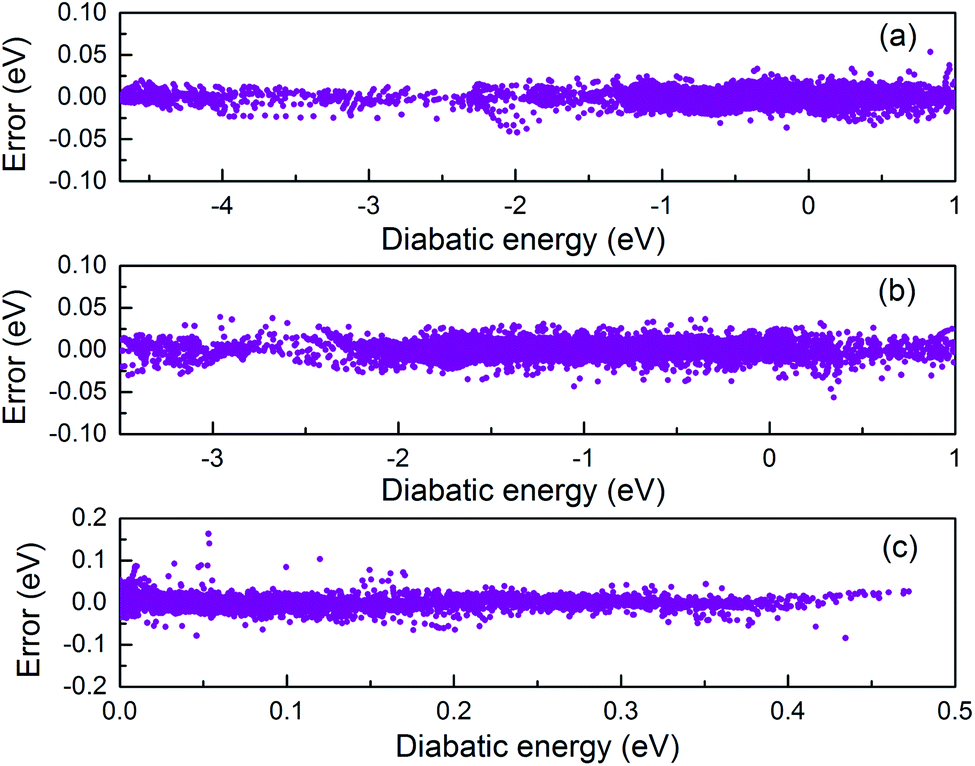
In present work, the V11, V22, and V12 terms in the diabatic potential were individually fitted using the PIP-NN method. The root-mean-square-errors (RMSE) for V11, V22, and V12 terms are 4.95, 6.45 and 9.17 meV, respectively. Fig. 1 shows the fitting errors for V11, V22, and V12 as a function of the diabatic energy. Because the sign of V12 in the PES abruptly changes with diabatic energy, the absolute values of V12 were used in fitting process. The energy derivatives using the absolute values were discontinuity and nonzero values emerged for high symmetry geometries (C2v, D2h); however, these effects on the final quantum dynamics results are negligible. The relatively large fitting error was mainly due to the complex morphology of V12. Due to the intrude of excited states, the fitting error for the PES of V22 is larger than the PES of V11.
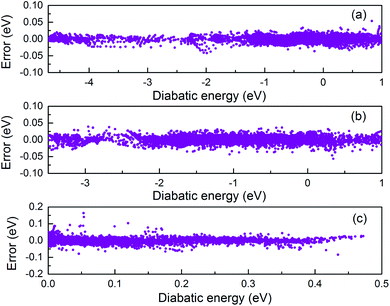 |
| | Fig. 1 The distributions of fitting errors as a function of diabatic energy for V11 (a), V22 (b) and V12 (c). | |
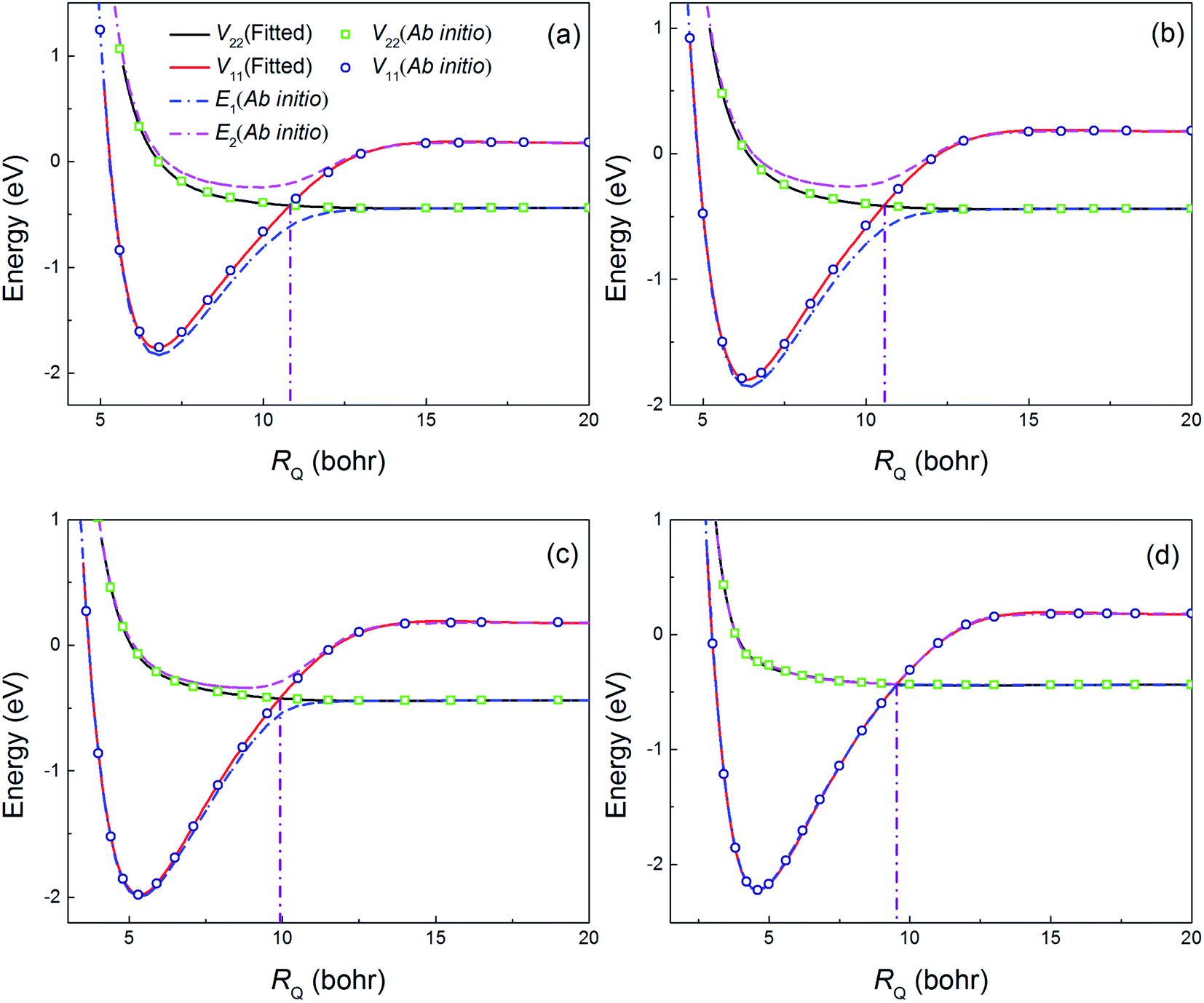
Fig. 2 shows a comparison of the fitting values and the ab initio calculation results as a function of RQ at several selected angles. The potential energy curves (PECs) in the diabatic representation crossed smoothly near the conical intersection area and the PECs in the adiabatic representation overlapped in the asymptotic region. These results indicated that potential coupling term V12 was zero in the asymptotic region. In addition, the position of crossing point becomes small with the increase of bond angle. The fitting values were in good agreement with the ab initio data which indicated that present PESs were well fitted.
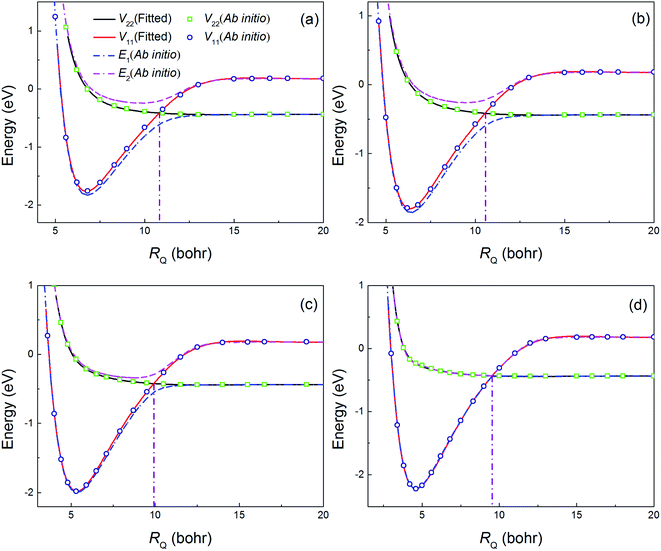 |
| | Fig. 2 Comparisons of the fitting values and the ab initio data at several selected angles ((a), (b), (c) and (d) correspond to 1°, 30°, 60°, and 89°, respectively) at a fixed H2 molecular bond length of 3.4 bohr. | |
The accuracy of long-range interaction potential will affect the dynamics results for the H + RbH reaction, especially in the low collision energy region. Therefore, long-range interaction potential of H close to RbH in collinear structures is shown in Fig. 3, when the RbH bond length is fixed at equilibrium. To test the accuracy of the fitting results for the long-range interaction potential, the ab initio results are also shown in Fig. 3. As shown in the figure, the fitting results are in good agreement with the ab initio values. However, when zoom in the long-range region, the differences between fitting results and ab initio values are more obvious. The fitting error is about 1 meV for the long-range region.
 |
| | Fig. 3 The long-range interaction potential as H atom approach to the RbH molecule in collinear structures. | |
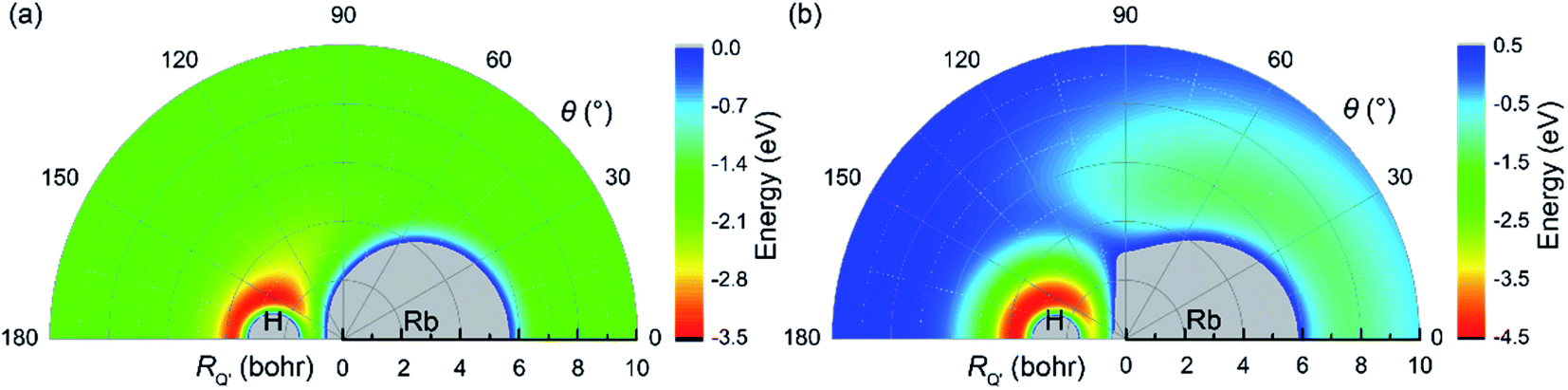
The potential energies for an H atom moving around an RbH molecule on the V22 and V11 PESs are displayed in Fig. 4(a) and (b), respectively, for a fixed RbH bond length of 4.78 bohr. RQ′ is the distance between the H atom and the center of the RbH molecule. As seen in the figure, a deep well was observed near the H atom in both Fig. 4(a) and (b). This well suggests that at low collision energies, if an H atom approaches the RbH molecule from the Rb side, it will move around the RbH molecule to form an H2 molecule. Thus, the sideward and backward scattering signals may be more obvious at low collision energies. Meanwhile, at high collision energies, the incoming H atom will be repelled and the H atom in RbH does not form chemical bond with the incoming H atom. If the H atom approach the RbH molecule from H atom side, the incoming H atom will be trapped in the deep well and an H2 molecule will form.
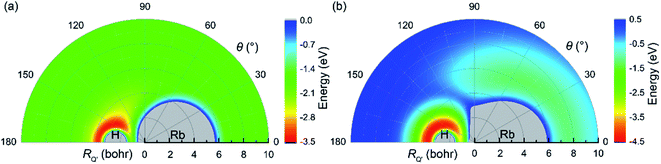 |
| | Fig. 4 Color plot of an H atom moving around an RbH molecule. The bond length of RbH is fixed at the equilibrium distance (4.78 bohr). (a) Denotes the V22 PES (b) denotes the V11 PES. | |
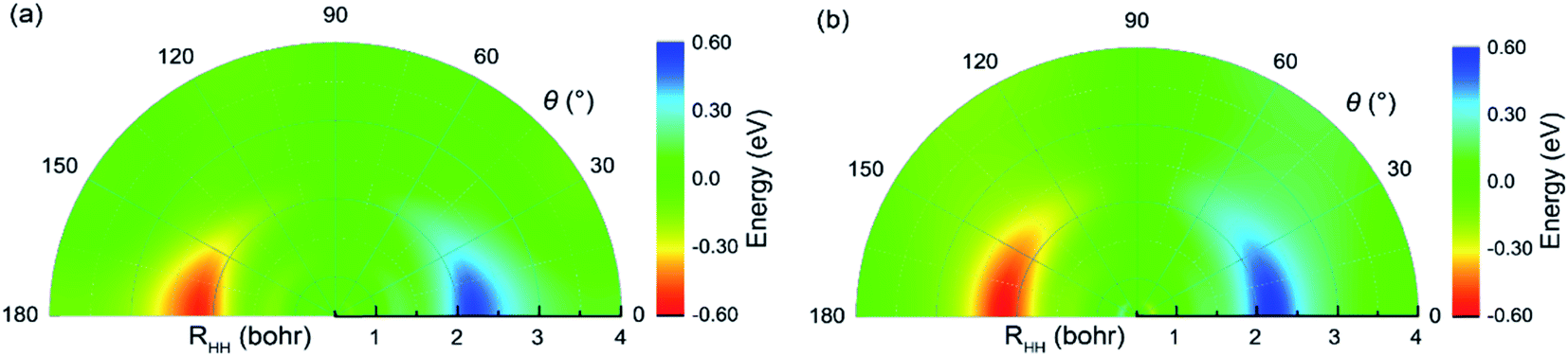
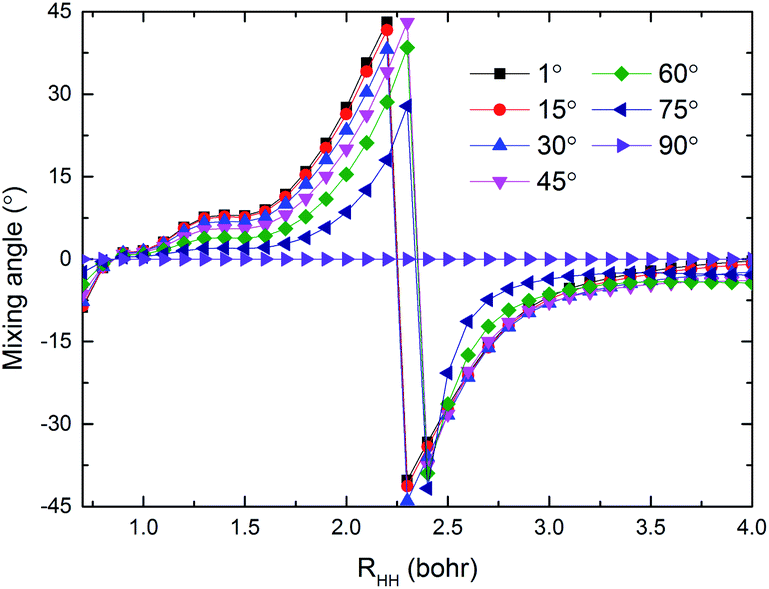
The potential energies of the coupling term V12 are shown in Fig. 5(a) and (b) for RQ equal to 4.4 and 5.4 bohr, respectively. As seen in the figure, the largest value for the V12 is about 0.6 eV in the collinear structure. In addition, V12 is almost equal to zero beyond the cross region. It is very clear that the largest values of V12 are mainly focused in the region 0 ≤ θ (°) ≤ 30, 1.8 ≤ RHH (bohr) ≤ 2.7 when RQ = 4.4 bohr and in the region 0 ≤ θ (°) ≤ 45, 1.8 ≤ RHH (bohr) ≤ 2.5 when RQ = 5.4 bohr. The regular shape of V12 indicates that the present diabatic PESs are a reasonable description of cross region. Fig. 6 presents the mixing angles at several selected angles for RQ = 5.4 bohr and correspond to the data in Fig. 5(b). The mixing angle is very small in the region beyond the cross area and increases in the vicinity of the cross region where it reaches a maximum value of 45°, which corresponds to the cross point. At the conical intersection at 90°, the mixing angle is equal to zero, because the coupling term V12 = 0.
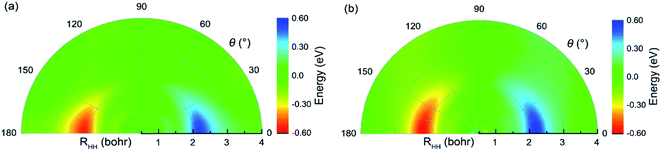 |
| | Fig. 5 Color plot for the coupling term V12 as a function of internal coordinates when the RQ equal to 4.4 bohr (a) and 5.4 bohr (b), respectively. | |
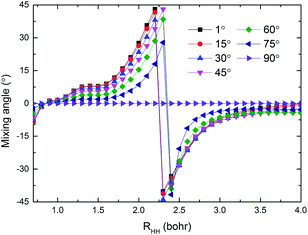 |
| | Fig. 6 The mixing angles at different selected angles when RQ = 5.4 bohr. | |
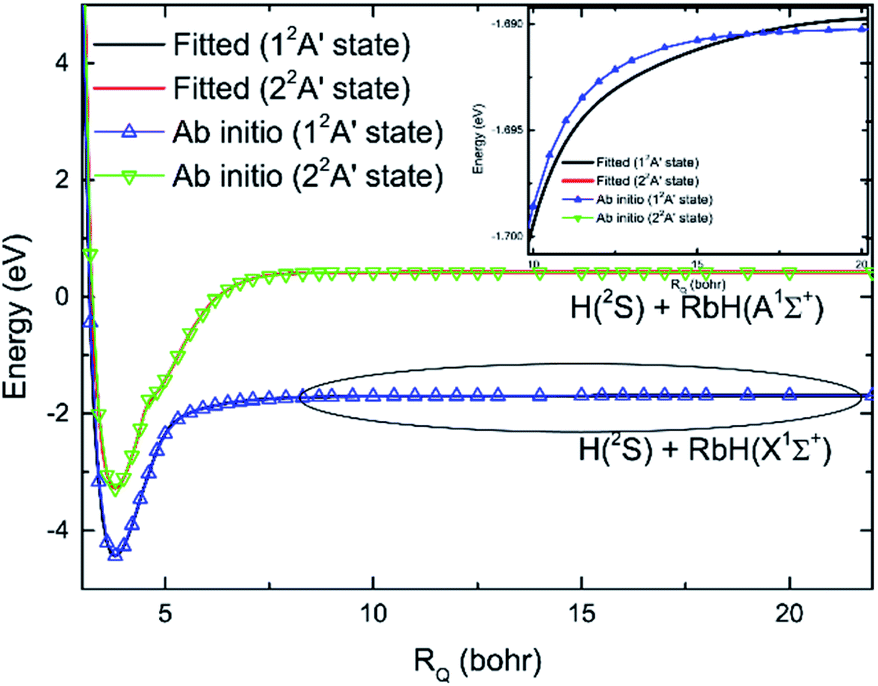
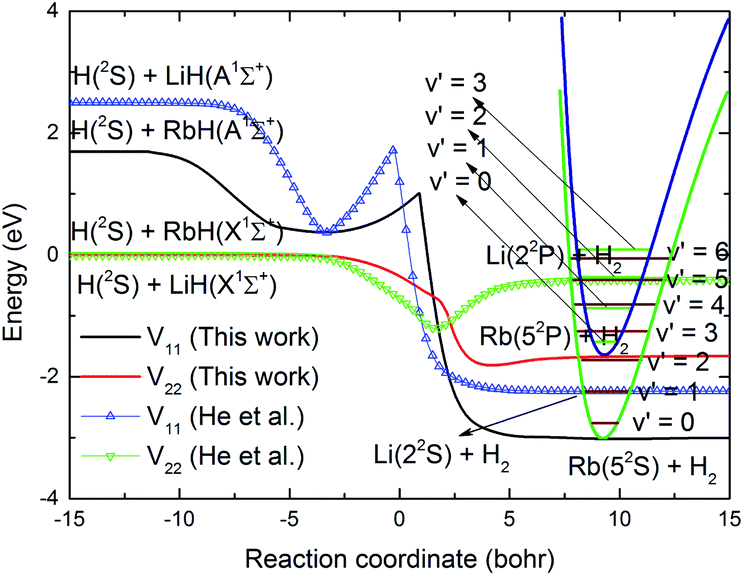
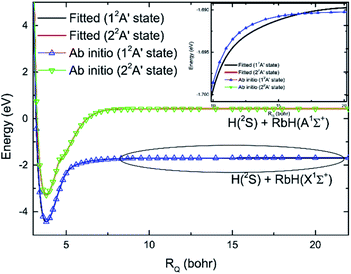
The Rb + H2 reaction studied here is very similar to the previous studied Li + H2 reaction as both are highly exothermic reactions between an alkali metal and hydrogen. The minimum energy paths from the RbH2 diabatic PESs calculated here and that from the LiH2 diabatic PESs reported by He et al.38 are compared in Fig. 7. As seen in the figure, the energy of the H(2S) + RbH(X1Σ+) channel was set as zero. For convenient comparison, the energy of the H(2S) + LiH(X1Σ+) channel was also set as zero. For the RbH2 system, it is very clear that the H(2S) + RbH(X1Σ+) reaction is highly exothermic and the energies are about −1.65 and −3.0 eV for the Rb(52P) + H2(X1Σg+) and Rb(52S) + H2(X1Σg+) channels, respectively. As the zero-point energy considered, the exoergic energies were −1.435 and −2.775 eV for these two channels, respectively. The H(2S) + LiH(X1Σ+) reaction is also an exothermic reaction and the exoergic energies were about −1.2 and −2.4 eV for the Li(22P) + H2(X1Σg+) and Li(22S) + H2(X1Σg+) channels, respectively. This comparison clearly shows that the exoergic energies of the H(2S) + RbH(X1Σ+) reaction are larger than that of H(2S) + LiH(X1Σ+) reaction. The Rb + H2 and Li + H2 reactions had other similar features including a well on the V22 PES and a well and a barrier on the V11 PES. Additional features of these stationary points on the RbH2 diabatic PESs, including the geometrical structure, energy, and harmonic vibrational frequency are presented in Table 2. As seen in the table, these stationary points belong to C2v symmetry.
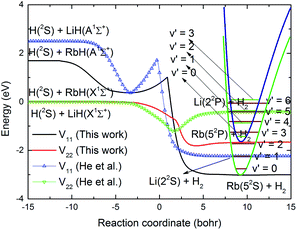 |
| | Fig. 7 Comparison of the global minimum energy paths of RbH2 diabatic PESs and that of the LiH2 diabatic PESs obtained from He et al.38 as a function of the reaction coordinate. | |
Table 2 The spectroscopic constants of the stationary pointsa
| RRbHa |
RRbHb |
RHH |
Angle (°) |
En (eV) |
ω1 (cm−1) |
ω2 (cm−1) |
ω3 (cm−1) |
| The unit is bohr for bond length. The angle is [HRbH]. Relative to the energy of Rb(52P) + H2(X1Σg+) asymptote. Relative to the energy of H + RbH(A1Σ+) channel. |
| Global minimum (V22 PES) |
| 5.611 |
5.611 |
1.435 |
14.7 |
−0.145b |
286.47 |
601.94 |
4014.17 |
|
| Local minimum (V11 PES) |
| 5.242 |
5.242 |
8.720 |
112.56 |
−1.316c |
171.40 |
572.42 |
810.67 |
|
| Transition state (V11 PES) |
| 5.761 |
5.761 |
4.128 |
42.0 |
−0.393c |
312.77 |
421.25 |
1847.46i |
3.2 Dynamics results and discussion
The optimal numerical parameters obtained from numerous numerical simulations of the reaction probabilities at total angular momentum J = 0 are shown in Table 3 and were used in all J > 0 nonadiabatic TDWP calculation.
Table 3 The optimal numerical parameters used in the nonadiabatic TDWP calculation (the atomic unit is used unless otherwise stated)a
| NtotR denotes the total grids in R direction, NintR denotes the grids in the interaction region of R direction, Ntotr denotes the total grids in r direction, Nasyr denotes the grids in the interaction region of r direction. |
| Grids range and size |
R ∈ [2.5, 22.0], NtotR = 179, NintR = 159 |
| r ∈ [2.5, 22.0], Ntotr = 179, Nasyr = 79 |
| jmin = 0 ∼ jmax = 259, Nj = 259 |
| Initial wave packet |
exp[−(R − Rc)2/2ΔR2]cos(k0R), Rc = 16.0 |
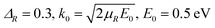 |
| Matching plane |
Rv = 16.0 |
| K-block |
K = 9 |
| Total propagation time |
45000 |
| Time step |
10 |
| Highest J value |
90 |
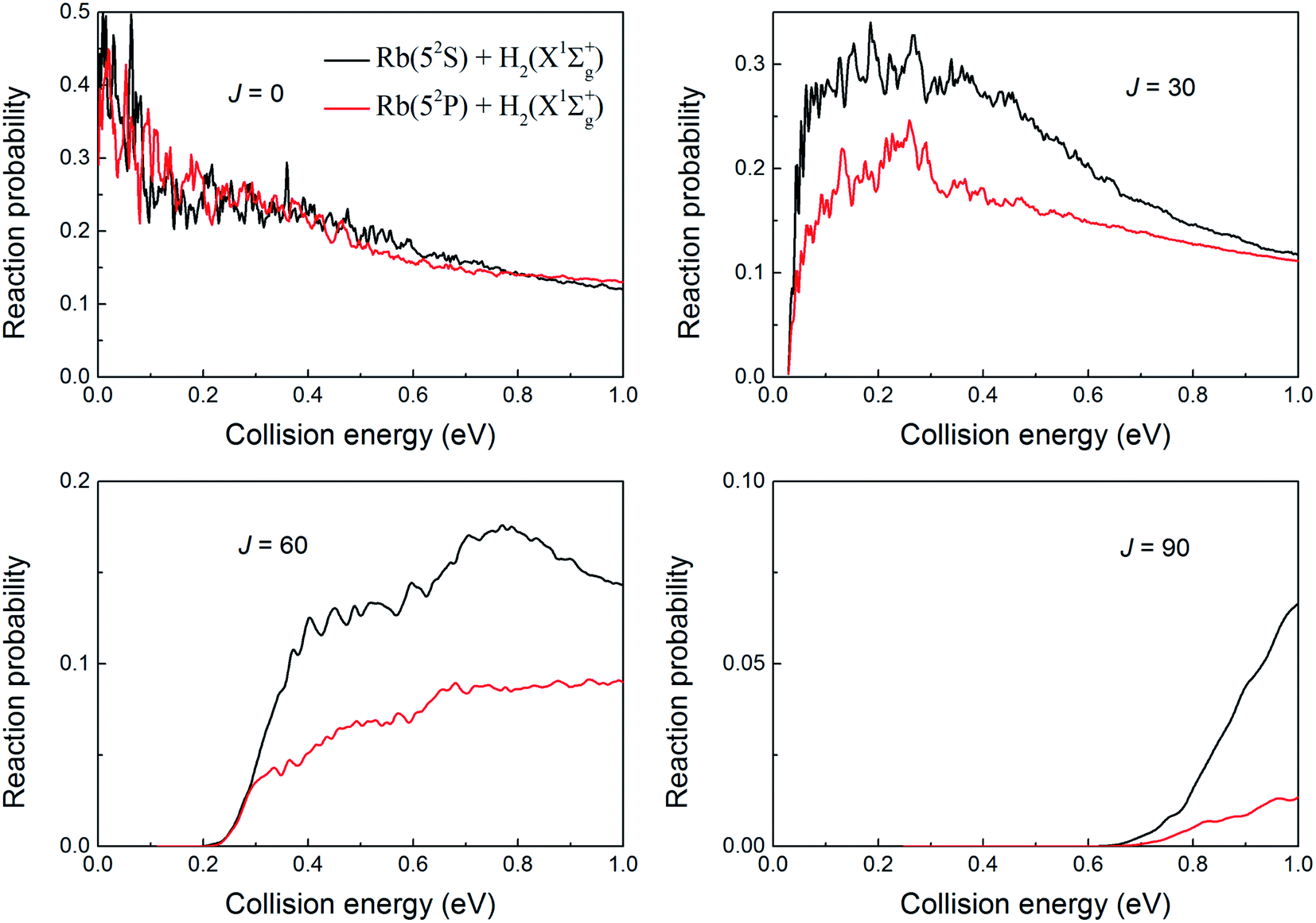
The total reaction probabilities of the H(2S) + RbH(X1Σ+) → Rb(52S) + H2(X1Σg+)/Rb(52P) + H2(X1Σg+) reaction for several selected total angular momentum of J = 0, 30, 60 and 90 are displayed in Fig. 8 over a collision energy range from 0.001 to 1.0 eV. As shown in the figure, the reaction probabilities of these two channels are similar at J = 0 and both decrease with an increase in the collision energy. This is a typical phenomenon seen in exothermic reaction without barrier. In fact, it is unusual that the reaction probabilities for these two reactions are almost equal at same collision energy. Before propagation, the initial wave packet is located in the H(2S) + RbH(X1Σ+) reactant channel. After propagation, the wave packets are distributed between the Rb(52S) + H2(X1Σg+) and Rb(52P) + H2(X1Σg+) channels according to the adiabatic-to-diabatic transformation matrix which is composed of the mixing angle. If the wave packet is evenly distributed between the two channels, the reaction probability of Rb(52P) + H2(X1Σg+) channel will be larger than that of Rb(52S) + H2(X1Σg+) channel. The exoergic energy of Rb(52P) + H2(X1Σg+) channel is less than that of Rb(52S) + H2(X1Σg+) channel, so the velocity of H in the corresponding Rb(52P) + H2(X1Σg+) channel is relatively low. Therefore, the reactivity of the Rb(52P) + H2(X1Σg+) channel will be higher than that of the Rb(52S) + H2(X1Σg+) channel. Here the results are inconsistent with the hypothesis. Therefore, we suppose that a large proportion of wave packet is distributed in the Rb(52S) + H2(X1Σg+) channel, which is the main channel of the H(2S) + RbH(X1Σ+) reaction. Moreover, the difference between the reaction probabilities of Rb(52S) + H2(X1Σg+) and Rb(52P) + H2(X1Σg+) channels increase with increasing J value. This shows that with the increase of J, the proportion of wave packet that goes into the Rb(52P) + H2(X1Σg+) channel becomes smaller and the nonadiabatic effect becomes less obvious. This conclusion is consistent with the conclusion of studies of the H + LiH reaction.39
 |
| | Fig. 8 Total reaction probabilities of the two channels of the H(2S) + RbH(X1Σ+) reactions at total angular momentums of J = 0, 30, 60 and 90. | |
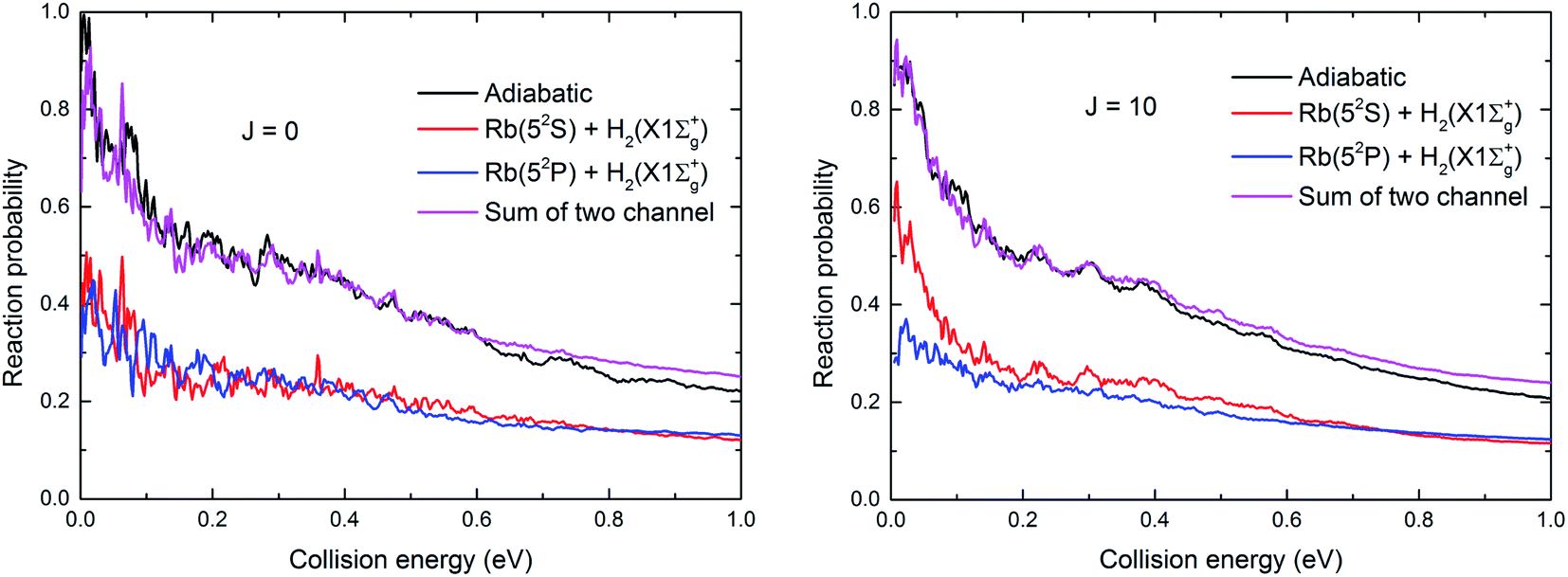
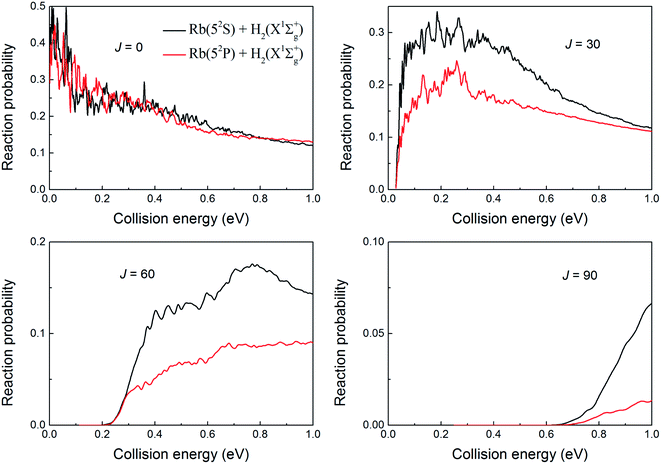
To further understand the nonadiabatic effects in the reaction, we also performed the adiabatic dynamics calculations based on the adiabatic PES which obtained by diagonalizing the diabatic PESs. The results are presented in Fig. 9 for J = 0 and 10. As seen in the figure, the reaction probabilities from the adiabatic results are larger, similar to the results seen for the H + LiH reaction.39 However, the sum of two channels in the nonadiabatic results is even larger than the adiabatic results, especially in the high collision energy range. This is very different from the results for the H + LiH reaction, in which the sum of two channels in the nonadiabatic values is smaller than the adiabatic values. We suppose such difference can be attributed to the different exothermic energies of these two reactions. However, it is important to note that the conclusions based on the adiabatic results are unreliable because the nonadiabatic effects were not considered for both H + RbH and H + LiH reactions.
 |
| | Fig. 9 The total reaction probabilities based on the adiabatic and diabatic PESs at J = 0 and 10, respectively. | |
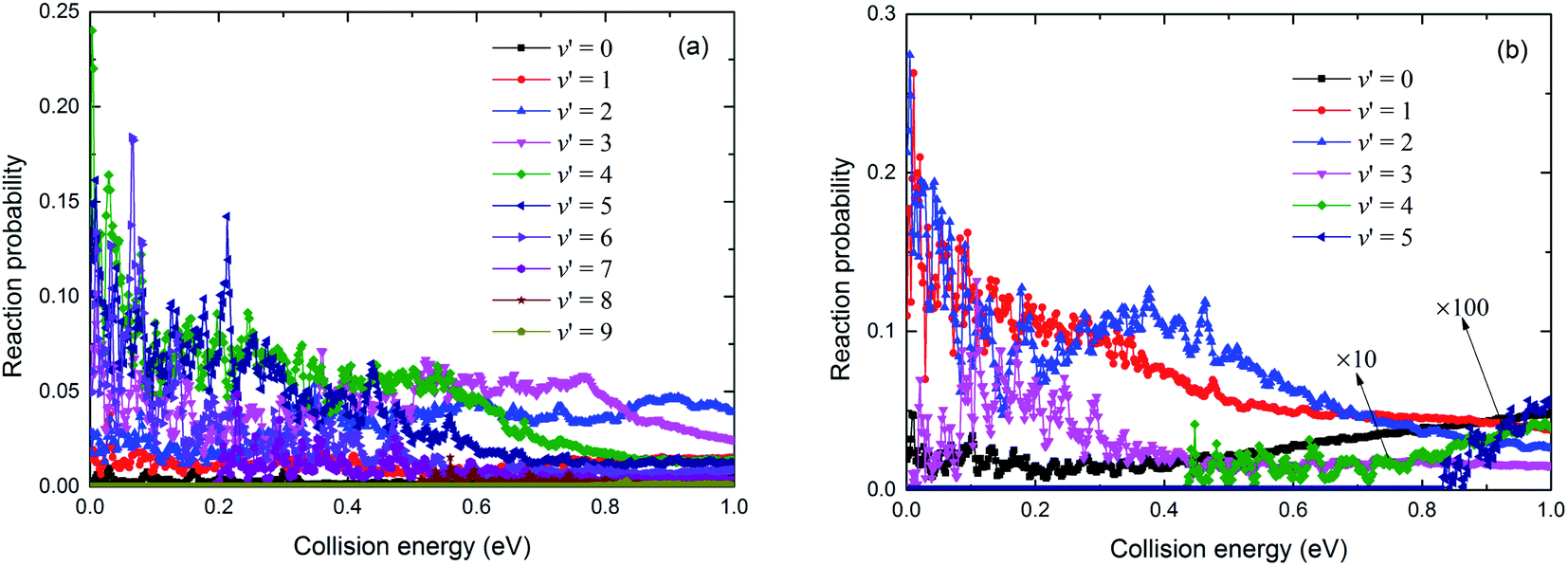
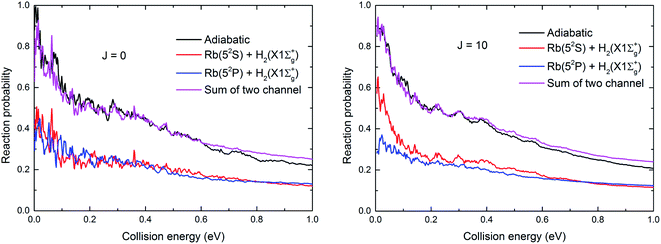
The resonance structures were observed on the reaction probability curves. Only a shallow well was found on the V22 PES (seen in Fig. 7). The vibrational state-resolved reaction probabilities at J = 0 of these two channels used to determine the reaction mechanism are shown in Fig. 10(a) and (b), respectively. Comparing the results in Fig. 10(a) with those in Fig. 10(b) shows that the Rb(52S) + H2(X1Σg+) channel had more vibrational states than the Rb(52P) + H2(X1Σg+) channel due to the larger exoergic energy of Rb(52S) + H2(X1Σg+) channel than that of the Rb(52P) + H2(X1Σg+) channel. As shown in Fig. 7, no well was observed in the Rb(52S) + H2(X1Σg+) channel, but some resonance structures were observed. We believe a potential well will be formed on the reaction path when the product is in the vibrational excited state, and the potential well deepens as the excited vibrational state energy level increases. Thus, the number of bound and quasi-bound states will be trapped in the well, and the resonance structures formed due to the coupling between the bound states in the well and the continuum states on the different vibrational states. We believe that these resonance structures can be classified as “Feschbach resonance”. As shown in Fig. 10(a), the amplitude of the reaction probability first increases with an increase in the number of vibrational states, then reaches a maximum value around v′ = 4, 5, and finally decreases with a further increase in the number of vibrational states. In addition, as the number of vibrational states increases, the resonance structure becomes more obvious as we speculated. For the Rb(52P) + H2(X1Σg+) channel, a shallow well was observed on the minimum energy path, however, as shown in Fig. 10(b), the situation was very similar to Fig. 10(a). The “Feschbach resonance” mechanism also plays an important role in the reaction.
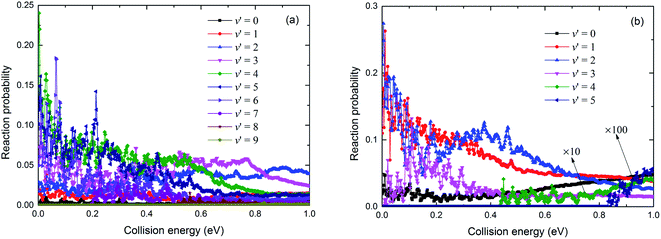 |
| | Fig. 10 The vibrational state-resolved reaction probabilities for the Rb(52S) + H2(X1Σg+) (a) and Rb(52P) + H2(X1Σg+) (b) channels at J = 0. | |
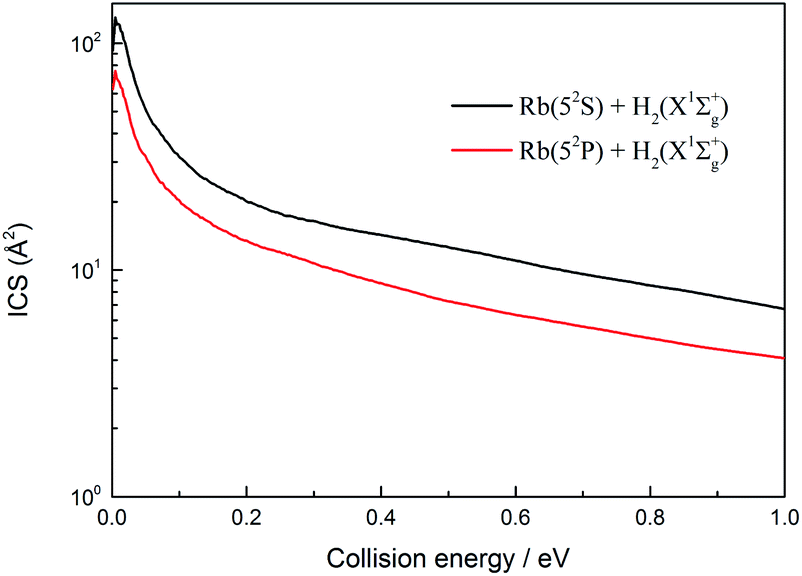
The total ICSs of the two channels of the H(2S) + RbH(X1Σ+) reaction are plotted in Fig. 11 as a function of collision energy. The ICSs of these two channels are very similar and both decrease with an increase of collision energy. In addition, the ICSs of the Rb(52S) + H2(X1Σg+) channel are larger than that of Rb(52P) + H2(X1Σg+) channel over the collision energies range studied. This is a predictable result. Due to the influence of nonadiabatic effect, the gap between each J partial wave constituting the ICSs of the Rb(52S) + H2(X1Σg+) channel and each J partial wave constituting the ICSs of the Rb(52P) + H2(X1Σg+) channel gradually increased with an increase in the J value.
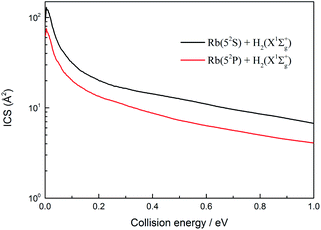 |
| | Fig. 11 Total integral cross sections of the two channels of the H(2S) + RbH(X1Σ+) reaction. | |
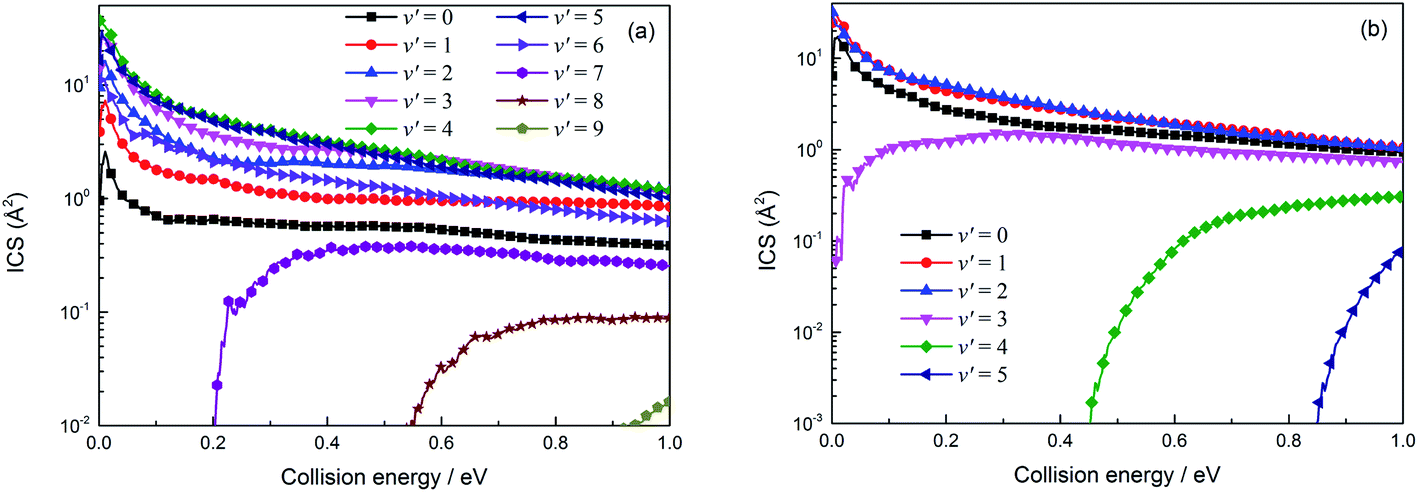
The vibrational state-resolved ICSs of the H(2S) + RbH (X1Σ+) reaction are displayed in the left (a) and right (b) panels of Fig. 12 and correspond to the Rb(52S) + H2(X1Σg+) and Rb(52P) + H2(X1Σg+) reactions, respectively. For the Rb(52S) + H2(X1Σg+) channel, the vibrational states below 7 are opened due to the large exoergic energy (seen in Fig. 7 approx. 2.775 eV) and the higher vibrational exited states (7, 8 and 9) are gradually opened as the collision energy further increases. This trend reflects that more energy is available below 7 and transformed into internal energy. The amplitude of the vibrational state-resolved ICS was very similar to the vibrational state distribution of the reaction probabilities (Fig. 10). The amplitude first increases, reaches the maximum value around a number of vibrational states of 4, and then decreases as the number of vibrational states further increases. The amplitude of the Rb(52P) + H2(X1Σg+) channel is very similar to the Rb(52S) + H2(X1Σg+) channel, but the number of vibrational states is relatively small, which can be attributed to the relatively small exoergic energy (approx. 1.435 eV).
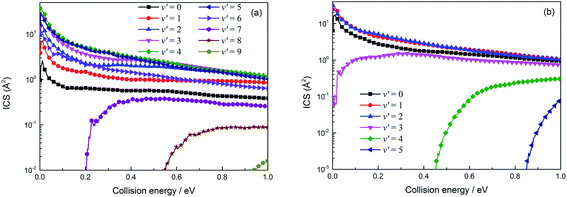 |
| | Fig. 12 Vibrational state-resolved integral cross sections for the two channels of the H(2S) + RbH (X1Σ+) reaction (panel (a) denotes the Rb(52P) + H2(X1Σg+) channel and panel (b) denotes the Rb(52S) + H2(X1Σg+) channel). | |
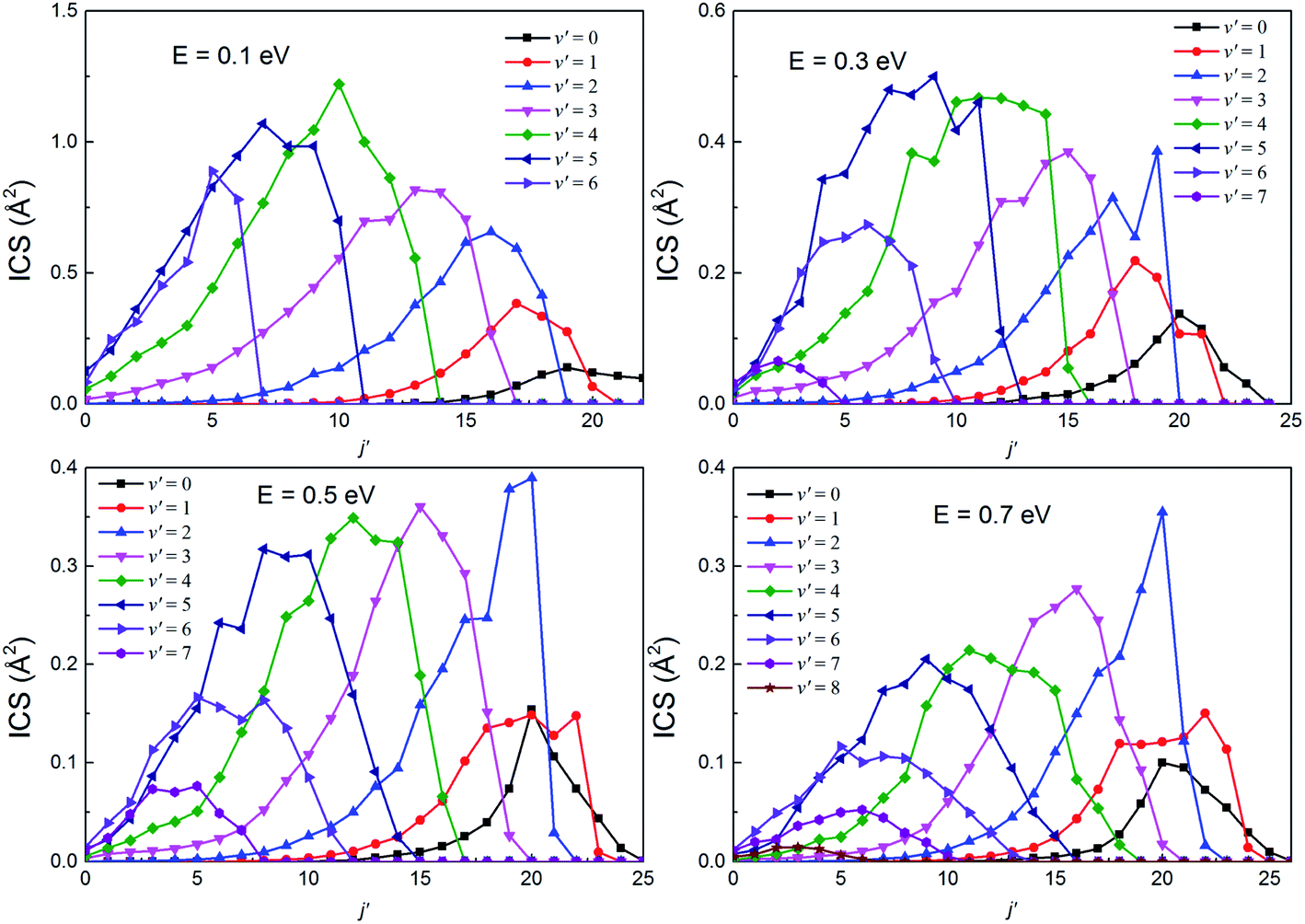
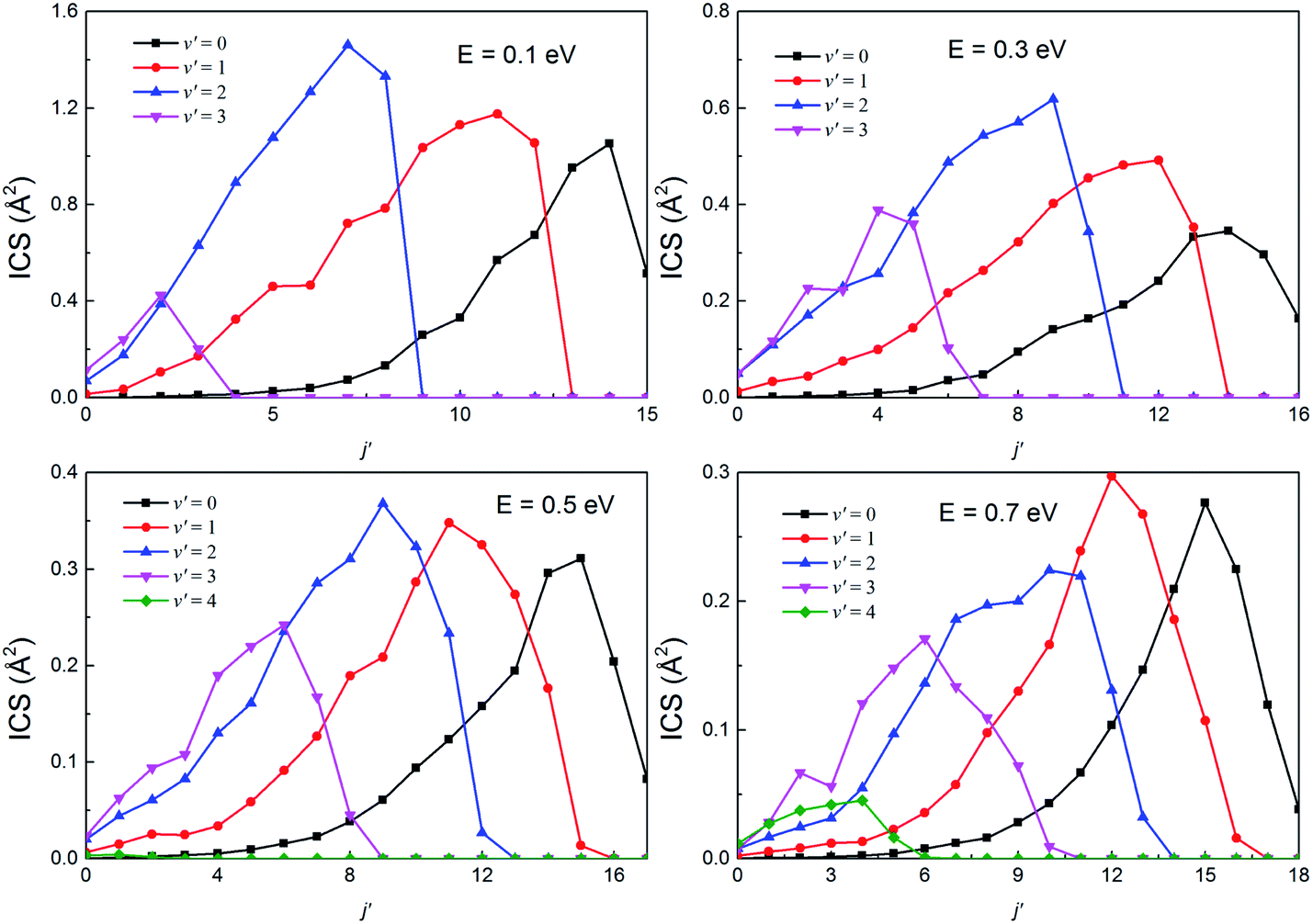
The rotational state-resolved DCSs of the H(2S) + RbH(X1Σ+) reaction at several selected collision energies of 0.1, 0.3, 0.5 and 0.7 eV are presented in Fig. 13 and 14 for the Rb(52S) + H2(X1Σg+) and Rb(52P) + H2(X1Σg+) channels, respectively. As seen in the figure, the shapes of each vibrational state-resolved ICSs are similar. The vibrational state-resolved ICSs first increase with j′, reach a maximum value, and then decrease as j′ further increases. In all cases, the rotational quantum number is the broadest in the ground state and narrows as v′ increases. At a given collision energy, the total energy is fixed. Thus, the internal energy is transformed from rotational to vibrational energy as v′ increases. In addition, the rational quantum number increases with increasing collision energy, although the increases are not very apparent. In all cases, the amplitude of each vibrational state-resolved ICS increases with increasing v′ before reaching a maximum value and then decreases as v′ further increases.
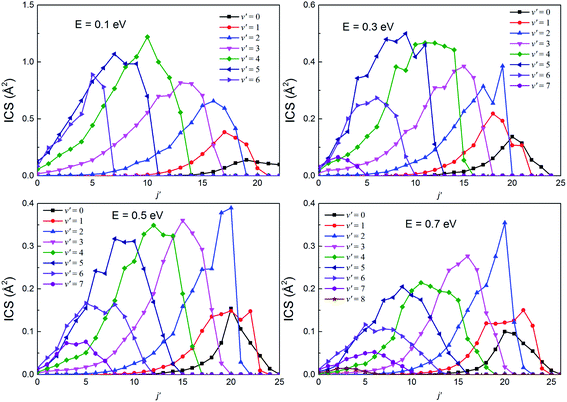 |
| | Fig. 13 The rotational state-resolved integral cross sections at several selected collision energies for the H(2S) + RbH(X1Σ+) → Rb(52S) + H2(X1Σg+) reaction. | |
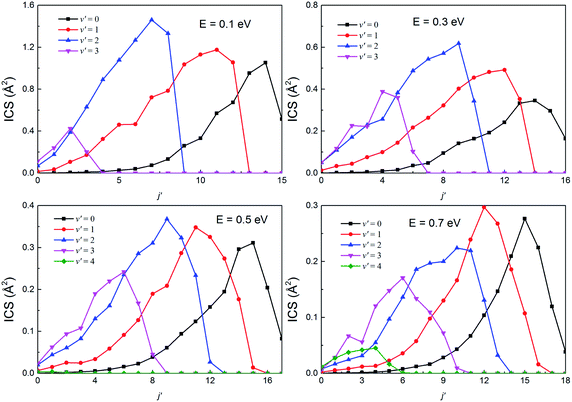 |
| | Fig. 14 The rotational state-resolved integral cross sections at several selected collision energies for the H(2S) + RbH(X1Σ+) → Rb(52P) + H2(X1Σg+) reaction. | |
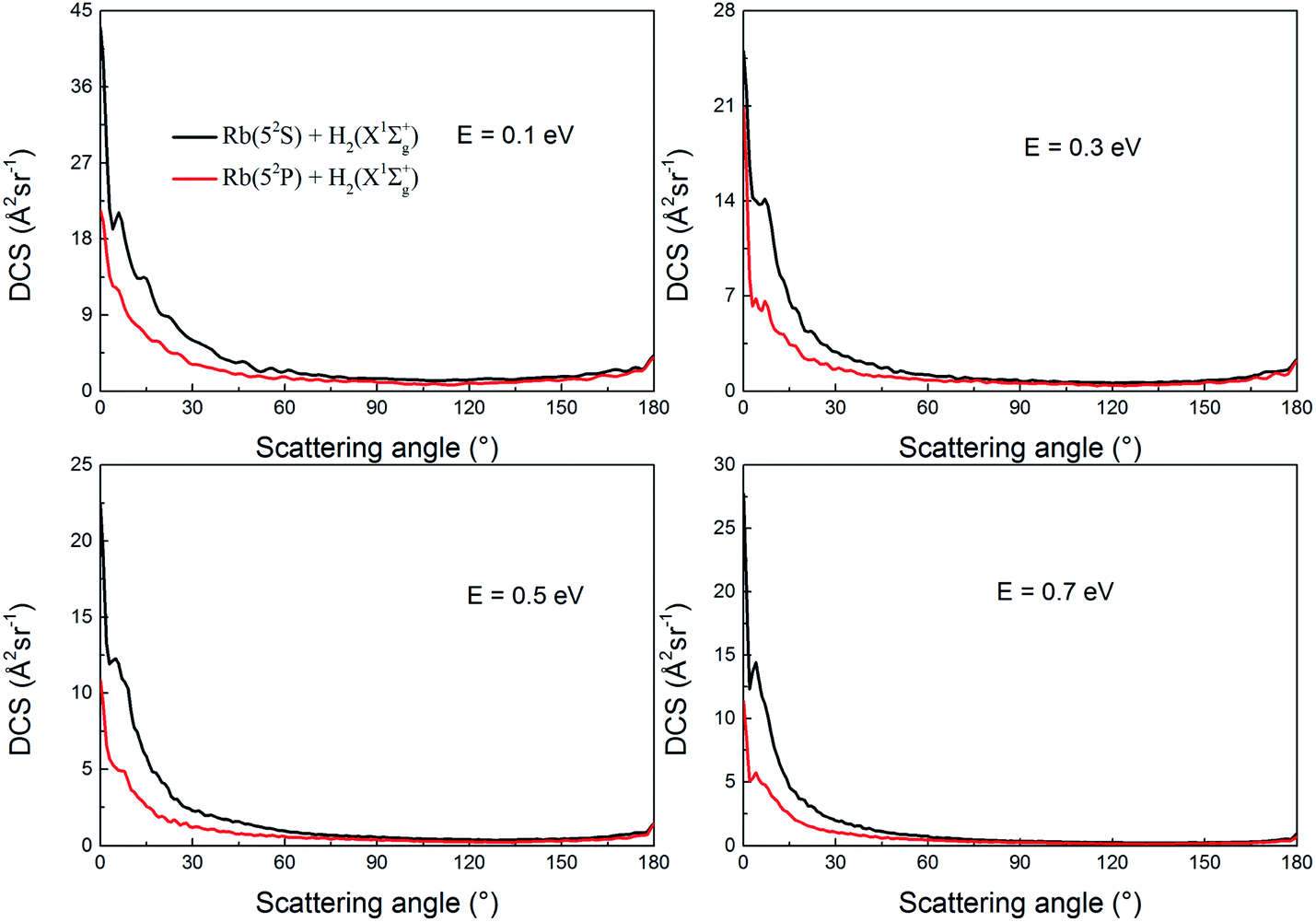
Fig. 15 shows the DCSs of the two channels at collision energies of 0.1, 0.3, 0.5 and 0.7 eV, respectively. As seen in the figure, the forward scattering signals are very apparent at all collision energies while the sideward and backward scattering signals are not obvious. For most scattering angles, the scattering intensity decreases as the collision energy increases which is consistent with trends in the ICSs shown in Fig. 11. In addition, the decrements in the scattering intensity are anisotropic. At high collision energies, the H2 molecule retain a higher forward scattering signal than sideward and backward scattering signals. For example, at a scattering angle of 1 degree, the DCSs are 25.1, 22.4 and 27.7 Å2 sr−1 for collision energies of 0.3, 0.5 and 0.7 eV, respectively. Whereas, at a scattering angle of 90°, the DCSs are 0.77, 0.50 and 0.37 Å2 sr−1 for 0.3, 0.5 and 0.7 eV, respectively. The DCSs are bias toward forward scattering, which indicates that the direct abstraction mechanism plays a dominant role in the reaction.
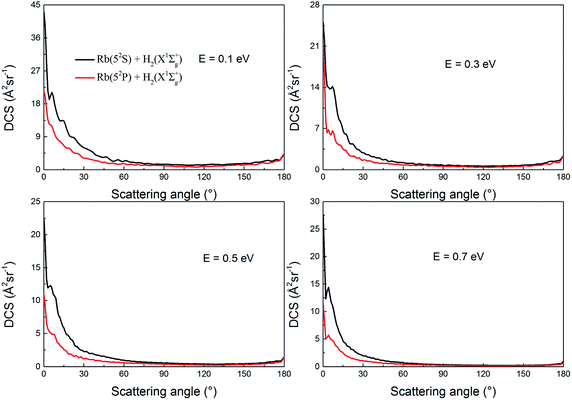 |
| | Fig. 15 Total differential cross sections of the two channels of the H(2S) + RbH(X1Σ+) reaction at several selected collision energies. | |
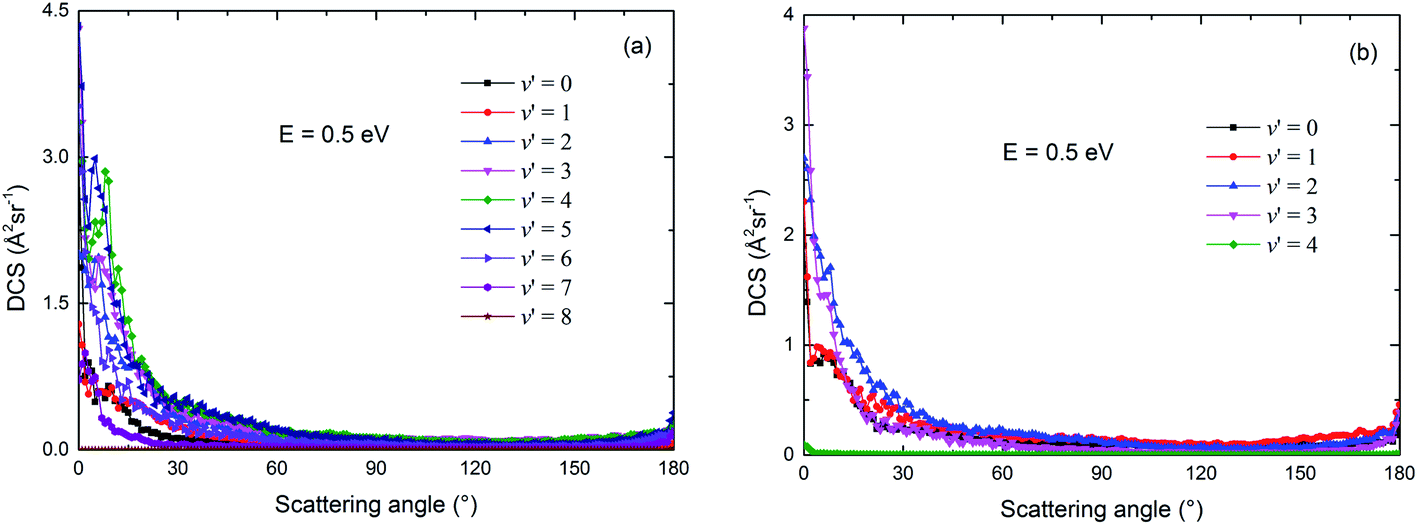
The vibrational state-resolved DCSs of the two channels of the title reaction are shown in Fig. 16(a) and (b), respectively, at a collision energy of 0.5 eV. Similar with the results in Fig. 10 and 12, more vibrational states were seen in the Rb(52S) + H2(X1Σg+) channel than in Rb(52P) + H2(X1Σg+) channel, due to the relatively larger exoergic energy of the Rb(52S) + H2(X1Σg+) channel. In addition, the shapes of the ICS for these vibrational states are very similar to the shape of the total ICS and are both biased towards forward scattering. This result also indicates that the direct abstraction mechanism dominates the reaction.
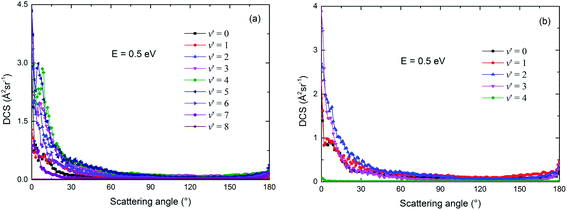 |
| | Fig. 16 The vibrational state-resolved differential cross sections for the Rb(52S) + H2(X1Σg+) (a) and Rb(52P) + H2(X1Σg+) (b) channels when the collision energy equal to 0.5 eV. | |
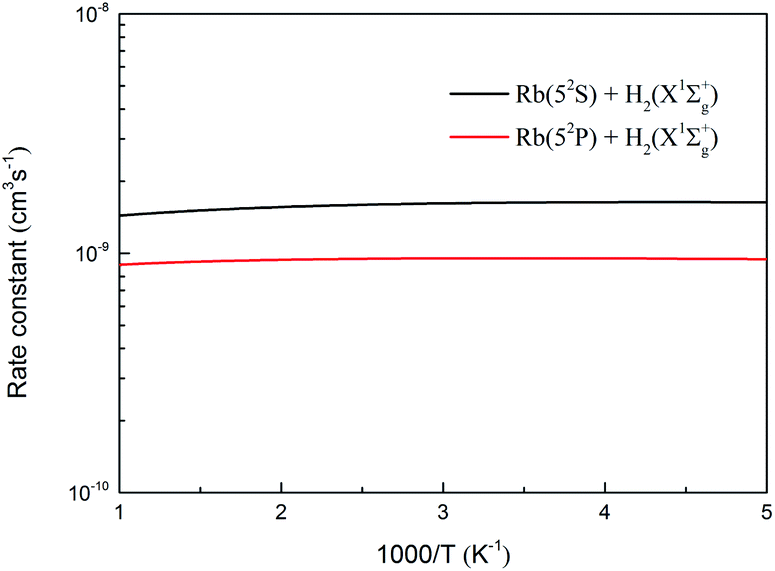
The specific initial state rate constants of the two channels of the H(2S) + RbH(X1Σ+) reaction are displayed in Fig. 17 over a temperature range from 200 to 1000 K. As seen in the figure, the rate constants for the initial state (v = 0, j = 0) of the two channels are not very sensitive to the temperature. The rate constants are around 1.6 × 10−9 and 9.4 × 10−10 cm3 s−1, for the Rb(52S) + H2(X1Σg+) and Rb(52P) + H2(X1Σg+) channels, respectively.
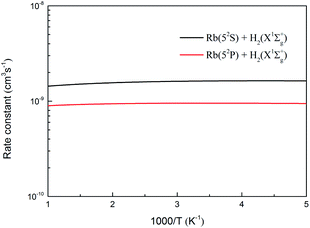 |
| | Fig. 17 Specific state rate constants of the H(2S) + RbH(X1Σ+) reaction over a temperature range from 200 to 1000 K. | |
4. Conclusion
In present work, new global diabatic PESs for the ground and first excited states of the RbH2 were constructed by fitting 17786 ab initio points. The SA-CASSCF and MRCI methods with aug-cc-pVQZ and Def2-QZVPP basis sets were used in the ab initio calculations. The mixing angle was obtained from the molecular properties of the dipole transition moment. The PIP-NN method was used to fit the diabatization terms, V11 and V22, as well as the coupling term V12 individually. These three terms were well fitted and the RMSEs were 4.95, 6.45 and 9.17 meV for V11, V22 and V12, respectively. The features of the new diabatic PESs were discussed in detail and dynamics studies of the H(2S) + RbH(X1Σ+) reaction were carried out based on the new diabatic PESs. The dynamics properties, including the reaction probabilities, ICSs, DCSs, rovibrational state distributions of the product, and rate constants are reported. The forward scattering signals indicate that the abstraction mechanism plays an important role in the reaction.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the 13th Five-Year Science and Technology Key Project of Education Department of Jilin Province (Grant No. JJKH20200482KJ).
References
- H. J. Werner, B. Follmeg and M. H. Alexander, J. Chem. Phys., 1988, 89, 3139–3151 CrossRef CAS.
- B. Jiang, C. J. Xie and D. Q. Xie, J. Chem. Phys., 2011, 135, 164311 CrossRef PubMed.
- B. Jiang and D. Q. Xie, Chin. J. Chem. Phys., 2009, 22, 601–604 CrossRef CAS.
- D. He, J. Yuan, H. Li and M. Chen, Sci. Rep., 2016, 6, 25083 CrossRef CAS PubMed.
- J. Yuan, D. He, S. Wang, M. Chen and K. Han, Phys. Chem. Chem. Phys., 2018, 20, 6638–6647 RSC.
- Z. J. Yang, J. C. Yuan, S. F. Wang and M. D. Chen, RSC Adv., 2018, 8, 22823–22834 RSC.
- T. Lenzen and U. Manthe, J. Chem. Phys., 2017, 147, 084105 CrossRef PubMed.
- J. Cuvellier, L. Petitjean, J. M. Mestdagh, D. Paillard, P. de Pujo and J. Berlande, J. Chem. Phys., 1986, 84, 1451–1458 CrossRef CAS.
- M. L. Chen, W. C. Lin and W. T. Luh, J. Chem. Phys., 1997, 106, 5972–5978 CrossRef.
- L. H. Fan, J. J. Chen, Y. Y. Lin and W. T. Luh, J. Phys. Chem. A, 1999, 103, 1300–1305 CrossRef CAS.
- J. Pascale, F. Rossi and W. E. Baylis, Europhys. Lett., 1987, 3, 183–188 CrossRef CAS.
- J. Pascale, F. Rossi and W. E. Baylis, Phys. Rev. A, 1987, 36, 4219–4235 CrossRef CAS PubMed.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1988, 89, 5803–5814 CrossRef CAS.
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1988, 145, 514–522 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- T. Leininger, A. Nicklass, W. Küchle, H. Stoll, M. Dolg and A. Bergner, Chem. Phys. Lett., 1996, 255, 274–280 CrossRef CAS.
- H. J. Werner and P. J. Knowles, J. Chem. Phys., 1985, 82, 5053–5063 CrossRef CAS.
- P. J. Knowles and H. J. Werner, Chem. Phys. Lett., 1985, 115, 259–267 CrossRef CAS.
- H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schutz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- D. W. Schwenke, S. L. Mielke, G. J. Tawa, R. S. Friedman, P. Halvick and D. G. Truhlar, Chem. Phys. Lett., 1993, 203, 565–572 CrossRef CAS.
- P. Halvick and D. G. Truhlar, J. Chem. Phys., 1992, 96, 2895–2909 CrossRef CAS.
- M. Peric, R. J. Buenker and S. D. Peyerimhoff, Mol. Phys., 1990, 71, 673–691 CrossRef CAS.
- J. Chen, Z. G. Sun and D. H. Zhang, J. Chem. Phys., 2015, 142, 024303 CrossRef PubMed.
- W. T. Li, J. C. Yuan, M. L. Yuan, Y. Zhang, M. H. Yao and Z. G. Sun, Phys. Chem. Chem. Phys., 2018, 20, 1039–1050 RSC.
- W. T. Li, D. He and Z. G. Sun, J. Chem. Phys., 2019, 151, 185102 CrossRef PubMed.
- B. Jiang, J. Li and H. Guo, Int. Rev. Phys. Chem., 2016, 35, 479–506 Search PubMed.
- B. Jiang and H. Guo, J. Chem. Phys., 2013, 139, 054112 CrossRef PubMed.
- M. T. Hagan and M. B. Menhaj, IEEE Transact. Neural Networks Learn. Syst., 1994, 5, 989–993 CrossRef CAS PubMed.
- Z. G. Sun, D. Q. Yu, W. B. Xie, J. Y. Hou, R. Dawes and H. Guo, J. Chem. Phys., 2015, 142, 174312 CrossRef PubMed.
- M. L. Yuan, W. T. Li, J. C. Yuan and M. D. Chen, Int. J. Quantum Chem., 2018, 118, e25493 CrossRef.
- W. B. Xie, L. Liu, Z. G. Sun, H. Guo and R. Dawes, J. Chem. Phys., 2015, 142, 064308 CrossRef PubMed.
- S. Liu, X. Xu and D. H. Zhang, J. Chem. Phys., 2012, 136, 144302 CrossRef PubMed.
- B. Zhao, Z. G. Sun and H. Guo, J. Chem. Phys., 2016, 144, 064104 CrossRef PubMed.
- M. D. Feit, J. A. Fleck and A. Steiger, J. Comput. Phys., 1982, 47, 412–433 CrossRef CAS.
- S. Gómez-Carrasco and O. Roncero, J. Chem. Phys., 2006, 125, 054102 CrossRef PubMed.
- Z. Sun, X. Lin, S.-Y. Lee and D. H. Zhang, J. Phys. Chem. A, 2009, 113, 4145–4154 CrossRef CAS PubMed.
- D. He, J. C. Yuan, H. X. Li and M. D. Chen, Sci. Rep., 2016, 6, 25083 CrossRef CAS PubMed.
- W. T. Li, J. X. Sun and D. He, Phys. Chem. Chem. Phys., 2020, 22, 17587–17596 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Jinghua Xub,
Haigang Yangb and
Jiaqiang Xu
ab,
Jinghua Xub,
Haigang Yangb and
Jiaqiang Xu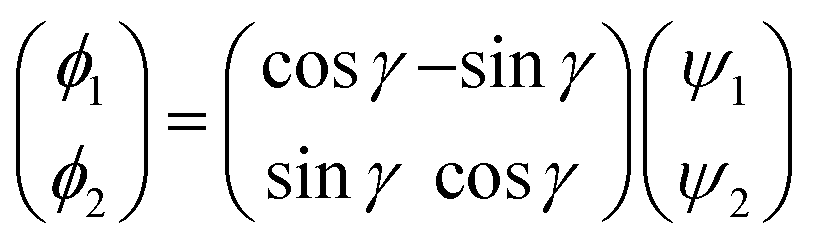
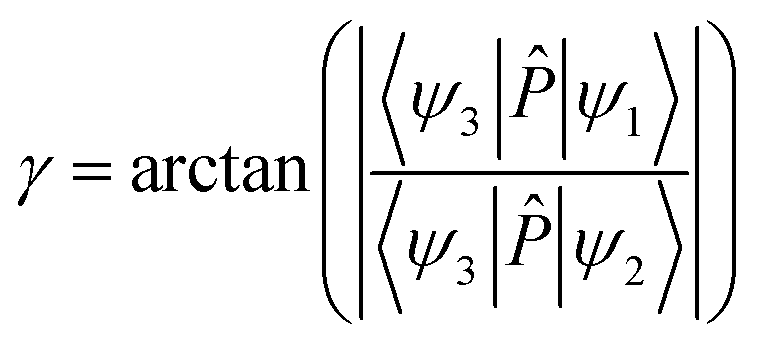
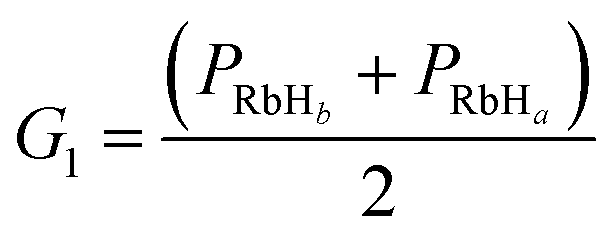
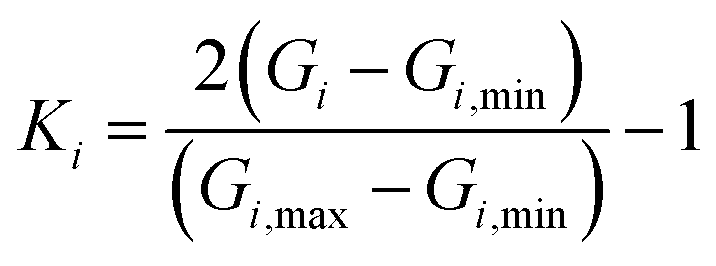
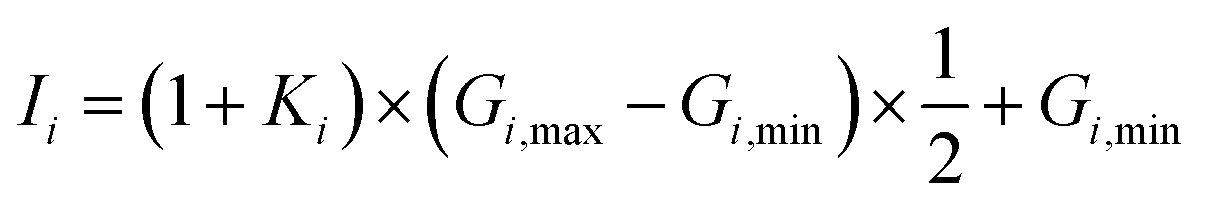
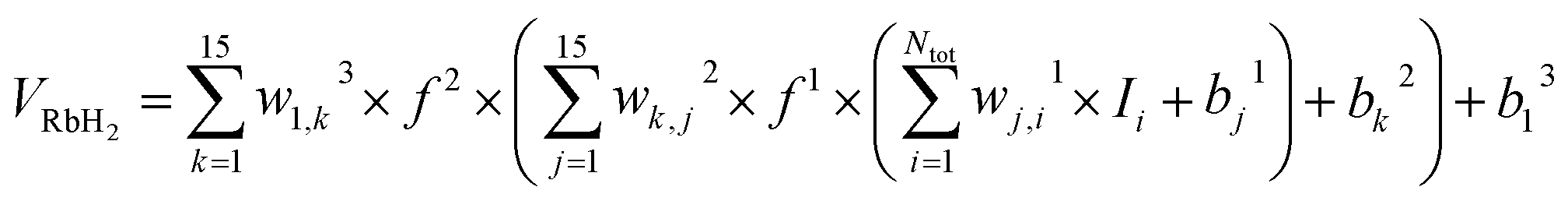
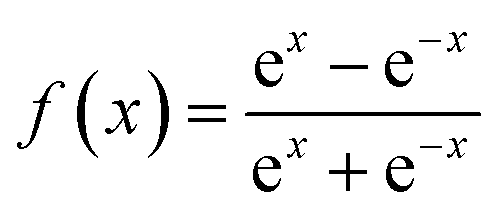
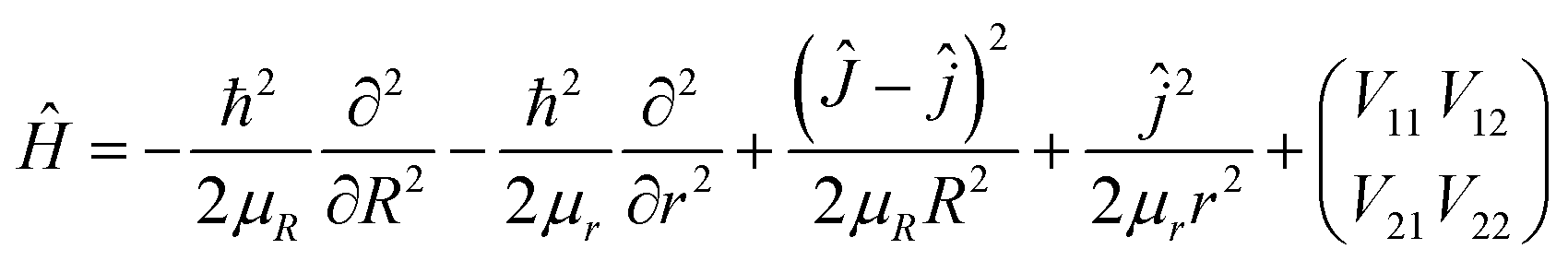
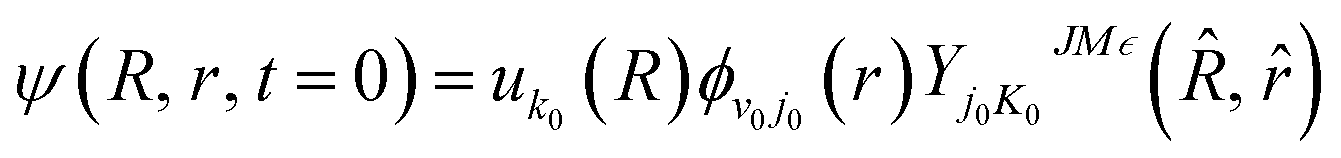
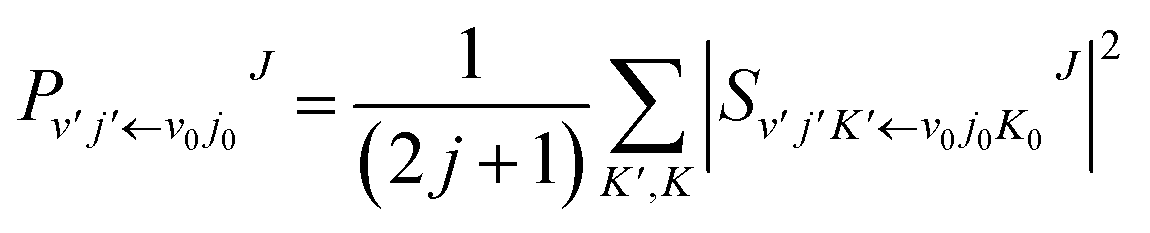
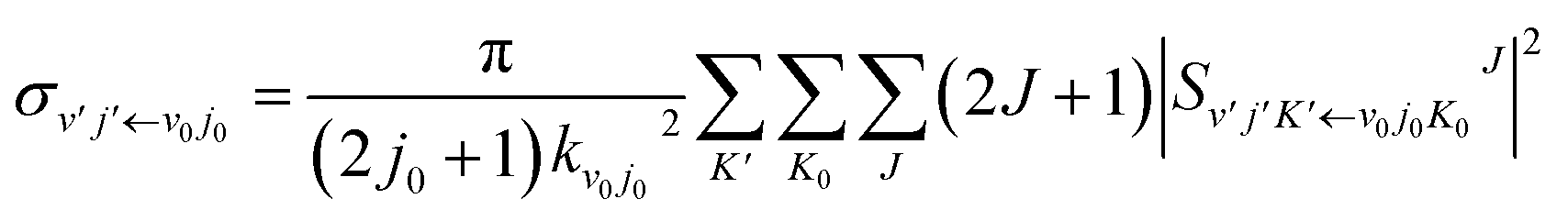
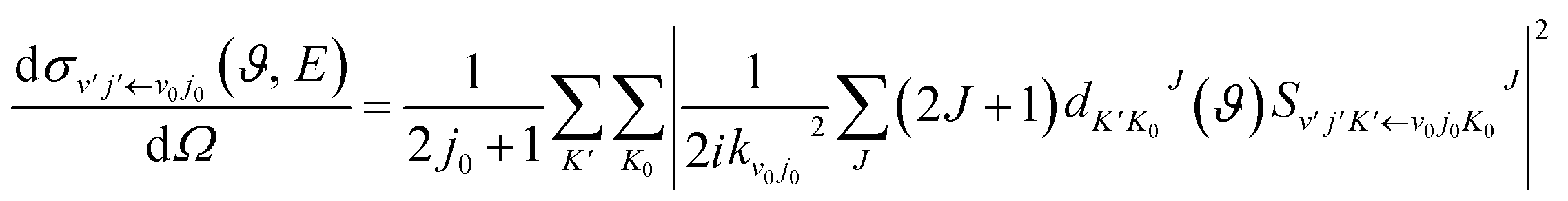
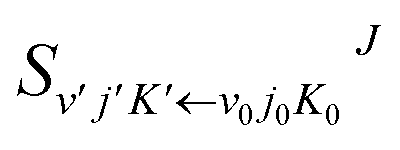 is the state-to-state S-matrix,
is the state-to-state S-matrix, 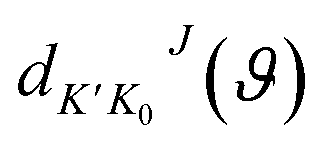 is the reduced rotation matrix element, and ϑ is the scattering angle.
is the reduced rotation matrix element, and ϑ is the scattering angle.