
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUracil derivatives as HIV-1 capsid protein inhibitors: design, in silico, in vitro and cytotoxicity studies†
Deepthi Ramesh a,
Amaresh Kumar Mohanty‡
b,
Anirban De‡a,
Balaji Gowrivel Vijayakumara,
Aiswarya Sethumadhavanc,
Suresh Kumar Muthuvelb,
Maheswaran Manic and
Tharanikkarasu Kannan*a
a,
Amaresh Kumar Mohanty‡
b,
Anirban De‡a,
Balaji Gowrivel Vijayakumara,
Aiswarya Sethumadhavanc,
Suresh Kumar Muthuvelb,
Maheswaran Manic and
Tharanikkarasu Kannan*a
aDepartment of Chemistry, Pondicherry University, Kalapet, Puducherry-605014, India. E-mail: tharani.che@pondiuni.edu.in
bDepartment of Bioinformatics, Pondicherry University, Kalapet, Puducherry-605014, India
cDepartment of Microbiology, Pondicherry University, Kalapet, Puducherry-605014, India
First published on 13th June 2022
Abstract
A series of novel uracil derivatives such as bispyrimidine dione and tetrapyrimidine dione derivatives were designed based on the existing four-point pharmacophore model as effective HIV capsid protein inhibitors. The compounds were initially docked with an HIV capsid protein monomer to rationalize the ideas of design and to find the potential binding modes. The successful design and computational studies led to the synthesis of bispyrimidine dione and tetrapyrimidine dione derivatives from uracil and aromatic aldehydes in the presence of HCl using novel methodology. The in vitro evaluation in HIV p24 assay revealed five potential uracil derivatives with IC50 values ranging from 191.5 μg ml−1 to 62.5 μg ml−1. The meta-chloro substituted uracil compound 9a showed promising activity with an IC50 value of 62.5 μg ml−1 which is well correlated with the computational studies. As expected, all the active compounds were noncytotoxic in BA/F3 and Mo7e cell lines highlighting the thoughtful design. The structure activity relationship indicates the position priority and lower log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values as the possible cause of inhibitory potential of the uracil compounds.
P values as the possible cause of inhibitory potential of the uracil compounds.
1. Introduction
The evolution and pandemic of the Acquired Immune Deficiency Syndrome (AIDS) have been one of the greatest challenges faced by humankind. The first report of the disease came in the early 1980s, though it is widely accepted to have originated in the 1920s in Kinshasa.1 The causative agent for the syndrome, the retrovirus Human T-lymphotropic virus type III (HTLV-III) was discovered in 1984 and was later renamed Human Immunodeficiency Virus (HIV) in 1986.1 To date, HIV-AIDS continues to be a major public health concern without any major cure for the disease. This is reflected in the 2020 World Health Organisation report as 680000 people died from AIDS and 1.5 million people were newly infected with HIV.2 Two types of HIV (HIV-1 and HIV-2) have been recognized based on molecular and antigenic differences and among them, HIV-1 is more prevalent across the world.1 AIDS is presently kept under control by highly active antiretroviral therapy (HAART) transforming the ailment from fatal infectious disease to controllable chronic affliction thereby reducing the mortality rate of HIV-positive patients. The current treatment for HIV through HAART targets diverse stages of the viral life cycle, especially two vital viral enzymes reverse transcriptase and protease. HAART has six major classes of drugs: Reverse Transcriptase (RT) inhibitors like Nucleoside RT inhibitors (NRTIs) and, non-nucleoside RT inhibitors (NNRTIs), integrase inhibitors (INIs), protease inhibitors (PIs), entry or fusion inhibitors (FIs), and chemokine receptor antagonists (CCR5 antagonists).3 The cornerstone of HAART treatment is the co-administration of various drugs inhibiting HIV via different mechanisms of action. However, virologic failure is observed in patients receiving HAART in low-income countries.4 Even with decreasing mortality rates, HAART is correlated with emerging drug resistance, which is attributed to high mutation rates of HIV-1 because of the error-prone nature of HIV-RT and its inability to proofread.5
Additionally, a numerous replication cycles occur in HIV-infected individuals thereby creating drug-resistant mutant strains of HIV.6 These drug-resistant strains are also being passed between individuals, thus limiting the treatment options for even newly afflicted persons. The extended HAART treatment is also associated with toxicities like neuropathy, myopathy, hepatic steatosis as seen in NRTIs, drug hypersensitivities related with NNRTIs, and metabolic syndromes and diarrhoea from using PIs.7 In this circumstance, there is an imperative need to find new anti-HIV drugs with an improved mechanism of action and novel targets in the HIV life cycle.
One suitable target is HIV-1 capsid protein (CA, p24) which is remarkably underexplored.
Capsid protein performs a vital role in HIV pathogenesis in the initial and late phases of the HIV lifecycle. The cone-shaped CA is released into cell cytosol after viral entry and disassembles, allowing reverse transcription and genome integration establishing HIV infection. The interaction of CA with Gag polyprotein during viral particle assembly results in the proteolytic cleavage, transforming the immature particles to mature virions forming viral RNA and other proteins.8 As the CA is involved in multiple stages of viral replication, it can be an attractive antiviral target. CA inhibitors can bind to the capsid perturbing the stability of the protein and are also capable of interfering with the infection by competing for host factor binding.9 CA is highly sensitive to mutations and has higher sequence conservation than other HIV proteins. The resistance of HIV to CA inhibitors requires mutations compromising the fitness of the virus, hence leading to a higher barrier to resistance and therapeutic efficiency.10,11 So far, only a handful of capsid inhibitors are reported with benzodiazepine,12 benzimidazole,13 pyrrolopyrazolones,14 furan,15 and indole16,17 scaffolds as in Fig. 1. However, there is no CA targeting compounds in clinical practice, and hence, a potential CA inhibitor holds promise for future drug development.
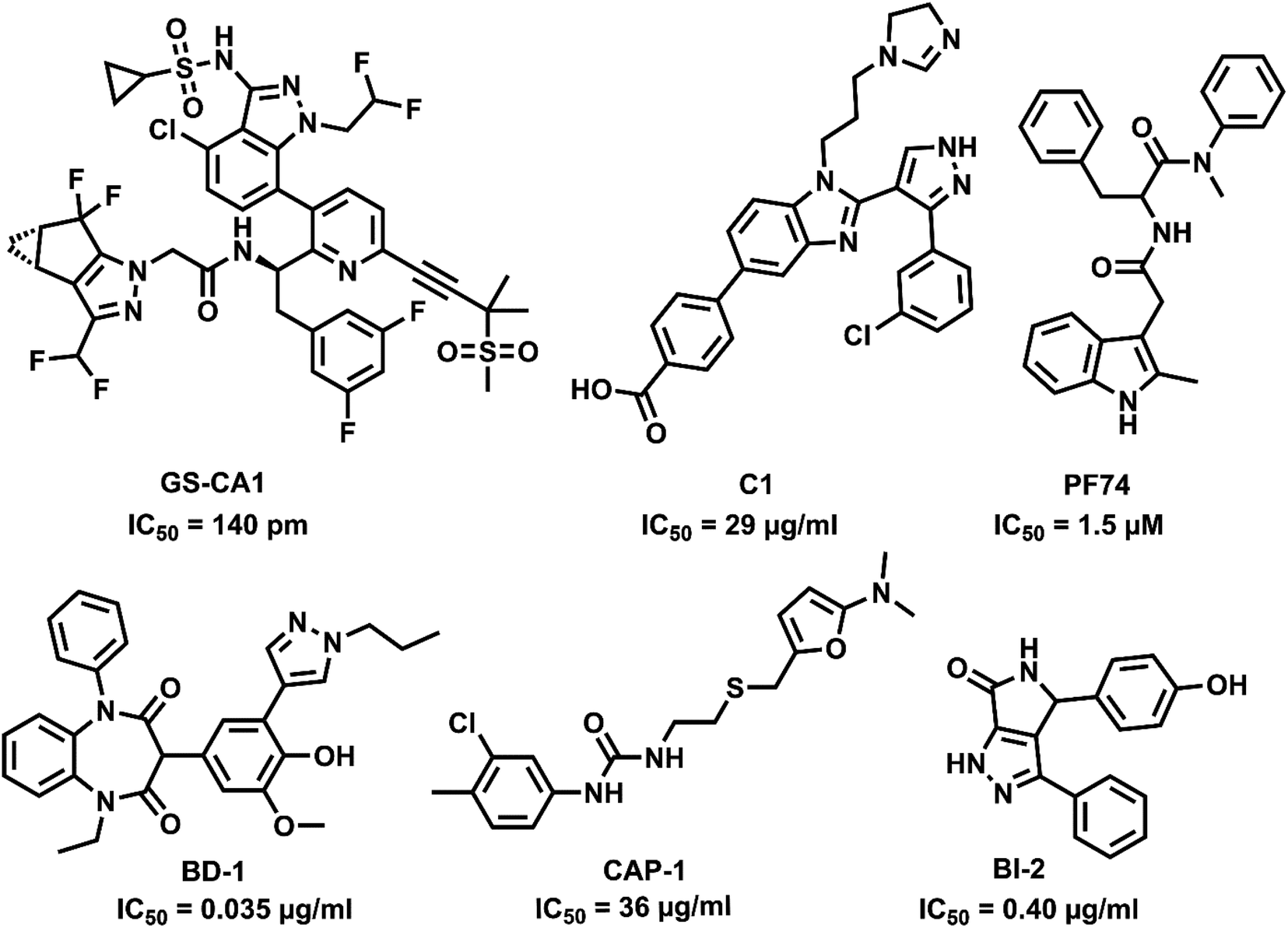 | ||
| Fig. 1 Capsid inhibitors reported in the literature. | ||
In drug development, the usage of small molecules is an efficient strategy considering the technical difficulties while employing other alternatives like short nucleotide segments or small peptides. Nucleosides and their corresponding nucleobases form major components in nucleic acids and bioactive systems, thus modifying their structure for drug development has a notable effect. Uracil is one such nucleobase, a naturally occurring pyrimidine, and can be a promising candidate as a CA inhibitor. Uracil can have a wide range of pharmaceutical properties depending on the substitution at N1, N3, C5, and C6 positions.18 Uracil pharmacophore is well known in anti-cancer (5-fluorouracil, tegafur),19 anti-herpes virus (trifluridine),20 and antidiabetic (linagliptin and alogliptin) drugs.21 The importance of the uracil scaffold in anti-HIV drugs is widely recognized with reports of uracil hybrid molecules as NNRTIs;22–25 however, there is no commercial uracil-based CA inhibitor.
Considering the factor of cross-resistance in HIV drugs, it is essential to introduce new drugs to which the virus has never been exposed before. A set of compounds were designed by incorporating flexible and polar di-uracil moieties capable of efficient hydrogen bonding to develop novel CA inhibitors with potent anti-HIV activity. Molecular modelling techniques like molecular docking and molecular dynamics simulation were used for the efficient design of uracil derived CA inhibitors. The interactions and binding affinities of the designed compounds with CA were determined by molecular docking. Molecular dynamics simulations were further performed on superior complexes to validate the binding affinities and interactions.
The drug likeness of the compounds was taken into account before synthesis by considering their pharmacokinetic properties. Lipinski's rule of five was taken as the parameter for screening out compounds with poor absorption and permeability. Additionally, the adsorption, distribution, metabolism, excretion, and toxicity (ADMET) properties were also considered as crucial pharmacokinetic features. The resulting compounds were successfully synthesized via novel synthetic methodology using moderate conditions and mild reagents. The biological assays like anti-HIV p24 assay and cytotoxicity studies of synthesized 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives were carried out for further confirmation of computational observations and the results are discussed here.
2. Experimental section
2.1. Materials and methods
NMR spectra were recorded using 400 MHz (1H NMR) and 101 MHz (13C NMR) on a Bruker Avance-400 spectrometer. The chemical shifts are reported in ppm units with trimethyl silane as internal standard and J values are in hertz. Mass spectra were recorded using AGILENT Technologies 6530B Accurate Mass QTOF-LC/MS spectrometer. Fourier transform infrared (FT-IR) spectra were recorded on a Thermo Nicolet 6700 FT-IR spectrometer. Single crystal X-ray data were collected on Xcalibur Eos, Rigaku Oxford Diffraction instrument X-ray diffractometer with Mo-Kα radiation (λ = 0.71073 Å). Empirical absorption was done using SCALE3 ABSPACK scaling algorithm. The refinement was performed using XL, in the Olex 2-1.2 package and the structural solution by using SHELXS-97. The reagents and solvents were obtained from commercial sources and used by its very nature.Dulbecco's Modified Eagle Medium (DMEM) media, Fetal Bovine Serum and antibiotic solution for p24 assay were obtained from Thermo Fischer Scientific, United States. German collection of microorganisms and animal cell cultures, DSMZ, Germany supplied Mo7e (ACC 104) and BA/F3 (ACC 300) cell lines. The cells were maintained in RPMI 1640 medium (Gibco, Waltham, MA USA) with 10% fetal calf serum (HiMedia, India) in the presence of 20 ng ml−1 human IL-3 and 10 ng ml−1 murine IL-3 (Peprotech Asia, Rehovot, Israel) respectively.
2.2. Docking studies
2.3. General procedure for the synthesis of bis(pyrimidine-2,4(1H,3H)-diones), 1a–28a
Uracil (1 mmol for synthesizing compounds 1a–26a and 28a and 2 mmol for 27a) in 5 ml water and 5 drops of conc. HCl (12 N) were taken in a round bottom flask and kept for stirring to initiate the reaction. After dissolution of uracil, the corresponding aldehyde was added slowly to the reaction mixture. The reaction mixture was refluxed for 6 hours at 70 °C and the progress of reaction was monitored by TLC (using ethyl acetate–hexane mixture). After completion, the mixture was cooled down to room temperature, poured into crushed ice, and neutralized by adding sodium bicarbonate solution (1 M) to allow precipitation. The resulting precipitate was filtered, washed with hot methanol to remove any aldehyde impurities and dried under vacuum to get compounds 1a–28a.2.4. General procedure for the synthesis of 5-(hydroxy(4-nitrophenyl)methyl)pyrimidine-2,4(1H,3H)-dione
Uracil (1 mmol) in 5 ml water and 5 drops of conc. HCl (12 N) were taken in an RB flask and kept for stirring to initiate the reaction. After dissolution of uracil, the corresponding aldehyde (1 mmol) was added slowly to the reaction mixture. The reaction mixture was refluxed for 4 hours at 70 °C and the progress of the reaction was monitored by TLC (using ethyl acetate–hexane mixture). After verifying the formation of the intermediate spot by TLC, the reaction mixture was cooled to room temperature and poured into crushed ice. The reaction mixture was neutralized by adding sodium bicarbonate solution (1 M) to allow precipitation. The resulting precipitate was filtered, washed with hot methanol to remove unreacted aldehyde impurities and dried under vacuum to get intermediate compound.2.5. In vitro anti-HIV activity using the HIV-1 gag p24 inhibition assay
TZM-bl cells were plated at 1 × 104 cells per well in a 96 well plate and were incubated at 37 °C for 24 hours. The cells were infected with HIV-1 viral stocks at TCID50 of 250 for preliminary screening and TCID50 of 400 for IC50 determination. The viral stocks were later incubated with various concentrations of the samples (1 μg ml−1 for preliminary screening and varying concentrations for IC50 determination). The virus – sample complex was incubated at 37 °C for 5 days in CO2 incubator. Following incubation, the cell supernatants were tested for the presence for HIV gap p24 core protein by HIV p24 ELISA kit (Xpress Bio, USA).2.6. Statistical analysis
All results are expressed as mean ± S.D. and experiments were performed in triplicate. Values were compared using ANOVA by GraphPad Prism, version 5 (GraphPad Software, Inc., USA).2.7. Cytotoxicity studies
Cell cytotoxicity assay was performed using WST-1 (Roche, Basel, Switzerland) according to manufacturer's protocol as given in ESI.†3. Results and discussion
3.1. Molecular design
The studies show that the diarylpyrimidine derivatives are highly potent anti-HIV compounds24,28 and hence we were curious if the introduction of alternate pharmacophores can produce compounds with compelling anti-HIV properties. The “four-point pharmacophore model” of existing diarylpyrimidines24 consists of the hydrophobic region, the hydrogen bond interaction region, tolerant regions I and II (open towards solvent protein interface and is the most flexible region). The design of novel bis(pyrimidine-2,4(1H,3H)-diones) was inspired from this model, and modifications were applied to all regions for effective hydrogen bonding with the target as given in Fig. 2. Flexibility is one of the prominent features of HIV drugs, and hence we have incorporated phenyl ring with changeable substitutions. The change in substituents in the phenyl ring can improve the physicochemical properties of the designed compounds. The direct linkage of differently substituted phenyl rings to uracil moieties in the current scheme is maintained to investigate their viral potency. The hydrogen bonding of uracil moieties can offer better membrane permeability than other standard HIV inhibitors. The structures of all the designed compounds are given in Fig. 3.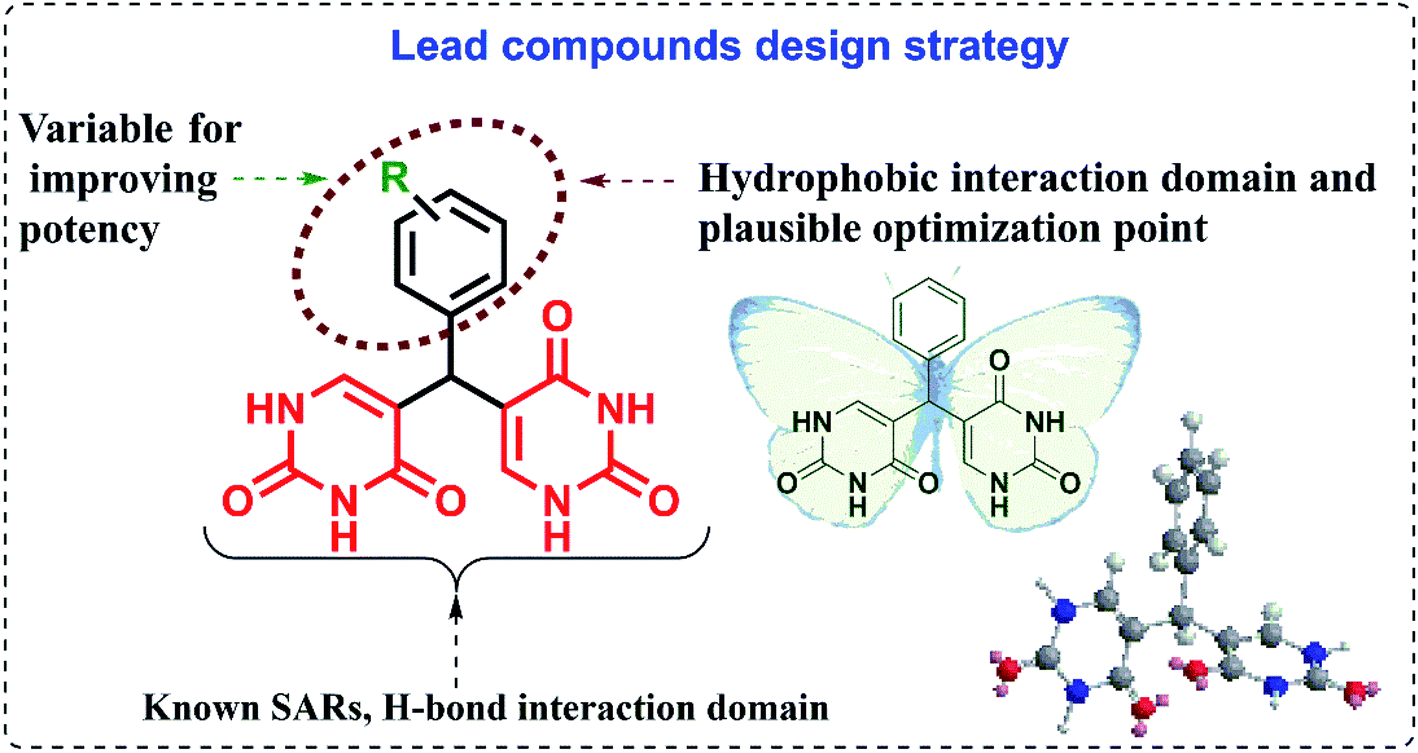 | ||
| Fig. 2 The design strategy of lead compounds. | ||
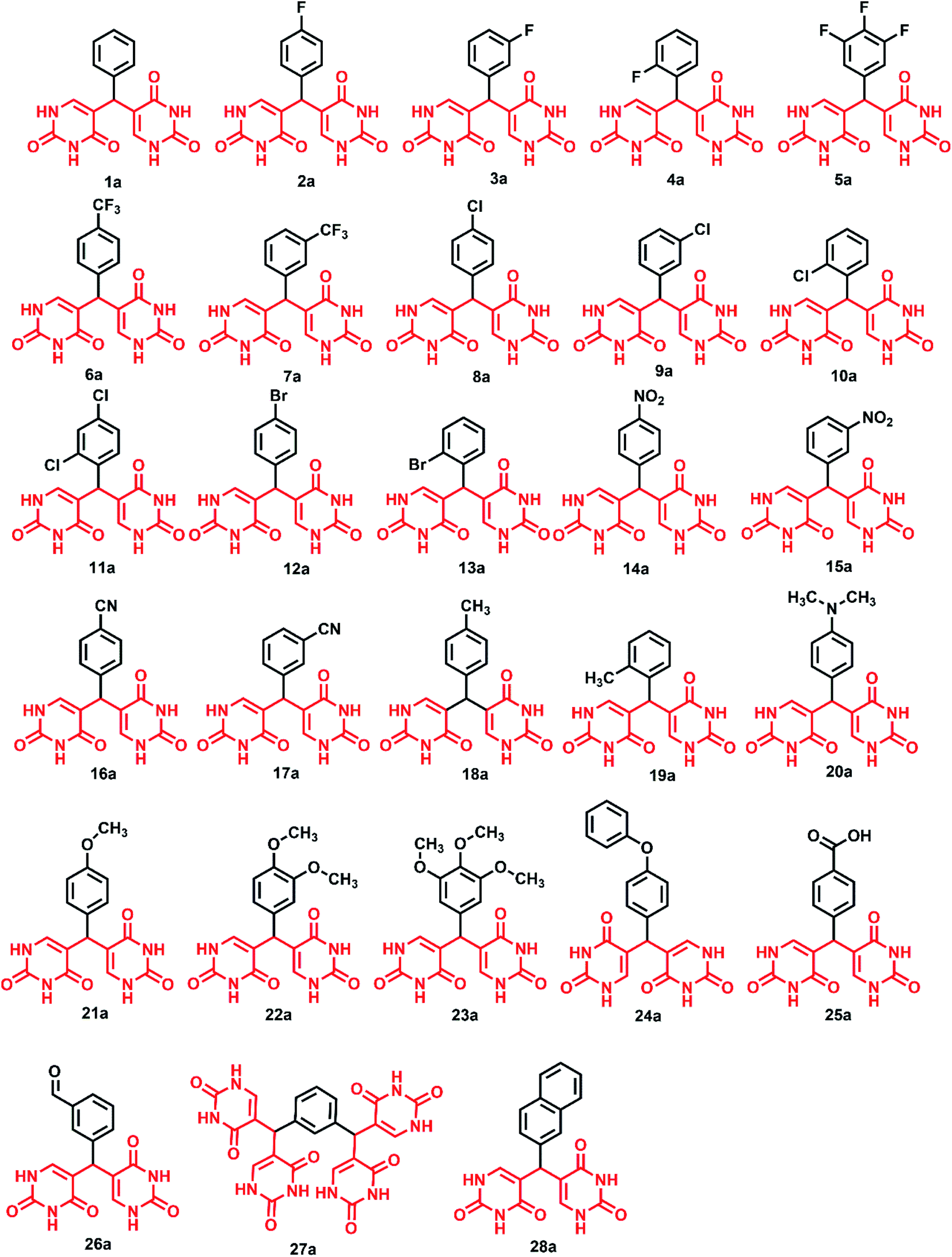 | ||
| Fig. 3 Library of designed 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives. | ||
The drug-like molecular properties of the compounds and ADMET properties were evaluated before molecular modelling studies for effectively screening out the compounds without drug like properties and the results are discussed in the following section.
3.2. Physicochemical properties of 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives
Drug likeness is an intricate balance of a wide range of molecular properties and structural features, namely, lipophilicity, H bonding, molecular size, electronic distribution, etc. determine if a compound is a drug or a non-drug. The presence of these pharmacophoric properties affects the behaviour of a compound within an organism in transportation, protein affinity, metabolic stability, and toxicity.29 Even when absorption and bioavailability are two different parameters of the compound, the information regarding the absorption can help in designing the compounds with higher bioavailability. Lipinski's rule of five is a preliminary effort that helps to screen out the compounds with poor absorption and permeability30,31 and the results are summarised in supplementary Table S1.†LogP value determines the solubility and permeability of the compound which in turn affects absorption, a crucial factor for drug likeness. Drug candidates must possess required solubility to travel via the membrane without being trapped in them. The logP values of the drugs should be in the clogP < 3 range for better in vivo potency as well as for solubility.32,33 All the designed compounds were found to have lower logP values indicating solubility and less toxicity. The positive logP values represents a liking for lipophilic environment and negative values denote a liking for hydrophilic or lipophobic environment.34 Out of 28 compounds, 17 compounds showed negative logP values indicating their lipophobic nature. Moving on to molecular weight as a crucial parameter, except 27a, the molecular weights of all the compounds were in the allotted range of below 500 falling in line with Lipinski's rules. However, with molecular weights below 1000 Da, all these compounds can also be classified under small molecules,35 having several advantages like predictable pharmacokinetics and pharmacodynamics properties, easy manufacture, higher stability and bioavailability.36 These compounds have plentiful NH and OH groups capable of donating H-bonds along with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups to accept incoming bonds and are expected to have a momentous impact on interacting proteins and nucleic acids via H-bonding. The compounds can thereby be stabilised within the binding pockets of the biological target due to H-bonding ability.37 Hence, 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives were designed by considering the number of H-donors and acceptors. As per Lipinski's rule, except 4 compounds, all the remaining derivatives showed H-bonding within the accepted range. The rotatable bond count is the measure of molecular flexibility which along with total polar surface area predicts good oral bioavailability. Higher bioavailability is the measure of designing compounds as good therapeutic agents.38 The increased number of rotatable bonds has negative effect on membrane permeability and surprisingly all the designed 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives have rotatable bonds in the 2–6 range which is acceptable by Veber's rule. Total polar surface area (TPSA) is correlated to the compound's physicochemical properties in terms of toxicities. Compounds with lower TPSA values have adverse toxicities as they can easily cross the cellular membranes and get distributed into tissues. Compounds with low-ClogP/high-TPSA are likely to be 2.5 times cleaner than toxic.33 All the compounds possess high TPSA values from 131.45 to 262.90, eradicating the hypothesis that low TPSA increases the binding of compounds to off-targets.33 As per the drug likeness rules, 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives showed drug like molecular properties and were further evaluated for their respective Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties.
O groups to accept incoming bonds and are expected to have a momentous impact on interacting proteins and nucleic acids via H-bonding. The compounds can thereby be stabilised within the binding pockets of the biological target due to H-bonding ability.37 Hence, 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives were designed by considering the number of H-donors and acceptors. As per Lipinski's rule, except 4 compounds, all the remaining derivatives showed H-bonding within the accepted range. The rotatable bond count is the measure of molecular flexibility which along with total polar surface area predicts good oral bioavailability. Higher bioavailability is the measure of designing compounds as good therapeutic agents.38 The increased number of rotatable bonds has negative effect on membrane permeability and surprisingly all the designed 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives have rotatable bonds in the 2–6 range which is acceptable by Veber's rule. Total polar surface area (TPSA) is correlated to the compound's physicochemical properties in terms of toxicities. Compounds with lower TPSA values have adverse toxicities as they can easily cross the cellular membranes and get distributed into tissues. Compounds with low-ClogP/high-TPSA are likely to be 2.5 times cleaner than toxic.33 All the compounds possess high TPSA values from 131.45 to 262.90, eradicating the hypothesis that low TPSA increases the binding of compounds to off-targets.33 As per the drug likeness rules, 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives showed drug like molecular properties and were further evaluated for their respective Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties.
3.3. ADMET properties of 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives
Most of the potential drug candidates in in vitro studies fail in further drug development stages due to their efficacy and safety profile, highlighting that ADMET properties (as given in Table S1†) play a vital role in drug design. Solubility is one of the basic requirements of an orally administrated drug, and higher lipophilicity impacts the aqueous solubility. The optimal drug solubility is higher than −4log mol L−1, and all the designed compounds showed solubility higher than −3log mol L−1. The ability for absorption determines the oral bioavailability and hence it is important to screen the compounds for intestinal permeability before synthesis. The human colon adenocarcinoma (Caco-2) cell line has morphological and functional similarities with human enterocytes, thereby making that a favoured model to estimate the permeability of drug candidates in high throughput screening. As required, all the derivatives showed higher permeability than the optimal value of −5.15.39 As the intestine is the primary absorption site, the percentage of absorption of the compounds was estimated. Except for two compounds (25a and 27a), all the derivatives showed absorption similar to or higher than C1.
The volume of distribution (VD) of the compounds was evaluated and higher VD shows drug distribution in tissues. LogVD < −0.15 is considered lower and except 28a, all other compounds are better distributed in plasma than tissues. The fraction unbound of all the compounds was lower and in the range of reference drugs indicating that the compounds can be diffused to cellular membranes. Like reference drugs, the designed compounds were blood–brain barrier (BBB) impermeable as they displayed logBBB < −0.1. The cytochromes are responsible for the biotransformation of most of the foreign objects and hence the metabolism of the designed compounds by cytochromes were evaluated in silico. Majority of designed compounds were identified as substrates and/or inhibitors of CYP3A4 and CYP1A2. The study is beneficial as it predicts whether parallel administration of compounds with a known CYP substrate can alter drug disposition, toxicity or efficacy.40 The total clearance, oral rat toxicity and hepatotoxicity, were also considered parameters for drug design.
3.4. In silico studies
Prior to synthesis, the compounds were explored via in silico analysis to validate the drug design for p24 protein. Molecular docking studies against HIV CA were carried out followed by molecular dynamic simulation studies for the most promising compounds.The binding pockets for both NTD and CTD are well documented.26,44–48,50 CTD binding pocket residues include Arg173, Gln179, Lys182, Tyr169 and Leu172.46–48,50 NTD binding pocket contains numerous important residues including, Asn53, Leu56, Asn57, Thr58, Val59, Gln63, Met66, Gln67, Leu69, Lys70, Ile73, Tyr130, Ala105 and Thr107.46–48,50 The results of the docking study were extremely promising as all members of the library had multiple interactions within the NTD binding pocket. All compounds showed hydrophobic interactions within the NTD pocket.
All the compounds except 1a, 27a and 17a, showed one or more H-bonds with residues from Asn57 to Lys70. Asn57 interaction in particular is a highly conserved residue for CA intra-hexamer NTD–NTD hydrogen bond network.41 GlideScores of compounds were compared to identify which is most promising, more negative glidescores correspond with better binding bearing in mind that there are no absolute good or Glidescores only relative comparisons to be made. 9a was most promising (as given in Fig. 4) with its glidescore of −6.41 kcal mol−1 but compounds 7a, 10a, 1a, 24a and 3a were the next most promising based on their binding scores between −5.52 kcal mol−1 to −4.99 kcal mol−1. The remaining compounds had glidescores between −4.02 kcal mol−1 and −4.91 kcal mol−1. Molecular dynamics simulations were performed to understand the stability of the protein-ligand complex of 9a and HIV CA monomer due to 9a having the highest GlideScore.
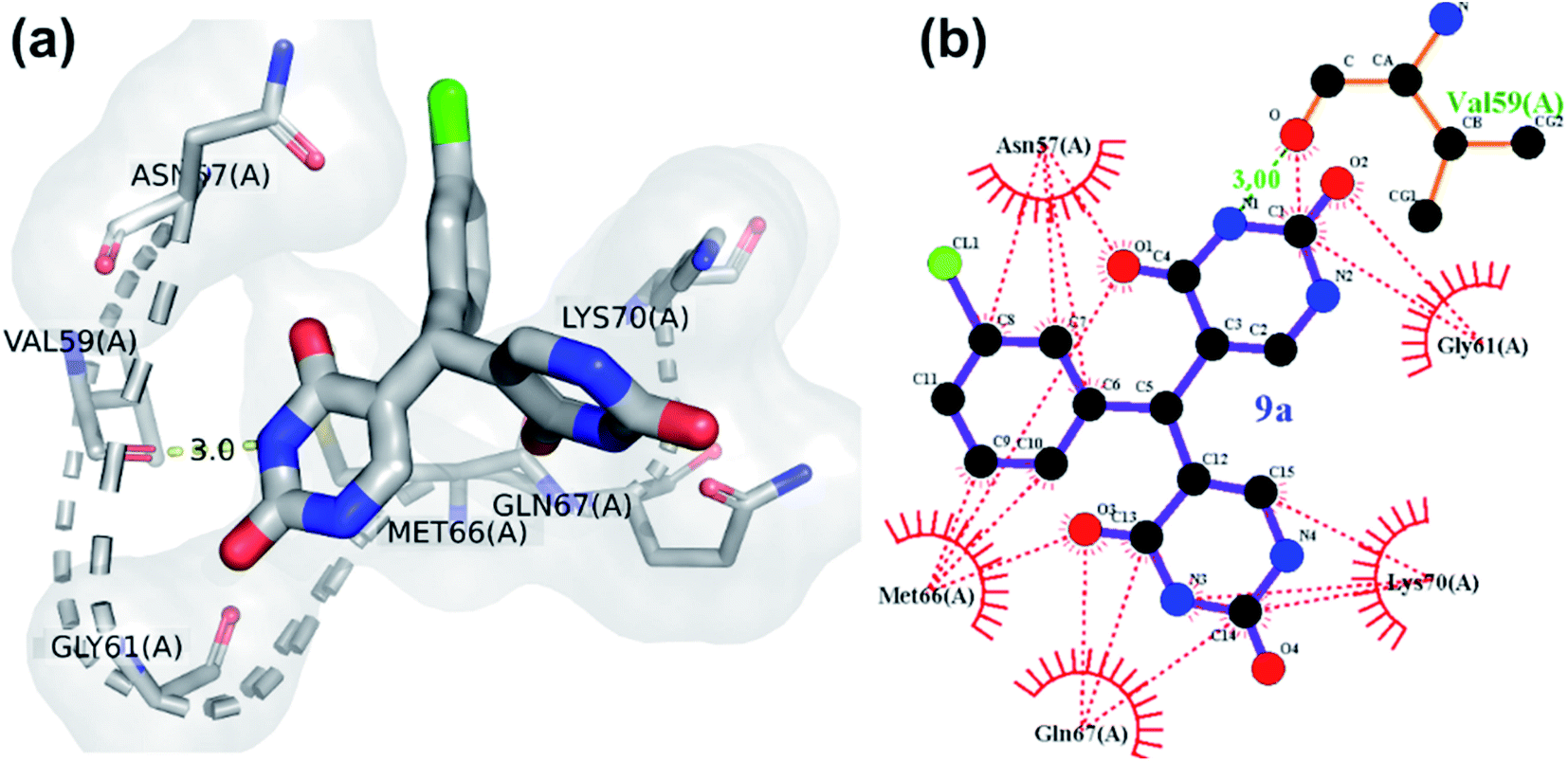 | ||
| Fig. 4 Binding interaction of compound 9a with HIV-CA (a) 3D diagram (b) ligplot diagram. | ||
| Compound code | Glidescore (kcal mol−1) | Interacting residuesa | |
|---|---|---|---|
| H bond interactions (Bond length in Å) | Hydrophobic interactions | ||
| a Key pocket residues are marked in bold letters. Residues capable of drug-resistance related mutations are given in italic and bold letters.48 H-bonds between 2.7 Å to 3.35 Å and hydrophobic contacts between 2.90 Å and 3.90 Å were considered. | |||
| 1a | −5.26 | — | Met66, Gln63, Ile73, Leu56, Lys70, Asn57 |
| 2a | −4.46 | Val59 (3.02) | Met66, Lys70, Gln67, Asn57, Gly61, Val59 |
| 3a | −4.99 | Val59 (2.85), Met66 (2.78) | Lys70, Leu56, Leu69, Ile73, Asn57, Gly61, Gln63, Val59, Met66 |
| 4a | −4.46 | Gly61 (3.06), Gln63 (2.83) | Met66, Asn57, Leu56, Gly60, Val59, Gln67, Gln63, Gly61 |
| 5a | −4.60 | Gln67 (2.80), Val59 (2.89) | Met66, Lys70, Asn57, Val59, Gln67 |
| 6a | −4.91 | Gly61 (2.84), Gln63 (2.96), Val59 (3.22) | Gln67, Asn57, Met66, Gly60, Gln63, Val59, Gly61 |
| 7a | −5.52 | Gln67 (3.03), Asn57 (3.01) | Met66, Asn57, Gln63, Gln67 |
| 8a | −4.61 | Gly61 (3.22, 2.96), Gln63 (2.88) | Met66, Leu56, Asn57, Gln67, Gly60, Gly61, Gln63 |
| 9a | −6.41 | Val59 (3.00) | Met66, Gln67, Lys70, Gly61, Asn57, Val59 |
| 10a | −5.31 | Asn57 (2.95), Val59 (2.94), Gly61 (2.85) | Lys70, Met66, Gly60, Gln63, Gln67, Val59, Gly61, Asn57 |
| 11a | −4.54 | Gly61 (2.87, 2.82) | Met66, Asn57, Leu56, Gln63, Gly60, Gly61 |
| 12a | −4.49 | Gly61 (2.76, 2.96), Gln63 (2.88) | Met66, Asn57, Leu56, Gln67, Gly60, Gln63, Gly61 |
| 13a | −4.32 | Asn57 (2.96), Val59 (2.96), Gly61 (2.88) | Met66, Gly60, Gln63, Lys70, Gln67, Asn57, Val59, Gly61 |
| 14a | −4.37 | Gly61 (2.76, 3.11), Gln63 (3.03) | Met66, Leu56, Asn57, Gly60, Gln67, Gly61, Gln63 |
| 15a | −4.58 | Val59 (2.99), Gly61 (3.02), Lys70 (3.05) | Met66, Gly60, Gly67, Asn57, Val59, Gly61, Gln63 |
| 16a | −4.30 | Val59 (3.22), Gly61 (2.86), Gln63 (3.03) | Met66, Gly60, Gly67, Leu56, Asn57, Val59, Gly61, Gln63 |
| 17a | −4.17 | Val59, Gly61, Gln63, Gln67, Lys70 | — |
| 18a | −4.53 | Gln57 (3.03), Val59 (2.86) | Gln57, Val59, Tyr130, Asn53, Leu56, Met66, Asn57, Lys70 |
| 19a | −4.66 | Val59 (2.98), Gly61 (2.90), Asn57 (3.01) | Gly60, Leu56, Lys70, Met66, Gln67, Gln63, Val59, Gly61, Asn57 |
| 20a | −4.43 | Lys70 (3.08), Gln67 (2.78) | Lys70, Gln67, Gly60, Val59, Gln63, Asn57, Asn53, Leu56, Ile73, Met66, Tyr130 |
| 21a | −4.61 | Gly61 (2.93, 3.32), Gln63 (2.83), Val59 (3.08) | Gln67, Leu56, Asn57, Met66, Gly60, Gly61, Gln63, Val59 |
| 22a | −4.43 | Val59 (3.00), Gln67 (3.02) | Gln63, Leu56, Asn57, Lys70, Asn53, Leu69, Met66, Gln67, Val59 |
| 23a | −4.21 | Gln67 (2.92) | Asn57, Val59, Asn53, Lys70, Leu69, Met66, Gln63, Gln67 |
| 24a | −5.15 | Gly61 (2.77, 3.14) | Gly60, Leu56, Asn57, Ile73, Asn53, Tyr130, Met66, Gln63, Gln67, Gly61 |
| 25a | −4.72 | Asn57 (3.12), Val59 (2.75), Gln63 (3.03), Met66 (2.87) | Leu56, Gly60, Gln63, Met66, Asn57, Val59 |
| 26a | −4.02 | Gly61 (2.99), Val59 (2.84), Lys70 (3.15) | Gly60, Gln63, Gln67, Asn57, Met66, Val59, Gly61 |
| 27a | −4.50 | — | Asn53, Asn57, Val59, Gly61, Gln63, Met66, Gln67, Lys70, Tyr130 |
| 28a | −4.20 | Gln67 (2.91), Val59 (2.72) | Met66, Leu56, Asn57, Lys70, Gln67, Val59 |
The hydrogen bond interaction of HIV-1 CA monomer with compound 9a is shown in Fig. 4. The interactions of 9a with HIV-1 CA monomer has a −6.41 kcal mol−1 glidescore which is significantly better than the second highest interacting agent compound 7a at −5.52 kcal mol−1. Residues Val59, Gln67, and Lys70 were observed to form hydrogen bonds with compound 9a. These amino acid residues are found in the R3 pocket of the protein molecule. The R3 pocket plays a vital role in forming a bridge between the NTD of one HIV CA monomer and the CTD of another monomer.41,42 This indicates that ligand molecule bound to active site amino acid residues, may hinder the HIV-1 capsid assembly. The conformations over time of protein and protein–ligand complex were compared by generating the root means square deviation (RMSD) plot as shown in Fig. 5c. RMSD is coarse method to note deviation of conformation or change in physical location in comparison to an initial state. In the current study, protein alone does not undergo significant conformational changes as indicated by the RMSD and finds relatively stability at an early stage and does not deviate much for the remainder of the simulation without much fluctuation. On the other hand, the protein-ligand complex shows higher fluctuation and is less rigid than the protein alone. The RMSD of the protein indicates relative stability from 25 ns to 100 ns. In the case of the protein–ligand complex, the RMSD value fluctuates markedly throughout the simulation. The protein–ligand complex achieves relative stability from 80 ns to 100 ns when compared to the first 80 ns wherein the fluctuations were much higher. This graph suggests the compound 9a has a profound impact on protein conformation and stability. The root means square fluctuation (RMSF) graph is a coarse method that allows us to observe which amino acid residues deviate from their position during simulation. The RMSF plot shows residue numbers 75 to 100 of protein–ligand complex deviate greatly as shown in Fig. 5d. This is significant as the active site of the protein exists between the residues 75 to 100. The ligand significantly disrupted the active site residues when compared with simulation of protein alone. The radius of gyration of only protein maintains a constant size throughout simulation, whereas, for the protein–ligand complex the radius of gyration fluctuates in size within the 0.5 nm range as shown in Fig. 5a. This result indicates compound 9a has a mild effect on change in overall protein size. The plot of solvent-accessible surface area (SASA) in Fig. 5b shows that in protein–9a complex SASA has overall decreased in comparison with simulation of plain protein. The lower SASA is a direct result of compound 9a being present in the binding site cavity. This lower accessibility of solvent to the binding cavity may be detrimental as water molecules present in the binding site are known to help stabilize HIV capsid conformations.41 The binding energy of 9a post dynamics simulation as obtained from PRODIGY server analysis was −6.4 kcal mol−1.51
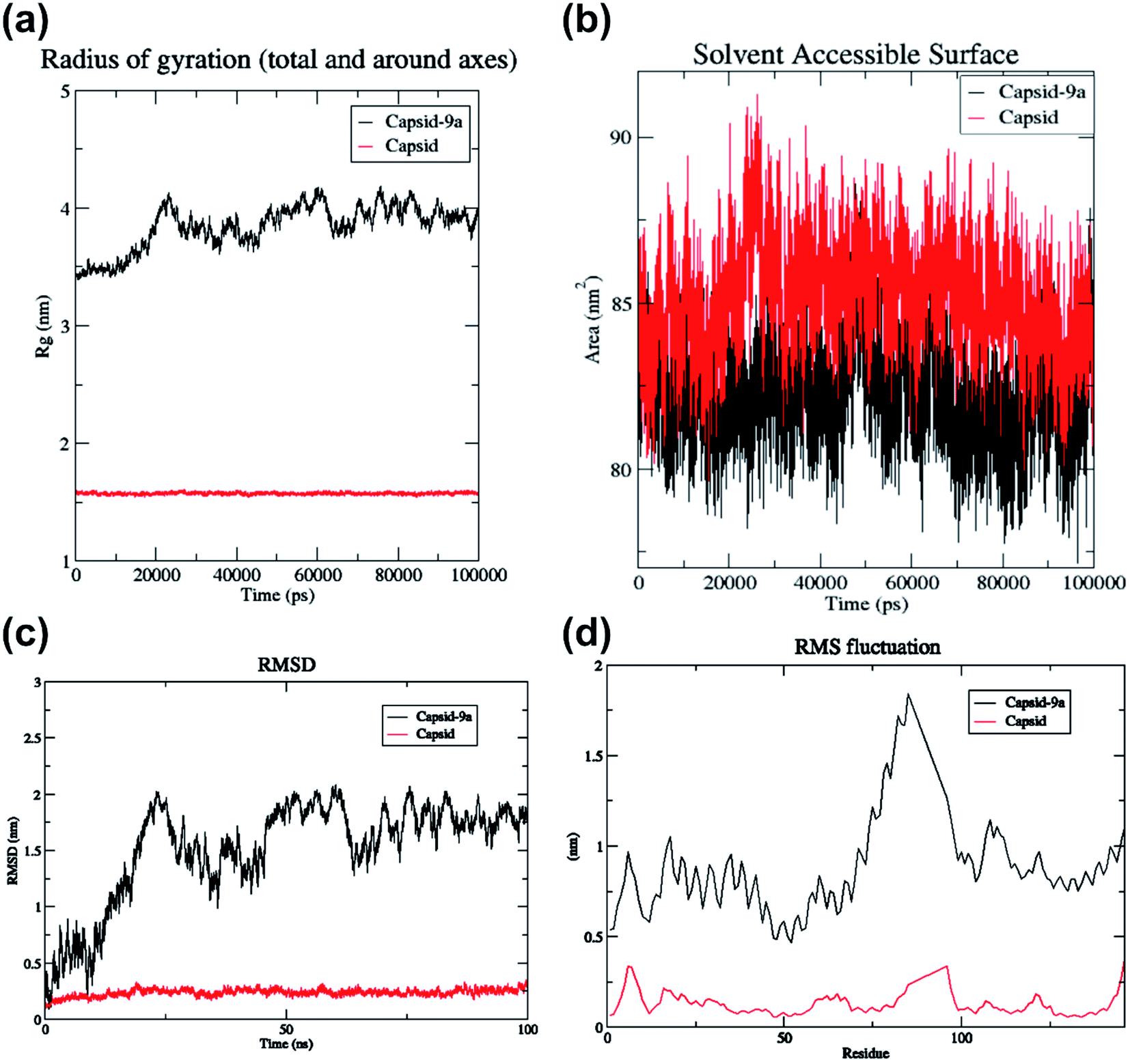 | ||
| Fig. 5 Data obtained from molecular dynamics simulation of HIV-1 CA monomer with compound 9a. | ||
3.5. Chemistry
The promising results from in silico studies led us to proceed with the synthesis of designed compounds. The synthesis involving uracil and benzaldehyde in literature ended up with a mixture of 5-(chloro(phenyl)methyl)pyrimidine-2,4(1H,3H)-dione and bis(pyrimidine-2,4(1H,3H)-diones).52 The isolation of bis(pyrimidine-2,4(1H,3H)-diones) as major product was unreported and this prompted us to delve deeper to design the synthetic strategy for these compounds as depicted in Scheme 1. Here, the reaction between differently substituted aldehydes and uracil in 1:1 ratio in H2O with conc. HCl as catalyst at 70 °C yielded novel 5,5′-(phenylmethylene)bis(pyrimidine-2,4(1H,3H)-dione) derivatives. The novel bis pyrimidinedione derivatives having both hydrophilic and lipophilic groups did not undergo any further reactions and were stable. The optimization of reaction conditions is given in Table S2.† For understanding the structure–activity relationships (SARs), numerous substituted bis pyrimidine derivatives (1a–28a) were prepared. In an analogous approach, 5,5′,5′′,5′′′-(1,3-phenylenebis(methanetriyl))tetrakis(pyrimidine-2,4(1H,3H)-dione) (27a) was synthesized. Here, 27a was synthesized using the same reaction conditions from isophthalaldehyde and uracil in 1:2 ratio.
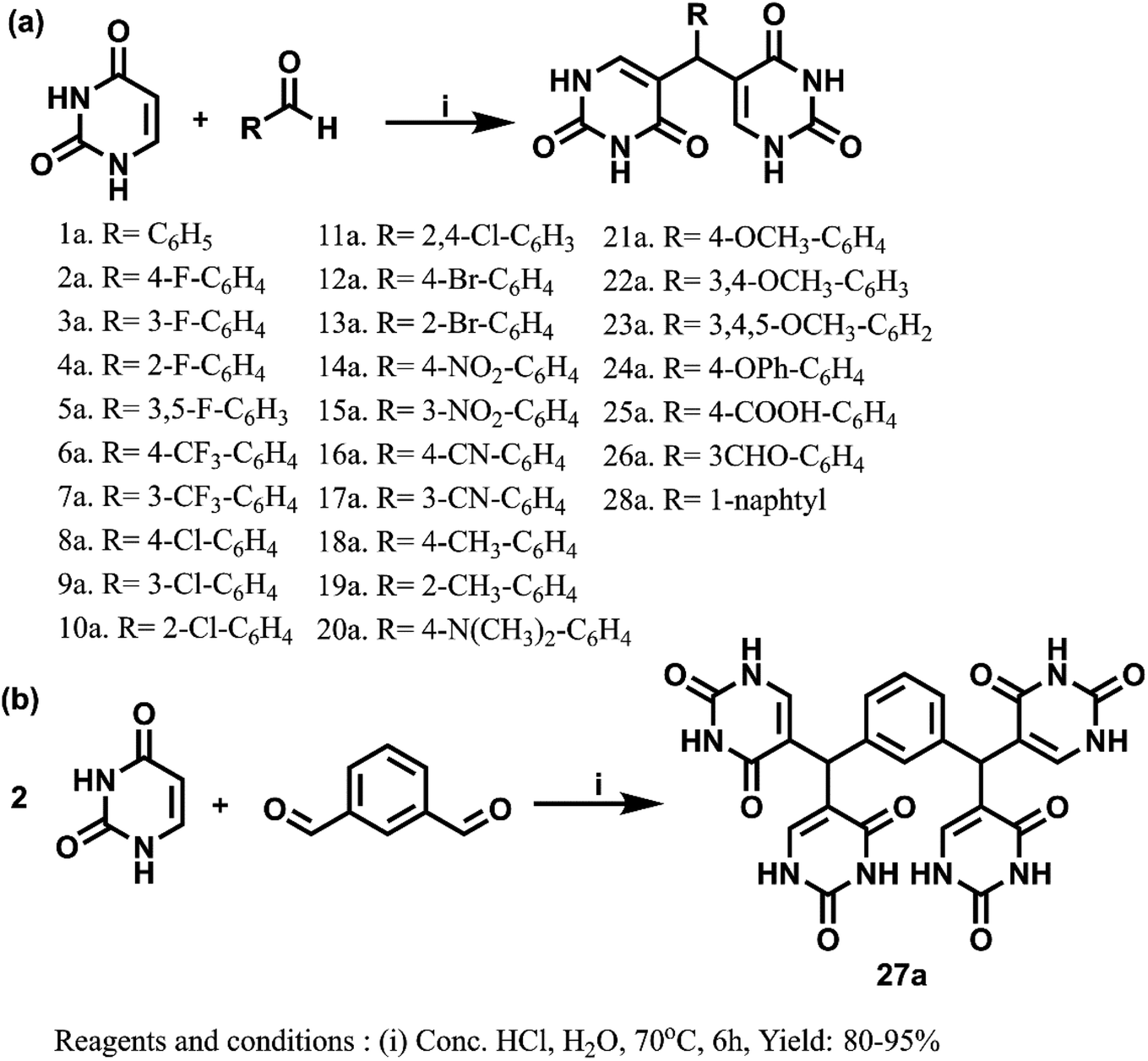 | ||
| Scheme 1 (a) Synthesis of bis(pyrimidine-2,4(1H,3H)-diones). (b) Synthesis of 5,5′,5′′,5′′′-(1,3-phenylenebis(methanetriyl))tetrakis(pyrimidine-2,4(1H,3H)-dione). | ||
The uracil derivatives so formed were characterised by spectroscopic techniques. In all the compounds, the characteristic peak of the bis(pyrimidine-2,4(1H,3H)-diones) derivatives appears as singlet around 5–6 ppm in 1H Nuclear Magnetic Resonance (NMR) spectra and around 35–50 ppm in 13C NMR spectra, occasionally merging with DMSO septet. The NH protons of uracil appear as singlets around 10–11 ppm in 1H NMR spectra and CO peaks of uracil appear at 160–163 ppm in 13C NMR spectra. The experimental and theoretical molecular weights show exact likeness in mass spectra indicating the formation of expected product.
3.6. Mechanistic considerations
In case of reaction between uracil and 4-nitro benzaldehyde (Scheme 1), initially 5-(hydroxy(4-nitrophenyl) methyl) pyrimidine-2,4(1H,3H)-dione was isolated along with the expected product. The structure of intermediate compound in Fig. 6b was confirmed by NMR spectra and single crystal XRD data. The characteristic hydroxy peak of the intermediate appears as a singlet at 4.5 ppm merging with solvent water peaks. The CH protons resonate at 5.59 ppm and the uracil NH peaks appear at 10–11 ppm in 1H NMR spectra. Moving on to 13C NMR spectra, C–OH peak appears at 67.12 ppm and CO peaks appear around 150–163 ppm. The experimental and theoretical molecular weights show exact likeness indicating the formation of intermediate compound. To verify the role of the isolated intermediate, further reaction of this compound with uracil was carried out as per standardised reaction conditions. The reaction proceeded to form 5,5′-((4-nitrophenyl) methylene) bis(pyrimidine-2,4(1H,3H)-dione), which was yet again characterised by spectroscopic techniques. The previously reported formation of 5-(chloro(phenyl)methyl)pyrimidine-2,4(1H,3H)-dione52 was not observed and this led us to develop a plausible mechanism for the reaction as given in Fig. 6a. The synthetic protocol involves the activation of carbonyl compound by the acid catalyst followed by initial nucleophilic addition of uracil to form the intermediate. After the formation of intermediate, the reaction may proceed via unimolecular substitution. The 5-(hydroxy(4-nitrophenyl) methyl) pyrimidine-2,4(1H,3H)-dione intermediate was isolated as the chance of formation of intermediate is higher since the benzylic carbocation is becoming less stable due to the presence of electron withdrawing nitro group at para position.
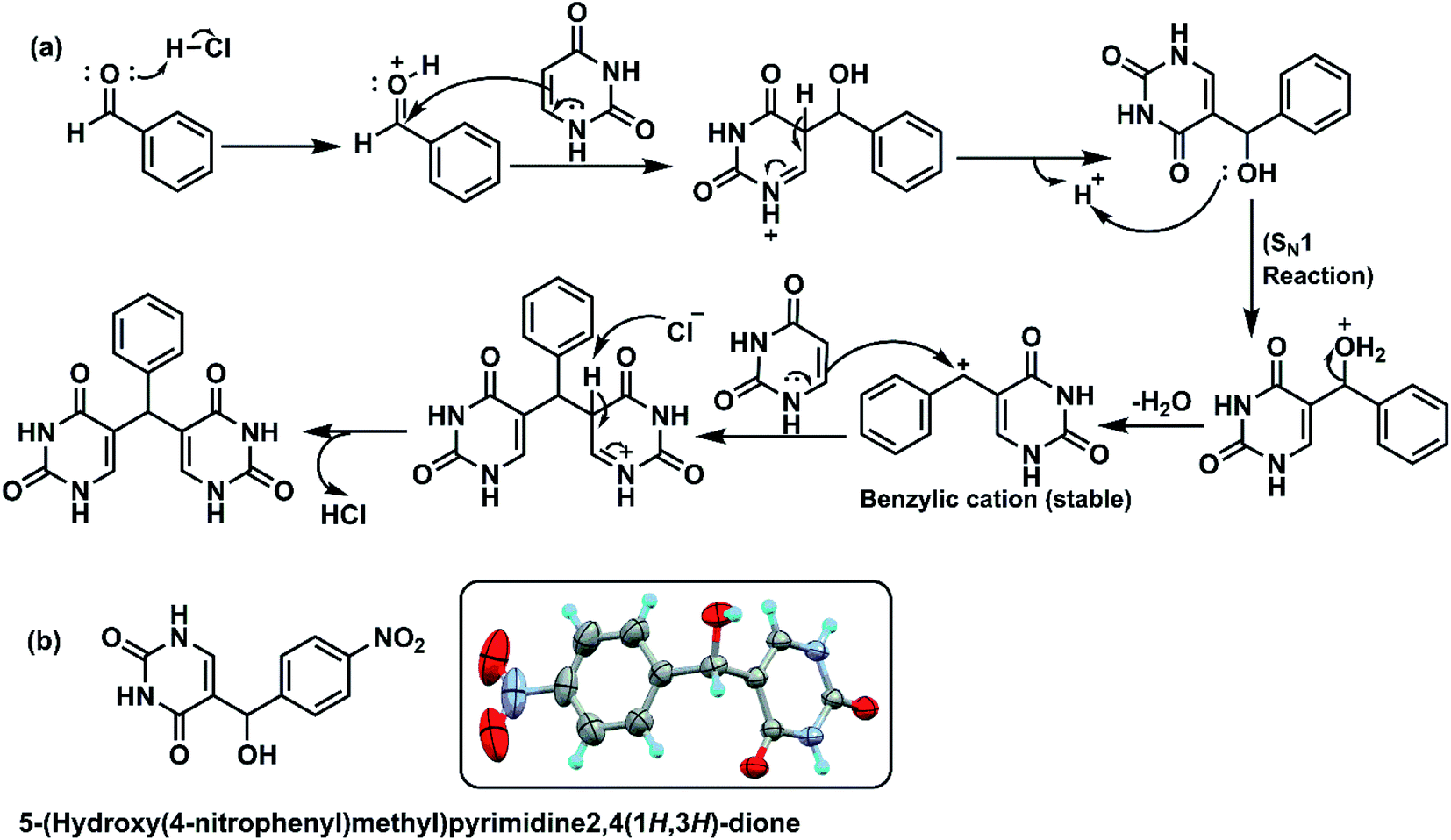 | ||
| Fig. 6 (a) Plausible reaction mechanism of formation of bis(pyrimidine-2,4(1H,3H)-dione) derivatives, (b) structure of an isolated intermediate. | ||
The protonation of the intermediate and subsequent SN1 reaction forms the resultant bis(pyrimidine-2,4(1H,3H)-diones). All the aforementioned derivatives were purified by washing with hot methanol resulting in the isolation of desired products, the structures were assigned based on spectral data and analytical evidences.
3.7. Crystallisation of 5-(hydroxy(4-nitrophenyl)methyl)pyrimidine-2,4(1H,3H)-dione and structure determination
The intermediate 5-(hydroxy(4-nitrophenyl)methyl)pyrimidine-2,4(1H,3H)-dione (given in Fig. 6b) was isolated in the reaction between uracil and 4-nitro benzaldehyde. The intermediate was washed with hot methanol to remove any unreacted aldehyde impurities before crystallization. The final pure compound was dissolved in minimal amount of DMSO and crystallized at room temperature. The XRD data are summarised in Table S3† and single crystal XRD diagram of intermediate (CCDC Number: 1946178) is given in Fig. 7. Here, two molecules of intermediate interacted via two CO and NH bonds of uracil moiety. This is in accordance with the reported H-bonding interactions in uracil.53 The H-bonding stabilizes the crystal structure. However, the free hydroxy group in intermediate compound did not participate in H bonding. The packing diagram reveals the presence of 5-(hydroxy(4-nitrophenyl)methyl)pyrimidine-2,4(1H,3H)-dione in the corners.
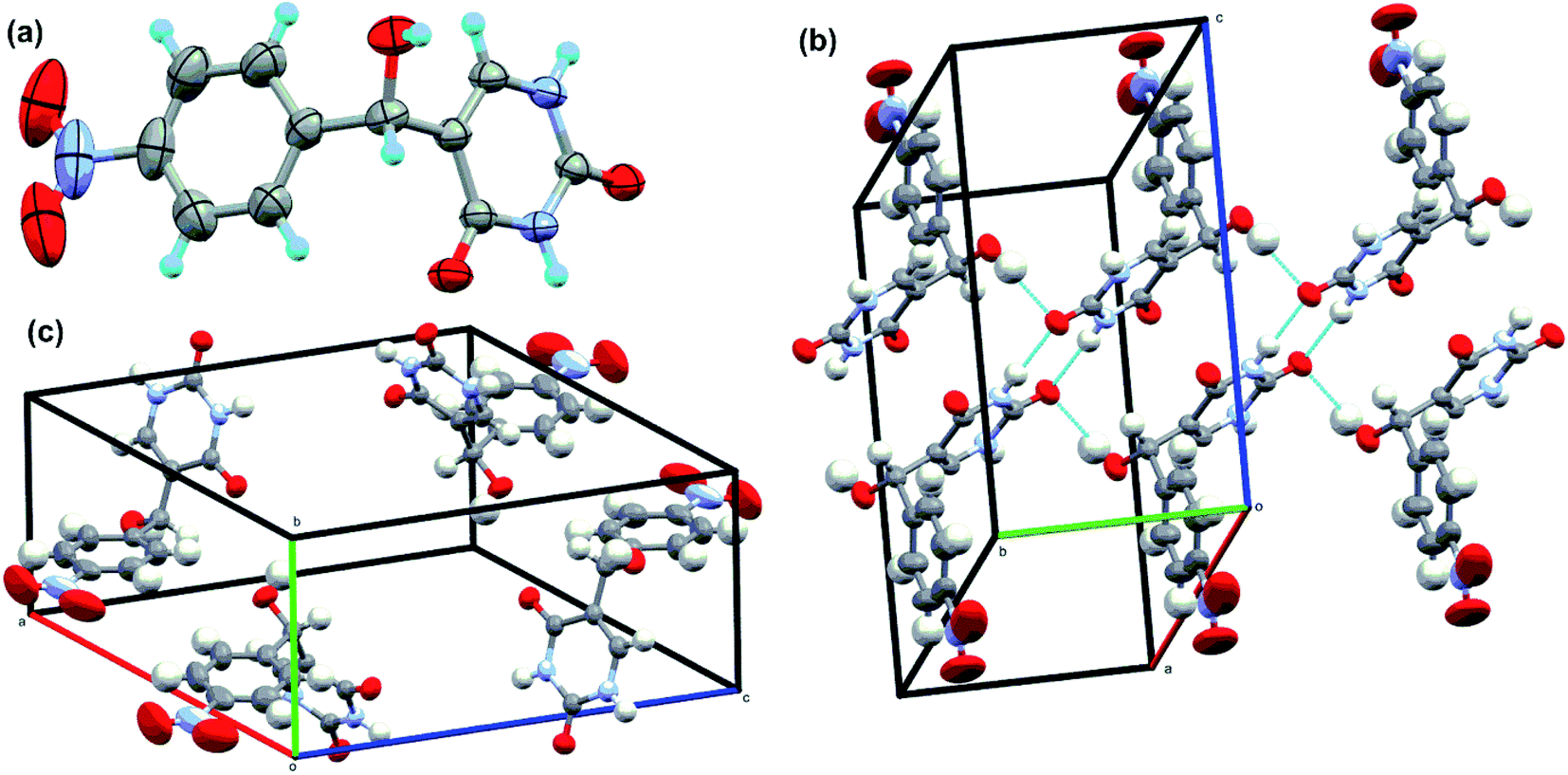 | ||
| Fig. 7 Single-crystal XRD data. (a) The ORTEP diagram, (b) hydrogen bond interaction diagram and (c) packing diagrams of intermediate compound in Fig. 6. | ||
3.8. Biological evaluation
To explore anti-HIV-1 activity, 1 μg ml−1 of bis(pyrimidine-2,4(1H,3H)-diones) were initially tested against activated PBMC infected with 250 TCID50 of a lab adapted strain of HIV-1C as given in Fig. 8. The detailed results are given in Table S4.† In the preliminary screening at 1 μg ml−1, five compounds showed promising activity with percentage inhibition varying from 58% (9a) to 74% (7a). Most of the compounds gave less than 20% inhibition and the modifications seem to render the bis(pyrimidine-2,4(1H,3H)-diones) inactive towards HIV-p24. However, out of 5 compounds showing promising activity, four compounds were halogen derivatives. Compared to other derivatives, the higher activity of halogens may be due to their high lipophilic nature which readily allows them to pass through the lipid layers of the viral membranes. This is well connected with the use of halogen bonding in HIV drug development as they can avoid the risks of emerging drug resistance.54 The preliminary active compounds were further screened for their cytotoxicity before with IC50 determination.
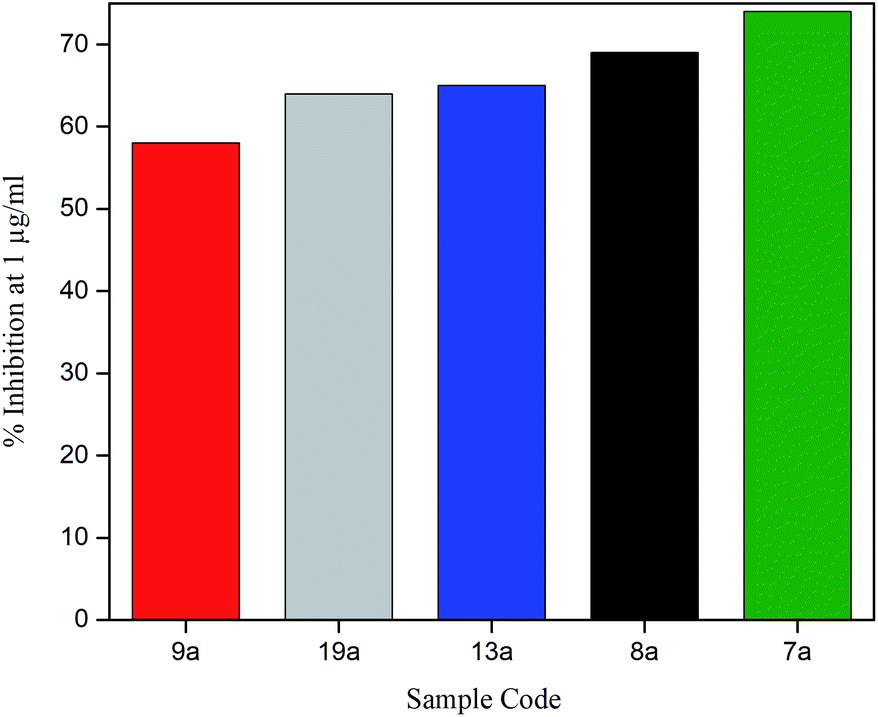 | ||
| Fig. 8 Effect of bis(pyrimidine-2,4(1H,3H)-diones) on HIV-1 p24 antigen production at 1 μg ml−1. | ||
3.9. Evaluation of cytotoxicity of promising compounds
Cytotoxicity of compounds 7a, 8a, 9a, 13a and 19a were tested in human megakaryocyte cell line (Mo7e) and Murine pro B cell line (BA/F3). Compounds inhibiting greater than 50% of cell growth were considered cytotoxic. When treated with 1 μg ml−1 of 7a, 8a, 9a, 13a and 19a, more than 90% of viability in Mo7e cells was observed. Similarly, we observed more than 90% viability in Mo7e cells treated with 5 μg ml−1 of all the compounds except 9a and 19a. Also, in BA/F3, cell viability of more than 90% was visible when treated with 1 μg ml−1 and 5 μg ml−1 of compounds 7a, 8a, 9a, 13a and 19a. Overall our results suggest that 1 μg ml−1 concentration of compounds 7a, 8a, 9a, 13a and 19a are not cytotoxic to Mo7e and BA/F3 cells as given in Fig. 9.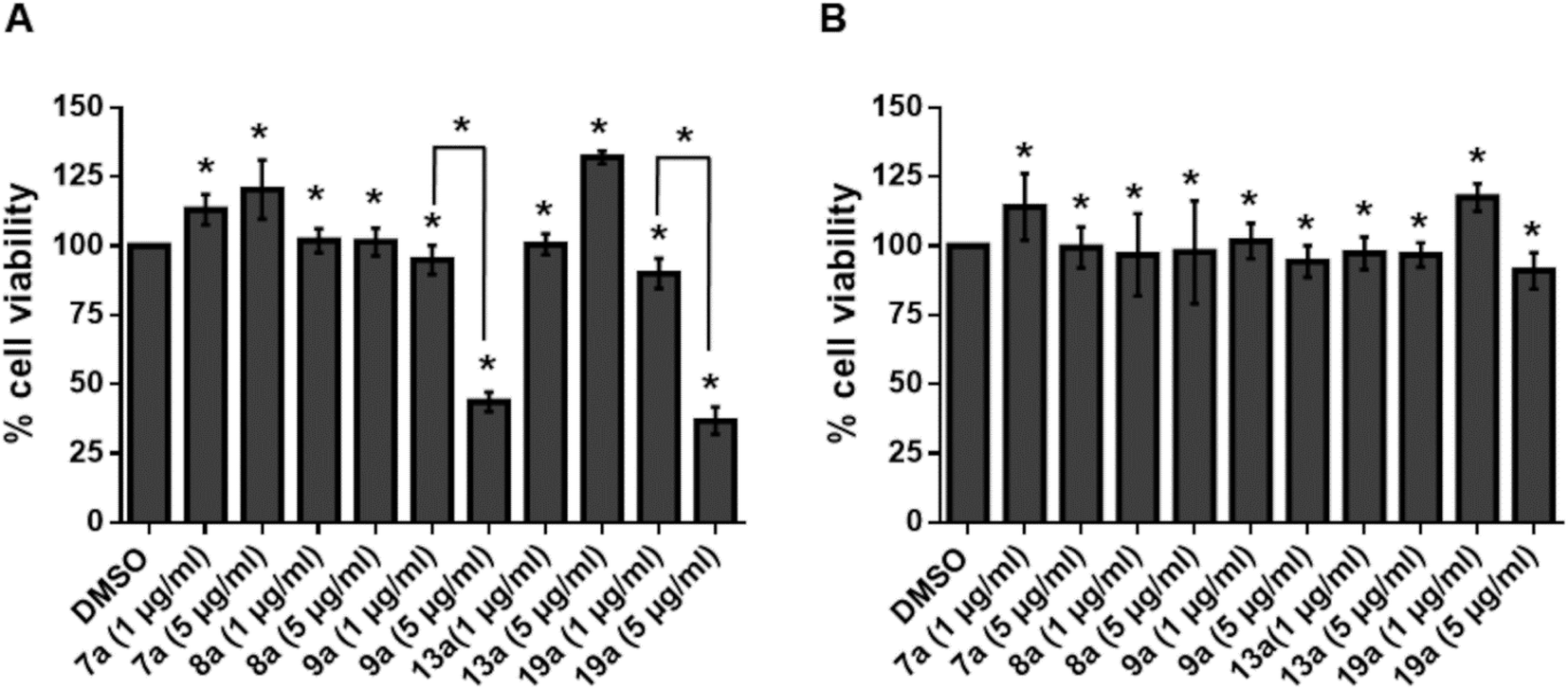 | ||
| Fig. 9 Percentage cell viability of Mo7e and BA/F3 against 7a, 8a, 9a, 13a and 19a. (A) Mo7e cells and (B) BA/F3 cells were stimulated with indicated concentrations of 7a, 8a, 9a, 13a and 19a for 24 hours. Data are represented as mean ± s.e.m. P value is calculated using Student's t-test. *P < 0.05. | ||
3.10. Evaluation of IC50 values of active compounds against p24 of HIV-1C
The inhibitory effect of the five active bis(pyrimidine-2,4(1H,3H)-diones) were further evaluated by HIV-p24 assay with 400 TCID50 of HIV-1C. The higher virus concentration was deliberately selected, and the IC50 of the compounds were evaluated in vitro and results are summarised in Fig. 10 and Table S5.†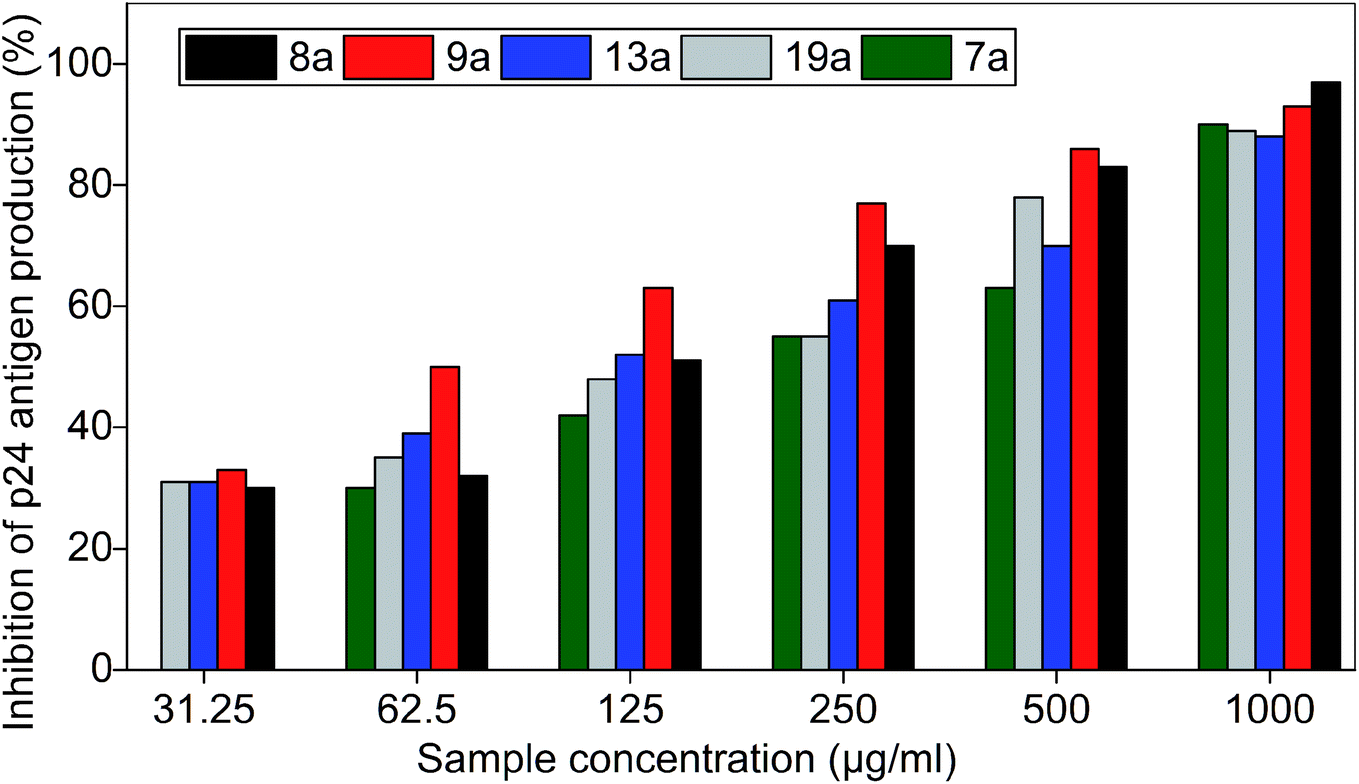 | ||
| Fig. 10 Inhibition of p24 of HIV-1C by bis(pyrimidine-2,4(1H,3H)-diones). Azidothymidine (AZT) was used as positive control and virus control was 1 × 104 PFU ml−1. The data shown is the mean ± S.D. of three independent experiments represented in percentage of inhibition. | ||
IC50 values of the compounds are as follows: 7a (191.5 μg ml−1), 8a (120.5 μg ml−1), 9a (62.5 μg ml−1), 13a (112.4 μg ml−1), and 19a (152.4 μg ml−1).
All the compounds which gave promising inhibition in preliminary studies are biologically and chemically diverse. The structure–activity relationships of the compounds are discussed in detail here. Most of the active compounds were halogen derivatives indicating that the functional group may play a role in the anti-HIV activity. However, the location of various halogens also influenced the anti-HIV activity with the compounds exhibiting multiple levels of potencies against HIV-1C with IC50 values ranging from 191.5 μg ml−1 to 62.5 μg ml−1. Compounds 8a, 9a and 13a were characterised by the presence of mono-halogen substituents like Cl and Br. Compared to the non-substituted 1a and other mono-halogen substitutions (2a, 3a, 4a, 10a, and 12a), the presence of Cl and Br substitutions is favourable for HIV-1C inhibition as seen from the in vitro studies. Surprisingly, F-substitutions (2a, 3a and 4a) made the compounds inactive against the pathogen, contrary to the reported works as most of the HIV drugs have F-derivatives. Notably, meta-Cl substitution (9a) with IC50 value of 62.5 μg ml−1 was necessary for the enhancement of anti-HIV activity when compared to the other substitutions. The higher activity of 9a may be due to lower logP value and meta effect of the substituent. Meta substitution with a non-flat substituent like Cl can enhance the aqueous solubility without affecting the lipophilicity, thereby improving the biological activity.55 The presence of para-Cl substitution in 8a drastically changed the IC50 value to 120.5 μg ml−1 while ortho-Cl in 10a was inactive against HIV-1C. However, reverse trend was observed for bromo substitutions as para-Br substitution in 12a was inactive when compared to ortho-Br substitution in 13a.The latter displayed an IC50 value of 112.4 μg ml−1. The better activity of 13a may be credited to its lower logP value indicating higher lipophilicity to permeate through viral cell membranes. Compounds 5a and 11a were characterised by disubstituted halogens and seemed to be unfavourable for anti-HIV activity as compared to monovalent analogues. For compounds bearing halogens, the position priority as observed from IC50 values are, meta > ortho > para as in the case of the compounds 9a > 13a > 8a.
For compounds 14a–17a, strong electron withdrawing groups like NO2 and CN seems to negatively affect the inhibitory potential of HIV-1C. In contrast, introducing an electron donating group like methyl derivative showed potent anti-HIV activity. However, considering the positions of the methyl derivatives, the IC50 values varied sharply with ortho-methyl substitution (19a) having an IC50 value of 152.5 μg ml−1 while para-methyl substitution (18a) remained inactive. Trifluoromethyl substitutions (6a and 7a) were also introduced as they are versatile groups in medicinal chemistry. However, it was not contributing much to the activity with 7a giving an IC50 value of 191.5 μg ml−1 while 6a was inactive. The compounds with N-methyl substitution (20a) and methoxy substitutions (21a, 22a and 23a) were inferior to methyl substitution. Replacing the methoxy substitution with phenoxy (24a), phenyl ring with naphthyl (28a) or the introduction of aldehyde substitution in various equivalence to form 26a and 27a or the use of carboxylic acid substituent (25a) did not affect the activity.
From the discussions it can be concluded that the position of the functional groups and logP values of the compounds decide the activity. Interestingly, all the active compounds were monovalent bioisostere of either H or Br. The hydrophobicity of the compounds also helped in bioactivity as they can easily penetrate through the lipid bilayer of HIV. The studies indicate the possibility of using halogen derivatives for further modifications of the pyrimidine for enhanced activity.
4. Conclusion
A series of uracil derivatives were carefully designed based on the existing four-point pharmacophore model as effective capsid protein inhibitors. The design strategy involves the incorporation of flexible and polar di-uracil moieties capable of efficient hydrogen bonding and considering lipophilic substituents for easier membrane penetration. This is in line with the emerging cross-resistance in HIV drugs. The compounds designed showed drug-like properties and were initially docked with HIV capsid protein monomer to find the potential binding modes. Molecular dynamic simulations were performed for one of the most promising compound 9a to explain structural changes of the systems. The RMSD and RMSF plots indicated significant structural alteration of HIV-1 CA monomer when complexed with compound 9a. The promising in silico results led us to the in vitro evaluation of compounds in HIV p24 assay which revealed five potential compounds with an IC50 values ranging from 191.5 μg ml−1 to 62.5 μg ml−1. The meta-chloro substituted 9a displayed promising activity with an IC50 value of 62.5 μg ml−1 which correlates well with the theoretical studies. The active compounds were noncytotoxic in BA/F3 and Mo7e cell lines highlighting the thoughtful design. The structure–activity relationship indicates the position priority and lower logP values as the possible cause of inhibitory potential of the compounds.
Author contributions
DR: conceptualization, methodology, investigation and writing – original draft AD: conceptualization, methodology and investigation. AKM and SKM: formal analysis, investigation and writing original draft (Carried out molecular docking and dynamic simulation studies for HIV CA protein). BVG: writing – review & editing (Data interpretation for computational studies), writing original draft and visualization. AS and MM: formal analysis, investigation, writing original draft (carried out the cytotoxicity studies for compounds). KT: resources, supervision, writing – review & editing, project administration and funding acquisition.Funding
Deepthi Ramesh is the recipient of DST for DST-INSPIRE fellowship (SRF). Prof. Tharanikkarasu Kannan received financial support from University Grants Commission (UGC), New Delhi under Special Assistance Program, stage DSA-I to Department of Chemistry, Pondicherry University (No. F.540/6/DSA-1/2016/(SAP-1) dated 31-10-2018).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Prof. M. Elanchezhiyan and Jerusha Daniel, Dept. of Microbiology, University of Madras, Chennai, India, for anti-HIV studies on consultancy basis. The authors are grateful for Central Instrumentation Facility (CIF), Pondicherry University for spectral analysis. The authors also thank Prof. Binoy K. Saha, Dr Arijit Saha and Dr Sharada Durgam, Department of Chemistry, Pondicherry University for XRD characterization. Authors thank Debayan Chattopadhyay, Department of Chemistry, Pondicherry University for timely help.Notes and references
- C. J. P. R. Ananthanarayan, Ananthanarayan and Paniker's Textbook of Microbiology, University Press, 10th edn, 2017 Search PubMed.
- Global progress report on HIV, viral hepatitis and sexually transmitted infections, Accountability for the global health sector strategies 2016–2021: actions for impact, World Health Organization, Geneva, 2021, Licence: CC BY-NC-SA 3.0 IGO Search PubMed.
- R. Romeo, D. Iannazzo, L. Veltri, B. Gabriele, B. Macchi, C. Frezza, F. Marino-Merlo and S. V. Giofrè, Molecules, 2019, 24, 1718 CrossRef CAS PubMed.
- J. C. Reed, D. Solas, A. Kitaygorodskyy, B. Freeman, D. T. B. Ressler, D. J. Phuong, J. V. Swain, K. Matlack, C. R. Hurt, V. R. Lingappa and J. R. Lingappa, J. Virol., 2021, 95, e00883-20 CrossRef PubMed.
- J. D. Roberts, K. Bebenek and T. A. Kunkel, Science, 1988, 242, 1171 CrossRef CAS PubMed.
- J. M. Coffin, Science, 1995, 267, 483 CrossRef CAS PubMed.
- S. Redshaw and M. Westby, Expert Opin. Emerging Drugs, 2001, 6, 209–224 CrossRef CAS PubMed.
- S. A. Ali, S.-Y. Teow, T. C. Omar, A. S.-B. Khoo, T. S. Choon and N. M. Yusoff, PLoS One, 2016, 11, e0145986 CrossRef PubMed.
- S. K. Carnes, J. H. Sheehan and C. Aiken, Curr. Opin. HIV AIDS, 2018, 13, 359–365 CrossRef CAS PubMed.
- G. Li, J. Verheyen, S. Y. Rhee, A. Voet, A. M. Vandamme and K. Theys, Retrovirology, 2013, 10, 126 CrossRef PubMed.
- S. J. Rihn, S. J. Wilson, N. J. Loman, M. Alim, S. E. Bakker, D. Bhella, R. J. Gifford, F. J. Rixon and P. D. Bieniasz, PLoS Pathog., 2013, 9, e1003461 CrossRef CAS PubMed.
- L. D. Fader, R. Bethell, P. Bonneau, M. Bös, Y. Bousquet, M. G. Cordingley, R. Coulombe, P. Deroy, A. M. Faucher, A. Gagnon, N. Goudreau, C. Grand-Maître, I. Guse, O. Hucke, S. H. Kawai, J. E. Lacoste, S. Landry, C. T. Lemke, E. Malenfant, S. Mason, S. Morin, J. O'Meara, B. Simoneau, S. Titolo and C. Yoakim, Bioorg. Med. Chem. Lett., 2011, 21, 398–404 CrossRef CAS PubMed.
- M. Tremblay, P. Bonneau, Y. Bousquet, P. DeRoy, J. Duan, M. Duplessis, A. Gagnon, M. Garneau, N. Goudreau, I. Guse, O. Hucke, S. H. Kawai, C. T. Lemke, S. W. Mason, B. Simoneau, S. Surprenant, S. Titolo and C. Yoakim, Bioorg. Med. Chem. Lett., 2012, 22, 7512–7517 CrossRef CAS PubMed.
- L. Lamorte, S. Titolo, C. T. Lemke, N. Goudreau, J. F. Mercier, E. Wardrop, V. B. Shah, U. K. von Schwedler, C. Langelier, S. S. Banik, C. Aiken, W. I. Sundquist and S. W. Mason, Antimicrob. Agents Chemother., 2013, 57, 4622–4631 CrossRef CAS PubMed.
- B. N. Kelly, S. Kyere, I. Kinde, C. Tang, B. R. Howard, H. Robinson, W. I. Sundquist, M. F. Summers and C. P. Hill, J. Mol. Biol., 2007, 373, 355–366 CrossRef CAS PubMed.
- S. Rankovic, R. Ramalho, C. Aiken, I. Rousso and F. Kirchhoff, J. Virol., 2018, 92, e00166-21 CrossRef PubMed.
- T. Kobayakawa, M. Yokoyama, K. Tsuji, M. Fujino, M. Kurakami, S. Boku, M. Nakayama, M. Kaneko, N. Ohashi, O. Kotani, T. Murakami, H. Sato and H. Tamamura, Biomolecules, 2021, 11, 208 CrossRef CAS PubMed.
- A. Pałasz and D. Cież, Eur. J. Med. Chem., 2015, 97, 582–611 CrossRef PubMed.
- K. Miura, M. Kinouchi, K. Ishida, W. Fujibuchi, T. Naitoh, H. Ogawa, T. Ando, N. Yazaki, K. Watanabe, S. Haneda, C. Shibata and I. Sasaki, Cancers, 2010, 2, 1717–1730 CrossRef CAS PubMed.
- A. A. Carmine, R. N. Brogden, R. C. Heel, T. M. Speight and G. S. Avery, Drugs, 1982, 23, 329–353 CrossRef CAS PubMed.
- N. Li, L.-J. Wang, B. Jiang, S.-J. Guo, X.-Q. Li, X.-C. Chen, J. Luo, C. Li, Y. Wang and D.-Y. Shi, Bioorg. Med. Chem. Lett., 2018, 28, 2131–2135 CrossRef CAS PubMed.
- P. Ordonez, T. Hamasaki, Y. Isono, N. Sakakibara, M. Ikejiri, T. Maruyama and M. Baba, Antimicrob. Agents Chemother., 2012, 56, 2581 CrossRef CAS PubMed.
- X. Wang, J. Zhang, Y. Huang, R. Wang, L. Zhang, K. Qiao, L. Li, C. Liu, Y. Ouyang, W. Xu, Z. Zhang, L. Zhang, Y. Shao, S. Jiang, L. Ma and J. Liu, J. Med. Chem., 2012, 55, 2242–2250 CrossRef CAS PubMed.
- H. Zhang, Y. Tian, D. Kang, Z. Huo, Z. Zhou, H. Liu, E. De Clercq, C. Pannecouque, P. Zhan and X. Liu, Eur. J. Med. Chem., 2017, 130, 209–222 CrossRef CAS PubMed.
- M. V. Putz, N. A. Dudaș and A. Isvoran, Int. J. Mol. Sci., 2015, 16, 19553–19601 CrossRef CAS PubMed.
- L. Wang, M. C. Casey, S. K. V. Vernekar, R. L. Sahani, J. Kankanala, K. A. Kirby, H. Du, A. Hachiya, H. Zhang, P. R. Tedbury, J. Xie, S. G. Sarafianos and Z. Wang, Eur. J. Med. Chem., 2020, 204, 112626 CrossRef CAS PubMed.
- A. K. Malde, L. Zuo, M. Breeze, M. Stroet, D. Poger, P. C. Nair, C. Oostenbrink and A. E. Mark, J. Chem. Theory Comput., 2011, 7, 4026–4037 CrossRef CAS PubMed.
- L.-Q. Sun, L. Zhu, K. Qian, B. Qin, L. Huang, C. H. Chen, K.-H. Lee and L. Xie, J. Med. Chem., 2012, 55, 7219–7229 CrossRef CAS PubMed.
- J. V. Turner and S. Agatonovic-Kustrin, in Comprehensive Medicinal Chemistry II, ed. J. B. Taylor and D. J. Triggle, Elsevier, Oxford, 2007, pp. 699–724, DOI:10.1016/B0-08-045044-X/00147-4.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- K. D. Freeman-Cook, R. L. Hoffman and T. W. Johnson, Future Med. Chem., 2013, 5, 113–115 CrossRef CAS PubMed.
- J. D. Hughes, J. Blagg, D. A. Price, S. Bailey, G. A. DeCrescenzo, R. V. Devraj, E. Ellsworth, Y. M. Fobian, M. E. Gibbs, R. W. Gilles, N. Greene, E. Huang, T. Krieger-Burke, J. Loesel, T. Wager, L. Whiteley and Y. Zhang, Bioorg. Med. Chem. Lett., 2008, 18, 4872–4875 CrossRef CAS PubMed.
- V. Khanna and S. Ranganathan, BMC Bioinf., 2009, 10(Suppl 15), S10 CrossRef PubMed.
- K.-H. Thierauch, in Encyclopedia of Cancer, ed. M. Schwab, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 3448–3451, DOI:10.1007/978-3-642-16483-5_5374.
- H. X. Ngo and S. Garneau-Tsodikova, MedChemComm, 2018, 9, 757–758 RSC.
- J. Kostal, in Advances in Molecular Toxicology, ed. J. C. Fishbein and J. M. Heilman, Elsevier, 2016, vol. 10, pp. 139–186 Search PubMed.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- N.-N. Wang, J. Dong, Y.-H. Deng, M.-F. Zhu, M. Wen, Z.-J. Yao, A.-P. Lu, J.-B. Wang and D.-S. Cao, J. Chem. Inf. Model., 2016, 56, 763–773 CrossRef CAS PubMed.
- S. F. Zhou, Curr. Drug Metab., 2008, 9, 310–322 CrossRef CAS PubMed.
- A. T. Gres, K. A. Kirby, V. N. Kewal Ramani, J. J. Tanner, P. Owen and S. G. Sarafianos, Science, 2015, 349, 99–104 CrossRef CAS PubMed.
- O. Pornillos, B. K. Ganser-Pornillos, B. N. Kelly, Y. Hua, F. G. Whitby, C. D. Stout, W. I. Sundquist, C. P. Hill and M. Yeager, Cell, 2009, 137, 1282–1292 CrossRef PubMed.
- B. K. Ganser-Pornillos, A. Cheng and M. Yeager, Cell, 2007, 131, 70–79 CrossRef CAS PubMed.
- B. N. Kelly, S. Kyere, I. Kinde, C. Tang, B. R. Howard, H. Robinson, W. I. Sundquist, M. F. Summers and C. P. Hill, J. Mol. Biol., 2007, 373, 355–366 CrossRef CAS PubMed.
- K. Singh, F. Gallazzi, K. J. Hill, D. H. Burke, M. J. Lange, T. P. Quinn, U. Neogi and A. Sonnerborg, Front. Microbiol., 2019, 10, 1227 CrossRef PubMed.
- X. Jiang, G. Wu, W. A. Zalloum, M. E. Meuser, A. Dick, L. Sun, C. H. Chen, D. Kang, L. Jing, R. Jia, S. Cocklin, K. H. Lee, X. Liu and P. Zhan, RSC Adv., 2019, 9, 28961–28986 RSC.
- L. Sun, T. Huang, A. Dick, M. E. Meuser, W. A. Zalloum, C. H. Chen, X. Ding, P. Gao, S. Cocklin, K. H. Lee, P. Zhan and X. Liu, Eur. J. Med. Chem., 2020, 190, 112085 CrossRef CAS PubMed.
- J. O. Link, M. S. Rhee, W. C. Tse, J. Zheng, J. R. Somoza, W. Rowe, R. Begley, A. Chiu, A. Mulato, D. Hansen, E. Singer, L. K. Tsai, R. A. Bam, C. H. Chou, E. Canales, G. Brizgys, J. R. Zhang, J. Li, M. Graupe, P. Morganelli, Q. Liu, Q. Wu, R. L. Halcomb, R. D. Saito, S. D. Schroeder, S. E. Lazerwith, S. Bondy, D. Jin, M. Hung, N. Novikov, X. Liu, A. G. Villasenor, C. E. Cannizzaro, E. Y. Hu, R. L. Anderson, T. C. Appleby, B. Lu, J. Mwangi, A. Liclican, A. Niedziela-Majka, G. A. Papalia, M. H. Wong, S. A. Leavitt, Y. Xu, D. Koditek, G. J. Stepan, H. Yu, N. Pagratis, S. Clancy, S. Ahmadyar, T. Z. Cai, S. Sellers, S. A. Wolckenhauer, J. Ling, C. Callebaut, N. Margot, R. R. Ram, Y. P. Liu, R. Hyland, G. I. Sinclair, P. J. Ruane, G. E. Crofoot, C. K. McDonald, D. M. Brainard, L. Lad, S. Swaminathan, W. I. Sundquist, R. Sakowicz, A. E. Chester, W. E. Lee, E. S. Daar, S. R. Yant and T. Cihlar, Nature, 2020, 584, 614–618 CrossRef CAS PubMed.
- F. Ternois, J. Sticht, S. Duquerroy, H. G. Krausslich and F. A. Rey, Nat. Struct. Mol. Biol., 2005, 12, 678–682 CrossRef CAS PubMed.
- H. A. A. Karim, T. Rungrotmongkol, S. M. Zain, N. A. Rahman, C. Tayapiwattana and V. Lee, J. Mol. Liq., 2019, 277, 63–69 CrossRef CAS.
- R. V. Honorato, P. I. Koukos, B. Jiménez-García, A. Tsaregorodtsev, M. Verlato, A. Giachetti, A. Rosato and A. M. J. J. Bonvin, Front. Mol. Biosci., 2021, 8, 729513 CrossRef PubMed.
- L. Spáčilová, P. Džubák, M. Hajdúch, S. Křupková, P. Hradil and J. Hlaváč, Bioorg. Med. Chem. Lett., 2007, 17, 6647–6650 CrossRef PubMed.
- L. Bencivenni, F. Ramondo, A. Pieretti and N. Sanna, J. Chem. Soc., Perkin Trans. 2, 2000, 1685–1693, 10.1039/B002342F.
- Z. Xu, Z. Yang, Y. Liu, Y. Lu, K. Chen and W. Zhu, J. Chem. Inf. Model., 2014, 54, 69–78 CrossRef CAS PubMed.
- Y. Ichikawa, M. Hiramatsu, Y. Mita, M. Makishima, Y. Matsumoto, Y. Masumoto, A. Muranaka, M. Uchiyama, Y. Hashimoto and M. Ishikawa, Org. Biomol. Chem., 2021, 19, 446–456 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1946178. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra02450k |
| ‡ Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |