
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(N-vinylcaprolactam) containing solid lipid polymer hybrid nanoparticles for controlled delivery of a hydrophilic drug gemcitabine hydrochloride†
Sai Geetika Surapaneniab and
Ashootosh V. Ambade *ab
*ab
aPolymer Science and Engineering Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune – 411008, India. E-mail: av.ambade@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad – 201002, India
First published on 14th June 2022
Abstract
Folic acid tagged and hydrophilic polymer containing solid lipid nanoparticles (SLNs) were formulated for the controlled and targeted delivery of gemcitabine, a hydrophilic drug. Drug loaded SLNs were prepared by double emulsion method and optimized by 32 level factorial design. Then, a hydrophilic polymer, namely, poly(N-vinylcaprolactam) (PVCL) was incorporated in the optimized SLN batch in the first aqueous phase (W1) to obtain solid lipid polymer hybrid nanoparticles (SLPHNs) that were further decorated with folic acid (F-SLPHNs). TEM analysis of SLNs and SLPHNs revealed the spherical shape with no aggregation while SLPHNs showed higher % EE. SLPHNs exhibited limited burst release of gemcitabine compared to SLNs as well as lower overall % release. All the formulations showed good cytocompatibility against MDA-MB-231 cell lines and folic acid-tagged hybrid particles (F-SLPHNs) showed remarkably higher cellular uptake.
Introduction
Cancer is one of the leading causes of death globally. Surgery, radiation therapy, chemotherapy, immune therapy, and hormonal therapy are a few of the treatment strategies currently available.1 Chemotherapy is the preferred method for post surgical supportive treatment however, upon intravenous (i.v.) administration, many routinely used chemotherapeutic agents do not accumulate well in tumors while their levels in certain healthy organs and tissues can be quite high due to their non specificity that leads to a disadvantageous balance between the efficacy and toxicity of systemic therapeutic interventions. To address this issue, nanocarriers that can selectively deliver the drug to cancer tissue have been extensively explored.Gemcitabine (Gem; 2′-deoxy-2′,2′-difluorocytidine) is a nucleoside pyrimidine analogue used to treat various solid tumors such as breast cancer, pancreatic cancer, ovarian cancer and lung cancer usually in combination with other drugs.2–5 Initially, it was developed as an antiviral drug and later investigated for cancer treatment.6 The major drawbacks of the gemcitabine use are its short half life due to rapid deamination into an inactive metabolite, 2′,2′-difluorodeoxyuridine (dFdU) and development of drug resistance over a period of time.7–9 These limitations hamper its usage as an effective treatment strategy and the development of novel and more efficient drug delivery systems for gemcitabine is necessary. In this direction, a variety of nanocarriers have been prepared to improve the pharmacokinetic profile of gemcitabine, among them solid lipid nanoparticles of gemcitabine have gained interest.10–12
Solid lipid nanoparticles (SLNs) are versatile drug delivery nanocarriers used in biomedical field for encapsulation of both hydrophilic and hydrophobic drugs.13,14 Among the other colloidal systems, SLNs have gained wide attention in the past decade owing to their superior properties such as longer circulation time, stability and easy scale up.15–17 SLNs are also amenable to easy surface functionalization thereby augmenting the pharmacokinetic profile of the therapeutic moieties.18,19 On the other hand, SLNs also exhibit burst release that leads to critical side effects and failure of treatment.20,21 Particularly, hydrophilic drugs like gemcitabine are more prone to leach out into the outer aqueous milieu during the manufacturing process that causes excessive burst release.
Recently, lipid polymer hybrid nanoparticles (LPHNs) have emerged as new generation of lipid carriers combining the advantages of liposomes and polymeric nanoparticles together for the delivery of gemcitabine.22–24 Though LPHNs possess prominent advantages, few drawbacks such as wide particle size distribution, tedious procedures and low yield are yet to be overcome. In case of hydrophilic drug gemcitabine, easy encapsulation and controlled delivery still remains a challenge. To address the above mentioned limitations of SLNs and LPHNs, we present here solid lipid polymer hybrid nanoparticles (SLPHNs) of gemcitabine.
There are few reports on the development of SLPHNs wherein SLN is taken as the core and coated with a polymer as outer shell to improve their properties and stability.25–27 However, we hypothesized that by integrating a hydrophilic polymer with the SLNs in the core can lead to improved encapsulation efficiency and controlled release of gemcitabine due to enhanced hydrophilic interaction with the drug.
In the present study, we have incorporated poly(N-vinylcaprolactam) (PVCL), a hydrophilic polymer along with the drug during the preparation of SLNs for stronger interaction with the drug. The thus obtained solid lipid polymer hybrid nanoparticles (SLPHNs) have been investigated for their controlled release profile. PVCL contains a cyclic amide group wherein the nitrogen of the amide group is attached to a hydrophobic polymer backbone owing to which, it does not produce toxic small molecules upon hydrolysis making it attractive candidate for biomedical applications.28 Although biopolymers are known to be superior to synthetic polymers in terms of biocompatibility and biodegradability, their production, isolation and purification involves highly complex and costly processes.29 On the other hand, synthetic polymers such as PVCL can be synthesized and scaled-up easily. Moreover, PVCL is known as thermoresponsive polymer that can be further explored to prepare formulations with temperature-dependent drug release. Furthermore, growing interest in biomedical applications of PVCL indicates its great potential for FDA approval.30
Folate receptors are over expressed in wide varieties of tumors. Folic acid tagged nanoparticles are trusted to overcome the multidrug resistance and improve the cellular internalization,31,32 hence we have employed folic acid conjugated F-127 as a stabilizer to obtain folic acid decorated SLPHNs for targeted delivery. In general, small molecule ligands are considered to have weak interactions with their targets, however among these folic acid possesses higher affinity (Kd = 10−7 mM) for its receptors and simplicity of chemistry involved in conjugation with surfactants makes it attractive for targeting strategy.33–35 Folic acid also has several advantages over macromolecular ligands, for example, non-immunogenicity, ease of handling, stability and low cost have been reported extensively.35
Experimental
Materials and methods
Glycerol monostearate (GMS), soy lecithin, pluronic® F-127, 1,1′-carbonyldiimidazole (CDI), 2-aminoethanethiol, calcein and folic acid were purchased from TCI Chemicals (India). N-vinylcaprolactam (NVCL) and azobisisobutyronitrile (AIBN) were procured from Sigma Aldrich and recrystallized from n-hexane and methanol, respectively. Gemcitabine (Gem) was obtained as a gift sample. DMEM media, FBS, DAPI and trypsin EDTA were purchased from Invitrogen. MDA-MB-231 breast cancer cells were obtained from National Center for Cell Science (NCCS), Pune. 1,4-Dioxane, methanol, N,N-dimethylformamide (DMF), dichloromethane (DCM), diethyl ether were of analytical grade.NMR spectra were recorded on Bruker AV200 and AV400 MHz spectrometers using CDCl3 and DMSO-d6 as the solvents, respectively; TMS was used as a reference. Gel permeation chromatography (GPC) was carried out on a Viscotek PL-GPC-220 with chloroform as an eluent at 1 mL min−1 flow rate. Drug concentration measurements were performed on a Specord 210 plus Analytikjena spectrophotometer. Agilent UV-vis spectrophotometer equipped with Agilent 89090A Peltier cooling system was used to determine cloud point (CP) of the polymer. FT-IR spectrum of polymer was recorded on PerkinElmer FTIR spectrometer on ATR mode. Differential scanning calorimetric (DSC) analysis was carried out on a TA Q10 differential scanning calorimeter. Mean diameter, polydispersity index (PDI), and zeta potential were determined by using Zetasizer Nano ZS, Malvern Instrument Ltd, UK at 25 °C. The SLN dispersion was diluted 10 times with DI water before measurement. Transmission electron microscopy (TEM) analysis was performed on FEI Technai F20 instrument. SLNs dispersion was diluted 10 times and drop cast on carbon coated copper grid and analyzed without any staining.
Synthesis procedures
Preparation of solid lipid nanoparticles (Gem-SLNs)
Gem loaded solid lipid nanoparticles were prepared by double emulsion solvent evaporation method. Gem was dissolved in DI water (W1) while soy lecithin and GMS were dissolved in dichloromethane (O). Aqueous phase (W1) was added to organic phase (O) slowly and emulsified with probe sonication for 1 minute (30% amplitude, 25 °C and 5 s lapse time) to form W1/O primary emulsion. This primary emulsion was further added slowly to the aqueous phase containing F-127 (W2) and re-emulsified using probe sonication for 3 minutes (30% amplitude, 25 °C and 5 s lapse time) to form W1/O/W2 secondary emulsion. Afterwards, organic solvent was removed under reduced pressure on rotary evaporator. The obtained SLNs were isolated by centrifugation at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes at 4 °C and unentrapped drug was removed by repeated washings with DI water. The SLN pellet was suspended in DI water under sonication and final volume was adjusted to 10 mL.
000 rpm for 30 minutes at 4 °C and unentrapped drug was removed by repeated washings with DI water. The SLN pellet was suspended in DI water under sonication and final volume was adjusted to 10 mL.
Preparation of solid lipid polymer hybrid nanoparticles (Gem-SLPHNs)
Gem-SLPHNs were prepared by same procedure as that of Gem-SLNs with minor modifications. Typically, gemcitabine (10 mg) and PVCL (100 mg) were dissolved in DI water (W1). Soy lecithin (100 mg) and GMS (150 mg) were dissolved in DCM (O). Aqueous phase (W1) was added slowly to the organic phase (O) and sonicated on probe sonicator for 1 minute at 30% amplitude and 25 °C with 5 s laps time. Obtained W1/O primary emulsion was added slowly to the aqueous phase containing F-127 (W2) and probe sonicated for 3 minutes at 30% amplitude and 25 °C with 5 s lapse time to obtain W1/O/W2 double emulsion. DCM was evaporated under reduced pressure on rotary evaporator and SLNs were separated by centrifugation at 18000 rpm for 30 minutes at 4 °C. SLN pellet was suspended in DI water under bath sonication and volume was adjusted to 10 mL.
Folic acid decorated SLPHNs (Gem-F-SLPHNs) were prepared by following the same procedure as that for Gem-SLPHNs and F-127 was replaced by FA-F127.
In vitro release studies
The amount of gemcitabine released from different formulations was determined by dialysis method at 37 °C and 25 °C. Solid lipid nanoparticles equivalent to 1 mg of gemcitabine were packed in dialysis bag (M.W. cut off = 2 KD) and submerged in a phosphate buffer (pH 7.4, 10 mM). Aliquots (1 mL) were collected at regular intervals (1, 2, 4, 6, 8, 10, 12, 24 h) and the same amount was replaced by fresh media to maintain the sink conditions. Collected aliquots were diluted suitably and analyzed using UV-visible spectrophotometer. In-house developed calibration curve was used to calculate the concentration of drug in the sample (Fig. S1†).Cellular uptake studies
Cellular uptake studies were conducted by following earlier reported procedure with minor modifications.37 Briefly, 50000 MDA-MB-231 cells per well in DMEM containing 10% FBS were seeded in a 24 well plate. Cells were allowed to adhere at 37 °C with 5% CO2 for 24 h. After 24 h incubation the media was replaced with samples. Dye (calcein) loaded SLNs, SLPHNs and F-SLPHNs samples were prepared in plain DMEM media without FBS. The cells were allowed to uptake the samples for 3 h at 37 °C with 5% CO2. After incubation, the cells were washed with PBS three times and cell fixation was done using 4% paraformaldehyde. The nucleus staining was done using DAPI. Images were acquired using an epifluorescence microscope from Carl Zeiss (Axio Observer Z1).
Results and discussion
Synthesis of PVCL and FA-F-127 conjugate
PVCL, selected as the hydrophilic polymer, was synthesized by free radical polymerization using AIBN as initiator (Scheme 1). This polymerization method is used for polymer synthesis on commercial scale. Aminoethanethiol was used as a chain transfer agent to control the molecular weight. 1H NMR spectrum of the polymer showed absence of the peaks at 7.2, 4.8 and 4.2 ppm corresponding to vinyl protons of the monomer (Fig. S2†). GPC analysis gave Mn of 25000 g mol−1 with dispersity of 1.9 (Fig. S3†). PVCL in this molecular weight range has been used in block copolymer nanocarriers designed for controlled drug delivery.38,39 The polymer was also characterized by FTIR spectroscopy (Fig. S4†). Characteristic bands for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CH and CH2 at 1654 cm−1, 3102 cm−1 and 962 cm−1, respectively, in the monomer spectrum were found to be absent in the polymer. As PVCL tends to absorb moisture, a broad peak corresponding to O–H stretching was observed at 3473 cm−1.40 PVCL is known to possess temperature-responsive property hence cloud point of the synthesized PVCL was determined by measuring the change in transmittance with temperature and was obtained as 34 °C (Fig. S5†). The conjugation of folic acid to F-127 was carried out as per the reported method (Fig. S6†).36 Carboxylic group of folic acid was activated by 1,1′-carbonyldiimidazole (CDI) and the activated ester was reacted with F-127. Formation of the conjugate was confirmed by 1H NMR spectroscopy analysis in D2O (Fig. S7†). Presence of the characteristic pteridine ring proton at 8.53 ppm and appearance of peaks corresponding to aromatic protons at 6.68 ppm and 7.32 ppm indicated the successful conjugation of folic acid with F-127.
C, CH and CH2 at 1654 cm−1, 3102 cm−1 and 962 cm−1, respectively, in the monomer spectrum were found to be absent in the polymer. As PVCL tends to absorb moisture, a broad peak corresponding to O–H stretching was observed at 3473 cm−1.40 PVCL is known to possess temperature-responsive property hence cloud point of the synthesized PVCL was determined by measuring the change in transmittance with temperature and was obtained as 34 °C (Fig. S5†). The conjugation of folic acid to F-127 was carried out as per the reported method (Fig. S6†).36 Carboxylic group of folic acid was activated by 1,1′-carbonyldiimidazole (CDI) and the activated ester was reacted with F-127. Formation of the conjugate was confirmed by 1H NMR spectroscopy analysis in D2O (Fig. S7†). Presence of the characteristic pteridine ring proton at 8.53 ppm and appearance of peaks corresponding to aromatic protons at 6.68 ppm and 7.32 ppm indicated the successful conjugation of folic acid with F-127.
 | ||
| Scheme 1 Synthesis of PVCL. | ||
Incompatibility studies
It is imperative to study the incompatibility between the chosen excipients and drug as interactions between them may hamper the therapeutic efficacy of active ingredient.41 Drug-excipients compatibility was studied using differential scanning calorimetry (DSC) analysis. Any appearance of extra peaks in the DSC curves indicates the possible drug-excipient interaction leading to drug-excipient incompatibility. The endotherm for gemcitabine was retained in DSC curves of all the drug-excipient mixtures indicating the absence of incompatibility (Fig. S8†). Sharp endothermic melting peak was observed for pure gemcitabine at 274 °C. High intensity endothermic peaks at 56.8 °C and 74 °C correspond to F-127 and GMS, respectively, demonstrating their crystalline nature. PVCL exhibited broad endothermic peak at 62 °C indicating the irregular arrangement of the polymeric chains. Soy lecithin shows melting point at 236 °C, however in the physical mixture of soy lecithin and gemcitabine the endothermic peak of soy lecithin was found to have shifted to 185 °C that signifies the interactions between soy lecithin and gemcitabine. Physical mixture of gemcitabine and F-127 as well as FA-F127 did not show any significant change in the drug peak shape or position indicating the absence of interaction of the drug with the stabilizers. Broadening and shifting of the peak of drug was observed in the physical mixtures of the drug with GMS, PVCL and soy lecithin. However, additional peaks corresponding to interactions between drug and excipients were not observed suggesting probable molecular rearrangements in the physical mixtures rather than formation of new chemical bonds.Preparation and optimization of formulation
Solid lipid nanoparticles (SLNs) have emerged as promising colloidal drug delivery systems owing to their interesting properties.42 Though SLNs have been investigated widely to deliver different classes of drugs such as anticancer drugs, Alzheimer's drugs, antimicrobial agents43–45 efficiently at the target site, encapsulation of hydrophilic drugs in the SLNs still remains challenging because of insufficient affinity between the drug and lipid matrix.Primarily, gemcitabine loaded SLNs were prepared by double emulsion method, which is a two step emulsification process that involves the use of lipids, surfactants, water and organic solvents. It has been reported that proper blend of the surfactants effectively reduces the particle size, polydispersity index (PDI) and increases the stability than a single surfactant.46 Usually the blend comprises one surfactant with low hydrophilic lipophilic balance (HLB) to stabilize the W1/O primary emulsion and the other with high HLB to stabilize the O/W2 secondary emulsion. In the present study, soy lecithin was chosen as lipophilic surfactant to stabilize W1/O emulsion and F-127 was selected as hydrophilic surfactant to stabilize O/W2 emulsion.
Further, optimization of SLNs was done by 32 level factorial design using Design-Expert® 13 software. Drug to lipid ratio (X1), concentration of F-127 (X2) were chosen as the independent variables and concentration of soy lecithin was kept constant at 1% w/v. The effect of X1 and X2 on the dependent variables, particle size (Y1) and % encapsulation efficiency (Y2) was studied. Nine batches of formulations were prepared with varied amounts of chosen variables; drug to lipid ratio ranging from 1:5 to 1:15 and F-127 concentration ranging from 0.25% w/v to 0.75% w/v and characterized for particle size and % EE (Table 1).
| Formulation code | Drug to lipid ratio (X1) | F-127 (% w/v) (X2) | Particle size (nm) (Y1) | PDI | Zeta potential (mV) | % EE (Y2) |
|---|---|---|---|---|---|---|
| a PDI: polydispersity index, % EE: percent encapsulation efficiency. Data are expressed as mean ± S.D. (n = 3). | ||||||
| F1 | 1:5 |
0.5 | 270 ± 10.7 | 0.20 ± 0.03 | −32 ± 0.4 | 32.5 ± 2.12 |
| F2 | 1:10 |
0.5 | 290 ± 14.14 | 0.22 ± 0.027 | −32.5 ± 0.2 | 36 ± 5.65 |
| F3 | 1:15 |
0.5 | 323 ± 11.07 | 0.24 ± 0.061 | −30.9 ± 2.6 | 43.5 ± 4.06 |
| F4 | 1:5 |
0.75 | 238 ± 12.13 | 0.16 ± 0.006 | −31.9 ± 1.2 | 28 ± 4.24 |
| F5 | 1:10 |
0.75 | 248 ± 15.31 | 0.2 ± 0.022 | −36.1 ± 0.4 | 32.5 ± 4.94 |
| F6 | 1:15 |
0.75 | 282 ± 14.97 | 0.18 ± 0.026 | −36 ± 0.6 | 39 ± 5.07 |
| F7 | 1:5 |
0.25 | 250 ± 11.7 | 0.20 ± 0.004 | −38 ± 0.4 | 34.5 ± 3.53 |
| F8 | 1:10 |
0.25 | 262 ± 12.82 | 0.25 ± 0.011 | −37.4 ± 1.2 | 38 ± 4.24 |
| F9 | 1:15 |
0.25 | 319 ± 12.47 | 0.27 ± 0.019 | −35.9 ± 1.2 | 46.5 ± 6.19 |
In addition, PDI was found to be <0.3 in all the formulations and zeta potential ranging from −30 to −38 mV was observed. The negative zeta potential can be attributed to the anionic nature of the lipid used in the formulation and also signifies the moderate stability of the formulations. Generally, formulations with zeta potential greater than +25 mV and lower than −25 mV are considered stable due to electrostatic repulsion between the particles, since lower zeta potential may cause aggregation of the particles through van der Waal interactions.47 Particle size in the range of 238 ± 12.13 to 319 ± 12.47 nm and % encapsulation efficiency (% EE) in the range of 28 ± 4.24 to 46.5 ± 6.19 were obtained in the designed nine formulations. It was suggested by the software that quadratic model was best fitted for all the responses studied. Drug to lipid ratio and concentration of F-127 were found to have significant effect (p < 0.05) on % EE and particle size. Polynomial response equation(s) for particle size and % EE are given below in which A is the drug to lipid ratio and B is the surfactant (F-127) concentration. Positive sign signifies the concurred effect and negative sign signifies the hindered effect.
| Particle size = 285.22 + 27.66A − 10.5B − 6.25A × B + 13.66A2 − 27.83B2 |
| % EE = 36.11 + 5.67A − 3.25B − 0.25A × B + 1.83A2 − 0.919B2 |
Drug to lipid ratio was found to have positive effect on the particle size and % EE. The increase in particle size with increase in lipid concentration can be attributed to the tendency of the oil globules to coalesce at higher concentration that leads to larger core and hence higher % EE.48 On the other hand, surfactant showed negative effect on the particle size and % EE. The decrease in particle size with increase in surfactant concentration may be due to stabilization of the interface by surfactant that prevents particle aggregation49 while the decrease in % EE is because of increase in solubilization of the drug with increase in the surfactant concentration.50
Three dimensional response surface plots were drawn to study the effects of pre-determined factors on the particle size (Fig. 1a) and % EE (Fig. 1b). Based on the highest % EE, F9 was chosen for further studies (in the following discussion, Gem-SLNs refers to F9 unless otherwise mentioned). TEM analysis of Gem-SLNs (F9) revealed spherical shape with no aggregation (Fig. 2a), however, particle size was smaller than average particle size measured by DLS due to drying effect in sample preparation.51
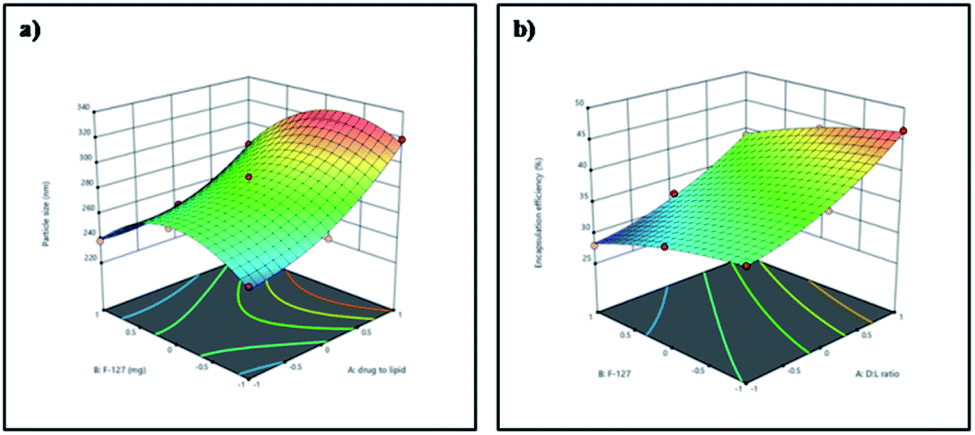 | ||
| Fig. 1 3D response surface plots for the (a) particle size and (b) % EE. | ||
 | ||
| Fig. 2 TEM images of (a) Gem-SLNs (b) Gem-SLPHNs; (c) DLS size distribution of Gem-SLNs, Gem-SLPHNs and Gem-F-SLPHNs (d) UV-vis spectrum of Gem-SLNs and Gem-FSLNs. | ||
The water contact angle of PVCL has been reported to be 25° at 25 °C52 suggesting that it is a hydrophilic polymer and hence suitable for our purpose. Gem-SLPHNs were prepared by incorporating PVCL (1% w/v) in the aqueous phase (W1) along with the drug, the composition and process parameters were kept constant as that of the Gem-SLNs (F9). PVCL was incorporated in the innermost aqueous layer (W1) along with the drug to achieve higher encapsulation through hydrophilic interactions. Gem-SLPHNs manifested slightly higher particle size of 344 ± 13.4 nm than the Gem-SLNs. This could be either due to swelling of the polymer chains incorporated within the formulation or due to formation of agglomerates.53 However, TEM images (Fig. 2b) showed no evidence of agglomeration. Hence, slight increase in the particle size is most probably on account of swelling of PVCL chains. Moreover, as PVCL was not present on the surface of particles it did not alter the zeta potential (Table 2). Higher % EE exhibited by Gem-SLPHNs than Gem-SLNs can be attributed to increase in the hydrophilic interactions between the polymer and gemcitabine.
| Sample | Particle size (nm) | PDI | Zeta potential (mV) | % EE |
|---|---|---|---|---|
| a Data are expressed as mean ± S.D. (n = 3). | ||||
| Gem-SLNs | 319 ± 12.4 | 0.27 ± 0.02 | −35.9 ± 1.2 | 46.5 ± 6.2 |
| Gem-SLPHNs | 344 ± 13.4 | 0.39 ± 0.01 | −35.5 ± 1.0 | 51.3 ± 4.2 |
| Gem-F-SLPHNs | 377 ± 11.2 | 0.34 ± 0.08 | −38 ± 3.25 | 49.6 ± 5.9 |
Further, folic acid conjugated F-127 (FA-F127) was used as an emulsifier in the preparation of nanoparticles to obtain the SLPHNs functionalized with targeting ligands. The schematic illustration of overall preparation of Gem-F-SLPHNs is shown in Fig. 3.
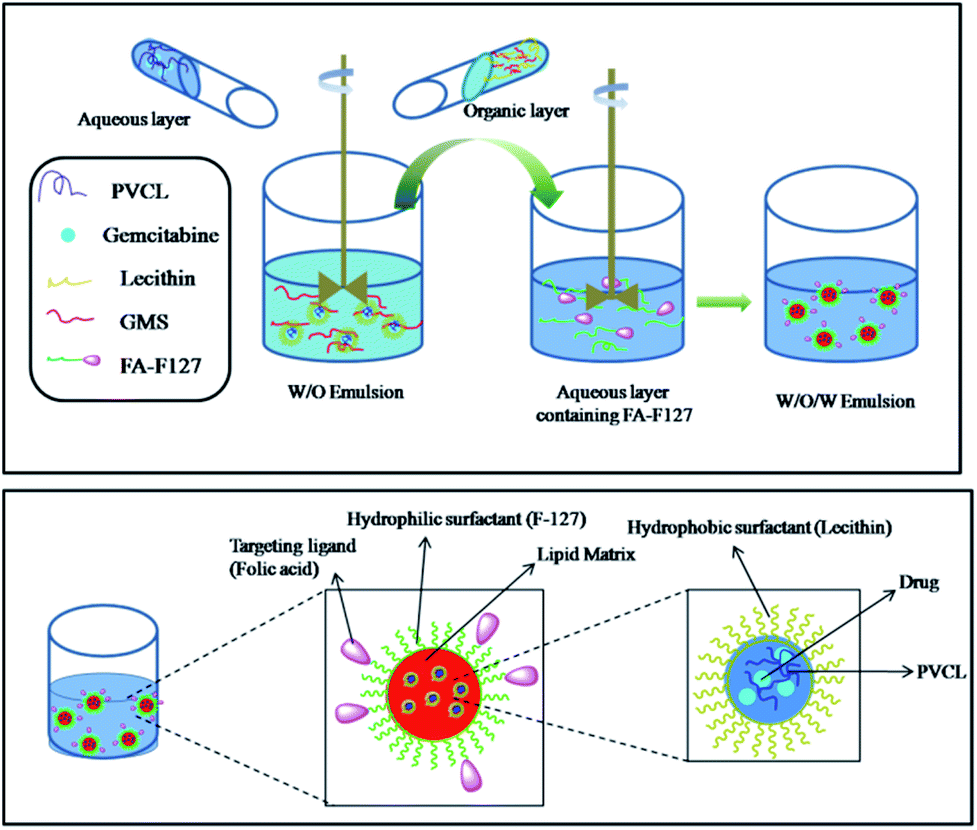 | ||
| Fig. 3 Schematic illustration of preparation of Gem-F-SLPHNs. | ||
The average particle size was found to be increased (∼377 nm) for Gem-F-SLPHNs prepared using FA-F127 as an emulsifier compared to Gem-SLPHNs probably due to the presence of folic acid on the surface of the SLNs (Fig. 2c). Similar observation has been made in the literature.19 The presence of folic acid on the surface was confirmed by UV-vis spectroscopy analysis, which showed the appearance of characteristic peak of folic acid at 365 nm (Fig. 2d). Schematic representation of the possible structures of Gem-SLNs, Gem-SLPHNs and Gem-F-SLPHNs is shown in Fig. S9.†
In vitro drug release studies
In vitro release of gemcitabine from the formulations was studied in 10 mM phosphate buffer (pH 7.4) at 37 °C. All the formulations showed biphasic drug release profiles, that is, immediate burst release for 2 h followed by sustained release. Immoderate burst release can cause dose related toxicities and needs to be minimized. Gem-SLNs exhibited maximum of 49.37 ± 6.3% of its drug release within 2 h, whereas formulations containing PVCL, that is, Gem-SLPHNs significantly reduced the release of drug in the burst phase and only 33.4 ± 4.6% of the drug was released (Fig. 4a). Burst release in the initial hours can be attributed to presence of drug in the outer stratum.54 Incorporation of PVCL within the SLN is expected to allow the drug to remain within the innermost aqueous compartment through hydrophilic interactions with PVCL and reduce its partitioning into the outer layer during the preparation process, thereby successfully reducing the % release in the burst phase. The PVCL concentration was changed from 0.5% w/v to 1.5% w/v and better control over the drug release was observed at the higher concentration of PVCL (Fig. S10†). Furthermore, Gem-F-SLPHNs showed slightly lower release than Gem-SLPHNs probably due to presence of folic acid on the surface of the particle that offered a barrier to the drug.18 A similar pattern of restricted release of gemcitabine from the mannose tagged solid lipid nanoparticles was reported in the literature.10 | ||
| Fig. 4 In vitro release of gemcitabine in (a) Gem-SLNs, Gem-SLPHNs and Gem-F-SLPHNs at physiological temperature (37 °C) and (b) Gem-SLNs and Gem-SLPHNs at 25 °C and at 37 °C (% CDR= % cumulative drug release, data is expressed as mean ± S.D. (n = 3)). | ||
Drug release studies were also performed at 25 °C (below CP) to probe the thermosensitivity of the prepared formulations (Fig. 4b). Gem-SLNs exhibited slightly lower drug release than at 37 °C. This can be attributed to the decrease in the diffusion rate of the drug at lower temperature.55 However, similar pattern, that is, negligible difference in drug release with change in temperature, was also observed in the formulation containing PVCL, which is a thermoresponsive polymer. This is in contrast to the significantly enhanced drug release that was expected above CP. This could be achieved by loading PVCL within the inner aqueous phase (W1), so that it would not show enough thermosensitivity by being shielded from the outer aqueous environment thereby limiting the drug release to the same levels as at 25 °C.
Further, drug release kinetics and drug release mechanism at 37 °C was studied by fitting the drug release profile data of SLNs, SLPHNs and F-SLPHNs in different mathematical models viz. zero order, first order, Higuchi and Korsmeyer–Peppas models. Correlation coefficient values and n values are given in Table S1.† Drug release data was best fitted in the Higuchi model with higher linearity coefficient than zero order and first order models. Additionally, n values in the Peppas model were found to be lower than 0.45 for all the three formulations, which indicates that the release of the drug from formulations follows the Fickian diffusion.56
Hemolysis study
To investigate the biocompatibility of polymers for in vivo applications hemolytic activity of blank SLNs, SLPHNs, F-SLNs and F-SLPHNs was investigated. All formulations were tested at different concentrations (1, 2, 3, 4, 5 mg mL−1). The % hemolysis for samples at the higher concentration (5 mg mL−1) is presented in Fig. 5a and data for other concentrations is given in ESI (Fig. S11†). The results suggested that all samples were safe to be used at tested concentration as they showed <5% hemolysis.57 Triton X-100 was used as positive control that showed strong damage to the red blood cells and PBS was used as negative control. | ||
| Fig. 5 (a) % Hemolysis of different formulations at concentration of 5 mg mL−1. (b) In vitro cell viability assay (MTT assay) with blank, Gem-SLN, Gem-F-SLN and Gem-F-SLPHN. | ||
Cell viability studies
Cytotoxicity of the blank (unloaded) F-SLPHNs, Gem-SLNs, Gem-F-SLNs and Gem-F-SLPHNs was investigated by MTT assay by using MDA-MB-231 cell line (Fig. 5b). Nearly 100% cell viability was observed with blank F-SLPHNs that ascertained the non-cytotoxic nature of the targeting nanoparticles developed here. Drug loaded F-SLNs showed higher cytotoxicity than all the other formulations. This can be explained as the result of better internalization due to presence of folic acid as well as due to lack of PVCL that caused maximum drug release in the burst phase. On the other hand, Gem-F-SLPHNs showed lower cytotoxicity than Gem-F-SLNs owing to the presence of polymer that helps in controlling the drug release. Gem-SLNs manifested lowest cytotoxicity among all the formulations due to poor internalization by virtue of the absence of folic acid groups on the surface of the particles. Taken together, these results indicate that presence of folic acid and hydrophilic polymer is essential for the efficient internalization in the cells and controlled release of the drug, respectively. However drug loaded in SLNs manifested higher cytotoxicity than the free drug in dose dependent manner, probably due to the lipophilic nature of the nanocarrier that facilitated the cellular internalization.58Cellular uptake
Drug uptake by cells was investigated by epifluorescence microscopy. As gemcitabine is a non-fluorescent drug, a hydrophilic dye viz. calcein was used to investigate the drug uptake by cells. Calcein was loaded in F-SLPHNs, SLPHNs and SLNs. Cells were incubated with dye loaded solid lipid nanoparticles for 3 h and fluorescence intensity was determined by epifluorescence microscopy. Cells incubated with F-SLPHNs exhibited relatively more dye uptake compared to SLNs and SLPHNs as expected due to the presence of folic acid on the surface (Fig. 6). These results are in good agreement with the MTT assay results. Average fluorescence intensity of 30 cells was determined (Fig. S12†). | ||
| Fig. 6 Epifluorescence images of MDA-MB-231 cells incubated for 3 h with calcein loaded F-SLPHN (panel A), SLPHN (panel B) and SLN (panel C), the images from left to right show cell nuclei stained by DAPI (blue), fluorescence of calcein (green), and overlay of the two images. All images were taken at same exposure time for calcein and the scale bar is 5 μm. | ||
Conclusions
To address the unresolved issue of burst release of hydrophilic drugs from SLNs, a hydrophilic polymer viz. PVCL was added during the formulation of SLNs containing hydrophilic drug viz. gemcitabine that would result in better encapsulation as well as controlled release due to hydrophilic–hydrophilic interactions between the polymer and drug. Addition of PVCL in the inner most aqueous phase (W1) along with the drug led to higher % EE. A prominent control over the burst release from 49% for SLNs to 33% for PVCL containing nanoparticles, (SLPHNs) was demonstrated. Further, decoration of the SLPHNs with folic acid led to remarkable increase in the cellular uptake. In the literature, coating of SLNs with a polymer has been used to improve their stability and properties in general. We have shown for the first time that by incorporation of a hydrophilic polymer in the core of the SLNs decrease in the burst release and improvement in the % EE of the hydrophilic drugs can be specifically achieved. Thus, it was demonstrated that folic acid decorated, hydrophilic polymer containing SLNs are potential nanocarriers of hydrophilic anticancer drugs such as gemcitabine for controlled release and targeted delivery.Author contributions
Sai Geetika Surapaneni (SGS): conceptualization, experimental work, data curation, analysis, and manuscript writing. Ashootosh V. Ambade (AVA): supervision, validation, funding acquisition, writing and editing of manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SGS would like to thank Indian Council of Medical Research (ICMR), New Delhi for the research fellowship. AVA acknowledges financial support from CSIR-National Chemical Laboratory (MLP300326).Notes and references
- C. Pucci, C. Martinelli and G. Ciofani, Ecancermedicalscience, 2019, 13, 1–26 CrossRef PubMed
.
- R. F. Ozols, Semin. Oncol., 2005, 32, 4–8 CrossRef PubMed
- D. A. Yardley, Semin. Oncol., 2005, 32, 14–21 CrossRef PubMed
- J. Carmichael, U. Fink, R. C. G. Russell, M. F. Spittle, A. L. Harris, G. Spiessi and J. Blatter, Br. J. Cancer, 1996, 73, 101–105 CrossRef CAS
- T. Hoang, K. M. Kim, A. Jaslowski, P. Koch, P. Beatty, J. McGovern, M. Quisumbing, G. Shapiro, R. Witte and J. H. Schiller, Lung Cancer, 2003, 42, 97–102 CrossRef
- V. Bianchi, S. Borella, F. Calderazzo, P. Ferraro, L. Chieco Bianchi and P. Reichard, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 8403–8407 CrossRef CAS PubMed
- R. Moog, A. Burger, M. Brandl, J. Schüler, R. Schubert, C. Unger, H. Fiebig and U. Massing, Cancer Chemother. Pharmacol., 2002, 49, 356–366 CrossRef CAS PubMed
- J. M. Reid, W. Qu, S. L. Safgren, M. M. Ames, M. D. Krailo, N. L. Seibel, J. Kuttesch and J. Holcenberg, J. Clin. Oncol., 2004, 22, 2445–2451 CrossRef CAS
- J. L. Abbruzzese, R. Grunewald, E. A. Weeks, D. Gravel, T. Adams, B. Nowak, S. Mineishi, P. Tarassoff, W. Satterlee, M. N. Raber and W. Plunkett, J. Clin. Oncol., 1991, 9, 491–498 CrossRef CAS PubMed
- N. Soni, N. Soni, H. Pandey, R. Maheshwari, P. Kesharwani and R. K. Tekade, J. Colloid Interface Sci., 2016, 481, 107–116 CrossRef CAS PubMed
- P. T. Nandini, R. C. Doijad, H. N. Shivakumar and P. M. Dandagi, Drug Delivery, 2015, 22, 647–651 CrossRef CAS PubMed
- S. Momin, S. Khan, D. M. Ghadge and K. S. Bhise, J. Drug Delivery Ther., 2017, 7, 1–12 Search PubMed
- C. Tapeinos, M. Battaglini and G. Ciofani, J. Controlled Release, 2017, 264, 306–332 CrossRef CAS PubMed
- Y. Mirchandani, V. B. Patravale and S. Brijesh, J. Controlled Release, 2021, 335, 457–464 CrossRef CAS PubMed
- L. Priano, D. Esposti, R. Esposti, G. Castagna, C. De Medici, F. Fraschini, M. R. Gasco and A. Mauro, J. Nanosci. Nanotechnol., 2007, 7, 3596–3601 CrossRef CAS
- K. Greish, Methods Mol. Biol., 2010, 624, 25–37 CrossRef CAS PubMed
- R. Shegokar, K. K. Singh and R. H. Müller, Int. J. Pharm., 2011, 416, 461–470 CrossRef CAS
- H. Pawar, S. K. Surapaneni, K. Tikoo, C. Singh, R. Burman, M. S. Gill and S. Suresh, Drug Delivery, 2016, 23, 1453–1468 CrossRef CAS PubMed
- K. Rajpoot and S. K. Jain, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1236–1247 CrossRef CAS
- R. H. Müller, K. Mäder and S. Gohla, Eur. J. Pharm. Biopharm., 2000, 50, 161–177 CrossRef
- H. Hajipour, H. Hamishehkar, M. Rahmati-Yamchi, D. Shanehbandi, S. N. Soltan Ahmad and A. Hasani, Int. J. Canc. Manag., 2018, 11, e9402 Search PubMed
- X. Zhao, F. Li, Y. Li, H. Wang, H. Ren, J. Chen, G. Nie and J. Hao, Biomaterials, 2015, 46, 13–25 CrossRef CAS PubMed
- T. E. Yalcin, S. Ilbasmis-Tamer and S. Takka, Int. J. Pharm., 2018, 548, 255–262 CrossRef CAS PubMed
- T. E. Yalcin, S. Ilbasmis-Tamer and S. Takka, Int. J. Pharm., 2020, 580, 119246 CrossRef CAS PubMed
- P. Fonte, T. Nogueira, C. Gehm, D. Ferreira and B. Sarmento, Drug Delivery Transl. Res., 2011, 1, 299–308 CrossRef CAS
- V. Piazzini, L. Cinci, M. D'Ambrosio, C. Luceri, A. R. Bilia and M. C. Bergonzi, Curr. Drug Delivery, 2018, 16, 142–152 CrossRef PubMed
- T. Wang, M. Bae, J. Y. Lee and Y. Luo, Food Hydrocolloids, 2018, 84, 581–592 CrossRef CAS
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS PubMed
- A. Mtibe, M. P. Motloung, J. Bandyopadhyay and S. S. Ray, Macromol. Rapid Commun., 2021, 42, 2100130 CrossRef CAS PubMed
- N. A. Cortez-Lemus and A. Licea-Claverie, Prog. Polym. Sci., 2016, 53, 1–51 CrossRef CAS
- Y. Huang, K. Mao, B. Zhang and Y. Zhao, Mater. Sci. Eng., C, 2017, 70, 763–771 CrossRef CAS PubMed
- H. Khatri, N. Chokshi, S. Rawal, B. M. Patel, M. Badanthadka and M. M. Patel, Int. J. Pharm., 2020, 583, 119386 CrossRef CAS PubMed
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2004, 56, 1649–1659 CrossRef CAS
- M. Srinivasarao and P. S. Low, Chem. Rev., 2017, 117, 12133–12164 CrossRef CAS PubMed
- Z. Zhao, A. Ukidve, J. Kim and S. Mitragotri, Cell, 2020, 181, 151–167 CrossRef CAS PubMed
- H. Vu-Quang, M. S. Vinding, T. Nielsen, M. G. Ullisch, N. C. Nielsen, D.-T. Nguyen and J. Kjems, Polymers, 2019, 11, 743 CrossRef CAS PubMed
- O. S. Muddineti, P. Kumari, B. Ghosh, V. P. Torchilin and S. Biswas, ACS Appl. Mater. Interfaces, 2017, 9, 16778–16792 CrossRef CAS PubMed
- V. Kozlovskaya, F. Liu, B. Xue, F. Ahmad, A. Alford, M. Saeed and E. Kharlampieva, Biomacromolecules, 2017, 18, 2552–2563 CrossRef CAS PubMed
- V. Kozlovskaya and E. Kharlampieva, ACS Appl. Polym. Mater., 2020, 2, 26–39 CrossRef CAS
- S. Halligan, K. Murray, M. Hopkins, I. Rogers, J. Lyons, O. Vrain and L. Geever, J. Appl. Polym. Sci., 2020, 137, 1–10 CrossRef
- K. Q. Nishath, F. Tirunagari, M. Husna, V. R. Nandagopal and A. Jangala, J. Appl. Pharm. Sci., 2011, 1, 66–71 Search PubMed
- S. Hamimed, M. Jabberi and A. Chatti, Naunyn-Schmiedeberg's Arch. Pharmacol., 2022, 4, 1–19 Search PubMed
- W. Wang, T. Chen, H. Xu, B. Ren, X. Cheng, R. Qi, H. Liu, Y. Wang, L. Yan, S. Chen, Q. Yang and C. Chen, Molecules, 2018, 23, 1578 CrossRef PubMed
- J. A. Loureiro, S. Andrade, A. Duarte, A. R. Neves, J. F. Queiroz, C. Nunes, E. Sevin, L. Fenart, F. Gosselet, M. A. N. Coelho, M. C. Pereira and N. Latruffe, Molecules, 2017, 22, 277 CrossRef PubMed
- J. He, S. Huang, X. Sun, L. Han, C. Chang, W. Zhang and Q. Zhong, Nanomaterials, 2019, 9, 1162 CrossRef CAS
- W. Mehnert and K. Mäder, Adv. Drug Delivery Rev., 2001, 47, 165–196 CrossRef CAS PubMed
- P. Senthil Kumar, A. Arivuchelvan, A. Jagadeeswaran, N. Punniamurthy, P. Selvaraj, P. N. Richard Jagatheesan and P. Mekala, Appl. Nanosci., 2015, 5, 661–671 CrossRef CAS
- M. A. Schubert and C. C. Müller-Goymann, Eur. J. Pharm. Biopharm., 2003, 55, 125–131 CrossRef CAS PubMed
- T. Helgason, T. S. Awad, K. Kristbergsson, D. J. McClements and J. Weiss, J. Colloid Interface Sci., 2009, 334, 75–81 CrossRef CAS PubMed
- M. Shah and K. Pathak, AAPS PharmSciTech, 2010, 11, 489–496 CrossRef CAS PubMed
- T. G. F. Souza, V. S. T. Ciminelli and N. D. S. Mohallem, J. Phys.: Conf. Ser., 2016, 733 Search PubMed
- B. C.-K. Tong, Physiol. Behav., 2017, 176, 139–148 CrossRef PubMed
- B. Sarmento, D. Mazzaglia, M. C. Bonferoni, A. P. Neto, M. Do Céu Monteiro and V. Seabra, Carbohydr. Polym., 2011, 84, 919–925 CrossRef CAS
- S. Parvez, G. Yadagiri, M. R. Gedda, A. Singh, O. P. Singh, A. Verma, S. Sundar and S. L. Mudavath, Sci. Rep., 2020, 10, 1–14 CrossRef PubMed
- T. Shirakura, T. J. Kelson, A. Ray, A. E. Malyarenko and R. Kopelman, ACS Macro Lett., 2014, 3, 602–606 CrossRef CAS PubMed
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Poloniae Pharmaceutica – Drug Research, 2010, 67, 217–223 CAS
- Z. Song, W. Zhu, N. Liu, F. Yang and R. Feng, Int. J. Pharm., 2014, 471, 312–321 CrossRef CAS
- W. Wang, L. Zhang, T. Chen, W. Guo, X. Bao, D. Wang, B. Ren, H. Wang, Y. Li, Y. Wang, S. Chen, B. Tang, Q. Yang and C. Chen, Molecules, 2017, 22, 1814 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02845j |
| This journal is © The Royal Society of Chemistry 2022 |