DOI:
10.1039/D2RA02682A
(Paper)
RSC Adv., 2022,
12, 22939-22945
Computational exploration for possible reaction pathways, regioselectivity, and influence of substrate in gold-catalyzed cycloaddition of cyanamides with enynamides†
Received
27th April 2022
, Accepted 18th July 2022
First published on 15th August 2022
Abstract
The current work focuses on the DFT calculation of the rational mechanism and catalytic activity of the gold(I)-catalyzed isotetradehydro-Diels–Alder cycloaddition of cyanamides and enamides to substituted 2,6-diaminopyridines. IPrAuCl is used as a model catalyst to catalyze cyanamide and enynamide reactants with different substituents in DCM as a research system. DFT data indicates that the catalytic cycle starts from the triple bond coordination between the catalyst's gold cation and the enamide to obtain the gold π-complex, and the cyanamide attacks the alkynyl carbon atom from different directions to generate two reaction channels of five-membered and six-membered heterocycles, respectively. The calculation results show that the 2,6-diaminopyridine compounds produced by this catalytic reaction have lower activation energy and higher reactivity, that is, the pyridine skeleton structure can be easily obtained under mild reaction conditions. At the same time, electron-withdrawing substituents in the reactants are more helpful for the reaction. In addition to being in good agreement with the experimental data, the calculated results also provide an important contribution to the further understanding of the mechanism of such reactions.
1. Introduction
Diaminopyridine is an important organic synthesis intermediate, which is mostly used in the synthesis of medicines. It is one of the important intermediates for the preparation of PDE4 inhibitors, anti-plasmodium falciparum drugs, iNOS/nNOS inhibitors and protein kinase inhibitors, especially phenazopyridine is produced as an analgesic. Furthermore, it can also be used as an organic dye.1–7 The Diels–Alder reaction is also recognized as an ideal synthetic method to form the six-membered ring structure of diaminopyridine. Although the tetradehydrogen-Diels–Alder (TDDA) reaction is affected by harsh conditions and functionalized substrates, it can still be completed by the [4 + 2] cycloaddition reaction between enynes and alkynes.8 In particular, it is a better synthesis idea to prepare aromatic heterocycles by hetero-TDDA reaction. There are also some related reports on the formation of pyridine compounds. Dubbaka and Xie's research team respectively explored the use of functionalized alkynyl lithium and aryl magnesium reagents and TfOH to promote the fusion of enynes and nitriles into pyridine.9,10 At the same time, Barluenga et al. synthesized 2-methoxypyridine from diacetylene and nitrile in a relatively mild environment under the catalysis of Et3PAuCl/AgSbF.11 The hetero-TDDA reaction as a new synthetic method under a single catalyst is more effective and has higher regioselectivity. Subsequently, some experimental and theoretical studies have been reported on the gold-catalyzed carbo-TDDA and hetero-TDDA reactions.12–17
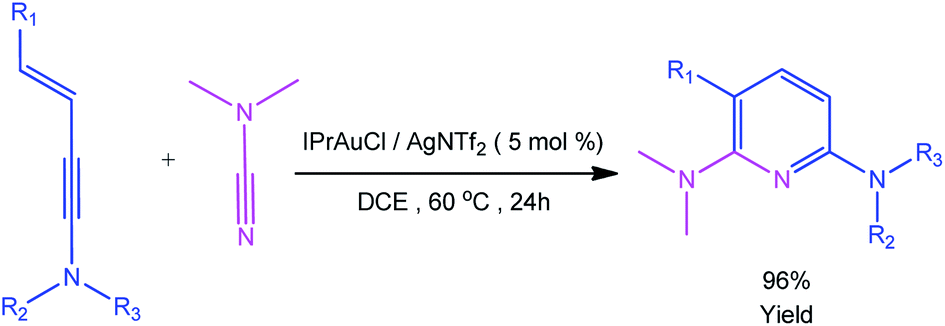
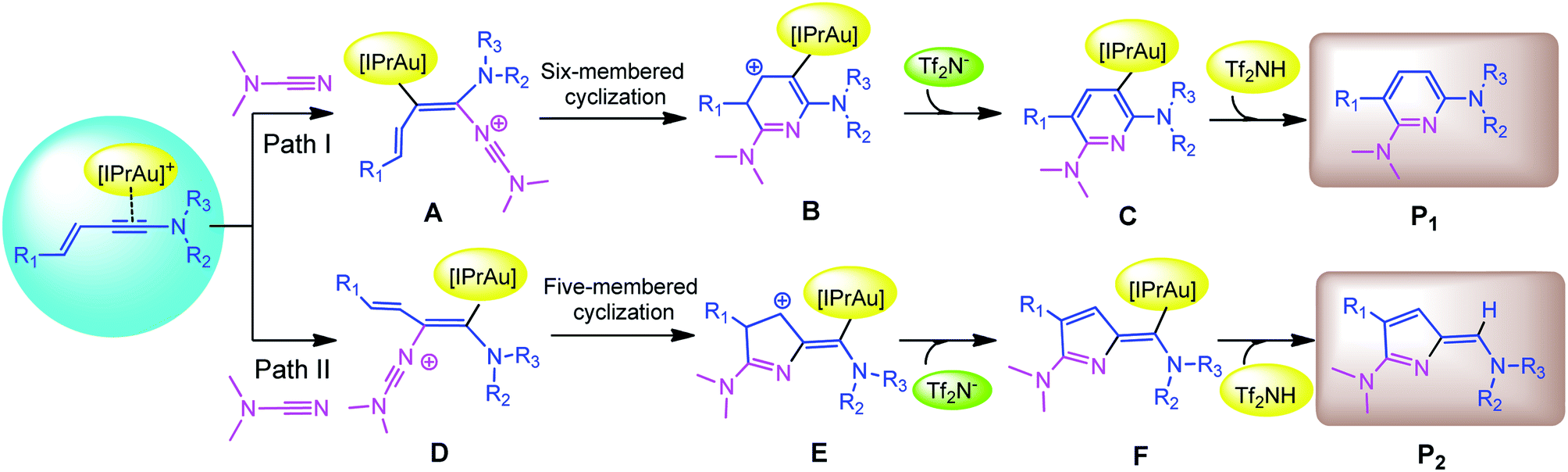
Recently, Kukushkin and Dubovtsev et al. reported that under the action of gold catalyst IPrAuCl/AgNTf2, enynamides and cyanamides can undergo hetero-TDDA cycloaddition reaction in DCM solvent to produce 2,6-diaminopyridine in higher yield (70–99%), in which gold contributed the main catalytic effect in this reaction system (Scheme 1).18 According to the experimental study of Dubovtsev and Kukushkin, a possible synthesis mechanism is given in Scheme 2. For the gold-catalyzed system, the enamide first reacts with the gold complex [IPrAu]+ to form the reaction precursor complex I. Subsequently, cyanamide attacks the amino terminal carbon atom of intermediate I to obtain intermediate II. Then, intermediate II undergoes hetero-TDDA cycloaddition reaction to generate gold pyridine complex III. Finally, the 2,6-diaminopyridine product is obtained through the elimination of the gold intermediate III and the gold catalyst is regenerated. In order to understand the overall reaction mechanism of the hetero-TDDA cycloaddition catalyzed by gold (Scheme 2), and to investigate the different attack modes of cyanamide on the activated triple bond, a set of more detailed steps are proposed as shown in Scheme 3. We conduct an in-depth systematic density functional theory (DFT) calculation study to study the experimentally observed mechanism, selectivity and activity influencing factors.
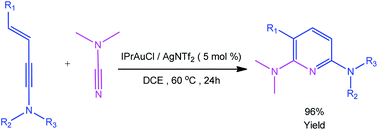 |
| | Scheme 1 Gold-catalyzed cycloadditions of cyanamides and enynamides. | |
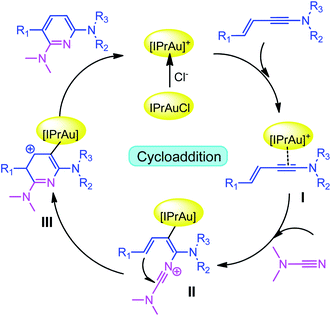 |
| | Scheme 2 Plausible mechanism proposed by Kukushkin and Dubovtsev et al. | |
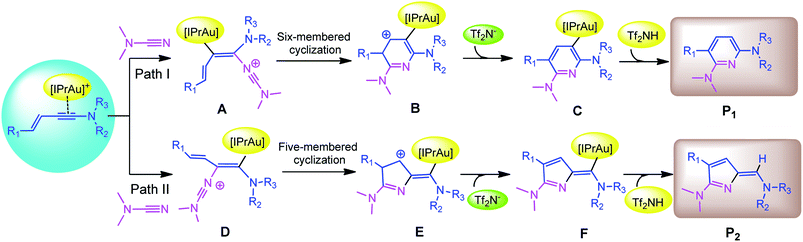 |
| | Scheme 3 Proposed reaction pathways on the basis of Kukushkin and Dubovtsev's study. | |
Therefore, in this work, a computational study is carried out: (1) determine the possible reaction pathways in the title reaction corresponding to the experiment; (2) compare the catalytic pathways and activities in detail by changing the different substituents in the substrate; (3) analyze the gold-catalyzed hetero-TDDA cycloaddition reaction mechanism and regioselectivity. It is believed that the data obtained by the computational research will help to better explain the different yields formed by the gold-catalyzed cycloaddition reaction system, and provide effective theoretical support for the gold-catalyzed hetero-TDDA cycloaddition reaction.
2. Computational details
All quantum calculations are done using Gaussian 09 (ref. 19) and B3LYP20,21 density functional theory (DFT).22,23 All geometric optimizations are performed using B3LYP/6-31G(d,p), SDD theoretical level. In the basis set, except for Au atoms that use effective core potential SDD,24 all main group atoms are 6-31G(d,p) basis set.25 All the minima and transition state structures are verified by the absence of negative eigenvalues or a negative eigenvalue in the vibration frequency analysis and the animation demonstration with negative vector coordinates. In order to obtain more accurate energy and consider the solvation effect, B3LYP-D3(BJ)26,27 is used to calculate the single point energy of the optimized geometry, which is based on B3LYP and includes the empirical D3 dispersion term proposed by Grimme with Becke–Johnson damping. This method is also a relatively recognized and reliable calculation method.28–33 Recently, there have also been some related reports on gold-catalyzed DFT studies.34–36 Moreover, in the recalculation, a larger basis set SDD with extra f37/outer p38 functions for gold, 6-311++G(2d,2p) basis set for other atoms, with the experimental system of dichloroethane (DCE) as the solvent based on (IEFPCM- SMD) solvation model39,40 is used. Intrinsic reaction coordinates (IRC)41–43 are calculated for each transition state to confirm the correct connection of the corresponding structures in the two directions on the reaction path. The geometric configurations in this study are all displayed and explained through the CYLview program.44
3. Result and discussion
Based on the original study by Kukushkin and Dubovtsev et al., we propose several potential pathways for catalytic reaction models (Scheme 3). In order to explore the more active enynamide reactants in this model reaction, the substituents Ms, Me, Ph and thienyl were used as enynamide reactants as several reaction models in the DFT study. The three systems correspond to A, B and C series, respectively.
3.1 Computational results for [IPrAu]+-catalyzed reaction (series A)
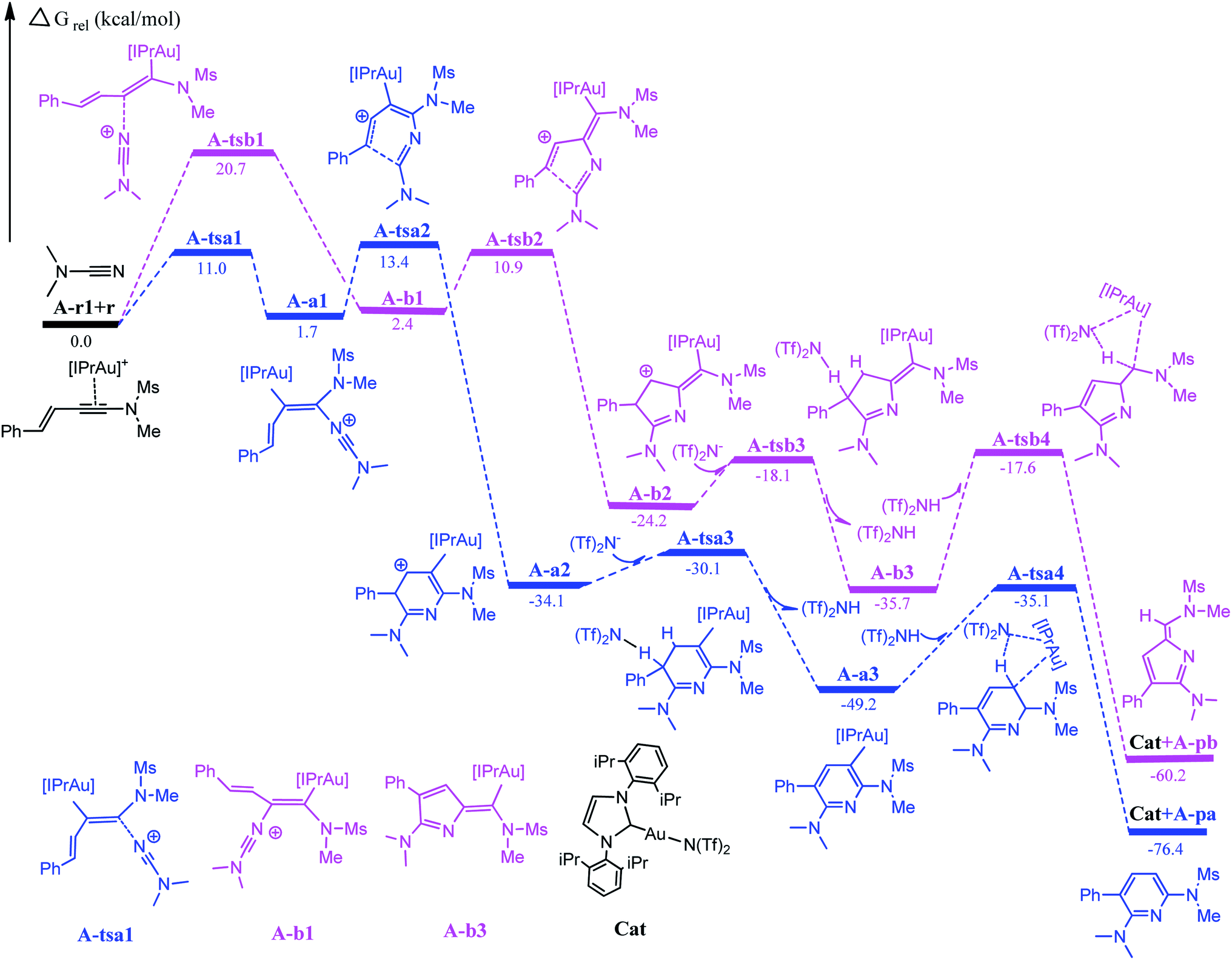
The first step in the series A starts with the coordination of the Au atom with the π bond of the alkyne moiety of enynamide to form a stable intermediate A-r1. The frontier molecular orbital diagram and orbital energy of A-r1 are shown in Fig. 1. The energy difference between the two molecular orbitals of HOMO and LUMO is 4.19 eV, and the coordination of the C–C triple bond with gold enhances the electrophilicity of this C–C bond. The Condensed Fukui functions (f+) of the C1 and C2 atoms of A-r1 are 0.057 and 0.085, respectively, where the C2 atom is 0.028 larger than the C1 atom, indicating that the C2 atom is the more easily attacked active site. Fig. 2 and 3 show the energy profiles of the A series and present the selected structure and key geometric parameters. Further details for all structures are provided in ESI.† From Fig. 3, where the bond lengths of the two Au–C bonds in A-r1 are 2.130 and 2.780 Å, respectively. Structural and electronic features show that C2 atoms are more prone to nucleophilic reactions. Then, the terminal N atom in dimethylcyanamide is nucleophilically attacked to the C2 atom of A-r1 through A-tsa1 to form the intermediate A-a1. The activation free energy of this step is 11.0 kcal mol−1 (Fig. 1 pathway a). Besides, for an intermediate A-a1, free energy of the reaction is 1.7 kcal mol−1 related to reactants A-r1 + r. In A-a1, the C1 and C2 atoms have fully characterized the hybridization mode of sp2. Subsequently, the sp-carbon atom (C4) of cyanamide and the alkenyl C3 atom in A-a1 undergo electrophilic hetero-TDDA cycloaddition to obtain a six-membered ring product. The activation free energy of A-TSa2 is 11.7 kcal mol−1, while the reaction energy of A-a2 is 35.8 kcal mol−1 relative to that of A-a1, and this step is also a strongly exothermic process. The next step is that (Tf)2N− in the solution attacks the hydrogen atom attached to C3 through A-tsa3 to obtain the skeleton geometry of gold-2,6-diaminopyridine compound (A-a3) and release the (Tf)2NH molecule. This step requires a relatively low activation energy of 4.0 kcal mol−1 and releases an energy of 15.1 kcal mol−1. Finally, The gold atom of the intermediate A-3a was attacked by (Tf)2NH through a catalytic reduction process (A-tsa4) to regenerate the catalyst (Cat: [IPrAu]N(Tf)2) and obtain the final product A-pa. The barrier for this step is 14.1 kcal mol−1, and the results show that the entire process is strongly exothermic with an energy of −76.4 kcal mol−1 relative to the reactants.
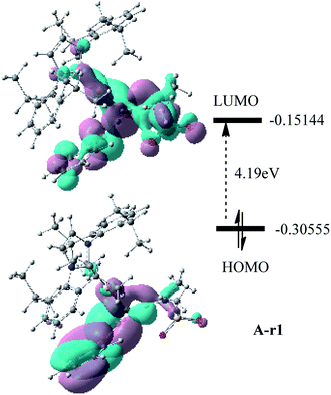 |
| | Fig. 1 Frontier molecular orbitals and orbital energies of A-r1. | |
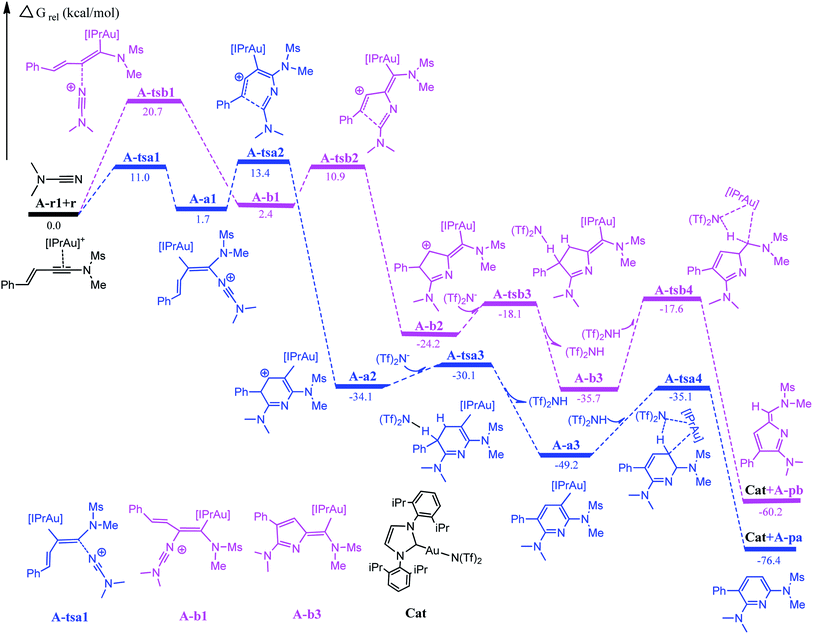 |
| | Fig. 2 Full energy profile for Au-catalyzed cycloaddition of dimethylcyanamide and enynamide A-r1 (R1 = Ph, R2 = Ms, R3 = Me). Relative energies are in kcal mol−1. | |
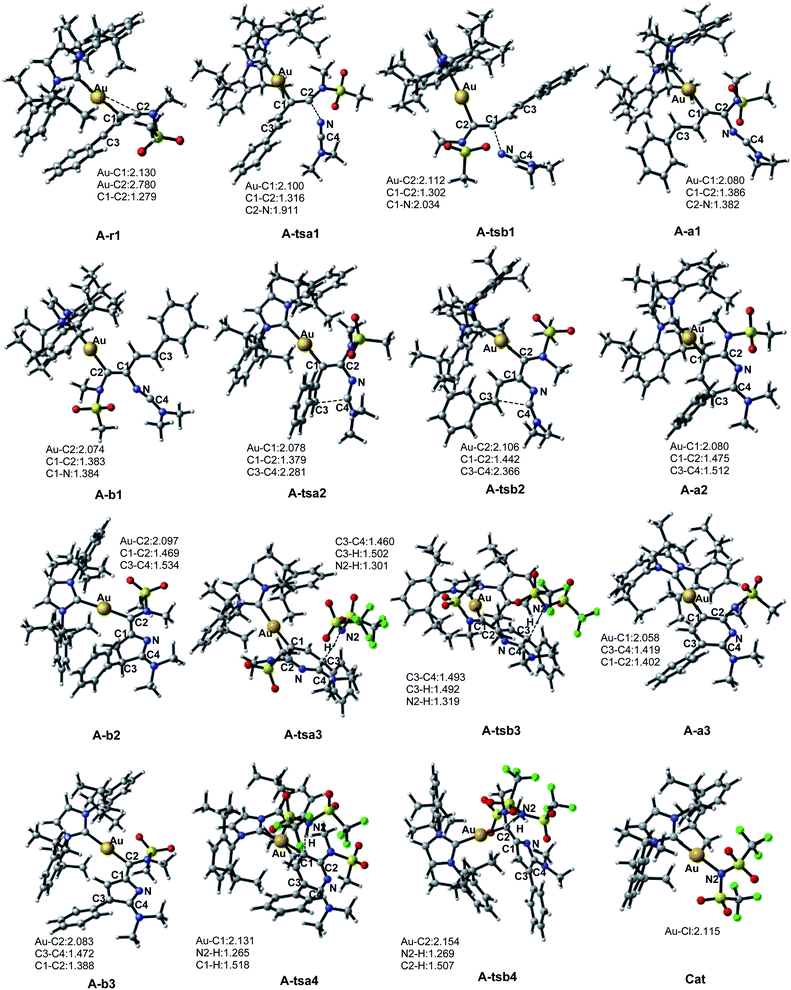 |
| | Fig. 3 Selected key intermediates and transition states for the series A (enynamide A-r1). Representative bond lengths are in Å. | |
Starting with the reactants, if the terminal N atom of dimethylcyanamide is nucleophilic to the C1 atom of A-r1, the reaction pathway b can be formed. Since N attacks another alkynyl carbon atom connected to the styryl group through A-tsb1 to generate the intermediate A-b1, its activation free energy is 20.7 kcal mol−1, while free reaction energy for A-b1 is 0.7 kcal mol−1 with regard to A-a1 (Fig. 1). Compared with A-tsb1, A-tsa1 has a relatively lower barrier indicating that cyanamide attacks the amino carbon atom of the carbon–carbon triple bond in A-r1 more easily than the alkenyl carbon atom. This fact also suggests that amino groups are more conducive to nucleophilic attack than alkenyl groups. For the next intramolecular electrophilic addition reaction, the subsequent step will be to generate the five-membered ring intermediate A-b2 with the free activation energy of 8.5 kcal mol−1. The free energy of reaction of A-b2 is −26.6 kcal mol−1 related to A-b1. The activation energies of the two channels in this step are close to each other, but the exothermic energy of pathway b is 20.9 kcal mol−1 less than that of pathway a. The activation free energy of (Tf)2N− attacking A-b2 to remove (Tf)2NH molecules through A-tsb3 is only 6.1 kcal mol−1, and the energy of generating 2H-pyrrole skeleton A-b3 is 11.5 kcal mol−1 lower than that of A-b2. The activation energy of the last step to generate the product A-pb and release the catalyst is 18.1 kcal mol−1, which is 4.0 kcal mol−1 higher than that of A-tsa4. The free reaction energy of A-pb is −60.2 kcal mol−1 related to the starting reactants.
Compared with pathways a and b, channel a has a lower activation free energy than channel b, indicating that this gold-catalyzed hetero-TDDA cycloaddition is more inclined to generate six-membered pyridines rather than five-membered 2H-pyrroles derivatives, the difference in the activation energy value of the first step reaction between the two is 10.7 kcal mol−1. According to the experimental results, the hetero-TDDA cycloaddition of enamide A-r1 and dimethylcyanamide catalyzed by gold catalyst IPrAuCl can generate 2,6-diaminopyridine derivatives in 81% yield. According to our results, the catalyst [IPrAu]Cl play an important role in obtaining the six-membered ring product in the title reaction. DFT calculations clearly show that in the gold-catalyzed hetero-TDDA cycloaddition, the barrier of path a is lower than that of path b. At the same time, the calculation results show that obtaining the pyridine derivative configuration is a strongly exothermic process. From these data we can conclude that if [IPrAu]Cl is used as the catalyst, this reaction prefers to give the cyclized product of pyridine.
3.2 Reaction for reactant enynamide B-r1 (series B)
Based on series A, the gold-catalyzed reaction in which the methyl substituent on the amino group in A-r1 is replaced by a phenyl group is series B, and the initial reactant is marked as B-r1. For the two different reaction pathways, the energy profiles are given in Fig. S1 (ESI†), while the geometry of the channel a selected for the different species is shown in Fig. S2 (ESI†). Similar to series A, The first is that the terminal N atom of cyanamide attacks two alkynyl carbon atoms in B-r1 in different directions to obtain intermediates B-a1 and B-b1, respectively. The activation free energies required by these two channels for B-tsa1 and B-tsb1 in this step are 13.9 and 23.5 kcal mol−1, respectively, and the energy difference between the two is 9.6 kcal mol−1. Computational results also indicate that the first step is high regioselectivity for the formation of two different five- and six-membered ring products. Then two reaction channels with a reaction trend similar to series a were found. Channel a generates six-membered ring pyridine derivatives with lower activation energy than channel b generates pyrrole derivatives, that is, gold-catalyzed reactions of this type are more active along channel a, tending to obtain pyridine core. The activation free energy for B-tsa2, B-tsa3, B-tsa4 is 11.5, 5.1 and 17.0 kcal mol−1, respectively. The products B-pa and B-pb are both strongly exothermic processes, and free energy values relative to the reactants are −80.2 and −65.4 kcal mol−1, respectively. Compared with series A, the two channels of series B are also strongly exothermic, and their values are increased by 3.8 and 5.2 kcal mol−1 compared with series A, respectively. Moreover, the free energies of B-tsa1 and B-tsb1 were increased by 2.9 and 2.8 kcal mol−1, respectively, compared with A-tsa1 and A-tsb1 of series A, and the reactivity is slightly decreased.
3.3 Reactivity and regioselectivity for enynamide
In order to further explore the influence factors of unsubstituted groups on the reactivity and stereoselectivity, the phenyl group in the reactant A-r1 is substituted with a thienyl group based on the experimental data to obtain the series C(C-r1). The reactant C-r1 first interacts with cyanamide to form two different reaction channels of five-membered ring and six-membered cycloaddition through two different attack modes (Fig. S3, ESI†). The activation free energies for nucleophilic attack and hetero-tetradehydro-Diels–Alder cycloaddition are 15.3 and 11.5 kcal mol−1, respectively, for the six-membered ring main product, while the by-product five-membered ring corresponds to the first-step nucleophilic reaction and the activation free energies of the second-step electrophilic addition were 26.7 and 8.1 kcal mol−1, respectively. The DFT results clearly show that the reaction energy of the two channels in the first step is higher than that of series A and series B, and their energy difference is 11.4 kcal mol−1. The free energies relative to the reactants along channel a and channel b are −76.7 and −60.4 kcal mol−1, respectively, during the entire catalytic reaction process. It can also be clearly seen from Fig. S3† that the pathway for generating pyridine derivatives from channel a is the main reaction channel, which is also consistent with the experimental facts and proves the rationality of the mechanism prediction of Scheme 2. From the structures of the ynamides in the three reaction systems, it can be seen that the reactants with electron-withdrawing N-formyl substituents have good reactivity and good yields. Also, alkyl reactants attached to N are more reactive than aryl reactants. The effect of different substituents on the vinyl group on the reactivity is investigated. Compared with the phenyl group, the thienyl group is not conducive to the reaction of the system, and only 71% of the yield is obtained in the experiment. Computational study found that the reaction series A is one of the most ideal reactants, that is, a phenyl group is attached to the vinyl group of the reactant, and the amine substituents are methylsulfonyl and methyl, respectively.
4. Conclusion
In conclusion, the gold(I)-catalyzed hetero-TDDA cycloaddition reaction mechanism and regioselectivity of cyanamides and enynamides are investigated by DFT calculations. Based on our calculation results, the following conclusions can be obtained: (1) it is easy to see from the calculation results that cyanamide attacks different alkynyl carbon atoms to form two different reaction channels. (2) The main product is to attack the carbon atom connected to the amino group to generate 2,6-diaminopyridine instead of the other direction to obtain five-membered ring pyrrole derivatives. (3) Reactants with electron-withdrawing substituents can further reduce the activation free energy of the reaction and increase its reactivity. (4) This computational study provides a good complement to the experimental report by Shcherbakov et al., which also provides an important reference for mechanistic studies of other gold(I)-catalyzed cycloaddition reaction systems.
Author contributions
Xinghui Zhang: problem selection, writing, and data analysis. Haixiong Shi: simulations, analysis, manuscript first draft. Caihong Ji: result analysis, manuscript editing. Jianyi Wang: methods, project management, result analysis, manuscript editing. Liping Jiang: data analysis, writing.
Conflicts of interest
We declare that there is no conflict of interest including any financial, personal or other relationships with other people or organizations.
Acknowledgements
We gratefully acknowledge the financial support from Natural Science Foundation of Gansu Province (20JR5RA479 of Prof. Xinghui Zhang), the Outstanding Youth Research Program of Lanzhou University of Arts and Sciences (2021SZZX06 of Prof. Xinghui Zhang), National Natural Science Foundation of China (22165017 of Prof. Jianyi Wang). We are grateful to the reviewers for their invaluable suggestions.
References
- S. D. Kuduk, C. Ng, D. M. Feng, J. M. C. Wai, R. S. L. Chang, C. M. Harrell, K. L. Murphy, R. W. Ransom, D. Reiss, M. Ivarsson, G. Mason, S. Boyce, C. Tang, T. Prueksaritanont, R. M. Freidinger, D. J. Pettibone and M. G. Bock, J. Med. Chem., 2004, 47, 6439–6442 CrossRef CAS PubMed.
- A. Gube, H. Komber, K. Sahre, P. Friedel, B. Voit and F. Böhme, J. Org. Chem., 2012, 77, 9620–9627 CrossRef CAS PubMed.
- C. Fehér, M. Papp, Á. Gömöry, L. Nagy, J. Wouters, G. Lendvay and R. S. Földes, Organometallics, 2016, 35, 4023–4032 CrossRef.
- S. Mondal, D. Dinda, B. K. Shaw and S. K. Saha, J. Phys. Chem. C, 2016, 120, 11085–11091 CrossRef CAS.
- I. Mellone, N. Gorgas, F. Bertini, M. Peruzzini, K. Kirchner and L. Gonsalvi, Organometallics, 2016, 35, 3344–3349 CrossRef CAS.
- S. Panja, S. Mondal, S. Ghosh, U. Ghosh and K. Ghosh, ACS Omega, 2020, 5, 13984–13993 CrossRef CAS PubMed.
- X. Xu, L. Lai, J. H. Jiang, Z. Q. He and S. Song, J. Phys. Chem. C, 2019, 123, 9702–9712 CrossRef CAS.
- W. Li, L. Zhou and J. Zhang, Chem.–Eur. J., 2016, 22, 1558–1571 CrossRef CAS PubMed.
- S. R. Dubbaka, M. Kienle, H. Mayr and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 9093–9096 CrossRef CAS PubMed.
- L. G. Xie, S. Niyomchon, A. J. Mota, L. González and N. Maulide, Nat. Commun., 2016, 7, 10914 CrossRef PubMed.
- J. Barluenga, M. Á. Fernández-Rodríguez, P. García-García and E. Aguilar, J. Am. Chem. Soc., 2008, 130, 2764–2765 CrossRef CAS PubMed.
- Q. Zhao, D. F. León Rayo, D. Campeau, M. Daenen and F. Gagosz, Angew. Chem., Int. Ed., 2018, 57, 13603–13607 CrossRef CAS PubMed.
- F. Zamani, R. Babaahmadi, B. F. Yates, M. G. Gardiner, A. Ariafard, S. G. Pyne and C. J. T. Hyland, Angew. Chem., Int. Ed., 2019, 58, 2114–2119 CrossRef CAS PubMed.
- A. Y. Dubovtsev, N. V. Shcherbakov, D. V. Darin and V. Y. Kukushkin, J. Org. Chem., 2020, 85, 745–757 CrossRef CAS PubMed.
- A. Y. Dubovtsev, D. V. Darin and V. Y. Kukushkin, Org. Lett., 2019, 21, 4116–4119 CrossRef CAS PubMed.
- A. Y. Dubovtsev, D. V. Darin, M. Krasavin and V. Y. Kukushkin, Eur. J. Org. Chem., 2019, 2019, 1856–1864 CrossRef CAS.
- Y. C. Hu, Y. Zhao, B. Wan and Q. A. Chen, Chem. Soc. Rev., 2021, 50, 2582 RSC.
- N. V. Shcherbakov, D. V. Dar'in, V. Y. Kukushkin and A. Y. Dubovtsev, J. Org. Chem., 2021, 86, 7218–7228 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda,O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussion 09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- D. Andrae, U. Haussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- L. Goerigkab and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- H. Kruse, L. Goerigk and S. Grimme, J. Org. Chem., 2012, 77, 10824–10834 CrossRef CAS PubMed.
- A. M. Duan, S. S. An, J. D. Xue, X. M. Zheng and Y. Y. Zhao, RSC Adv., 2020, 10, 13442–13450 RSC.
- D. Yu, D. Wu, J. Y. Liu, S. Y. Li and Y. Li, RSC Adv., 2020, 10, 34413–34420 RSC.
- Y. X. Zhu, W. X. Ding, J. F. Hu, J. Xiong and J. Y. Li, RSC Adv., 2022, 12, 10534–10539 RSC.
- H. W. Zhang, W. Z. Li, Y. X. Wang, Y. P. Tao, Y. Wang, F. Yang and Z. Q. Gao, RSC Adv., 2021, 11, 25795–25800 RSC.
- R. B. Alnoman, E. Nabil, S. Parveen, M. Hagar and M. Zakaria, RSC Adv., 2022, 12, 11420–11435 RSC.
- G. X. Zhu, A. You, H. C. Song and Z. G. Li, RSC Adv., 2022, 12, 10014–10019 RSC.
- R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714–11724 CrossRef CAS PubMed.
- Y. Wang, P. J. Cai and Z. X. Yu, J. Am. Chem. Soc., 2020, 142, 2777–2786 CrossRef CAS PubMed.
- K. Lv and X. G. Bao, Org. Chem. Front., 2022, 9, 693–702 RSC.
- M. Couty and M. B. Hall, J. Comput. Chem., 1996, 17, 1359–1370 CrossRef CAS PubMed.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- C. Gonzalex and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- C. Y. Legault, CYLview, 1.0b Université de Sherbrooke, Sherbrooke, Quebec, Canada, 2009, https://www.cylview.org Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Haixiong Shi,
Caihong Ji,
Jianyi Wang and
Liping Jiang
*,
Haixiong Shi,
Caihong Ji,
Jianyi Wang and
Liping Jiang