
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganobase-catalyzed 1,1-diborylation of terminal alkynes under metal-free conditions†
Junchen Lia,
Min Anb,
Zhenhua Gaoa,
Yongbiao Guo a,
Haibo Liua,
Peichao Zhaoa,
Xiaojing Bi*a,
Enxue Shi*a and
Junhua Xiao*a
a,
Haibo Liua,
Peichao Zhaoa,
Xiaojing Bi*a,
Enxue Shi*a and
Junhua Xiao*a
aState Key Laboratory of NBC Protection for Civilian, Beijing, P. R. China. E-mail: xiao.junhua@pku.edu.cn; exshi@sina.com; xiaojingbimail@yeah.net
bCollege of Chemistry and Environmental Engineering, Sichuan University of Science and Engineering, Zigong, P. R. China
First published on 1st June 2022
Abstract
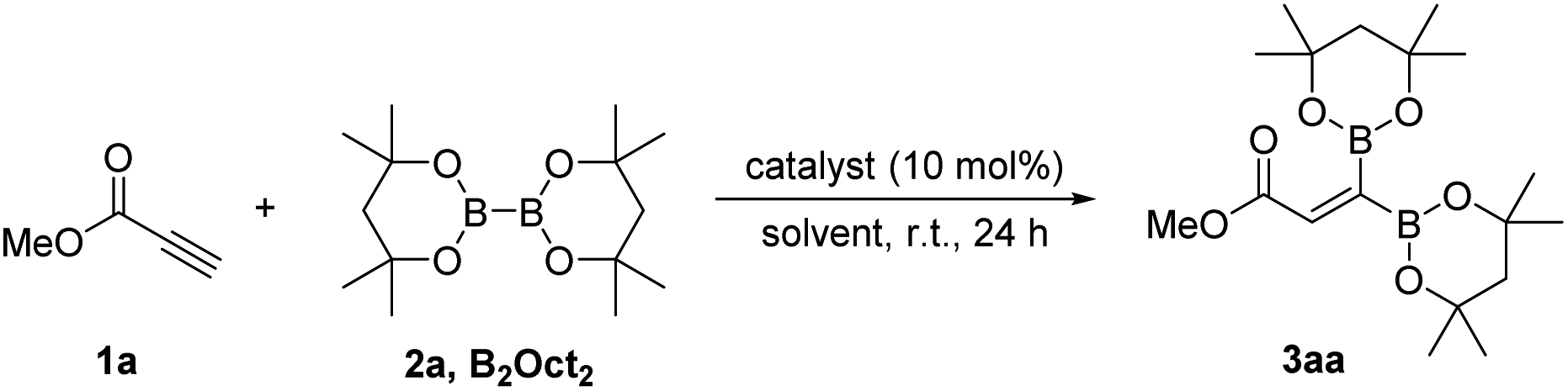
An organobase-catalyzed 1,1-diborylation of terminal alkynes from propargylic derivatives with bis(2,4-dimethylpentane-2,4-glycolato)diboron (B2oct2) is first reported, regioselectively providing 1,1-diborylalkene products with high efficiency. The catalytic pathway is well postulated on the basis of DFT calculations.
Organoboron compounds have attracted much attention as versatile building blocks in the synthesis of carbon–carbon and carbon heteroatom bonds,1 which main features are their stability and ease of handling. Among this important class of reagents, 1,1-diborylalkenes are of particular interest, in part due to that the two geminal boron moieties can be differentiated and transformed via Pd-catalyzed cross-coupling reactions in a progressive manner to provide the bioactive and functional polysubstituted alkene motifs.2,3 Sequent manipulations of 1,1-diborylalkenes with different electrophiles allow for the stereoselective synthesis of challenging unsymmetrically substituted alkenes.4 What's noteworthy, is that the presence of the geminal boron moiety always has a positive effect on the first cross-coupling reaction. Owing to the significance of 1,1-diborylalkenes as general reaction intermediates in organic synthesis, developing efficient and practical methods for their synthesis is highly desirable.
In comparison with previous multi-step routes, 1,1-diborylation of terminal alkynes with diboron reagents represents one of the most step-economical and atom-economical strategies for synthesis of 1,1-diborylalkenes.5 In recent years, several elegant works were continuously discovered.6–9 Sawamura reported a seminal 1,1-diborylation of terminal propargylic derivatives with bis(pinacolato)diboron (B2pin2) catalyzed by a strong inorganic base (LiOtBu) for the preparation of 1,1-diborylalkenes (Scheme 1, eqn (1)).6 A mild 1,1-diborylation of terminal alkynes with B2pin2 catalyzed by a cobalt complex was explored by Chirik and co-workers (Scheme 1, eqn (2)).7 What's more, stereoselective 1,1-diborylation products with two different boron substituents could be realized using an unsymmetrical diboron reagent pinB–Bdan (dan = naphthalene-1,8-diaminato). After that, Ingleson successfully developed a low-coordinate NHC–zinc hydride complex-catalyzed 1,1-diborylation of terminal alkynes using pinacolborane (HBpin) via C–H borylation and hydroboration process (Scheme 1, eqn (3)).8 In 2020, Engle and co-workers explored a copper-catalyzed protocol for the stereodefined 1,1-diborylation of terminal alkynes, involving a sequential dehydrogenative borylation of the alkyne substrate with 1,8-diaminonaphthalatoborane (HBdan), followed by hydroboration with HBpin (Scheme 1, eqn (4)).9 Although having achieved such progress, relatively harsh reaction conditions, expensive ligand or preformed catalyst were always involved. Therefore, development of user-friendly and environmental benign protocols for preparation of 1,1-diborylalkenes is still highly desirable to meet the requirements of sustainable development in the field of scientific research and industrial production. In this context, we report on an environmental benign and metal-free protocol for 1,1-diborylation of terminal alkynes with bis(2,4-dimethylpentane-2,4-glycolato)diboron (B2oct2) under the catalysis of organobase 2-tert-Butyl-1,1,3,3-tetramethylguanidine (BTMG) at room temperature.
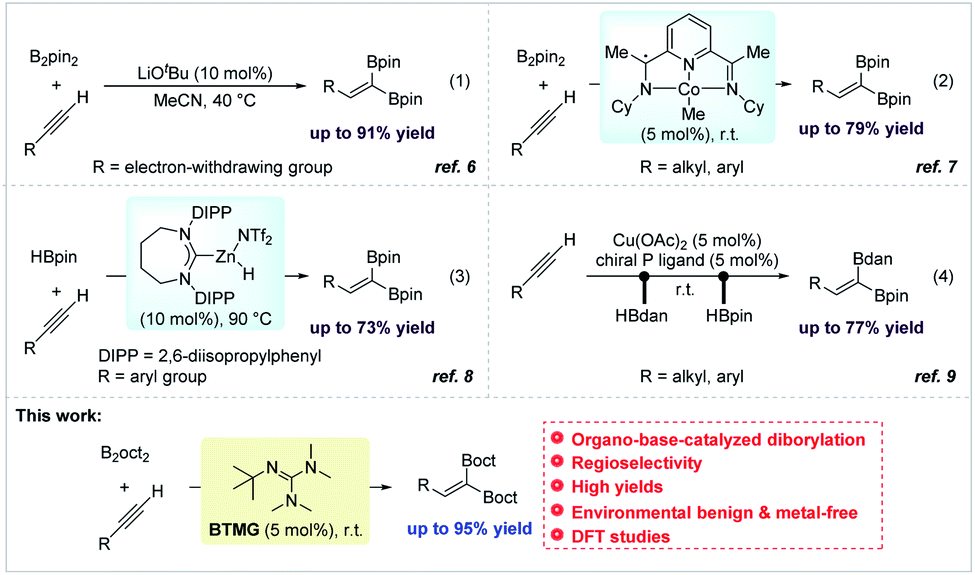 | ||
| Scheme 1 Previous work on 1,1-diborylation and our work. | ||
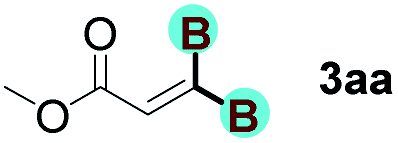
Inspired by Sawamura's strong inorganic base catalyzed 1,1-diborylation strategy6 and the rapid development of organocatalysis in recent years,10 we focused on the role of organic base as catalyst in this type of reaction. At the outset, a commercially strong organic base 1,1,3,3-tetramethylguanidine (TMG) was selected as the catalyst to verify its catalytic reactivity in the reaction between methyl propiolate 1a and B2oct2 2a. The effect of solvent was initially evaluated and the results indicated that hexane, dichloromethane, 1,2-dichloroethane and toluene all failed to afford the desired gem-diborylated product (Table 1, entries 1–4). To our delight, when CCl4 was used as the solvent, 17% yield of product 3aa was obtained (entry 5). Whereas, further solvent screening experiments with THF, Et2O, and EtOAc gave only inferior results (entries 6–8). A slightly higher yield of 35% was obtained utilizing DMF as the solvent (entry 9). Gratifyingly, the yield of 3aa was dramatically increased to 79% when MeCN was employed as the solvent (entry 10). Then, organobase catalyst screening was performed (entries 11–19), among which organobase like Et3N, BTMG and TMEDA all proved being effective catalysts, producing 3aa in yields of 95%, 98% and 77%, respectively (entries 11–13). However, other organobases like TBD, DBU, pyridine and quinine all showed nearly no catalytic activity (entries 14–17). DMAP and 2,6-lutidine were not the proper catalyst because the yields were less than 20% (entries 18–19). Several kinds of inorganic base were also tested to catalyze our reaction, carbonates (K2CO3, Na2CO3, and Li2CO3) all led to the failure of the reaction (entries 20–22), nevertheless, Cs2CO3 and NaOH showed excellent performance with formation of product 3aa in 95% yield (entries 23–24). The strong inorganic base (LiOtBu), which was employed in Sawamura's work,6 exhibited no catalytic activity (entry 25) and the high catalytic activity of Et3N and Cs2CO3 indicated that the alkalinity of the base may be not the critical factor for this reaction (entries 11 and 23). When the transformation was conducted in the absence of catalyst, no product was observed (entry 26). It is noteworthy that further lowering the loading of BTMG to 5 mol%, a still high yield of 96% was obtained (entry 27). However, when the loading of other bases (Et3N, Cs2CO3 and NaOH) was also lowered to 5 mol%, an obvious reduction of yields was observed (entries 28–30). Therefore, the catalytic system consisting of 5 mol% BTMG as the catalyst in MeCN performed well as the optimal conditions.
|
|||
|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Yieldb (%) |
| a Reaction conditions: methyl propiolate 1a (0.1 mmol), B2oct2 2a (0.1 mmol), catalyst (10 mol%), solvent (2 mL), rt., 24 h.b Yields determined by GC-MS.c Isolated yields in parenthesis.d 5 mol% of catalyst was employed. | |||
| 1 | Hexane | TMG | 0 |
| 2 | DCM | TMG | 0 |
| 3 | DCE | TMG | 0 |
| 4 | Toluene | TMG | 0 |
| 5 | CCl4 | TMG | 17 |
| 6 | THF | TMG | 7 |
| 7 | Et2O | TMG | 5 |
| 8 | EtOAc | TMG | 4 |
| 9 | DMF | TMG | 35 |
| 10 | MeCN | TMG | 79 |
| 11 | MeCN | Et3N | 95 |
| 12 | MeCN | BTMG | 98 (95)c |
| 13 | MeCN | TMEDA | 77 |
| 14 | MeCN | TBD | 0 |
| 15 | MeCN | DBU | 0 |
| 16 | MeCN | Pyridine | 0 |
| 17 | MeCN | Quinine | 0 |
| 18 | MeCN | DMAP | 12 |
| 19 | MeCN | 2,6-Lutidine | 19 |
| 20 | MeCN | Li2CO3 | 0 |
| 21 | MeCN | Na2CO3 | 0 |
| 22 | MeCN | K2CO3 | 0 |
| 23 | MeCN | Cs2CO3 | 95 |
| 24 | MeCN | NaOH | 95 |
| 25 | MeCN | LiOtBu | 0 |
| 26 | MeCN | — | 0 |
| 27d | MeCN | BTMG | 96 (94)c |
| 28d | MeCN | Et3N | 65 |
| 29d | MeCN | Cs2CO3 | 21 |
| 30d | MeCN | NaOH | 86 |
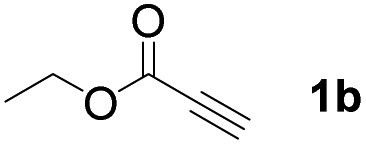
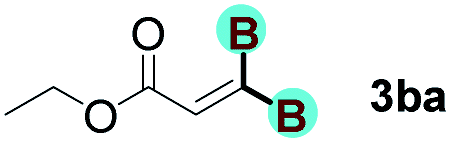
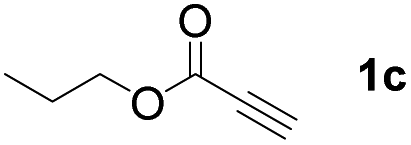
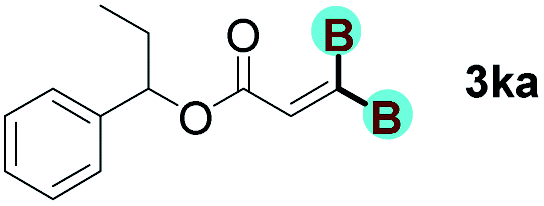
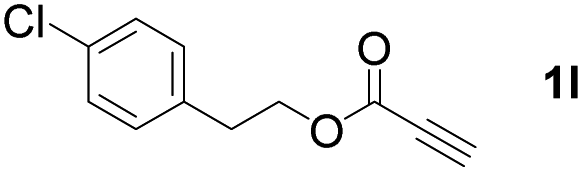
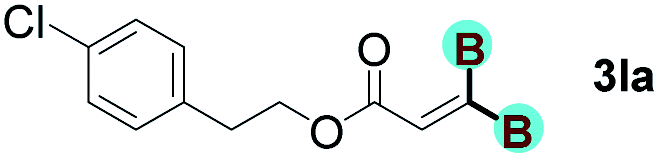
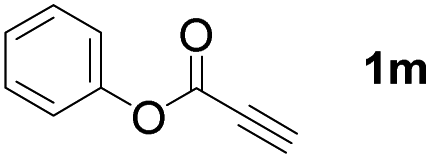
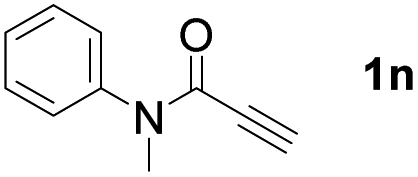
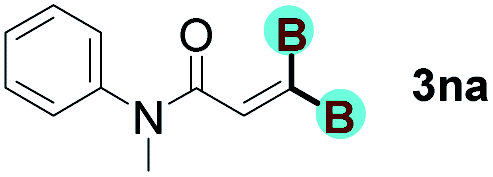
With optimized conditions in hand, we next sought to investigate the scope of this regioselective 1,1-diborylation with respect to the alkyne component (Table 2). A broad array of propargylic derivatives can effectively serve as coupling partners. Linear alkyl ester group of propargyl acid such as methyl, ethyl, n-propyl, n-butyl and n-pentyl were well-tolerated to furnish 1,1-diborylated products in excellent yield (entries 1–5, 88–94% yield). Steric bulk proximal to the ester functionality is compatible, as exemplified by the presence of isopropyl, isobutyl, tert-butyl and isopentyl substituents (entries 6–9, 86–91 yield%). We found that benzyl and 1-phenylpropyl propiolate also participated readily to give the corresponding products in a yield of 92% and 78%, respectively (entries 10–11). Moreover, 4-chlorophenethyl propiolate was found to be a competent substrate with the chloro-group remaining unreacted in this transformation (entry 12, 93% yield). Phenyl propionate reacted well to form the desired diborylated product 3ma in 78% yield (entry 13). Notably, N-methyl-N-phenylpropiolamide also showed good reactivity towards such diborylation reaction, which provided the desired product 3na in 53% yield (entry 14).
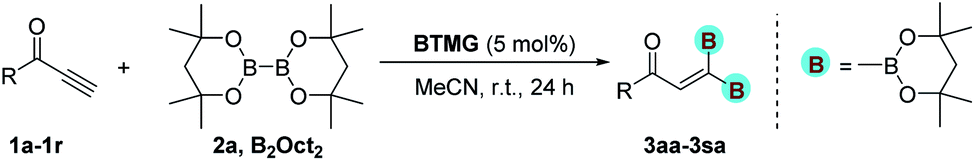
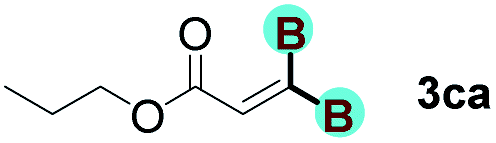
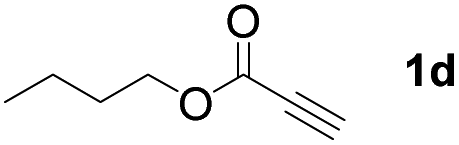
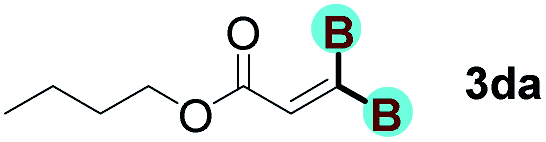
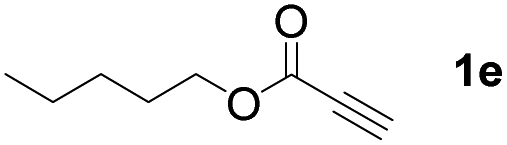
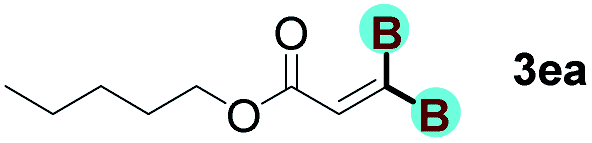
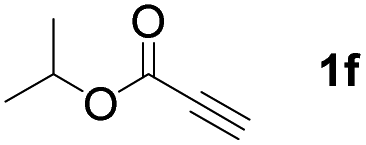
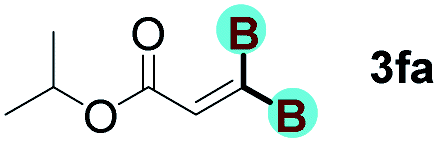
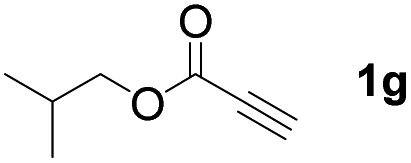
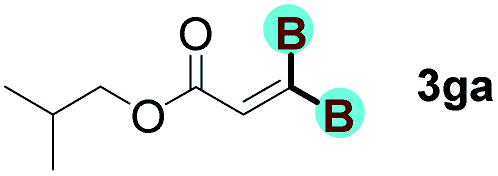
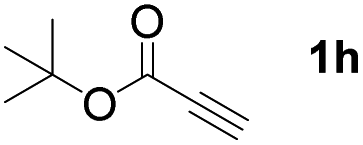
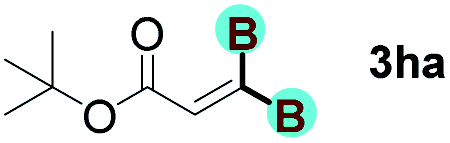
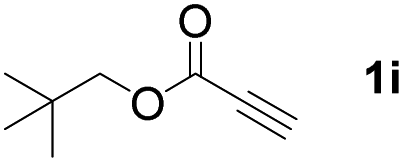
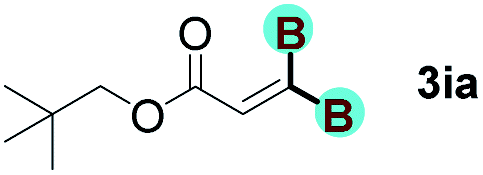
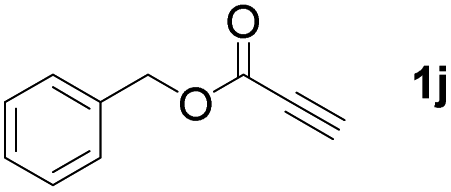
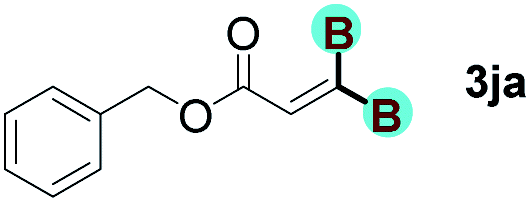
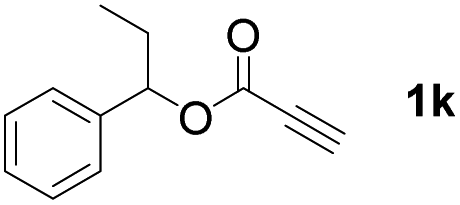
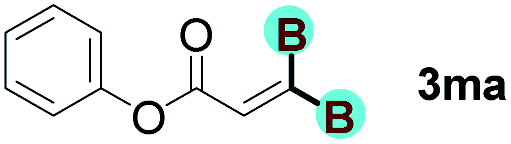
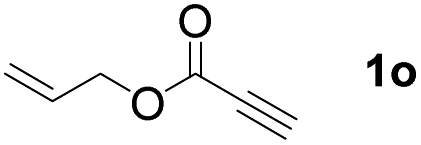
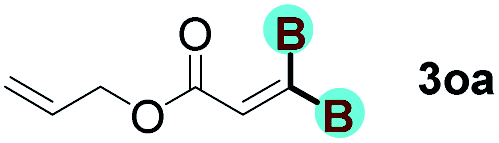
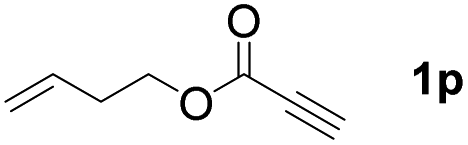
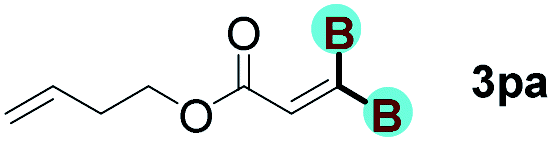
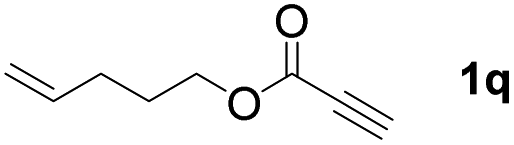
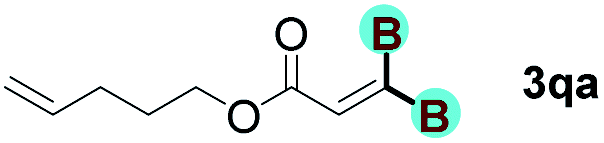
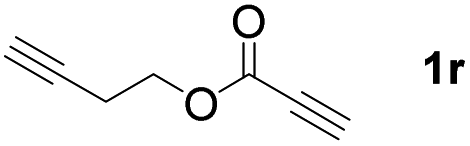
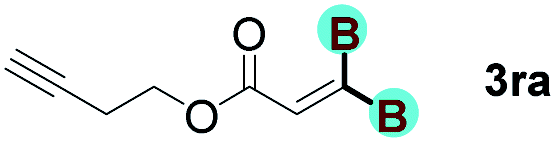
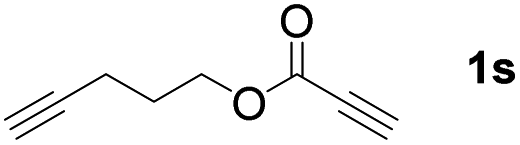
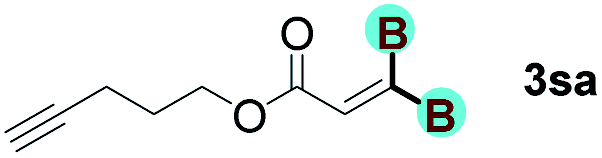
The diborylation of propiolate esters bearing a terminal alkenyl group all proceeded well to give the diborylated products and kept the alkenyl group untouched (entries 15–17, 89–93% yield). It should be mentioned that diyne substrates demonstrate exceptional chemoselectivity and undergoes 1,1-diborylation exclusively at the site of propiolic acid (entries 18–19, 82–88% yield). Unfortunately, attempts to run the diborylation reaction with other types of alkynes resulted in frustrating results (for more details, please see ESI†).
We also examined the gem-diborylation reaction of methyl propiolate 1a with other types of diboron reagents (Scheme 2). The 1,1-diborylation reaction proceeded well when B2pin2 (2b) was used as the diborylating reagent, affording the title diborylated product 3ab in 80% isolated yield. It should be noted that a comparable yield of 95% was achieved when another six-membered partner B2hex2 (2f) was employed. However, other diboron compounds, such as Bis[(−)-pinanediolato]diboron (2c), B2cat2 (2d), pinBBdan (2e), B2(NMe2)4 (2g), and B2(OH)2 (2h) proved to be ineffective under this 1,1-diborylation reaction.
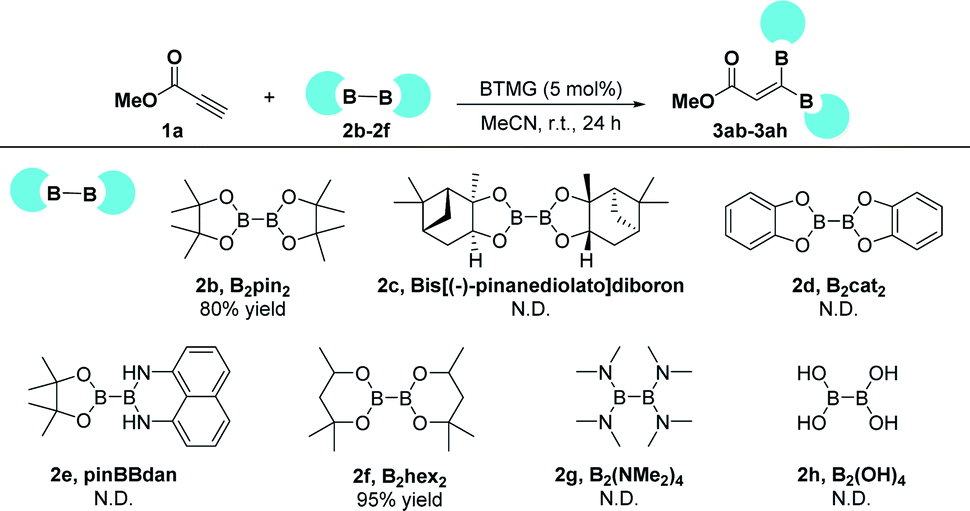 | ||
| Scheme 2 1.1-Diborylation reaction using other diboron reagents. Reaction conditions: methyl propiolate (0.1 mmol), diboron reagent (0.1 mmol), BTMG (5 mol%), MeCN (2 mL), rt., 24 h. N.D. = not detected. | ||
To demonstrate the practicability and scalability of our methodology, the gram-scale 1,1-diborylation of methyl propiolate 1a with B2oct2 was carried out on 12 mmol to produce 3aa in 90% yield (3.95 g) (Scheme 3).
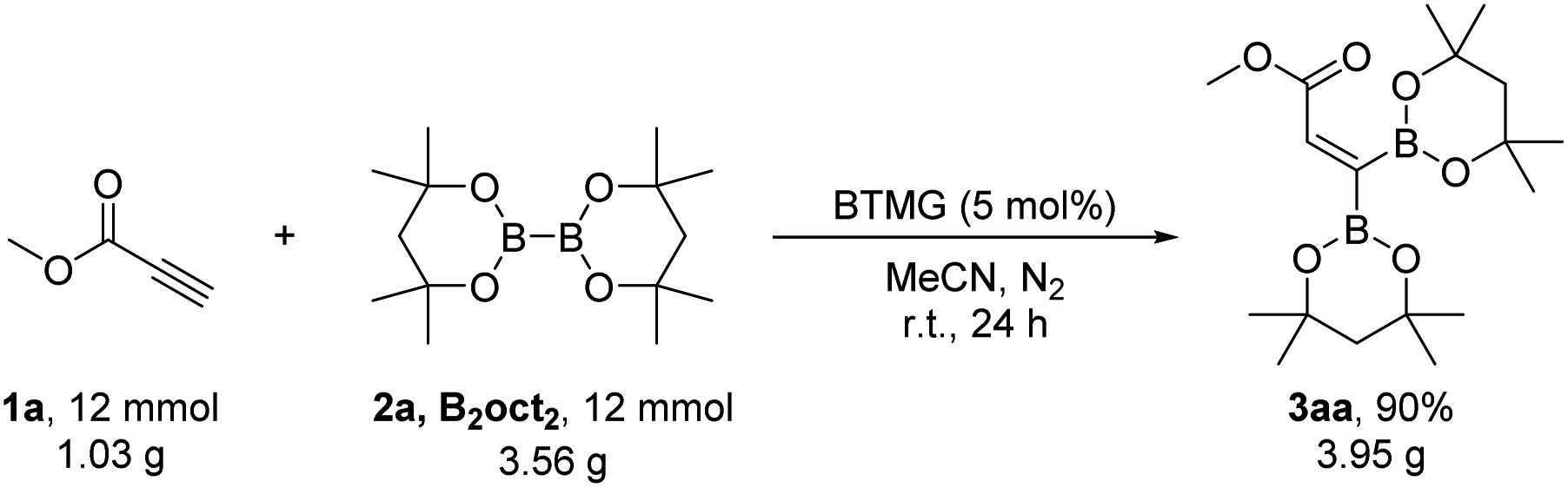 | ||
| Scheme 3 Scale up synthesis. | ||
To better understand the reaction pathway for organobase-catalyzed 1,1-diborylation of terminal alkynes, DFT studies are performed using methyl propiolate 1a and B2oct2 2a as the model substrate and BTMG as the organobase. The calculated energy profile is shown in Fig. 1. The initial step is a concerted three-component proton transfer/borylation process (TS1), the base (BTMG) helps to abstract a proton from the terminal alkyne 1a to give alkynyl anion I and protonated base. At the same time, the negatively charged terminal carbon of the alkyne could attack the boron center of the diboron species, leading to a two-component intermediate II. Then the negatively charged diboron species would undergo a 1,2-boryl migration process (TS2) to generate a 1,1-diborylallene species III. Finally, another proton transfer (TS3) from protonated base generated in the previous step to the negative-charged oxygen atom of the allene species II to give an allene hemiacetal IV, which then tautomerizes to the 1,1-diborylated product 3aa.
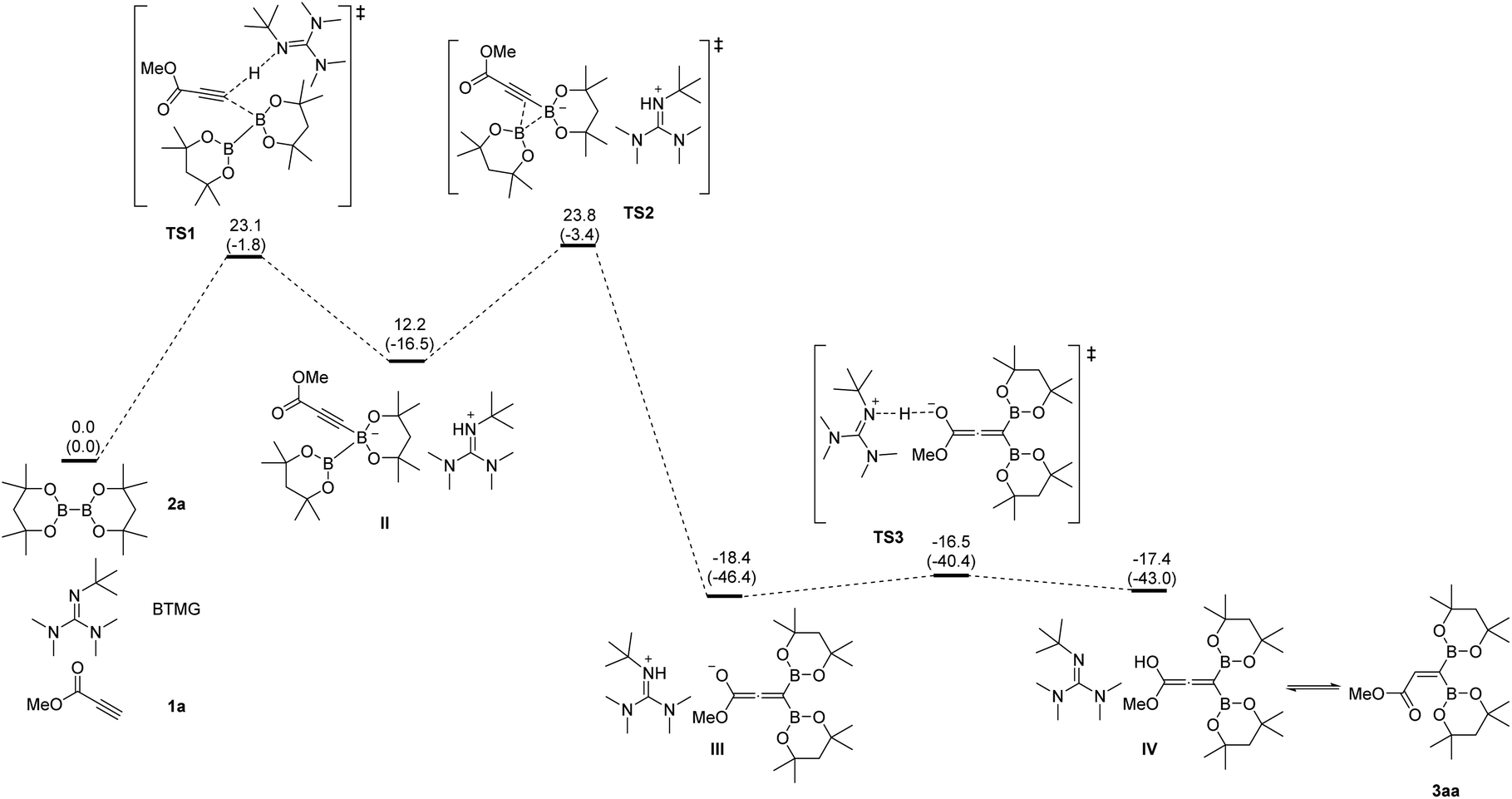 | ||
| Fig. 1 Energy profile calculated for the organobase-catalyzed 1,1-diborylation of methyl propiolate. Relative free energies and electronic energies (in parentheses) are given in kcal mol−1. | ||
Based on the results of previous work6 and our DFT studies, a plausible reaction pathway was postulated as in Scheme 4. Methyl propiolate 1a is firstly deprotonated in the presence of an organobase to give an alkynyl anion I, which then nucleophilically attacks the diboron reagent 2a to form an alkynyl diboron complex II. Complex II occurs a 1,2-boryl migration process to produce a gem-diborylallene intermediate III, followed by a protonation to afford the 1,1-diborylalkene product 3aa with releasing the organobase catalyst to complete the catalytic cycle.
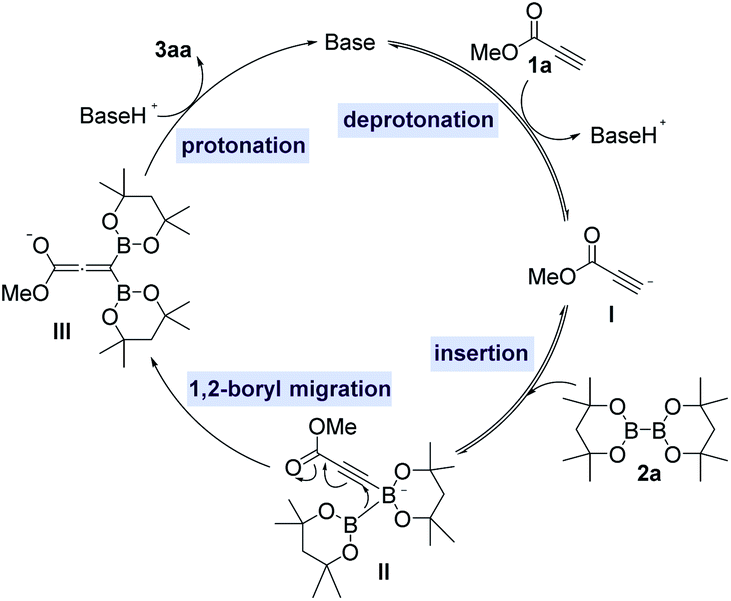 | ||
| Scheme 4 Proposed catalytic mechanism. | ||
Conclusions
In conclusion, an efficient and practical methodology for 1,1-diborylation of terminal alkynes were well developed by the catalysis of a commercially available organobase under mild conditions. Employing B2oct2 as the diborylating reagent, a series of propargylic derivatives was confirmed to be of high efficiency towards this regioselective 1,1-diborylation reaction. DFT calculations were introduced to well demonstrate the catalytic mechanism. This work provides an alternative approach for the preparation of synthetically important 1,1-diborylalkenes.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Synthesis and Application of Organoboron Compounds, ed. E. Fernandez and A. Whiting, Springer, Berlin, 2015 Search PubMed; (b) Boronic Acids, ed. D. G. Hall, Wiley-VCH, Weinheim, 2011 Search PubMed; (c) N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (d) A. J. J. Lennox and G. C. Lloyd-Jones, Selection of Boron Reagents for Suzuki–Miyaura Coupling, Chem. Soc. Rev., 2014, 43, 412–443 RSC; (e) S. J. Geier and S. A. Westcott, Dehydrogenative Borylation: The Dark Horse in Metal-Catalyzed Hydroborations and Diborations?, Rev. Inorg. Chem., 2015, 35, 69–79 CAS.
- For transformations of 1,1-diborylalkenes, please see: (a) M. Shimizu, C. Nakamaki, K. Shimono, M. Schelper, T. Kurahashi and T. Hiyama, Stereoselective Cross-Coupling Reaction of 1,1-Diboryl-1-alkenes with Electrophiles: A Highly Stereocontrolled Approach to 1,1,2-Triaryl-1-alkenes, J. Am. Chem. Soc., 2005, 127, 12506–12507 CrossRef CAS PubMed; (b) M. Shimizu, K. Shimono, M. Schelper and T. Hiyama, Stereocontrolled Approach to 1,3,4,6-Tetraarylated 1,3,5-Hexatrienes, Synlett, 2007, 2007, 1969–1971 CrossRef; (c) X. Feng, H. Jeon and J. Yun, Regio- and Enantioselective Copper(I)-Catalyzed Hydroboration of Borylalkenes: Asymmetric Synthesis of 1,1-Diborylalkanes, Angew. Chem., Int. Ed., 2013, 52, 3989–3992 CrossRef CAS PubMed; (d) N. Kumar, N. Eghbarieh, T. Stein, A. I. Shames and A. Masarwa, Photoredox-Mediated Reaction of gem-Diborylalkenes: Reactivity Toward Diverse 1,1-Bisborylalkanes, Chem. - Eur. J., 2020, 26, 5360–5364 CrossRef CAS PubMed; (e) I. Beletskaya and C. Moberg, Element–Element Additions to Unsaturated Carbon–Carbon Bonds Catalyzed by Transition Metal Complexes, Chem. Rev., 2006, 106, 2320–2354 CrossRef CAS PubMed.
- For applications of polysubstituted alkenes, please see: (a) V. C. Jordan, Antiestrogens and Selective Estrogen Receptor Modulators as Multifunctional Medicines. 1. Receptor Interactions, J. Med. Chem., 2003, 46, 883–908 CrossRef CAS PubMed; (b) S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, Simple, Efficient, and Modular Syntheses of Polyene Natural Products via Iterative Cross-Coupling, J. Am. Chem. Soc., 2008, 130, 466–468 CrossRef CAS PubMed; (c) G. Petruncio, Z. Shellnutt, S. Elahi-Mohassel, S. Alishetty and M. Paige, Skipped Dienes in Natural Product Synthesis, Nat. Prod. Rep., 2021, 38, 2187–2213 RSC; (d) R. G. S. Berlinck, C. M. Crnkovic, J. R. Gubiani, D. I. Bernardi, L. P. Iócaa and J. I. Quintana-Bullaa, The Isolation of Water-soluble Natural Products – Challenges, Strategies and Perspectives, Nat. Prod. Rep., 2022, 39, 596–669 RSC; (e) C.-L. Li, S.-J. Shieh, S.-C. Lin and R.-S. Liu, Synthesis and Spectroscopic Properties of Finite Ph2N-Containing Oligo(arylenevinylene) Derivatives That Emit Blue to Red Fluorescence, Org. Lett., 2003, 5, 1131–1134 CrossRef CAS PubMed.
- H. Wen, L. Zhang, S. Zhu, G. Liu and Z. Huang, Stereoselective Synthesis of Trisubstituted Alkenes via Cobalt-Catalyzed Double Dehydrogenative Borylations of 1-Alkenes, ACS Catal., 2017, 7, 6419–6425 CrossRef CAS.
- (a) N. Xu, Y. Hu and C. Liu, Recent Advances on Catalytic Synthesis and Application of 1,1-Diborylalkenes, J. Mol. Catal., 2020, 34, 87–96 CAS; (b) X. Wang, Y. Wang, W. Huang, C. Xia and L. Wu, Direct Synthesis of Multi(boronate) Esters from Alkenes and Alkynes via Hydroboration and Boration Reactions, ACS Catal., 2021, 11, 1–18 CrossRef CAS; (c) J. Royes, A. B. Cuenca and E. Fernández, Access to 1,1-Diborylalkenes and Concomitant Stereoselective Reactivity, Eur. J. Org. Chem., 2018, 2018, 2728–2739 CrossRef CAS.
- A. Morinaga, K. Nagao, H. Ohmiya and M. Sawamura, Synthesis of 1,1-Diborylalkenes through a Brønsted Base Catalyzed Reaction between Terminal Alkynes and Bis(pinacolato)diboron, Angew. Chem., Int. Ed., 2015, 54, 15859–15862 CrossRef CAS PubMed.
- S. Krautwald, M. J. Bezdek and P. J. Chirik, Cobalt-Catalyzed 1,1-Diboration of Terminal Alkynes: Scope, Mechanism, and Synthetic Applications, J. Am. Chem. Soc., 2017, 139, 3868–3875 CrossRef CAS PubMed.
- R. J. Procter, M. Uzelac, J. Cid, P. J. Rushworth and M. J. Ingleson, Low-Coordinate NHC–Zinc Hydride Complexes Catalyze Alkyne C–H Borylation and Hydroboration Using Pinacolborane, ACS Catal., 2019, 9, 5760–5771 CrossRef CAS.
- Y. Gao, Z.-Q. Wu and K. M. Engle, Synthesis of Stereodefined 1,1-Diborylalkenes via Copper-Catalyzed Diboration of Terminal Alkynes, Org. Lett., 2020, 22, 5235–5239 CrossRef CAS PubMed.
- (a) S. Bertelsen and K. A. Jørgensen, Organocatalysis—after the Gold Rush, Chem. Soc. Rev., 2009, 38, 2178–2189 RSC; (b) B. List, Introduction: Organocatalysis, Chem. Rev., 2007, 107, 5413–5415 CrossRef CAS; (c) W. Cao, X. Liu and X. Feng, Chiral Organobases: Properties and Applications in Asymmetric Catalysis, Chin. Chem. Lett., 2018, 29, 1201–1208 CrossRef CAS; (d) V. A. Larionov, B. L. Feringa and Y. N. Belokon, Enantioselective “Organocatalysis in Disguise” by the Ligand Sphere of Chiral Metal-templated Complexes, Chem. Soc. Rev., 2021, 50, 9715–9740 RSC; (e) B. Han, X.-H. He, Y.-Q. Liu, G. He, C. Peng and J.-L. Li, Asymmetric Organocatalysis: an Enabling Technology for Medicinal Chemistry, Chem. Soc. Rev., 2021, 50, 1522–1586 RSC; (f) X. Xiao, C. Guan, J. Xu, W. Fu and L. Yu, Selenium-catalyzed Selective Reactions of Carbonyl Derivatives: State-of-the-art and Future Challenges, Green Chem., 2021, 23, 4647–4655 RSC; (g) X. Xiao, Z. Shao and L. Yu, A Perspective of the Engineering Applications of Carbon-based Selenium-containing Materials, Chin. Chem. Lett., 2021, 32, 2933–2938 CrossRef CAS; (h) H. Cao, R. Qian and L. Yu, Selenium-catalyzed Oxidation of Alkenes: an Insight into the Mechanisms and Developing Trend, Catal. Sci. Technol., 2020, 10, 3113–3121 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02638d |
| This journal is © The Royal Society of Chemistry 2022 |