
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis, structure revision and cytotoxic activity of Sch 53825 and its derivatives†
Leichuan Xu ,
Haoyun Ma,
Xinkun An,
Yihao Li,
Qian Zhang,
Xinlei Liu and
Mingan Wang*
,
Haoyun Ma,
Xinkun An,
Yihao Li,
Qian Zhang,
Xinlei Liu and
Mingan Wang*
Department of Applied Chemistry, College of Science, China Agricultural University, Beijing 100193, People's Republic of China. E-mail: wangma@cau.edu.cn
First published on 14th June 2022
Abstract
The first total synthesis of Sch 53825 (14) was achieved in 12 steps from 5-hydroxy-1-tetralone in 16% overall yield through N-benzyl cinchoninium chloride-catalyzed asymmetric epoxidation and a Mitsunobu reaction as the key steps. On this basis, the synthesis of palmarumycin B6 was improved using the same raw material with 6 steps and 32% overall yield. Also, three new analogues with two chlorine atoms were synthesized. Their structures were characterized by 1H, 13C NMR, HR-ESI-MS and X-ray diffraction data. The structure of natural Sch 53825 was revised as an epimer of compound 1 with the anti-hydroxy epoxide at C-4. Their cytotoxic activities against several tumor cell lines (HCT116, U251, BGC823, Huh-7 and PC9) showed that compound 11 exhibited excellent cytotoxicity against above mentioned cancer cell lines with IC50 < 0.5 μM.
Introduction
Spirobisnaphthalenes (spirodioxynaphthalenes) belong to a novel family of natural products isolated from fungi and plants. Generally, they are divided into four subtypes, types A, B, C and D, according to the different linkages of the two naphthalene rings.1,2 Spirobisnaphthalenes have attracted much attention because of their unique structures and broad spectrum of biological activities including antibacterial, antifungal, herbicidal, insecticidal, cytotoxic and antitumor activities.1,2 In the past thirty years, many reports related to the total synthesis and bioactivity evaluation of the spirobisnaphthalene natural products have appeared, such as palmarumycin CP1, CP2, CP17, preussomerin G, I, K and L.3,4 Recently, the biomimetic synthesis of rhytidenone A and elucidation of mode of action of the cytotoxic rhytidenone F were reported, the result showed that rhytidenone F had potent antitumor activity against several cancer cell lines with IC50 values of 0.01–0.82 μM.5 Spiroxins A–E were isolated from a marine-derived fungus, and are a type of DNA cleaving antitumor antibiotics.6 Spiroxins A, C and D have also been synthesized in three groups.7 The derivatives of spiromamakone A, isolated in 2006, showed excellent cytotoxicity against cervical carcinoma HeLa cells.8 Spiromamakone A was totally synthesized by Doi and co-workers in 2018 and revised the structure of spiro-preussione A, which was isolated in 2009.9 Spiroaxillarone A, an unique spirobisnaphthalene natural product, which was isolated from Cyanotis axillaris in 2019, has been synthesized via 5 steps in overall yield of 10%.10 Besides, it was reported that plecmillin A has excellent growth inhibition effect against human colorectal HCT116 cell line with IC50 2.08 μM and could induce both cell cycle arrest at G2/M phase and cell death through the P53–P21 pathway.11 Rhytidenone F also exhibited excellent growth inhibition effect against several cancer cell lines with IC50 0.01–0.82 μM, and the pulldown assay coupled with mass spectrometry analysis revealed the target protein PA28γ is covalently attached to rhytidenone F at the Cys92 residue. The interactions of rhytidenone F with PA28γ lead to the accumulation of P53. Consequently, the Fas-dependent signaling pathway is activated to initiate cellular apoptosis.5 These results indicated that these types of compounds are promising natural product leads for the discovery and development of antitumor drugs.In our previous work, the total synthesis of several spirobisnaphthalenes such as palmarumycin B6 (2, Fig. 1a) and C1 (3, Fig. 1b) have been finished, and also revised the structure of palmarumycin B6.12a,b The bioassay results showed that palmarumycin B6 had the larvicidal activity with an LC50 value of 32.7 μM,12b and also palmarumycin C1 and guignardin E exhibited significant cytotoxicity against several tumor cell lines with an IC50 in the range of 2.45–15.39 μM towards HCT116, U87-MG, HepG2, BGC823 and PC9.12c
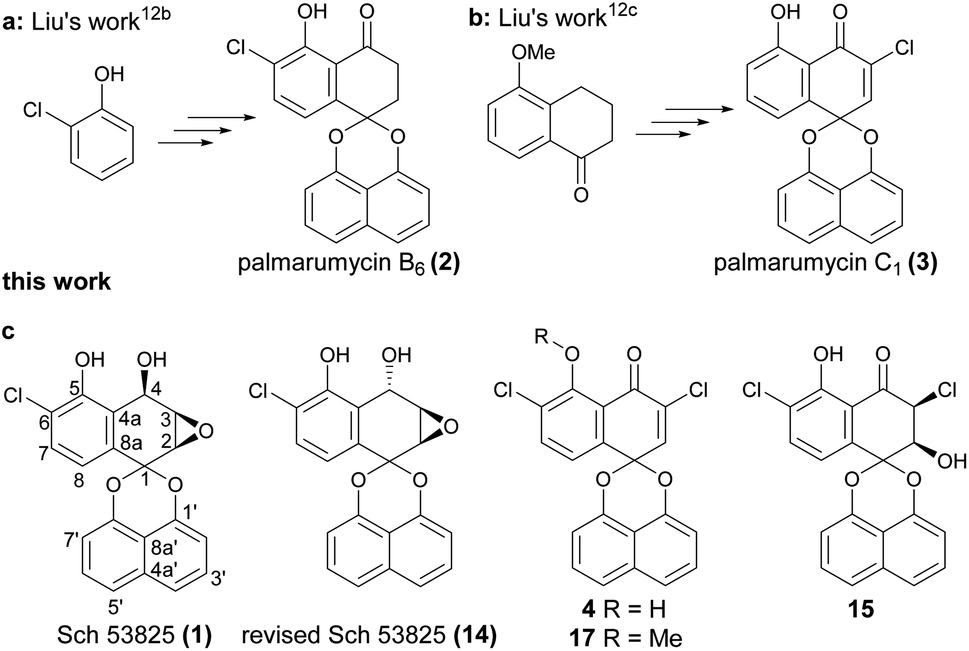 | ||
| Fig. 1 The structures of Sch 53825, palmarumycin B6 (a), C1 (b), and three new derivatives (c). | ||
In 1996, Chu and co-workers identified two novel spirobisnaphthalene compounds Sch 53823 and Sch 53825 (1, Fig. 1c) from the fermentation broth of an unidentified fungus collected from the dead leaves of Ruercus virginiana Miller growing in Tamalupas, Mexico.13 Sch 53823 was easily obtained from reduction of palmarumycin C2. Interestingly, Sch 53825, which has a chlorine atom on the ortho position of hydroxyl group, exhibited in vitro inhibitory activity in the formylmethionyl leucyl phenylalanine (fMLP)-stimulated phospholipase (PLD) assay with an IC50 value of 19 μM.
From the structure point, Sch 53825 and palmarumycin B6 should have the same biosynthetic pathway, and to the best of our knowledge, total synthesis of Sch 53825 or its structure modification has not been reported to date. Also, the absolute configuration of Sch 53825 has not been verified. Based on these and the excellent phospholipase D inhibitory activity of Sch 53825, it attracted our attention after we completed the synthesis of palmarumycin B6.12b So we launched a project to investigate the synthesis and druggability of the target compound. Chlorinated spirobisnaphthalenes generally have better biological activity in this kind of natural products. For example, Spiroxin A exhibited a mean cytotoxicity (IC50 = 0.09 μg mL−1), which was stronger than spiroxin C and D.6a Moreover, due to the excellent larvicidal and antitumor activity of palmarumycin B6 and C1 respectively, the three new Sch 53825 derivatives with two chlorine atoms (4, 15, 17, Fig. 1c) were designed based on the character of the two structures and synthesized to further improve their larvicidal or antitumor activities. In this context, a feasible route for the total synthesis of Sch 53825 and its derivatives were designed, and their cytotoxic activities were also evaluated.
Results and discussion
The retro-synthetic analysis of Sch 53825 was shown in Scheme 1. Sch 53825 can be obtained through asymmetric reduction of 13, which can be synthesized by stereoselectivity epoxidation of 12. Compound 12 can be prepared from a ketalization of 5-methoxy-6-chlorotetralone (7) and 1,8-dihydroxynapthalene (DHN) in a similar process used in the synthesis of palmarumycin B6.12b Following the same procedure, we could obtain enough amount of compound 10 easily, the final assembly of Sch 53825 was carried out as summarized in Scheme 2. | ||
| Scheme 1 Retrosynthetic analysis of Sch 53825. | ||
 | ||
| Scheme 2 Total synthesis of Sch 53825 and palmarumycin B6 (2). Reagents and conditions: (a) diisopropylamine hydrochloride, toluene, −10 °C, 93%; (b) CH3I, K2CO3, acetone, reflux, 97%; (c) (CH3O)3CH, PPTs, MeOH, reflux; (d) 1,8-dihydroxynaphthalene, TsOH, toluene, reflux, 69% for two steps; (e) PDC, Celite, t-BuOOH, benzene, rt, 77%; (f) (I) TMSOTf, Et3N, 0 °C, (II) Pd(OAc)2, CH3CN, rt, 82% for two steps; (g) B-bromocatecholborane, CH2Cl2, 0 °C, 77%; (h) N-benzylcinchoninium chloride, t-BuOOH, NaOH (0.1 M), toluene/H2O, 0 °C, 77%; (i) NaBH4, THF/MeOH, rt, 71%; (j) (I) p-nitrobenzoic acid, PPh3, DEAD, THF, 0 °C, (II) LiOH, THF/H2O, rt, 95% for two steps; (k) TMSI, CHCl3, 0 °C, 68%. | ||
First, starting with the intermediate ketone 10 of palmarumycin B6, we attempted direct α,β-dehydrogenative oxidations by utilizing DDQ,12c IBX, or iodic acid.14 However, both DDQ and IBX failed to introduce double bonds at the α,β-position in 10. Although the use of iodic acid successfully introduced the double bonds, one of the hydrogen atom on naphthalene ring of the target molecule will be substituted by iodine in the meantime since the oxidation reactions tend to produce varying amounts of detrimental iodine as a by-product.14 After extensive experiments, a ketone–enone conversion was conducted using Saegusa conditions in 82% yield.15 Then, compound 11 was demethylated with B-bromocatecholborane to afford compound 12 as the literature described.12c Next, the stereoselective epoxidation of 12 with t-BuOOH catalyzed with N-benzyl cinchoninium chloride at 0 °C yielded epoxide 13 in 97.4% ee value.4c,12b Two methods were tried to reduce the carbonyl of compound 13. First, palmarumycin CP2 could be efficiently converted into its corresponding dihydro derivative (93% ee) by asymmetric reduction using (+)-B-chlorodiisopinocampheylborane.3a But it was inactive with our substrate. In the synthesis of palmarumycin C11, syn-hydroxy epoxide was achieved in one step using sodium borohydride.3d Thus, Sch 53825 (1) was obtained using this method, and confirmed the syn-hydroxy epoxide structure with X-ray crystallographic data. It could also be found that the proton on C-4 is spatially correlated with the protons on C-2 and C-3 positions from NOESY spectra of compound 1 (Fig. S17†).
After completion the reported structure of Sch 53825, it was found that its 1H, 13C NMR and optical rotation data were not consistent with those of the reported natural product (Fig. 2).13 The chemical shifts of protons on C-2, C-3 and C-4 lie in a higher-field region than those of the natural product, the chemical shifts of carbon on C-2, C-3 and C-4 lie in a lower-field region than those of the natural product (Table 1). These differences in the chemical shifts may be related to the relative configuration of hydroxy at C-4. Thus, it was hypothesized that the correct structure of Sch 53825 possibly was the epimer of compound 1 at C-4 with an anti-hydroxy epoxide. To verify the real structure of natural Sch 53825, Mitsunobu reaction was employed to transfer 1 to its p-nitrobenzoate, then hydrolyzed with LiOH solution to afford the epimer 14, the spectroscopic and optical rotation data of 14 is in accordance with that reported by Chu and co-workers.13 So, the total synthesis of Sch 53825 was first furnished in 12 steps and 16% overall yield from commercially available 5-hydroxy-1-tetralone (5), and the structure of Sch 53825 was revised to be the anti-hydroxy epoxide of 1.
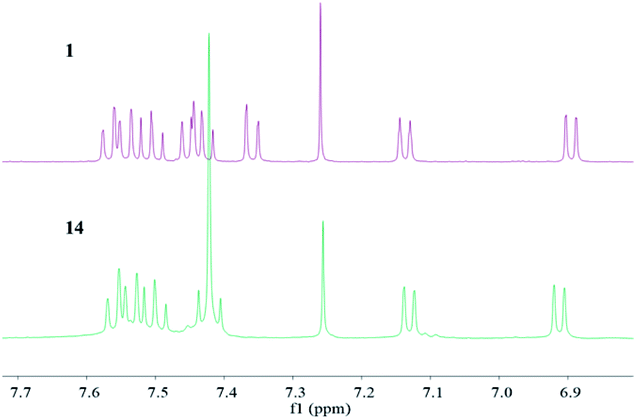 | ||
| Fig. 2 Comparison of 1H NMR spectra of compounds 1 and 14 in the low-field region. | ||
| Position | Sch 53825 (CDCl3) | Compound 1 (CDCl3) | Compound 14 (CDCl3) | |||
|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | — | 97.5 | — | 96.53 | — | 97.65 |
| 2 | 3.81 (d, 4.0) | 50.5 | 3.74–3.76 (m) | 52.59 | 3.81 (d, 3.9) | 50.73 |
| 3 | 3.72 (dd, 2.3, 4.0) | 52.9 | 3.42–3.45 (m) | 54.09 | 3.72 (dd, 2.4, 3.9) | 52.75 |
| 4 | 5.66 (brd, 2.3) | 61.5 | 5.47 (dd, 2.5, 10) | 66.44 | 5.66 (brs) | 62.03 |
| 4a | — | 122.3 | — | 121.22 | — | 121.94 |
| 5 | — | 150.2 | — | 151.70 | — | 149.72 |
| 6 | — | 123.4 | — | 123.60 | — | 123.02 |
| 7 | 7.44–7.48 (m) | 130.0 | 7.42–7.46 (m) | 130.76 | 7.45–7.49 (m) | 129.76 |
| 8 | 7.44–7.48 (m) | 120.1 | 7.42–7.46 (m) | 120.00 | 7.45–7.49 (m) | 120.24 |
| 8a | — | 131.3 | — | 130.90 | — | 131.64 |
| 1′ | — | 147.3 | — | 147.12 | — | 147.21 |
| 2′ | 7.42–7.63 (m) | 121.2 | 7.51 (d, 7.5) | 121.14 | 7.45–7.49 (m) | 121.29 |
| 3′ | 7.42–7.63 (m) | 127.6 | 7.57 (d, 7.8) | 127.48 | 7.53–7.62 (m) | 127.62 |
| 4′ | 6.97 (d, 7.3) | 109.2 | 6.90 (d, 8.0) | 109.11 | 6.97 (d, 7.4) | 109.31 |
| 4a′ | — | 134.3 | — | 134.17 | — | 134.31 |
| 5′ | 7.18 (d, 7.3) | 110.0 | 7.14 (d, 7.4) | 110.07 | 7.18 (d, 7.4) | 110.12 |
| 6′ | 7.42–7.63 (m) | 127.9 | 7.55 (d, 8.2) | 127.79 | 7.53–7.62 (m) | 127.88 |
| 7′ | 7.42–7.63 (m) | 121.2 | 7.37 (d, 8.4) | 120.41 | 7.53–7.62 (m) | 121.25 |
| 8′ | — | 147.4 | — | 147.16 | — | 147.24 |
| 8a′ | — | 112.9 | — | 112.80 | — | 112.94 |
With the successful total synthesis of Sch 53825, its three analogues were explored. Compound 4 could be afforded conveniently by the usage of CeCl3·7H2O in CH3OH/H2O at reflux temperature from epoxide 13 using the same method described in our previous work,12c while compound 15 with hydroxy at β-position was obtained when CeCl3 was used in CH3CN at room temperature. In addition, compound 17 was synthesized from 11 through the same method to investigate the differences of hydroxy and methoxy at C-5 position of these compounds on cytotoxicity (Scheme 3).
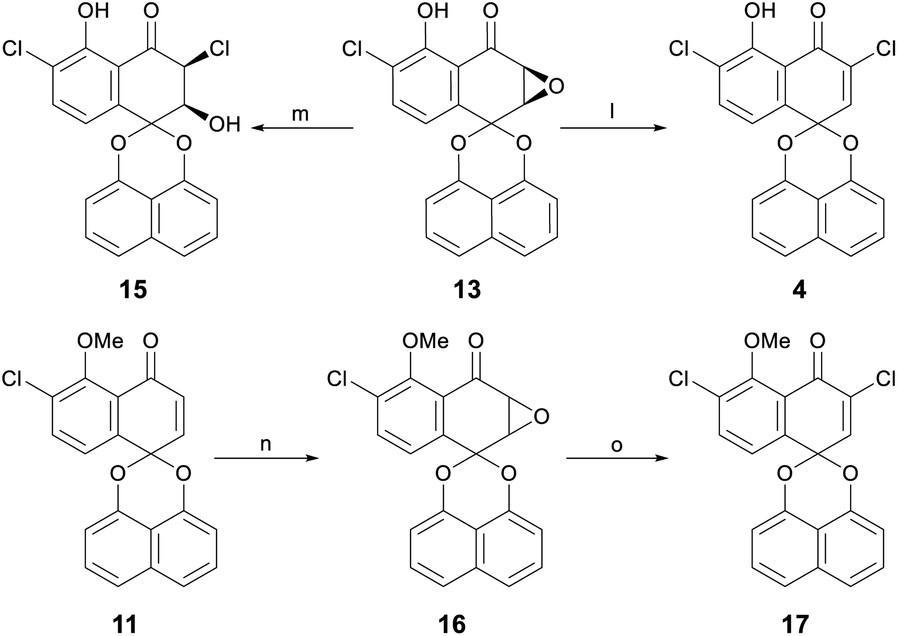 | ||
| Scheme 3 Synthesis of three derivatives of Sch 53825. Reagents and conditions: (l) CeCl3·7H2O, MeOH/H2O, reflux, 92%; (m) CeCl3, MeCN, rt, 47%; (n) t-BuOOH, TBD, toluene, rt, 87%; (o) CeCl3·7H2O, MeOH/H2O, reflux, 78%. | ||
After finishing the synthesis of the desired compounds, all structures were characterized with the 1H, 13C NMR, HR-ESI-MS data. In order to determine the absolute configuration of Sch 53825, the X-ray diffraction analysis of compound 1 was performed using Cu Kα radiation and its structure was depicted in Fig. 3, which unambiguously showed that the absolute configuration of C-2, C-3 and C-4 were 2R, 3R and 4R in Flack parameter 0.002(15) (CCDC ID 2169161,† Fig. 3). Based on this result, the actual absolute configuration of C-2, C-3 and C-4 of Sch 53825 (14) should be 2R, 3R and 4S. The C-2 and C-3 absolute configuration of compound 15 were 2S and 3S.
 | ||
| Fig. 3 ORTEP drawing of compound 1. | ||
In addition, it should be noted that in our previous synthetic route of palmarumycin B6,12b compound 7 was prepared from 2-chlorophenol through 9 steps with a total yield of 10%. This was very inefficient in preparing compound 7, so regioselective chlorination and then methylation of commercially available 5-hydroxy-1-tetralone (5) were attempted. The improved synthetic route of palmarumycin B6 was delineated in Scheme 2. First, there are many novel reports about the aromatic chlorination,16 but only 1,3-dichloro-5,5-dimethylhydantoin (DCDMH, 1.2 eq.) was convenient to this substrate.16b The key intermediate 5-methoxy-6-chlorotetralone (7) was afforded successfully through two steps on this base. It is worth mentioning that there would appear an 6,8-dichlorinated by-product when the temperature was risen to room temperature. The latter experimental procedures were followed our previous reported methods.12b Here, a convenient synthetic route of palmarumycin B6 from 5-hydroxy-1-tetralone was finished in 32% total yield within 6 steps.
The cytotoxic activities of these compounds against the tumor cell lines (HCT116, U251, BGC823, Huh-7 and PC9) were evaluated using a MTT assay11,12a and the results are shown in Table 2. These results indicated that compound 11 exhibited excellent cytotoxicity against above mentioned cancer cells (IC50 < 0.5 μM), while its demethylation product 12 had a reduced activity against these five cancer cells. In addition, compound 10 exhibited weaker cytotoxic activity than compound 11 having a α,β-unsaturated double bond. Comparison with the IC50 data of compounds 1, 13 and 14, we found that the reduction products of carbonyl group at C-4 (1 and 14) were inactive against these cancer cells (IC50 > 50 μM). The new target (4), bearing two chlorine atoms at the C-3 and C-6 positions, respectively, didn't have an increased cytotoxicity compared with palmarumycin C11 (ref. 2c) as we expected, while its synthetic precursor (15) which is not dehydrated exhibited significant cytotoxicity with an IC50 in the range of 3.21–13.57. Furthermore, compound 17 with a methoxy exhibited stronger cytotoxic activity (IC50 = 4.69 μM) against BGC823 than compound 4 (IC50 = 11.03 μM) with a hydroxyl at C-5 position. These results indicated that the carbonyl at C-4, methoxy at C-5 and the α,β-unsaturated double bond play a critical role for cytotoxicity. This may provide some inspiration for further research on this kind of nature products.
| Compounds | HCT116 | U251 | BGC823 | Huh-7 | PC9 |
|---|---|---|---|---|---|
| 1 | >50.0 | >50.0 | >50.0 | >50.0 | >50.0 |
| 2 | 47.11 | >50.0 | 32.12 | >50.0 | >50.0 |
| 4 | 19.71 | 14.11 | 11.03 | 13.64 | 19.65 |
| 9 | 31.10 | >50.0 | 4.02 | >50.0 | >50.0 |
| 10 | 24.56 | >50.0 | 49.33 | >50.0 | 24.16 |
| 11 | <0.5 | 0.38 | <0.5 | <0.5 | <0.5 |
| 12 | 23.65 | 16.69 | 9.82 | 12.79 | 17.34 |
| 13 | 18.60 | 5.39 | 13.05 | 1.42 | 7.14 |
| 14 | >50.0 | >50.0 | >50.0 | >50.0 | >50.0 |
| 15 | 4.07 | 13.27 | 3.22 | 12.91 | 3.21 |
| 17 | >50.0 | 21.46 | 4.69 | 13.86 | 16.97 |
| Taxol | 0.000037 | 1.667 | 0.000605 | 0.007930 | 0.000044 |
Conclusions
In summary, Sch 53825 was first totally synthesized with a 16% overall yield for 12 steps and the structure was revised, also the absolute configuration of Sch 53825 was confirmed. On the base, the synthesis of palmarumycin B6 was improved greatly with a 32% total yield for 6 steps. Further, three new analogues (4, 15, 17) with two chlorine atoms in the molecule were synthesized. These compounds were characterized by 1H, 13C NMR, HR-ESI-MS data and X-ray diffraction. Compound 11 displayed excellent cytotoxic activity against HCT116, U251, BGC823, Huh-7 and PC9 cell lines. Further biological studies and action mechanism of compound 11 are currently in progress in our lab.Experimental
General experimental procedures
Unless otherwise indicated, all reagents were purchased from commercial suppliers and used without further purification. Organic solutions were concentrated under reduced pressure using a rotary evaporator or oil pump. Flash column chromatography was performed using Qingdao Haiyang silica gel (200–300 mesh). Melting points (uncorrected) were measured on a WRX-4 microscopic melting point apparatus (Shanghai Yi Ce Instrument Factory). All 1H and 13C NMR spectra were obtained on Bruker DPX 300, 400 and 500 MHz spectrometer with CDCl3 as solvents and TMS as an internal standard. Chemical shifts are given in δ relative to residual solvent peak (usually chloroform: δ 7.26 for 1H NMR or 77.16 for 13C NMR), and coupling constants (J) in Hz. Signals are reported as follows: s (singlet), d (doublet), t (triplet), q (quartet), dd (double doublet), m (multiplet). High-resolution mass spectrometry data were acquired using an Agilent 6520 Q-Tof analyzer. Optical rotation values were measured with a JASCO P-2000 Polarimeter. The crystal structure was analyzed with a Thermo Fisher ESCALAB 250 four-circle X-ray diffractometer (Xcalibur, Eos, Gemini). HPLC analyses were performed on an Agilent 1100 instruments, UV detection was monitored at 254 and 220 nm, an IB N-5 column (5 μm, 4.6 × 250 mm) was used as the chiral stationary phase, and hexane/i-PrOH (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) was used as the mobile phase at a flow rate of 1.0 mL min−1.
:1) to give compound 6 (7.323 g, 93%) as a white powder. Mp 129–130 °C; (lit. 16 mp 133–135 °C); 1H NMR (400 MHz, CDCl3): δ = 7.61 (d, J = 8.5 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 5.88 (s, 1H), 2.96 (t, J = 6.2 Hz, 2H), 2.64 (t, J = 8 Hz, 2H), 2.17–2.11 (m, 2H); 13C NMR (101 MHz, CDCl3): δ = 197.82, 148.84, 132.70, 132.55, 126.64, 124.72, 119.77, 38.66, 23.51, 22.56. The data are consistent with that previous reported.17:1) to give compound 7 (7.373 g, 97%) as a white solid. Mp 80–81 °C; (lit. 12b mp 82–84 °C); 1H NMR (400 MHz, CDCl3): δ = 7.77 (d, J = 8.5 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 3.85 (s, 3H), 2.99 (t, J = 6.2 Hz, 2H), 2.62 (t, J = 6.2 Hz, 2H), 2.15–2.09 (m, 2H). The data are consistent with that previous reported.12b
:10) was used as the mobile phase at a flow rate of 1.0 mL min−1.
:1) to give compound 6 (7.323 g, 93%) as a white powder. Mp 129–130 °C; (lit. 16 mp 133–135 °C); 1H NMR (400 MHz, CDCl3): δ = 7.61 (d, J = 8.5 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 5.88 (s, 1H), 2.96 (t, J = 6.2 Hz, 2H), 2.64 (t, J = 8 Hz, 2H), 2.17–2.11 (m, 2H); 13C NMR (101 MHz, CDCl3): δ = 197.82, 148.84, 132.70, 132.55, 126.64, 124.72, 119.77, 38.66, 23.51, 22.56. The data are consistent with that previous reported.17:1) to give compound 7 (7.373 g, 97%) as a white solid. Mp 80–81 °C; (lit. 12b mp 82–84 °C); 1H NMR (400 MHz, CDCl3): δ = 7.77 (d, J = 8.5 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 3.85 (s, 3H), 2.99 (t, J = 6.2 Hz, 2H), 2.62 (t, J = 6.2 Hz, 2H), 2.15–2.09 (m, 2H). The data are consistent with that previous reported.12bSynthesis of intermediates 9 and 10
The intermediates 9 and 10 were synthesized following the literature procedures, and their analytical data were consistent with the reported data.12b:1) to give compound 11 (0.072 g, 82% for two steps) as a yellow solid. Mp 180–182 °C; 1H NMR (300 MHz, CDCl3): δ = 7.80 (d, J = 8.5 Hz, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.58 (d, J = 8.3 Hz, 2H), 7.47 (t, J = 7.9 Hz, 2H), 6.98 (d, J = 7.5 Hz, 2H), 6.90 (d, J = 10.5 Hz, 1H), 6.31 (d, J = 10.5 Hz, 1H), 3.99 (s, 3H); 13C NMR (75 MHz, CDCl3): δ = 182.12, 156.07, 147.30, 139.27, 136.32, 135.30, 134.29, 132.60, 131.76, 127.74, 124.94, 124.87, 121.54, 113.16, 110.03, 93.10, 61.71; HRMS (ESI), m/z [M + H]+ calcd for C21H14ClO4: 365.0575; found: 365.0570.:1) to give compound 12 (0.081 g, 77%) as a yellow solid. Mp 165–167 °C; 1H NMR (400 MHz, CDCl3): δ = 12.72 (s, 1H), 7.76 (d, J = 8.3 Hz, 1H), 7.60 (d, J = 8.3 Hz, 2H), 7.51–7.41 (m, 3H), 7.05 (d, J = 10.5 Hz, 1H), 6.99 (d, J = 7.6 Hz, 2H), 6.40 (d, J = 10.5 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 188.80, 157.57, 147.11, 140.44, 137.45, 136.58, 134.30, 129.56, 127.79, 124.42, 121.67, 119.78, 114.74, 113.05, 110.10, 92.74; HRMS (ESI), m/z [M + H]+ calcd for C20H12ClO4: 351.0419; found: 351.0416.:1) to afford compound 13 (0.282 g, 77%) as a yellow solid. Mp 238–240 °C; the ee value of compound 13 was analysed by HPLC to be 97.4%; [α]25D +325.0° (c = 0.65, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 11.84 (s, 1H), 7.74 (d, J = 8.3 Hz, 1H), 7.61 (d, J = 8.3 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.41 (t, J = 8.1 Hz, 1H), 7.19 (d, J = 7.4 Hz, 1H), 6.92 (d, J = 7.5 Hz, 1H), 4.10 (d, J = 4.0 Hz, 1H), 3.72 (d, J = 4.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 196.64, 157.35, 146.72, 146.47, 137.43, 135.40, 134.17, 127.79, 127.66, 124.73, 121.58, 121.46, 119.36, 113.13, 112.68, 110.22, 109.38, 95.75, 53.27, 53.18; HRMS (ESI), m/z [M + H]+ calcd for C20H12ClO5: 367.0368; found: 367.0371.:1) to produce compound 1 (0.047 g, 71%) as a yellow solid. Mp 172–174 °C; [α]25D +120.0° (c = 0.16, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 8.44 (s, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.51 (t, J = 7.5 Hz, 1H), 7.46–7.42 (m, 2H), 7.37 (d, J = 8.4 Hz, 1H), 7.14 (d, J = 7.4 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 5.47 (dd, J = 10 Hz, 2.5 Hz, 1H), 3.85 (d, J = 4.4 Hz, 1H), 3.76–3.74 (m, 1H), 3.45–3.42 (m, 1H); 13C NMR (125 MHz, CDCl3): δ = 151.70, 147.16, 147.12, 134.17, 130.90, 130.76, 127.79, 127.48, 123.60, 121.22, 121.14, 120.41, 120.00, 112.80, 110.07, 109.11, 96.53, 66.44, 54.09, 52.59; HRMS (ESI), m/z [M + H]+ calcd for C20H14ClO5: 369.0524; found: 369.0528.:1) to afford Sch 53825 (14, 0.336 g, 95% for two steps) as a white solid. Mp 126–127 °C; [α]25D +165.5° (c = 0.15, CHCl3); (lit. 13 mp 110–112 °C); [α]25D +167.0° (c = 0.1, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 7.62–7.53 (m, 3H), 7.49–7.45 (m, 3H), 7.18 (d, J = 7.4 Hz, 1H), 6.97 (d, J = 7.4 Hz, 1H), 6.10 (s, 1H), 5.66 (s, 1H), 3.81 (d, J = 3.9 Hz, 1H), 3.72 (dd, J = 3.9 Hz, 2.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 149.72, 147.24, 147.21, 134.31, 131.64, 129.76, 127.88, 127.62, 123.02, 121.94, 121.29, 121.25, 120.24, 112.94, 110.12, 109.31, 97.65, 62.03, 52.75, 50.73; HRMS (ESI), m/z [M + Na]+ calcd for C20H13ClO5Na: 391.0344; found: 391.0341. The data are consistent with that previous reported.13:1) to afford compound 2 (0.098 g, 68%) as a yellow solid. Mp 186–188 °C; (lit.12b mp 190–192 °C); 1H NMR (300 MHz, CDCl3) δ 13.02 (s, 1H), 7.72 (d, J = 8.1 Hz, 1H), 7.56 (dd, J = 8.4, 1.0 Hz, 2H), 7.46 (t, J = 8.1 Hz, 2H), 7.44 (d, J = 8.1 Hz, 1H), 6.99 (dd, J = 7.2, 1.0 Hz, 2H), 2.88 (t, J = 6.6 Hz, 2H), 2.49 (t, J = 6.6 Hz, 2H). The data are consistent with that previous reported.12b:1) to give compound 4 (0.071 g, 92%) as a yellow solid. Mp 183–185 °C; 1H NMR (500 MHz, CDCl3): δ = 12.42 (s, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.62 (d, J = 8.3 Hz, 2H), 7.50 (t, J = 7.9 Hz, 2H), 7.44 (d, J = 8.2 Hz, 1H), 7.20 (s, 1H), 7.00 (d, J = 7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ = 182.19, 157.81, 146.71, 137.22, 137.09, 136.41, 134.86, 134.31, 127.86, 124.98, 121.96, 120.02, 114.08, 112.84, 110.32, 93.32; HRMS (ESI), m/z [M + H]+ calcd for C20H11Cl2O4: 385.0029; found: 385.0024.:1) to produce compound 15 (0.056 g, 47%) as a yellow solid. Mp 121–122 °C; 1H NMR (500 MHz, CDCl3): δ = 12.36 (s, 1H), 7.75 (d, J = 8.2 Hz, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.53–7.43 (m, 4H), 7.15 (d, J = 7.5 Hz, 1H), 6.94 (d, J = 7.5 Hz, 1H), 5.32 (d, J = 1.7 Hz, 1H), 4.69 (s, 1H), 2.74 (s, 1H); 13C NMR (125 MHz, CDCl3): δ = 194.34, 157.58, 146.43, 145.74, 137.72, 135.90, 134.20, 127.84, 127.76, 124.98, 121.90, 121.73, 118.95, 114.97, 112.83, 110.08, 109.19, 98.21, 72.19, 62.36; HRMS (ESI); m/z [M + H]+ calcd for C20H14ClO5: 403.0135; found: 403.0134.:1) to afford compound 16 (0.133 g, 87%) as a yellow solid. Mp 164–165 °C; 1H NMR (500 MHz, CDCl3): δ = 7.69 (d, J = 8.5 Hz, 1H), 7.64 (d, J = 8.5 Hz, 1H), 7.60 (d, J = 8.3 Hz, 1H), 7.56–7.51 (m, 2H), 7.44 (t, J = 7.9 Hz, 1H), 7.19 (d, J = 7.4 Hz, 1H), 6.90 (d, J = 7.5 Hz, 1H), 4.08 (d, J = 4.4 Hz, 1H), 4.01 (s, 3H), 3.73 (d, J = 4.4 Hz, 1H); 13C NMR (125 MHz, CDCl3): δ = 191.17, 155.50, 146.91, 146.69, 136.35, 135.27, 134.30, 132.26, 127.87, 127.82, 124.94, 123.50, 121.64, 121.51, 112.75, 110.22, 109.50, 96.67, 62.92, 54.17, 53.24; HRMS (ESI), m/z [M + H]+ calcd for C21H14ClO5: 381.0524; found: 381.0529.:1) to give compound 17 (0.046 g, 78%) as a yellow solid. Mp 189–191 °C; 1H NMR (500 MHz, CDCl3): δ = 7.82 (d, J = 8.5 Hz, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.49 (t, J = 8.0 Hz, 2H), 7.10 (s, 1H), 6.99 (d, J = 7.5 Hz, 2H), 4.02 (s, 3H); 13C NMR (125 MHz, CDCl3): δ = 175.19, 156.62, 146.88, 138.80, 136.89, 135.95, 134.30, 133.09, 132.85, 127.83, 124.85, 124.16, 121.85, 112.95, 110.26, 93.54, 61.82; HRMS (ESI), m/z [M + H]+ calcd for C21H13Cl2O4: 399.0185; found: 399.0186.X-ray diffraction analysis of compound 1
Colourless plate-like crystals of compound 1 were obtained from a slowly evaporating mixed chloroform and methanol solution. A 0.22 × 0.19 × 0.17 mm3 crystal was selected for analysis. The parameters and structure information for compound 1 have been deposited at the Cambridge Crystallographic Data Centre. CCDC ID 2169161 contains the supplementary crystallographic data for this paper.†Evaluation of cytotoxic activity of title compounds
The cytotoxicity of Sch 53825 and its derivatives was evaluated against five human carcinoma cell lines (HCT116, U251, BGC823, Huh-7 and PC9) by MTT assay described in the literature.11,12 Taxol was selected as the positive control. All of these cell lines (colon cancer cell HCT116, human glioma cell U251, gastric cancer cell BGC823, human hepatoma cells Huh-7 and lung carcinoma cell PC9), obtained from the American Type Culture Collection (ATCC) and purchased from the Cell Culture Center of Institute of Basic Medical Sciences, Peking Union Medical College, Chinese Academy of Medical Sciences, were cultured with 10% fetal bovine serum (FBS) and penicillin (100 U mL−1)-streptomycin (100 μg mL−1) in Dulbecco's minimum essential medium (DMEM). The cell culture medium, serum and antibiotics were purchased from Invitrogen. All the cells were maintained at 37 °C in 5% CO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Natural Science Foundation of China (No. 21772229) was appreciated for the financial supports. Prof. Dai Jungui, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, was appreciated for the cytotoxic activity evaluation.Notes and references
- K. Miyashita and T. Imanishi, Chem. Rev., 2005, 105, 4515 CrossRef CAS PubMed
.
-
(a) L. Zhou, J. Zhao, T. Shan, X. Cai and Y. Peng, Mini-Rev. Med. Chem., 2010, 10, 977 CrossRef CAS PubMed
-
(a) A. G. M. Barrett, D. Hamprecht and T. Meyer, Chem. Commun., 1998, 8, 809 RSC
-
(a) S. Chi and C. H. Heathcock, Org. Lett., 1999, 1, 3 CrossRef CAS PubMed
- Z. Yue, H. C. Lam, K. Chen, I. Siridechakorn, Y. Liu, K. Pudhom and X. Lei, Angew. Chem., Int. Ed., 2020, 59, 4115 CrossRef CAS PubMed
-
(a) L. A. McDonald, D. R. Abbanat, L. R. Barbieri, V. S. Bernan, C. M. Discafani, M. Greenstein, K. Janota, J. D. Korshalla, P. Lassota, M. Tischler and G. T. Carter, Tetrahedron Lett., 1999, 40, 2489 CrossRef CAS
-
(a) K. Miyashita, T. Sakai and T. Imanishi, Org. Lett., 2003, 5, 2683 CrossRef CAS PubMed
-
(a) S. A. v. d. San, J. W. Blunt and M. H. G. Munro, Org. Lett., 2006, 8, 2059 CrossRef PubMed
-
(a) X. Chen, Q. Shi, G. Lin, S. Guo and J. Yang, J. Nat. Prod., 2009, 72, 1712 CrossRef CAS
-
(a) A. Wisetsai, R. Lekphrom, J. Boonmak, S. Youngme and F. T. Schevenels, Org. Lett., 2019, 21, 8344 CrossRef CAS PubMed
- J. Li, R. Ding, H. Gao, L. Guo, X. Yao, Y. Zhang and J. Tang, RSC Adv., 2019, 9, 39082 RSC
-
(a) T. Shan, J. Tian, X. Wang, Y. Mou, Z. Mao, D. Lai, J. Dai, Y. Peng, L. Zhou and M. Wang, J. Nat. Prod., 2014, 77, 2151 CrossRef CAS
- M. Chu, M. G. Patel, J.-K. Pai, P. R. Das and M. S. Puar, Bioorg. Med. Chem. Lett., 1996, 6, 579 CrossRef CAS
- K. C. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 1386 CrossRef CAS
- Y. Ito, T. Hirao and T. Saegusa, J. Org. Chem., 1978, 43, 1011 CrossRef CAS
-
(a) S. Song, X. Li, J. Wei, X. Shi, Y. Zhang, X. Zhang, L. Ai, Y. Zhu, X. Shi, X. Zhang and N. Jiao, Nat. Catal., 2020, 3, 107 CrossRef CAS
- C. F. Schwender, R. E. Pike and J. Shavel, J. Med. Chem., 1973, 16, 254 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2169161. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra02898k |
| This journal is © The Royal Society of Chemistry 2022 |