
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microfluidic electrochemical cell for studying the corrosion of uranium dioxide (UO2)†
Jennifer Yao ,
Nabajit Lahiri,
Shalini Tripathi,
Shawn L. Riechers,
Eugene S. Ilton,
Sayandev Chatterjee and
Edgar C. Buck*
,
Nabajit Lahiri,
Shalini Tripathi,
Shawn L. Riechers,
Eugene S. Ilton,
Sayandev Chatterjee and
Edgar C. Buck*
Pacific Northwest National Laboratory, 902 Battelle Blvd. P.O. Box 999, Richland, WA 99354, USA. E-mail: Edgar.Buck@pnnl.gov
First published on 4th July 2022
Abstract
We have developed a specialized microfluidic electrochemical cell that enables in situ investigation of the electrochemical corrosion of microgram quantities of redox active solids. The advantage of downscaling is the reduction of hazards, waste, expense, and greatly expanding data collection for hazardous materials, including radioactive samples. Cyclic voltammetry was used to monitor the oxidation–reduction cycle of minute quantities of micron-size uraninite (UO2) particles, from the formation of hexavalent uranium (U(VI)), U3O7 and reduction to UO2+x. Reaction progress was also studied in situ with scanning electron microscopy. The electrochemical measurements matched those obtained at the bulk-scale and were consistent with ex situ characterization of the run products by X-ray photoelectron spectroscopy, scanning transmission electron microscopy, and atomic force microscopy; thus, demonstrating the utility of the microfluidic approach for studying radioactive materials.
1 Introduction
The release of most radionuclides from spent nuclear fuel (SNF) in a geologic repository will be controlled by the dissolution rate of the uranium oxide (UO2) fuel matrix.1–3 Understanding the dissolution rate of UO2 is therefore of prime importance for prediction of waste form performance. Repository conditions, however, are projected to be complicated, whereas expected bulk anoxic conditions should strongly suppress the aqueous solubility of UO2,4,5 the self-generated radiolytic field from SNF can result in localized oxidizing conditions at the fuel–water interface. This research is aiming to address the issues and challenges raised in the Spent Fuel and Waste Science and Technology (SFWST) road map.6Shoesmith7 has shown the value of measuring the electrochemical corrosion potential of 238UO2. Indeed, the UO2 corrosion rate can be measured by electrochemical means even under reducing conditions where UO2 is only sparingly soluble. As radiolytic oxidants will lead to SNF dissolution and radionuclide release, there is great interest in investigating processes that slow this effect. Goldik and co-workers using bulk electrochemical techniques have shown how the formation of corroded surfaces blocked further reaction.8,9 Broczkowski et al.,5 again using electrochemical methods, examined the role of noble metal particles (NMP) and dissolved H2 with simulated SNF in an effort to simulate the role of H2 generation from anoxic corrosion of iron and the impact of NMP. The corrosion potential was found to be suppressed in the presence of H2 both with and without NMP present. The role of NMP as well as possible catalytic actions at the UO2 surface are an area of continued research.10 Nevertheless, electrochemical testing has proven to be an effective methods for investigating these systems.
Experiments with bulk amounts of SNF can be difficult owing to the need to use shielded hot cell facilities to protect against the intense β/γ radiation field.11 If the amount of SNF in a single test could be reduced several orders of magnitude, it might be possible to run experiments and other analytical characterization outside of a shielded facility. This would vastly reduce the overall difficulty of running experiments. To facilitate corrosion studies at microscale, we designed a microfluidic electrochemical cell, PAMEC (particle-attached microfluidic electrochemical cell), which was inspired by the System for Analysis at the Liquid Vacuum Interface (SALVI) microfluidic device that was developed at PNNL by Yu and co-workers.12–15 PAMEC is a three-electrode microfluidic electrochemical cell (E-cell) that has been modified by switching to a thinner but non-perforated Si3N4 widow that allows in situ scanning electron microscopy (SEM)/energy dispersive spectroscopy (EDS) analysis and maintenance of anoxic conditions as needed. PAMEC uses composite electrode made of particle materials as the working electrode. In this study, we applied UO2 powder to make the composite electrode in PAMEC device that enables in situ observation of the interfacial chemistry and morphological changes between UO2 electrode and electrolyte at microscale. Fig. 1 shows the experimental setup of the PAMEC, in conjunction with the employment of multiple characterization techniques including in situ SEM/EDS, X-ray photoelectron spectroscopy (XPS), scanning transmission electron microscopy (STEM), atomic force microscopy (AFM), and an electrochemical workstation.
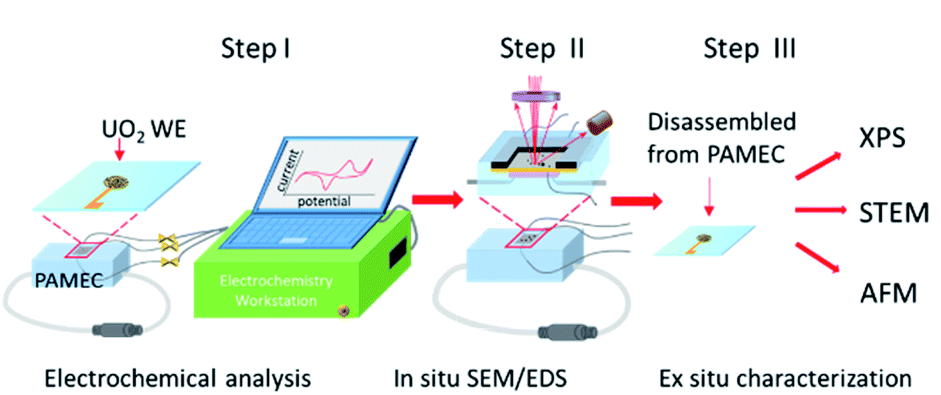 | ||
| Fig. 1 Schematic of the PAMEC and sequence of characterization steps. Step I: Highlights of the electrochemical analysis of the UO2 particle composite electrode. Step II: In situ SEM/EDS imaging and spectral analysis of the UO2 electrode. Step III: Ex situ characterization of the UO2 WE for oxidation state verification, surface oxidation imaging at atomic scale, and surface topography measurement. | ||
In this contribution, we validate the utility of the PAMEC for studying the corrosion of micrograms of materials, using UO2 particles as a case study. The metric for success is whether corrosion measurements are consistent with prior bulk studies.
2 Experimental section
2.1 Microfluidic electrochemical cell fabrication
The PAMEC is a three-electrode microfluidic E-cell, consisting of UO2 particles as the working electrode (WE), platinum wires as the reference electrode (RE) and counter electrodes (CE). The Si3N4 membrane (Norcada) was first coated with 10 nm titanium (Ti) and 40 nm gold (Au) using sputter coater (Cressington 208) to form a conductive path between the detection window (0.5 × 0.5 mm2) and the edge of the Si3N4 membrane (Fig. 2a). Most critically, the UO2 particle electrode was prepared by mixing the polyvinylidene fluoride (PVDF) binder and conductive carbon black (CB) in N-methyl-2-pyrrolidone solvent to form a slurry with a concentration of 8 mg mL−1 UO2. Earlier attempts by our group using a Nafion binder were less useful for radioactive materials because of the requirement for spin-coating.16 1 μL of this slurry was deposited on the 50 nm thick Si3N4 membrane window to form the UO2 WE (Fig. 2b). Later, it was bound with a polydimethylsiloxane (PDMS) reservoir (2 × 3 × 1.5 mm3) with pre-inserted CE and RE (Fig. 2c) to complete the rest of the assembly (Fig. 2d).13 The inspiration for using PVDF/CB as the electrode binding material comes from Grillet and co-workers17 who used it to test the performance of lithium-ion battery electrodes. The PVDF/CB binder exhibits excellent resistance to large electrochemical potentials typical of lithium batteries and is sufficiently porous and durable to enable the migration of both electrons and lithium between anode and cathode over long time periods. These factors made it ideal for our purposes where it was essential to maintain both electric and electrolyte contact between all the particles. | ||
| Fig. 2 Fabrication process of the PAMEC device, including (a) Au coating to form the conductive path on the Si3N4 membrane, (b) depositing UO2 PVDF/CB slurry on the detection window in the center of the Si3N4 membrane to form the WE, (c) bonding the reservoir made of PDMS with UO2-attached Si3N4 membrane, and (d) the parts assembly e.g., inserting Teflon tubing. (a) and (b) were imaged by an optical microscope (c) and (d) were taken by a digital camera. | ||
Fig. 3 provides a closer view of UO2 composite electrode. Fig. 3a shows a backscattered electron (BSE) image from a selected area on the UO2 WE. The secondary electrons (SE) image of the same area (Fig. 3b) highlights the porous microstructure of PVDF/CB network, which not only provides the conductive path but also allows the dispersion of the electrolyte into the UO2 WE. These UO2 particles originated from a single crystal UO2 which was milled to micron-sized particles. The average size of the UO2 particles is 1.6 μm (Fig. S1†).
 | ||
| Fig. 3 (a) BSE image of UO2 particles (bright particles) bound by PVDF/CB (dark area), and (b) SE image from the same area as (a), demonstrating the porous microstructure of the PVDF/CB network. | ||
2.2 Electrochemical testing
The electrochemical workstation (CHI660E, CH Instruments, Inc.) was employed to perform the electrochemical testing on multiple PAMEC devices. Cyclic voltammetry (CV) and amperometric current–time (i–t) technique were used to study the corrosion behavior of UO2 in a 0.1 M NaClO4 (pH 9.5) solution, the same solution used in bulk analysis.18–202.3 Sample characterization
In situ imaging of the PAMEC WEs was performed in the chamber of a FEI Quanta 250FEG Environmental SEM, in the low vacuum condition with an accelerating voltage of 20 keV and the spot size of 4.0. Ex situ XPS was performed on a Kratos AXIS Ultra DLD system using a monochromatic Al-Kα source (hν = 1486.7 eV) operating at an analysis chamber pressure of <2 × 10−9 torr. The intact PAMEC devices were transferred into an inert glove box attached to the fast entry port of the XPS instrument after electrochemical and in situ SEM/EDS analyses. The cells were disassembled under an argon atmosphere (O2 at ∼3 ppm and 0.2 ppm moisture) and mounted onto conductive copper tape for analysis. The energy scale was calibrated to the aliphatic C1s line set at 285.0 eV. Curve fitting followed the methodology outlined in Ilton et al.21Post electrochemical testing analysis on the UO2 electrode required making lift-outs using a FEI Helios 660 SEM-Focused Ion Beam (FIB). The lift-outs were examined on a JEOL 300F GrandARM probe-corrected scanning TEM (STEM) equipped with a high angle annular dark field (HAADF) detector. STEM-HAADF was used to look for evidence of oxidation through contrast changes. Similar approaches to investigating UO2 corrosion under anoxic conditions have been used by our group.22,23
Imaging with AFM was performed in tapping mode using probes with relatively long and soft cantilevers and settings to follow the tall and abrupt contours of the particles (Oxford/Asylum AC240TSA-R3, Setpoint 1.5 V, Drive amplitude 0.9 V, scan rate 0.3 Hz).
3 Results and discussion
3.1 Electrochemical testing
The electrochemical testing conditions and analyses methods for each UO2-attached PAMEC device and blank control are tabulated in Table 1. Due to the possible oxidation of UO2 particles during the device fabrication, the UO2 electrode was cathodically cleaned at −1.0 V vs. saturated calomel electrode (SCE) for 400 s to remove surface films formed from reaction with O2 prior to deaeration with N2 purging.| Device ID | Electrode | Electrolyte | Electrochemical analysis method | Imaging and spectral analysis | |
|---|---|---|---|---|---|
| Cyclic voltammetry | Amperometry | ||||
| PAMEC-1 | UO2 and PVDF/CB composite electrode | 0.1 M NaClO4 (pH 9.5), purged with N2 for 30 minutes | CV scan range −1.0 V to 0.5 V vs. SCE at various scan rates from 10 mV s−1 up to 200 mV s−1 | Applied −1.0 V vs. SCE for 400 s prior to CV scans; applied 0.5 V vs. SCE for 13 h after CV scans and followed with −1.0 V vs. SCE for 400 s | In situ SEM/EDS, XPS (E-cell was disassembled in XPS glove box) |
| PAMEC-2 | UO2 and PVDF/CB composite electrode | 0.1 M NaClO4 (pH 9.5), purged with N2 for 30 minutes | CV scan range −1.0 V to 0.5 V vs. SCE at 20 mV s−1 | Applied −1.0 V vs. SCE for 400 s prior to CV scans; applied 0.5 V vs. SCE for 4 h after CV scans | Ex situ XPS (E-cell was disassembled in XPS glove box) |
| PAMEC-3 | UO2 and PVDF/CB composite electrode | 0.1 M NaClO4 (pH 9.5), purged with N2 for 30 minutes | NA | Applied −1.0 V vs. SCE for 400 s prior to anodic oxidation; applied 0.5 V vs. SCE for 4 h | STEM (E-cell was disassembled in atmosphere) |
| PAMEC-4 | UO2 and PVDF/CB composite electrode | 0.1 M NaClO4 (pH 9.5), purged with N2 for 30 minutes | NA | Applied −1.0 V vs. SCE for 400 s prior to anodic oxidation; applied 0.5 V vs. SCE for 2.5 h | AFM (E-cell was disassembled in atmosphere) |
| PAMEC-5 | PVDF/CB composite electrode | 0.1 M NaClO4 (pH 9.5), purged with N2 for 30 minutes | CV scan range −1.0 V to 0.5 V vs. SCE at 20 mV s−1 | Applied −1.0 V vs. SCE for 400 s prior to CV scans | NA |
| PAMEC-6 | PVDF/CB composite electrode | 10 mM K4Fe(CN)6 and 10 mM K3Fe(CN)6 in 1.0 M aqueous KNO3 | CV scan range −0.3 V to 0.8 V vs. platinum RE at various scan rates from 5 mV s−1 up to 100 mV s−1 | Applied −1.0 V vs. SCE for 400 s prior to CV scans | NA |
PAMEC-1 WE were analyzed by electrochemical workstation, in situ SEM/EDS and then XPS, as listed in Table 1. After being loaded with 0.1 M NaClO4 (pH = 9.5), the U(IV)/U(VI) redox process for the UO2 WE was probed electrochemically using a combination of cyclic voltammetry (CV) and amperometry experiments, as summarized in Fig. 4. To better compare with the results which were based on the SCE RE,18–20 we have conducted the potential shift determination testing, as described in ESI (Fig. S2†), by testing the redox potentials of the ferrocyanide–ferricyanide redox couple using PAMEC-6 (Table 1). The potential reading based on Pt RE was negatively shifted ∼0.2 V comparing to results using the SCE RE. To facilitate comparison to electrochemical measurements from UO2 bulk analyses, potentials in this work were converted to the SCE scale in Fig. 4 and S3.†
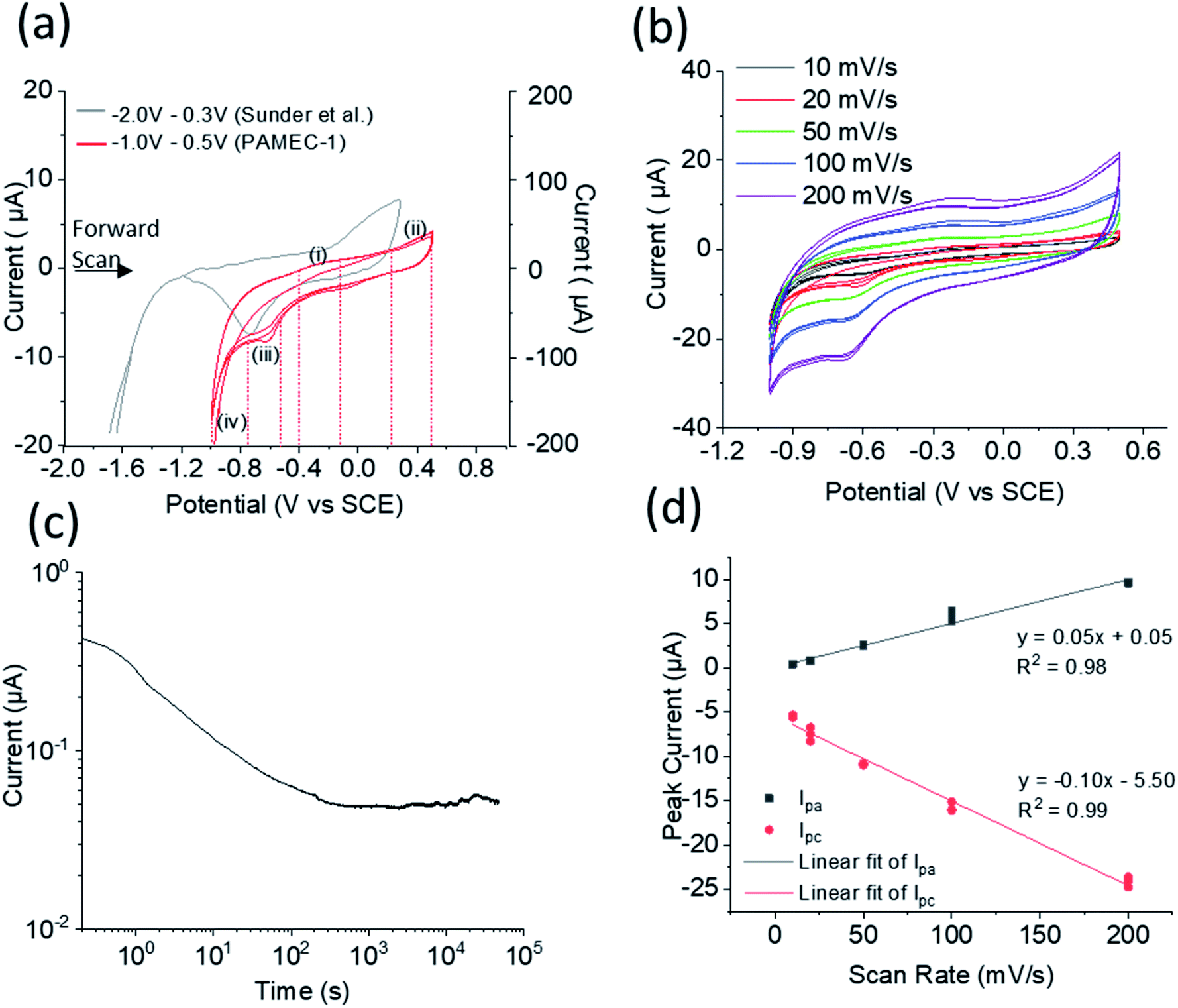 | ||
| Fig. 4 (a) Cyclic voltammograms comparison at 20 mV s−1 scan rate, with left Y-axis corresponding to our result using PAMEC-1 (red line) and right Y-axis corresponding to bulk analysis result reported by Sunder et al. (grey line),19 (b) cyclic voltammograms acquired at various scan rates using the same PAMEC-1 (b), and (c) the current–time (log–log) curve recorded for 13 hours at the potential of 0.5 V vs. SCE, and (d) anodic and cathodic peak current vs. scan rates. | ||
In the voltammetry experiments, the redox system was examined as a function of varying scan rates, namely 10 mV s−1, 20 mV s−1, 50 mV s−1, 100 mV s−1, and 200 mV s−1, within an electrochemical potential window ranging from −1.0 V to 0.5 V vs. SCE. For each scan rate, the electrochemical potentials were initially swept in the positive direction initiating at −1.0 V all the way up to 0.5 V vs. SCE, which was subsequently followed by a reverse sweep back to −1.0 V vs. SCE. These were followed by two more sweeps in the positive directions and reverse sweeps, totaling an overall six sweep segments. The multiple sweep segments were conducted to understand the chemical and electrochemical reversibility of the electrochemical processes.
A subset of cyclic voltammograms (20 mV s−1 over six sweep segments) were compared to the bulk UO2 WE conducted over similar conditions (Fig. 4a).18,19 It showed two anodic processes in the forward sweep and two cathodic processes in the reverse sweep, namely, an anodic process started at −0.24 V vs. SCE (marked as process (i)), a second anodic process at 0.38 V vs. SCE (marked as process (ii)), a cathodic process at −0.61 V vs. SCE (marked as process (iii)), and finally, a large reduction at −1.0 V vs. SCE (process (iv)). The processes observed in our microelectrochemical system approximately mirror the processes albeit with slightly shifted electrochemical potentials, as previously reported for bulk U(IV)/U(VI) redox processes that employed an UO2 WE under similar electrochemical testing and matrix conditions.18–20
The anodic process (i) in Fig. 4a is attributed to an oxidation of the non-stoichiometric area of the UO2+x surface on the PAMEC WE. A similar process was observed by Sunder et al.20 for their bulk UO2 electrode, but were absent or insignificant in the cases where the WE were constructed of more oxidative resistant UO2 materials, such as synthetic SNF,4 which further supported this process being a result of the surface oxidation. Further evidence of the UO2 surface in our PAMEC being prone to partial oxidation, was obtained from XPS studies on the ‘as prepared’ samples as described later (Fig. 5). As can be seen from the cyclic voltammograms, the peak current intensity for this particular process was observed to undergo a more significant drop from the first segment to the following segments, compared to the other processes (ii)–(iv). This indicates that as the surface monolayer gets oxidized, less of it is available for oxidation in subsequent segments, leading to a notable drop in current intensity. This observation is as what we expected, as the electrode surface undergoes irreversible oxidation when potential scanned over −0.4 V vs. SCE.24 The process (ii) is probably attributed to oxidation of the UO2 surface.20 This process has been observed in the bulk UO2 WE system and has been attributed to the oxidation of the surface to UO2.33.20,25,26 The slight increase in peak intensity suggests that the process is limited to surface oxidation, as opposed to extensive oxidative dissolution of the surface, which would be expected to be reflected in a more pronounced enhancement in the current intensity. The oxidative nature of this process is further corroborated by in situ SEM analysis (see Fig. 6) and direct evidence from XPS for U(VI) (see Fig. 5), as discussed later.
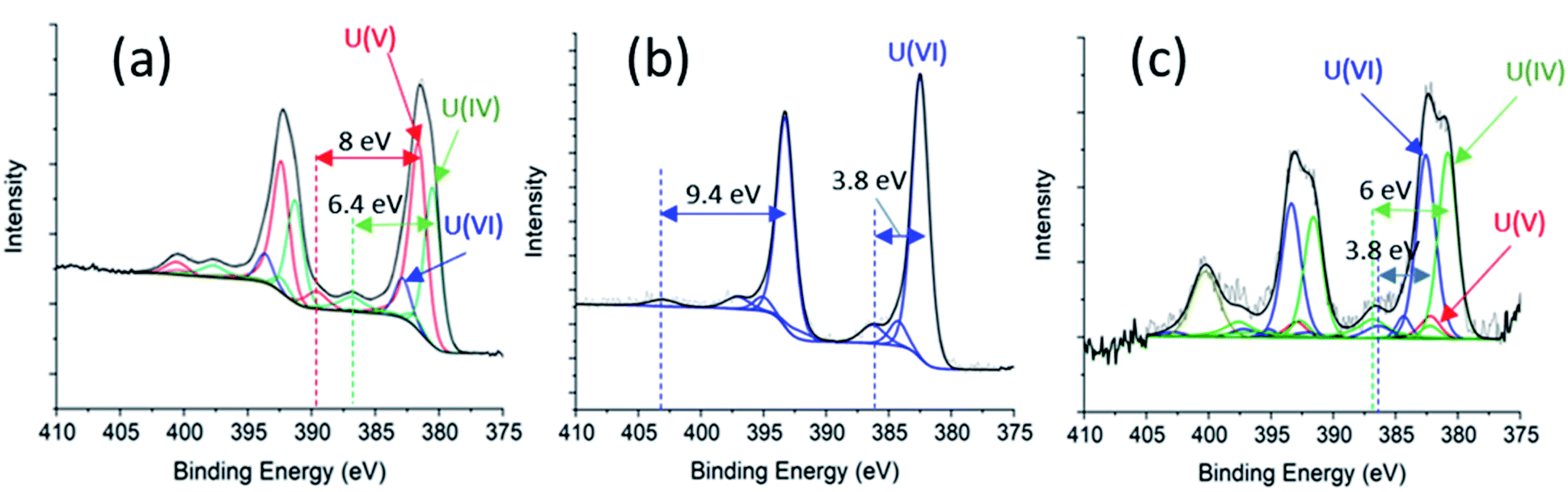 | ||
| Fig. 5 (a) U 4f spectral fitting for initial ‘as-received’ UO2 sample. (b) U 4f spectral deconvolution for PAMEC-2 showing clear evidence of electrochemical oxidation of U(IV) and U(V) to U(VI), and (c) U 4f spectral fitting for sample PAMEC-1 WE. | ||
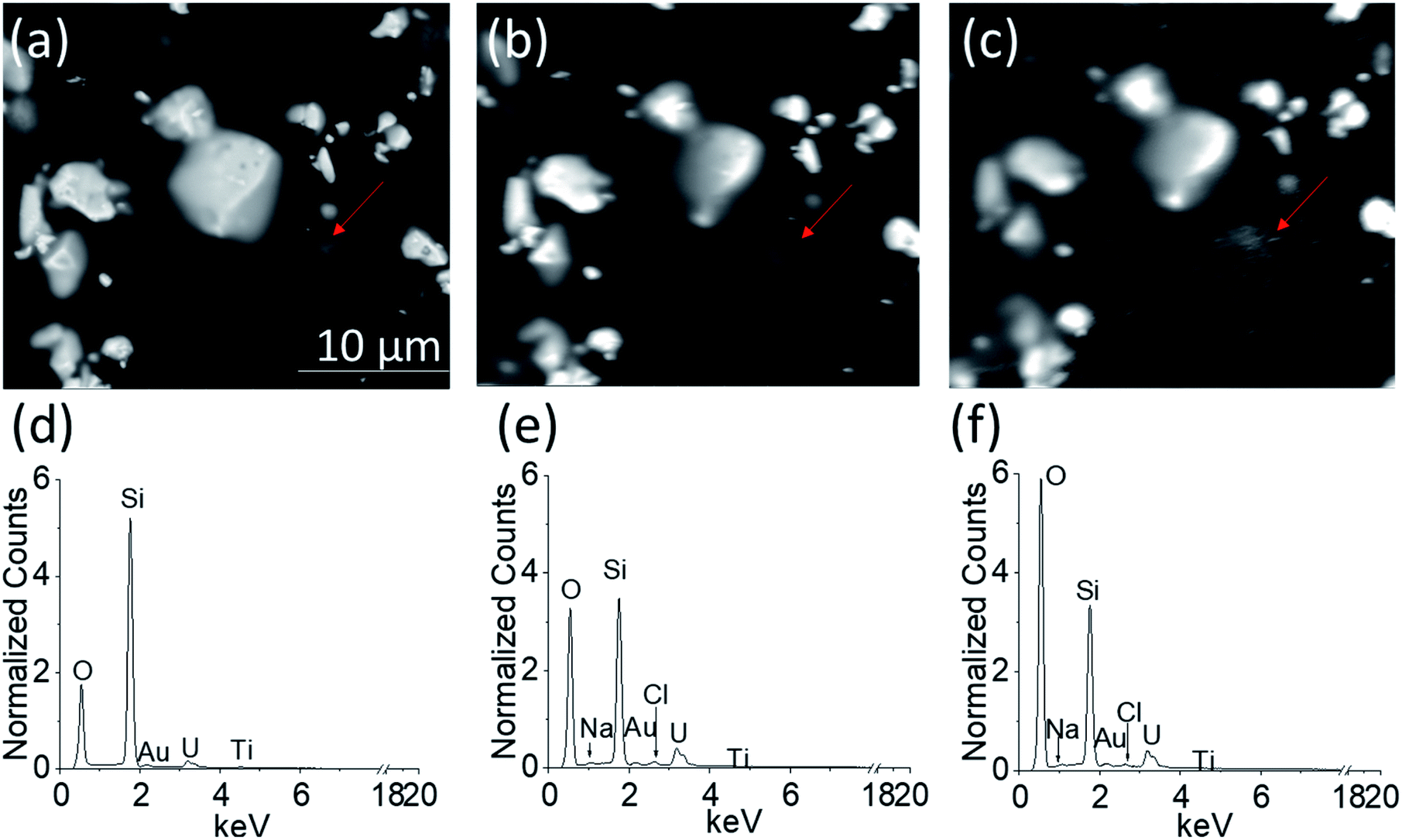 | ||
| Fig. 6 The BSE images of (a) empty PAMEC-1, (b) loaded with electrolyte before anodic corrosion, and (c) after 13 hours of corrosion, with corresponding EDS spectra (d, e, f) acquired from the area shown in (a, b, c), respectively. EDS spectra were all normalized to respective Si intensity. | ||
On the return cathodic scan, the peak at −0.61 V vs. SCE, (Fig. 4a, process (iii)) is thought to be the reduction of the UO2.33 (U3O7) which compares well to the peak potential observed in the bulk UO2 WE system,18–20 and supported by XPS results (Fig. 5c).
Finally, the process (iv) is attributed to the reduction of water (with H2 evolution process). The potential at which water reduction occurs is heavily dependent on the UO2 surface (i.e., SIMFUEL vs. UO2).27
Multiple successive cyclic voltammograms acquired at various scan rates (10 mV s−1 up to 200 mV s−1) showed consistent results, highlighting the durability of the PAMEC. Fig. S3a† also shows the reproducibility of cyclic voltammograms acquired from two separate experiments (PAMEC-1 and -2) and Fig. S3b† shows the cyclic voltammograms recorded in a wider potential range (−1.3 V to 0.7 V vs. SCE). In addition, cyclic voltammograms comparison between PAMEC w/- and w/o UO2 WE can be seen in Fig. S3,† verifying the potential peaks observed in the UO2-attached PAMEC devices were resulting from the UO2 redox rather than the Si3N4 substrate or binding materials, e.g., PVDF/CB.
The steady loss of current over time up to ∼1000 s (Fig. 4c) is possibly due to the formation of a protective U(VI) alteration layer, especially in the first 100 seconds. This presumably changes the electroactive surface which is reflected in the current response. In contrast, the slight increase of the current after 1000 s might indicate loss of this phase by, for instance, surface spallation that exposed fresh oxide.
Fig. 4d presents the anodic peak currents (Ipa in process i) and cathodic peak currents (Ipc in process iii) as a function of scan rates. Both Ipa and Ipca are observed to be linearly proportional to scan rate. This observed behavior is in strict contrast to a diffusion limited electrochemical process where the peak-current is proportional to the square-root of scan rate. On the contrary, the behavior is consistent with the electroactive specie being confined to the electrode surface without any diffusion, indicating the redox is an adsorption/surface-controlled process.
3.2 XPS analysis
Spectroscopic analysis XPS can be used to quantify the oxidation state of uranium at the near surface of solids.21,28,29 Sunder and co-workers30 have used XPS to investigate the corrosion of UO2 and simulated UO2 fuel.30 We employed XPS to analyse three samples for comparison, including the as-prepared ‘UO2’ WE (Fig. 5a), oxidized UO2 WE of PAMEC-2 (Fig. 5b), and oxidized UO2 WE of PAMEC-1 (Fig. 5c). The ‘as-prepared’ UO2 refers to the UO2 WE that had not been treated with electrochemical oxidation or reduction. After running CV scans (Fig. S3†) to demonstrate the reproducibility of the redox process among PAMEC devices, PAMEC-2 was anodically oxidized at the constant potential 0.5 V vs. SCE for approximately 4 hours. PAMEC-1 was oxidized at the same constant potential but for longer time (approximately 13 hours.) after CV scans shown in Fig. 4a and b. After 13 hours of oxidation, the electrode of PAMEC-1 was reduced at −1.0 V vs. SCE for 400 seconds, as described in Table 1. Both E-cells (PAMEC-1 and -2) were dissembled under argon gas in the XPS glove box prior to the analysis.We used the corroded sample (i.e., PAMEC-2) for our U(VI) standard and a previously analyzed, well-characterized, UO2 sample as our U(IV) standard to fit the U4f of the ‘as-prepared’ UO2 sample (see ESI,† XPS Analysis and Fitting Method). The initial fits were poor, particularly around the shoulder of the U 4f5/2 peak at ∼390 eV and the satellite feature at ∼400 eV. Introducing a U(V) component, along with U(IV) and U(VI), led to a good fit, as seen in Fig. 5a. The satellite features at 7.9 eV above the U 4f main peaks (at 381.6 eV and 392.4 eV) are characteristic of a U(V) species, while those at 6.4 eV above the main peaks (at 380.5 eV and 391.3 eV) are well within expected values for a U(IV).21 Thus, for the ‘as prepared’ UO2 WE, U(IV) and U(V) were the primary components at 45.1% and 44.8% of total near-surface uranium, respectively. U(VI) had only about 10% weight in the fit. The higher oxidation states (U(V) and U(VI)) were seen in the ‘as-prepared’ UO2 electrode, which is likely attributed to the UO2 oxidation when exposed to the atmosphere during the sample handling.
Modeling the U 4f spectrum of the oxidized sample (PAMEC-2), yielded a good fit with satellite features at 3.7 eV and 9.4 eV above the main peaks at 382.5 eV and 393.3 eV for U 4f7/2 and U 4f5/2 respectively (Fig. 5b). The satellites are diagnostic of U(VI), indicating that PAMEC-2 WE is dominantly U(VI).
Fitting the U 4f spectrum of PAMEC-1 WE yielded U(VI)/U(total) and U(IV)/U(total) equal to 50% and 45.3%, respectively (see Fig. 5c). The overall concentration of uranium in the solid sample was quite low possibly due to the mass loss after 13 hours of anodic dissolution, hence the low signal-to-noise ratio. The feature at ∼400 eV is too intense to be the satellite associated with U(V) 4f5/2 and is assigned to N 1s presumably arising from a nitride species due to the Si3N4 membrane of PAMEC. The existence of U(IV) is attributed to 400 s of reduction after 13 hours of oxidation, confirming the reversible redox reaction occurred in the PAMEC-1.
The above XPS results conform well to that observed in the CV scans and amperometry experiments. For instance, anodic oxidation of UO2, as seen in process (ii) of the cyclic voltammogram (Fig. 4a), results in the presence of U(VI) as the primary species. Whereas subsequent reduction (at −1.0 V vs. SCE for 400 s) resulted in the reduction of U(VI) to U(IV) as seen in Fig. 5c.
3.3 In situ SEM/EDS
Fig. 6a–c show the BSE images of the UO2 WE obtained through the Si3N4 detection window when PAMEC-1 was not loaded with electrolyte (Fig. 6a), loaded with electrolyte but before anodic corrosion (Fig. 6b) and after 13 hours of anodic corrosion (Fig. 6c), separately. All three images (Fig. 6a–c) show that the UO2 particles remained at the same position, demonstrating that the PVDF/CB network adhered well to the UO2 particles despite a long period of electrochemical corrosion.Importantly, the comparison of EDS spectra acquired from the corresponding areas shown in Fig. 6a–c, confirmed that the electrolyte solution (0.1 M NaClO4) was well dispersed throughout the UO2 WE (Fig. 4d–f, showing differences in normalized counts of O, Cl, and Na), owing to the porous structure of PVDF/CB structure.31,32 Detectable Au, Ti, and silicon (Si) are part of the PAMEC fabrication (see Section 2.1). The entire detection window of the PAMEC with the UO2 WE can be seen in Fig. S4.†
It is worth noting that the BSE image taken after 13 hours of oxidation (Fig. 6c) has more white spot features in the area (pointed by red arrow) than before electrochemical corrosion (Fig. 6b). Indeed, after corrosion, EDS shows a significant increase of O intensity compared to Fig. 6d and e, suggesting the formation of U3O7 or hydrated phases (UO22+ → UO3·xH2O),20 consistent with the precipitation of a U(VI) phase as confirmed with XPS (Fig. 5c) and AFM (Fig. 8). To our knowledge, it is the first study that is able to provide the comparison of UO2 electrode before and after corrosion by utilizing in situ SEM/EDS.
3.4 Structural and morphological analysis
The STEM-HAADF images exhibit bright contrast at the edge of the specimen indicating surface oxidation. The bright contrast (higher average atomic number) occurs in UO2+x with introduction of additional oxygen into the uraninite lattice with little or no volume expansion. The ‘as-prepared’ UO2 WE (Fig. 7a) and corroded one from PAMEC-3 (Fig. 6b) were oxidized as manifest by ∼10–20 and 50–150 nm thick high contrast layers, respectively. The STEM-HAADF images again exhibit bright contrast from the insertion of oxygen into the structure but in the oxidized specimen (UO2 lift-out from PAMEC-3), this region is much thicker. Although the orientations are different in the two images (the insert colorized images show atoms projected along the B[110] and B[111] crystal directions), the thick oxide layer can be clearly seen. The preparation of UO2 lift-outs can be found in Fig. S5.†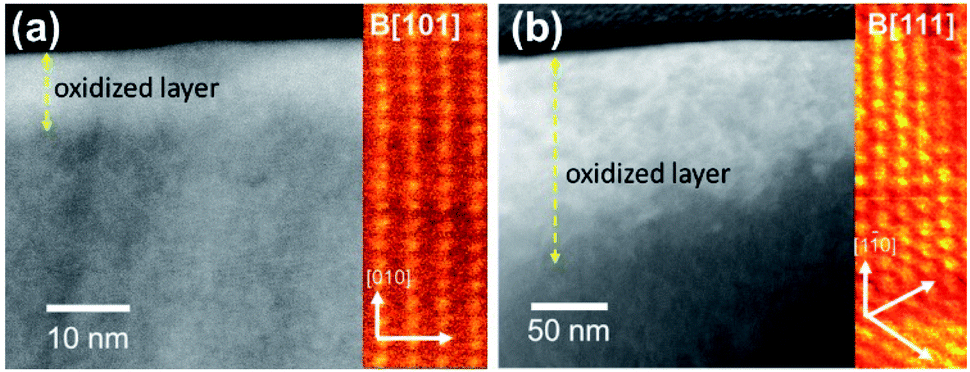 | ||
| Fig. 7 STEM-HAADF images of the (a) ‘as-prepared’ surface and (b) surface following corrosion in the PAMEC-3 showing the oxidation at the surface of the material. | ||
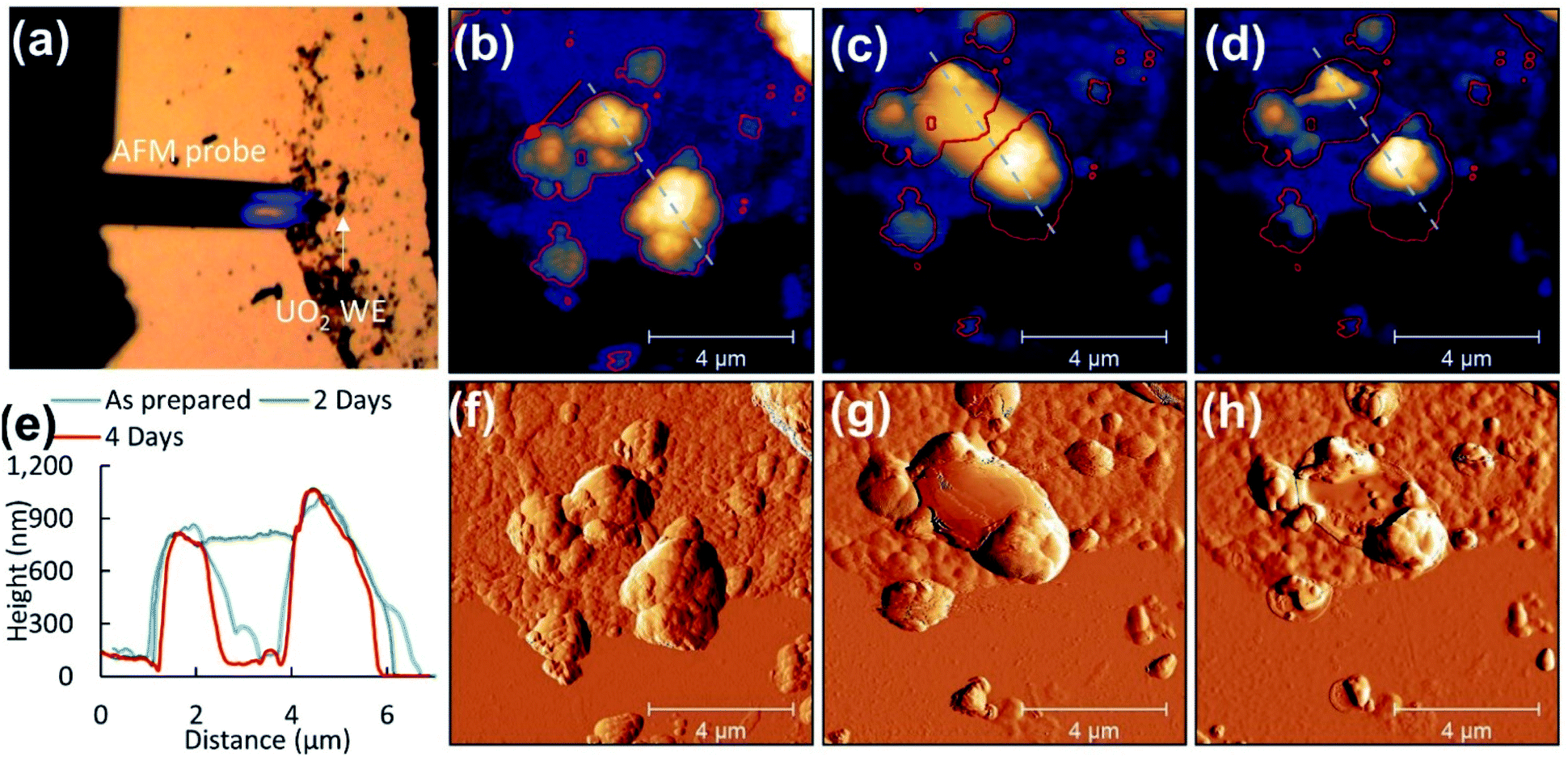 | ||
| Fig. 8 (a) Optical image of disperse UO2 WE particles, topography of (b) selected UO2 particles as prepared WE on the substrate, (c) two days, and (d) four days after corrosion. The corresponding amplitude images are included (f–h). Particle evolution from initial morphology is denoted by changes in lateral dimensions (red outline) and height given by cursor profiles (e) from the same region (grey lines). | ||
3.5 AFM
Previous studies have observed surface roughening around faceted grain structures and at grain boundaries of electrochemically corroded UO2 surfaces.33 Here AFM was employed to determine morphological changes of UO2 particles after corrosion in the PAMEC.AFM analysis of the ‘as prepared’ UO2 WE on the substrate was carried out on dispersed particles to measure individual particles (see Fig. 8a). ‘As prepared’ UO2 WE particles that were fabricated as PAMEC-4 were rough and were composed of aggregates of smaller particles (Fig. 8b and f). Two days after the corrosion was completed the identical disperse particle region was located (Fig. 8c and g). Large masses were observed as smooth coatings dispersed within and on top of the clustered particles suggestive of a hydrated phase that adhered to the surface. Four days after corrosion these domed features appeared to have either partially or completely collapsed (see Fig. 8d and h). A comparison of the identical region two and four days after corrosion details this dynamic process. Outlines of particles and cursor profiles (Fig. 8e) highlight the initial increase (grey line) followed by a decrease in volume (red line).
Based on the combined results of CV, XPS, in situ SEM/EDS and AFM measurements we observed the following progressional oxidative behavior (see Fig. 9). We observed clear oxidation to U(VI) after the application of a corrosion potential at 0.5 V vs. SCE for 13 hours (Fig. 4c), with the appearance of precipitates as shown by in situ SEM (Fig. 6c). The morphologies changed before and after drying (Fig. 8), as evidenced by AFM. This suggests that these could have been hydrated U(VI) alteration phases.
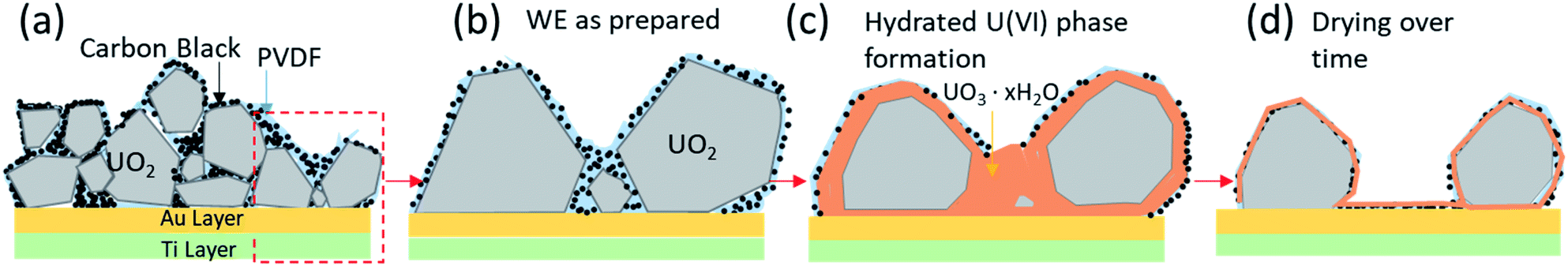 | ||
| Fig. 9 Schematic of oxidation behaviour based on CV, XPS, in situ SEM/EDS and AFM results: (a) attachment of UO2 particles using PVDF/CB on the conductive layer (Au and Ti) of the PAMEC, (b) “zoomed-in” schematic of UO2 particles as prepared on the substrate, (c) anodic oxidation of UO2 forming U(VI) phase and precipitates and corrosion dissolution, and (d) surface U(VI) alteration phases that are undergoing dehydration. | ||
4 Conclusion
We have shown that the cyclic voltammograms obtained with microgram quantities of UO2 in the PAMEC match the cyclic voltammogram of those obtained by bulk electrochemical methods.18–20After applying an electrochemical potential to reduce the surface and then oxidize the surface, in situ SEM imaging of the progress of the reaction showed clear evidence of a newly formed precipitate. PAMEC applied with fixed voltage (amperometry) responded to changes in oxidation state of the UO2 working electrode. These changes were confirmed by XPS to be oxidation of U(IV) to U(VI) through an intermediate U(V) species). Ex situ STEM-HAADF images clearly showed the oxidation layer at atomic scale. These changes were confirmed by XPS to be oxidation of U(IV) to U(VI) through an intermediate U(V) species. Further, AFM characterization revealed smooth rounded features that disappeared over time, suggesting that these could have been hydrated U(VI) alteration phases that eventually underwent dehydration when exposed to atmosphere. Indeed, it is well known that the loss of structural water from the interlayers of the uranyl oxide phases, such as schoepite, results in the progressive modification of the phase.34–36
In summary, PAMEC enables studying redox active hazardous materials at the microscale, which will greatly reduce the cost and risk. In particular, the results demonstrate the utility of this approach for studying the redox properties of microgram quantities of both fresh and spent nuclear fuels, with the promise of providing much needed data on the behavior of SNF in geological repository environments in an efficient way.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Electrochemical experiments and PVDF/CB modification by JY, XPS analysis and interpretation by NL and ESI, microscopy analysis JY, ST, SLR, and ECB, manuscript review and revision by SC. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Wes Lawrence, Dr Mark Nutt, Ms Wendy Erikson, Pacific Northwest National Laboratory (PNNL) for programmatic oversight. This work was supported by the DOE-NE (Nuclear Energy) Spent Fuel and Waste Science and Technology (SFWST) Campaign. UO2 materials were supplied by Dr Dallas Reilly. Mr Kyle Maloy and Mr Alan Cantrell provided radiological support. Dr Prasad Nair (DOE NE-81) and Dr Carlos Jove-Colon (Sandia National Laboratory) are thanked for technical discussions. Work was conducted at the PNNL Radiochemical Microscopy Q-suite. PNNL is operated by Battelle under contract DE-AC05-76RL01830.References
- J. Bruno, E. Cera, L. Duro, T. E. Eriksen and L. O. Werme, J. Nucl. Mater., 1996, 238, 110–120 CrossRef CAS.
- J. Bruno and R. C. Ewing, Elements, 2006, 2, 343–349 CrossRef CAS.
- R. C. Ewing, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
- L. Wu and D. W. Shoesmith, Electrochim. Acta, 2014, 137, 83–90 CrossRef CAS.
- M. E. Broczkowski, J. J. Noël and D. W. Shoesmith, J. Nucl. Mater., 2005, 346, 16–23 CrossRef CAS.
- D. Sassani, presented in part at the Conference: Proposed for presentation at The 7th International Symposium on Safety Improvement & Stakeholder Confidence in Radioactive Waste Management held December 2, 2020 in Busan, Republic of Korea – Virtual meeting, United States, 2020 Search PubMed.
- D. W. Shoesmith, J. Nucl. Mater., 2000, 282, 1–31 CrossRef CAS.
- J. S. Goldik, H. W. Nesbitt, J. J. Noël and D. W. Shoesmith, Electrochim. Acta, 2004, 49, 1699–1709 CrossRef CAS.
- J. S. Goldik, J. J. Noël and D. W. Shoesmith, Electrochim. Acta, 2006, 51, 3278–3286 CrossRef CAS.
- A. B. Fidalgo and M. Jonsson, J. Nucl. Mater., 2016, 477, 85–87 CrossRef.
- R. J. Finch, E. C. Buck, P. A. Finn and J. K. Bates, MRS Online Proc. Libr., 1999, 556, 431 CrossRef CAS.
- L. Yang, X.-Y. Yu, Z. H. Zhu, T. Thevuthasan and J. P. Cowin, J. Vac. Sci. Technol., A, 2011, 29, 061101 CrossRef.
- X.-Y. Yu, L. Yang, J. Cowin, M. Iedema and Z. Zhu, US Pat., 8555710, 2011 Search PubMed.
- X.-Y. Yu, B. Liu and L. Yang, Microfluid. Nanofluid, 2013, 15, 725–744 CrossRef CAS.
- X.-Y. Yu, J. Yao, E. C. Buck and Z. Zhu, Surf. Interface Anal., 2020, 52, 454–459 CrossRef CAS.
- J. Son, E. C. Buck, S. L. Riechers and X.-Y. Yu, Micromachines, 2021, 12, 60 CrossRef PubMed.
- A. M. Grillet, T. Humplik, E. K. Stirrup, S. A. Roberts, D. A. Barringer, C. M. Snyder, M. R. Janvrin and C. A. Apblett, J. Electrochem. Soc., 2016, 163, A1859–A1871 CrossRef CAS.
- S. Sunder, L. K. Strandlund and D. W. Shoesmith, Electrochim. Acta, 1998, 43, 2359–2372 CrossRef CAS.
- S. Sunder, L. K. Strandlund and D. W. Shoesmith, Anodic dissolution of UO2 in slightly alkaline sodium perchlorate solutions, Canada, 1996 Search PubMed.
- S. Sunder, N. H. Miller and D. W. Shoesmith, Corros. Sci., 2004, 46, 1095–1111 CrossRef CAS.
- E. S. Ilton and P. S. Bagus, Surf. Interface Anal., 2011, 43, 1549–1560 CrossRef CAS.
- A. J. Popel, S. R. Spurgeon, B. Matthews, M. J. Olszta, B. T. Tan, T. Gouder, R. Eloirdi, E. C. Buck and I. Farnan, ACS Appl. Mater. Interfaces, 2020, 12, 39781–39786 CrossRef CAS PubMed.
- S. R. Spurgeon, M. Sassi, C. Ophus, J. E. Stubbs, E. S. Ilton and E. C. Buck, Proc. Natl. Acad. Sci., 2019, 201905056, DOI:10.1073/pnas.1905056116.
- F. King and D. W. Shoesmith, Electrochemical studies of the effect of H2 on UO2 dissolution, Report 1404-0344, Sweden, 2004 Search PubMed.
- S. Sunder, D. W. Shoesmith, M. G. Bailey, F. W. Stanchell and N. S. McIntyre, J. Electroanal. Chem. Interfacial Electrochem., 1981, 130, 163–179 CrossRef CAS.
- H. Christensen, S. Sunder and D. W. Shoesmith, J. Alloys Compd., 1994, 213–214, 93–99 CrossRef.
- D. W. Shoesmith, W. H. Hocking, S. Sunder, J. S. Betteridge and N. H. Miller, J. Alloys Compd., 1994, 213–214, 551–553 CrossRef.
- M. Schindler, F. C. Hawthorne, M. S. Freund and P. C. Burns, Geochim. Cosmochim. Acta, 2009, 73, 2471–2487 CrossRef CAS.
- S. Sunder, D. W. Shoesmith, H. Christensen and N. H. Miller, J. Nucl. Mater., 1992, 190, 78–86 CrossRef CAS.
- S. Sunder, N. H. Miller, W. H. Hocking and P. G. Lucuta, J. Alloys Compd., 1994, 213, 503–505 CrossRef.
- R. Ram, V. Soni and D. Khastgir, Composites, Part B, 2020, 185, 107748 CrossRef CAS.
- V. A. Nguyen and C. Kuss, J. Electrochem. Soc., 2020, 167, 065501 CrossRef CAS.
- H. He, R. K. Zhu, Z. Qin, P. Keech, Z. Ding and D. W. Shoesmith, J. Electrochem. Soc., 2009, 156, C87 CrossRef CAS.
- R. J. Finch, M. L. Miller and R. C. Ewing, Radiochim. Acta, 1992, 58–59, 433–444 CrossRef CAS.
- D. J. Wronkiewicz, J. K. Bates, S. F. Wolf and E. C. Buck, J. Nucl. Mater., 1996, 238, 78–95 CrossRef CAS.
- D. J. Wronkiewicz and E. Buck, Rev. Mineral. Geochem., 1999, 38, 475–497 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of device fabrication, analyses methods and more electrochemical measurements. See https://doi.org/10.1039/d2ra02501a |
| This journal is © The Royal Society of Chemistry 2022 |