
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of cyclic α-pinane carbonate – a potential monomer for bio-based polymers†
Valentine C. Ezea,
Abdul Rehman *ab,
Manthan Patela,
Sajjad Ahmadb and
Adam P. Harveya
*ab,
Manthan Patela,
Sajjad Ahmadb and
Adam P. Harveya
aSchool of Engineering, Newcastle University, Newcastle upon Tyne NE1 7RU, UK
bDepartment of Chemical and Polymer Engineering, University of Engineering and Technology Lahore, Faisalabad Campus, Pakistan. E-mail: a.rehman2@uet.edu.pk
First published on 13th June 2022
Abstract
This work reports the first known synthesis of α-pinane carbonate from an α-pinene derivative. Pinane carbonate is potentially useful as a monomer for poly(pinane carbonate), which would be a sustainable bio-based polymer. α-Pinene is a major waste product from the pulp and paper industries and the most naturally abundant monoterpene in turpentine oil. α-Pinene is routinely converted to pinene oxide and pinanediol, but no study has yet demonstrated the conversion of pinanediol into α-pinane carbonate. Here, α-pinane carbonate was synthesised via carboxylation of α-pinanediol with dimethyl carbonate under base catalysis using triazabicyclodecene guanidine (TBD). 81.1 ± 2.8% α-pinane carbonate yield was achieved at 98.7% purity. The produced α-pinane carbonate was a white crystalline solid with a melting point of 86 °C. It was characterised using FTIR, NMR, GCMS and a quadrupole time-of-flight (QTOF) mass spectrometer. The FTIR exhibited a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak at 1794 cm−1 confirming the presence of a cyclic carbonate. GCMS showed that the α-pinane carbonate fragments with loss of CO2, forming pinene epoxide. Base hydrolysis of the α-pinane carbonate using NaOH/ethanol/water regenerated the pinanediol with formations of Na2CO3.
O peak at 1794 cm−1 confirming the presence of a cyclic carbonate. GCMS showed that the α-pinane carbonate fragments with loss of CO2, forming pinene epoxide. Base hydrolysis of the α-pinane carbonate using NaOH/ethanol/water regenerated the pinanediol with formations of Na2CO3.
1. Introduction
Monoterpenes are naturally occurring unsaturated hydrocarbons obtained as waste products from the pinewood pulping industry. The most commonly available monoterpenes are pinene and limonene, which are cyclic monoterpene isomers comprising two iso-propene units with a molecular formula of C10H16 (Scheme 1). Approximately 3.5 × 105 tons per year of turpentine oil is produced from the pulping industry globally,1,2 with the global supply of turpentine oil expected to increase as the pulping industry grows. The pulping process produces between 0.3 and 1.0 kg of turpentine oil per tonne of pulp,3 of which the predominant monoterpene is α-pinene (∼70%) when using the current sulphate processes.3,4 Limonene accounts for ∼8.6% (0.3 × 105 tons per annum) of the worldwide turpentine oil production.1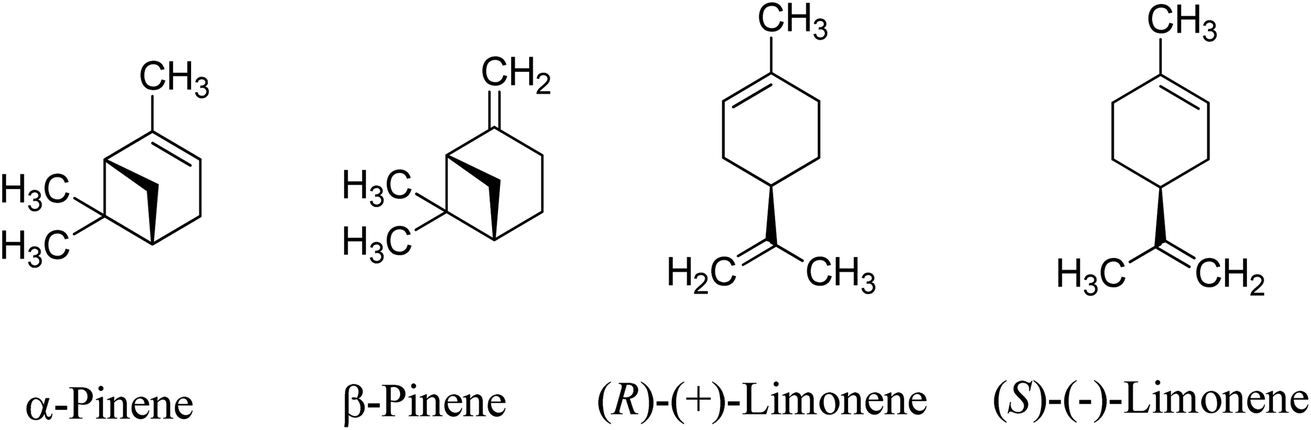 | ||
| Scheme 1 Common monoterpenes found in turpentine oil. | ||
Crude turpentine oil from steam distillation of pine is typically 75–85% α-pinene, and the remainder were 0–3% β-pinene and 5–15% limonene.5 A major source of limonene is the peel of citrus fruits which contains about 90 wt% limonene.6 Limonene is mainly obtained from the waste products of orange harvesting and peel from orange juice production.7 The large global tonnage of α-pinene and limonene could provide substantial amounts of sustainable platform chemicals for bio-based polymer productions.8 There have been extensive studies of limonene derivatives such as limonene epoxide9–11 and limonene bis-epoxides,12,13 which can be copolymerised with CO2 to obtain poly(limonene carbonates),9,11,14–16 as shown in Scheme 2(a). Limonene oxides and carbonates can be used as highly valuable platform chemicals for biopolymer synthesis.17–20 Conversely, despite α-pinene being the most abundant naturally occurring monoterpene,17 there are limited studies on transformations of α-pinene into essential derivatives, such as α-pinene epoxide and α-pinane carbonate.
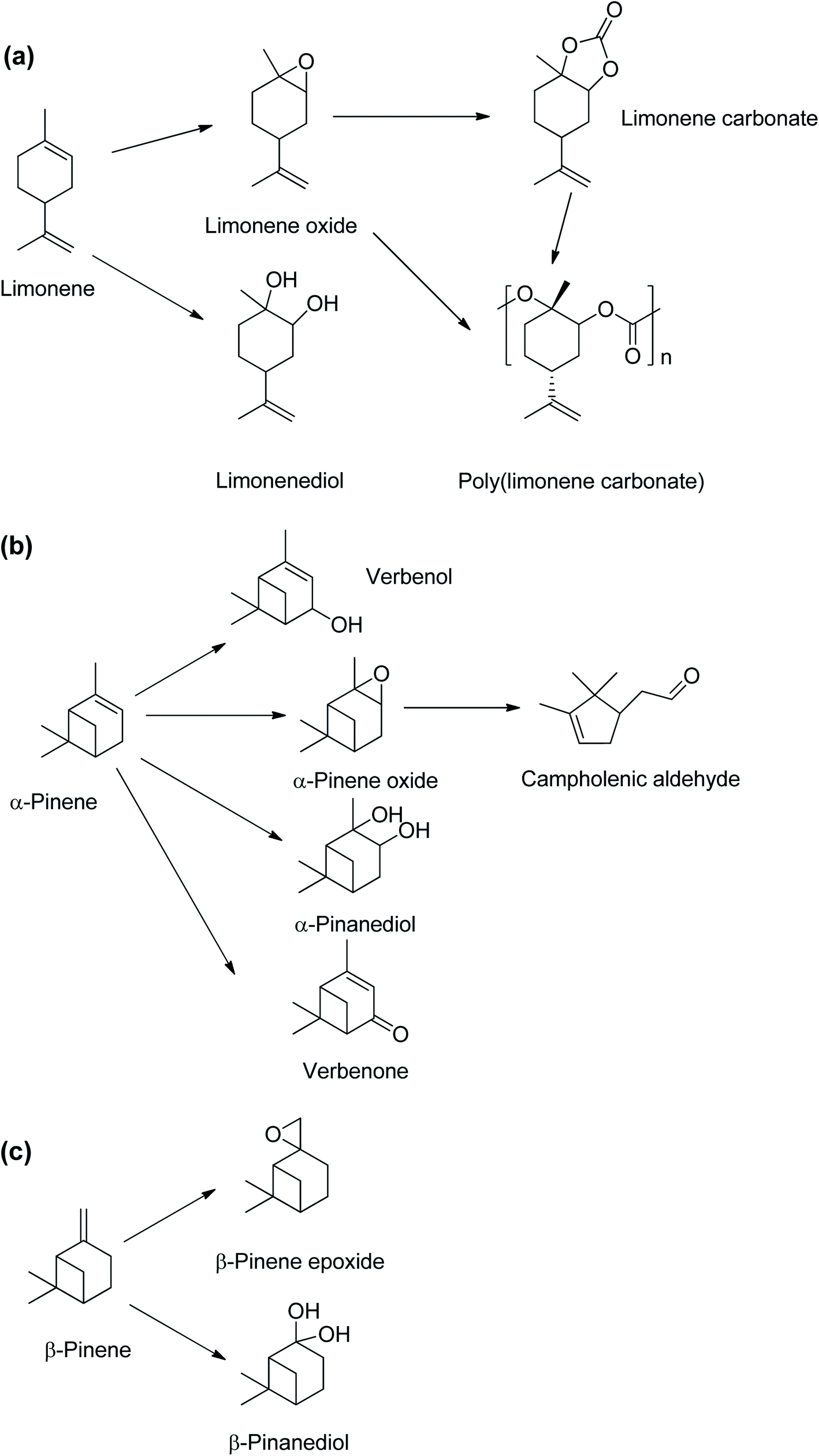 | ||
| Scheme 2 Chemical compounds derived from monoterpenes found in turpentine oil, (a) limonene, (b) α-pinene and (c) β-pinene. | ||
The α-pinane carbonate is potentially a monomer via a ring-opening copolymerisation to produce poly(α-pinane carbonate). The α-pinene epoxide is an important intermediate for other chemicals including campholenic aldehyde used in fragrances8,21,22 and pinanediol.23 α-Pinene can also be oxidised to produce essential chemicals such as verbenol and verbenone (Scheme 2(b)).8,24,25 Unlike limonene epoxide, which can be readily converted to limonene carbonate, for potential industrial productions of poly(limonene carbonate), the applications of α-pinene oxide have been mainly limited to its isomerisation products such as campholenic aldehyde,22 as no route from α-pinene oxide to the carbonate has yet been established.
Various attempts to synthesise polyether via homo-polymerisation of α-pinene oxide or poly(α-pinene carbonate) from α-pinene epoxide and CO2 have been unsuccessful. No poly(α-pinane carbonate) or α-pinane carbonate was obtained in the reactions of α-pinene epoxide with CO2.26 A recent review on the copolymerisation of sustainable epoxides with CO2 also reported that there are currently no available publications on the successful copolymerisation of α-pinene oxide with CO2.27 However, an existing patent claims that poly(pinene carbonate) can be produced by the reaction of pinene epoxide and CO2 in the presence of a (salen)CrCl/PPNCl catalyst.27 The lack of reactivity of the α-pinene epoxide in ring-opening polymerisation has been attributed to a higher reaction barrier of the α-pinene epoxide in the ring-opening process due to steric hindrance.26 The lack of reactivity is unfortunate, as there is potentially a significant market for pinane carbonate, given that the current production of cyclic carbonates is 4 million tons per year.28 This could add value to the paper and pulp industries, whilst introducing a new sustainable platform chemical. The cyclic pinane carbonate could be polymerised using established methods for ring-opening polymerisation of cyclic organic carbonates.29–31
This study reports the first synthesis of α-pinane carbonate from α-pinanediol. The synthesis route reported here involved a novel and eco-friendly process via reactions of α-pinanediol with dimethyl carbonate (DMC). Reactions of diols with DMC to obtain a cyclic carbonate have been reported for some other polyhydric alcohols,32 but have not previously been applied for α-pinanediol. The only existing report of pinane carbonate synthesis was via the reaction of 2,10-β-pinanediol (Scheme 2(c)) with ethyl chloroformate in pyridine, followed by a molecular rearrangement.33 Although 2,10-β-pinanediol could be obtained from hydroxylation of β-pinene, the content of β-pinene in crude turpentine oil is very low, typically about 0–3%, as compared to 75–85% for α-pinene.5 The DMC route was chosen for this synthesis because it is less toxic compared to the phosgene process.34 It is anticipated that the use of α-pinanediol would reduce the reaction barrier associated with α-pinene epoxide and overcome the steric hindrance,26 allowing for formations of α-pinane carbonate. α-Pinanediol can be readily obtained via osmium tetraoxide catalysed hydroxylation of naturally available α-pinene,35 or from hydrolysis of the α-pinene epoxide.23 Furthermore, DMC is a green solvent, which has been synthesised by CeO2 – catalysed reactions of CO2 and methanol (MeOH) to achieve >95% methanol conversions and >99% selectivity to DMC.36
Similarly, some other main methodologies for DMC preparation currently applied in the industry are oxidative carbonylation of MeOH, urea mediated synthesis and transesterification of ethylene carbonate and propylene carbonate.37
2. Materials and methods
2.1 Materials
Materials used in the experiments were pinane-2,3-diol (99%, Sigma-Aldrich), dimethyl carbonate (99%, Sigma-Aldrich), anhydrous methanol (99.8%, Sigma-Aldrich), absolute ethanol (99.5%, Fisher Scientific), sodium hydroxide (97%, Sigma-Aldrich), acetic acid (99%, Sigma-Aldrich), n-hexane (95%, Sigma-Aldrich), methyl heptadecanoate (99%, Sigma-Aldrich), and TBD guanidine (98%, Sigma-Aldrich), these chemicals were used as supplied.2.2 Experimental procedures
The pinane carbonate synthesis was performed in a 150 mL 2-neck batch reactor equipped with a condenser and a thermocouple/sampling unit, by reactions of pinanediol and DMC, as shown in Scheme 3. Synthesis of α-pinane carbonate was carried out using 0.18 mol of DMC, 0.03 mol of pinanediol, and 2 mmol of TBD guanidine catalyst. The reaction was carried in the round bottom flask. Pinanediol (5 g) and DMC (15.88 g), corresponding to a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMC to pinanediol molar ratio, were added to the reactor and heated to the reaction temperature of 90 °C using a heater-stirrer (IKA® Basic). This was followed by additions of 250 mg of TBD guanidine catalyst (5 wt% based on the pinanediol) and mixing at 600 rpm. The reaction mixture was refluxed at 90 °C for 6 h in the reactor. 0.25 mL samples were collected every 1 h and analysed by gas chromatography (GC) to monitor the progress of the reaction until the pinanediol peak disappeared.
:1 DMC to pinanediol molar ratio, were added to the reactor and heated to the reaction temperature of 90 °C using a heater-stirrer (IKA® Basic). This was followed by additions of 250 mg of TBD guanidine catalyst (5 wt% based on the pinanediol) and mixing at 600 rpm. The reaction mixture was refluxed at 90 °C for 6 h in the reactor. 0.25 mL samples were collected every 1 h and analysed by gas chromatography (GC) to monitor the progress of the reaction until the pinanediol peak disappeared.
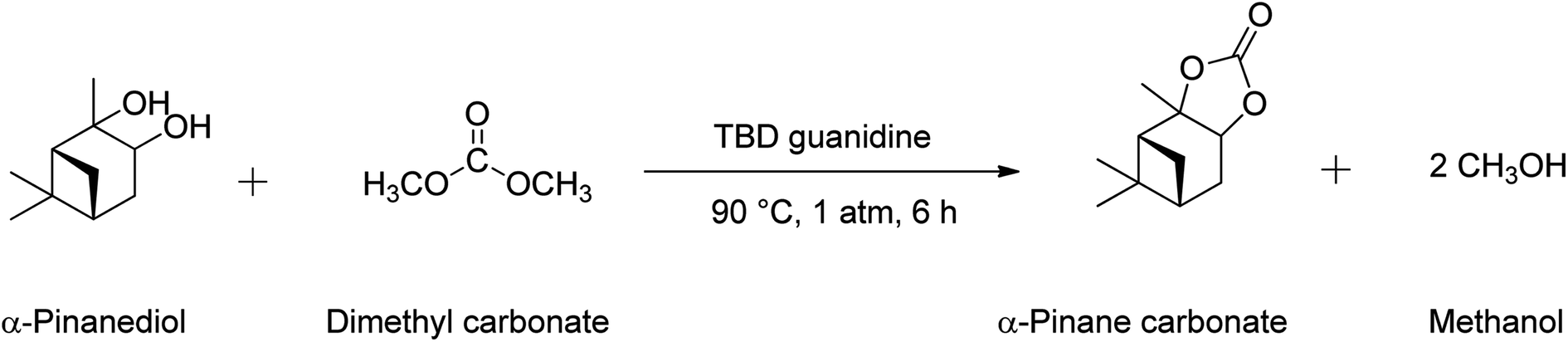 | ||
| Scheme 3 Reaction equation for the synthesis of α-pinane carbonate using 0.18 mol of DMC, 0.03 mol of pinanediol, and 2 mmol of TBD guanidine catalyst. | ||
The reaction mixture was then neutralized using 110 μL of acetic acid. Excess DMC and residual methanol by-products in the reaction were removed by rotary evaporation at 60 °C at 120 mmHg pressure. An amber-coloured solid was obtained, and this was purified by re-crystallization 3 times in methanol (25 mL). The recrystallized product was dissolved in a mixture of 25 mL n-hexane and 10 mL methanol with vigorous shaking. The hexane extract was a colourless viscous solution, from which white crystals of α-pinane carbonate appeared as the excess hexane evaporated at room temperature. The α-pinane carbonate was filtered out and dried at room temperature. The reaction mixture was recrystallized three times to remove any traces of the catalyst. About 4 g of α-pinane carbonate was obtained, with >98.7 wt% purity as confirmed by the GC. The product was characterised using NMR (1H and 13C), FTIR, GCMS, a differential scanning calorimeter (DSC) and a quadrupole time-of-flight (QTOF) mass spectrometer.
The reactivity of the synthesised α-pinane carbonate was investigated, especially to find a route for regenerating the pinanediol. This is important in ensuring that a suitable process exists for recycling pinanediol from the α-pinane carbonate at the product's end of life. About 0.5 g of the dried α-pinane carbonate was hydrolysed in the batch reactor with 25 mL of 0.5 M NaOH in an ethanol–water solution containing 10 (v/v)% deionised water. The 25 mL 0.5 M NaOH solution was heated in the reactor to 60 °C, followed by the transfer of 0.5 g α-pinane carbonate into the reactor and vigorous mixing at 600 rpm. The ratio of α-pinane carbonate to NaOH solution (w/v) in this study was adapted from the commonly used method for saponification of fats and oils.38–40 About 0.5 mL of the reaction sample was collected using a micropipette at various time intervals from 0–60 min. The sample was transferred into a pre-weighed 2 mL vial containing 14 μL of acetic acid to quench the reaction immediately.
2.3 Sample analysis
FTIR analysis of the samples was performed using a Mettler Toledo ATR-FTIR spectroscopy (React IR 4000) equipped with a DiComp diamond K6 conduit 16 mm probe. The spectra of the product samples were collected over the range 4000–650 cm−1. The instrument was initialized by collecting 256 scan background spectra for air and water vapours before collecting the spectra of the samples. 1H-NMR and 13C-NMR spectroscopy of the samples were recorded on a Bruker Avance III HD spectrometer at 700 MHz using a nitrogen-cooled cryoprobe prodigy™, with methanol-d (CD3OD) as a solvent. Melting points of the pinanediol and the produced α-pinane carbonate were determined using a Reichert platform melting point apparatus. DSC analysis was performed using (TA Instruments, DSC Q20 V24.11 Build 124), with a TA Universal analysis software to do the DSC analysis using 3.1–3.4 mg of samples that were carefully weighed into Tzero aluminium pans and sealed with Tzero aluminium lids. The samples were scanned over the temperature range of 25–400 °C at a heating rate of 5 °C min−1 and a 50 mL min−1 flow of oxygen-free nitrogen for the sample purge.A 6890 Hewlett Packard Series GC equipped with a Nukol™ fused silica column of 30 mm length, 0.32 mm ID and 0.25 μm film thickness was used to quantify the α-pinanediol and α-pinane carbonate in the samples. The GC oven temperature was programmed from 120 °C initial temperature held for 5 min and ramped at 15 °C min−1 to a final temperature of 210 °C and held for a further 10 min (total time of 21 min). The flame ionisation detector (FID) and injector temperatures were set at 260 and 250 °C, respectively, while a helium carrier gas was used at 10 PSI pressure. Quantifications of the α-pinanediol and α-pinane carbonate were based on a calibration curve using standard reagents where correlation data (R2 > 0.995) was obtained while methyl heptadecanoate was a GC internal standard. The calibration curve is drawn following concentration ranges from 1.0 to 50 mg kg−1 of the reference standard. About 100 mg of each sample was measured into a 2 mL GC vial and mixed with 500 μL of methyl heptadecanoate (10 mg mL−1 of ethanol), and 1 μL of the sample mixture was injected into the GC using a 5 μL GC syringe (SGE). The sample is run against this calibration curve to estimate its concentration. The α-pinanediol and α-pinane carbonate were also analysed using a GCMS: 7890B Agilent GC coupled to a 5977B mass selective detector (MSD) in full scan mode from 50–520 amu and 70 eV ionisation energy. The 7890B Agilent GC program was held at 50 °C for 2 min initially and ramped to 310 °C at 5 °C min−1 (total time of 75 min). Molecular ions of the analytes were determined with a QTOF mass spectrometer. The chromatogram showed a single peak with a major ion on Rt 11.084 with a maximum area covering the purity content. The exact mass measurement is followed by QTOF with the least error and uncertainty incorporation as the findings correlated with other analysis techniques and no impurity is highlighted.
3. Results and discussion
3.1 Productions and characterisation of the α-pinane carbonate yield
An α-pinane carbonate was synthesised from an α-pinene derivative for the first time, via reactions of α-pinanediol and DMC. The cyclic α-pinane carbonate yield for the process was 81.1 ± 2.8%, and the pinane carbonate was isolated as a white crystalline solid at 98.7% purity as confirmed by the GC. The melting points of the α-pinanediol and the α-pinane carbonate were determined by DSC, using methods reported elsewhere,41 and these values were 86 °C for the α-pinane carbonate, and 55 °C for the 2,3-pinanediol (ESI Fig. 1†). FTIR data for the α-pinane carbonate and the α-pinanediol are shown in Fig. 1. Fig. 1(a) shows an FTIR peak around 3200–3300 cm−1, which is assigned to the O–H stretching vibration for the α-pinanediol. This peak was conspicuously absent in the α-pinane carbonate product. The disappearance of the O–H peak resulted in the formation of a new peak at 1795 cm−1, corresponding to CO stretching vibration for cyclic carbonates, as shown in Fig. 1(b) and (c), which is significantly higher than the absorption wavenumber of about 1750 cm−1 for linear organic carbonate (DMC). The CO stretching vibration for the α-pinane carbonate compares well with 1800 cm−1, which has been reported for limonene cyclic carbonate.11 A previous study has also demonstrated that CO stretching for cyclic propylene carbonate occurs at a higher wavenumber (1799 cm−1) than the CO stretch for linear poly(propylene carbonate) with wavenumber at 1747 cm−1.42 Apart from the characteristic FTIR peaks at 3200–3300 cm−1 for the O–H and 1795 cm−1 for the CO functionalities, other FTIR peaks observed at 2980–2850 cm−1 for C–H stretching vibrations in methyl and methylene groups are similar for both species, as would be expected. The FTIR data clearly shows that the α-pinanediol was converted to α-pinane cyclic carbonate during the reaction.
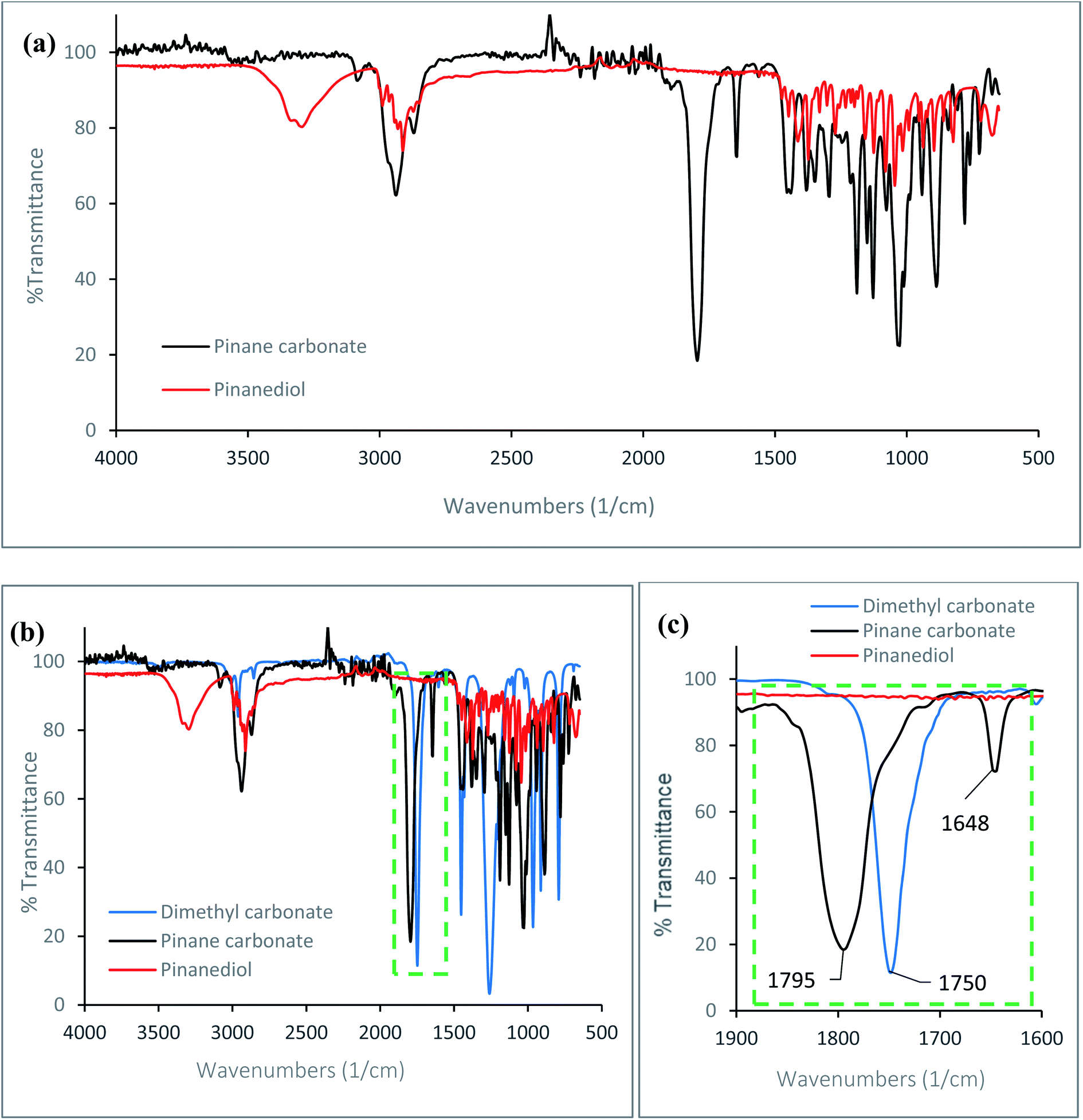 | ||
| Fig. 1 FTIR data collected using a Mettler Toledo React IR 4000 ATR-FTIR spectroscopy equipped with a DiComp diamond probe, (a) for the α-pinanediol and the pinane carbonate, (b) for α-pinanediol and pinane carbonate, with FTIR of DMC inserted as a typical linear carbonate, (c) shift in absorption wavelengths for cyclic and linear organic carbonates. | ||
The 1H NMR data for the α-pinanediol and the α-pinane carbonate are shown in Fig. 2. The α-pinanediol has 1H NMR peaks (700 MHz, CD3OD), as shown in Fig. 2(a), at the following chemical shifts (δ/ppm): 1.30 (3H, s), 3.98 (1H, dd), 1.89 (1H,t) for proton that points out of the α-pinane ring (exo-proton) and 1.97 (1H, dtd) for the proton that points inside the α-pinane ring (endo-proton) which is more shielded, 2.18 (1H, dtd), 0.98 (3H, s), 1.27 (3H, s), 2.43 (1H, dddd), 1H (1.47, m) for exo-proton and 1H (2.43, dddd) for the more shielded endo-proton, and 4.88 (2H, s) for the –OH groups. The 1H NMR peak at 4.88 ppm (2H, s) corresponds to the O–H protons of the pinanediol, which indicates an overlap in the 1H NMR signals for the two hydroxyl groups on the pinanediol. A total of 9 hydrogen environments corresponding to a total of 18 hydrogen atoms were observed for the α-pinanediol (C10H18O2).
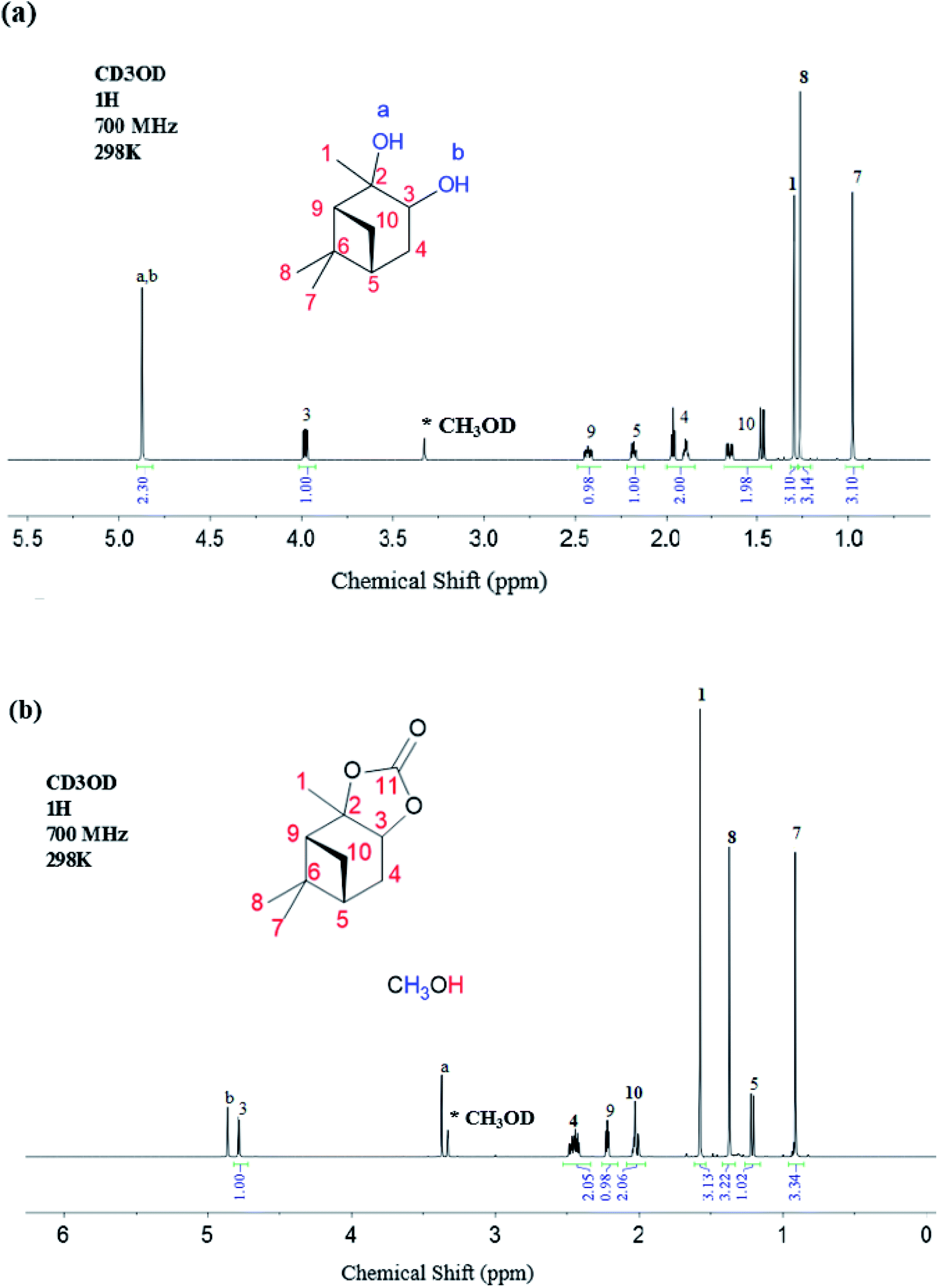 | ||
| Fig. 2 1H-NMR spectroscopy recorded on a Bruker Avance III HD spectrometer with nitrogen-cooled cryoprobe prodigy™, with methanol-d (CD3OD) as a solvent and operating at 700 MHz (a) α-pinanediol and, (b) α-pinane carbonate. | ||
The formation of α-pinane carbonate resulted in the disappearance of the proton peak at a chemical shift of 4.88 ppm, and slight variations in the chemical shift for some of the proton peaks, as shown in Fig. 2(b). The 1H NMR peaks (700 MHz, CH3OD) for the α-pinane carbonate were observed at the following chemical shifts (δ/ppm): 1.58 (3H, s), 4.78 (1H, d), 2.43 (2H, m), 1.21 (1H, d), 0.91 (3H, s), 1.37 (3H, s), 2.22 (1H, d) and 2.02 (2H, m). 1H NMR peaks were also observed at 3.4 ppm (3H, s) and 4.9 ppm (1H, s), due to remaining methanol impurities. Overall, 8 hydrogen environments were observed, corresponding with a total of 16 hydrogen atoms for the C11H16O2 in the α-pinane carbonate. The enantiotropic effect was observed in some of the 1H-NMR peaks due to the different chemical environment of protons. For instance, the two protons attached to C4 were in different chemical environments as endo and exo protons, likewise, the two protons attached to C10. Therefore, the enantiotropic effect accounts for the differences in chemical shifts for the C4 and the C10 protons shown in Fig. 2(a). Similarly, protons on the two methyl groups (C7 and C8) attached to carbon number 6 experience different chemical environments due to the enantiotropic effect. Protons on C7 are more shielded (δ = 0.98 ppm), indicating an endo-position of the C7 methyl group, while the protons on C8 are more deshielded (δ = 1.27 ppm) which suggests that the C8 methyl group occupies an exo-position on the α-pinane ring.
The 13C NMR data for the α-pinanediol and α-pinane carbonate are shown in Fig. 3. The α-pinanediol has 13C NMR peaks (700 MHz, CD3OD) at the following chemical shifts (δ/ppm): 23.2 (C7), 27.1 (C8), 27.7 (C1), 28.7 (C10), 37.3 (C6), 38.4 (C5), 40.4 (C4), 53.9 (C9), 68.5 (C3), 73.1 (C2). The carbon-13 NMR results in Fig. 3(a) show 10 different carbon environments, corresponding to the various carbon atoms on the C10H18O2 for the α-pinanediol. Mass spectra data (ESI Fig. 2†) from the GCMS analysis of α-pinanediol showed major m/z peaks at 55.0, 71.0, 99.1 (100%), 111.1, and 126.1, confirming the starting material as (1S,2S,3R,5S)-(+)-pinanediol (#7655 in NIST98.1 MS database). No molecular ion peak for pinanediol was observed at 170.3 m/z, indicating low stability of its molecular ions.
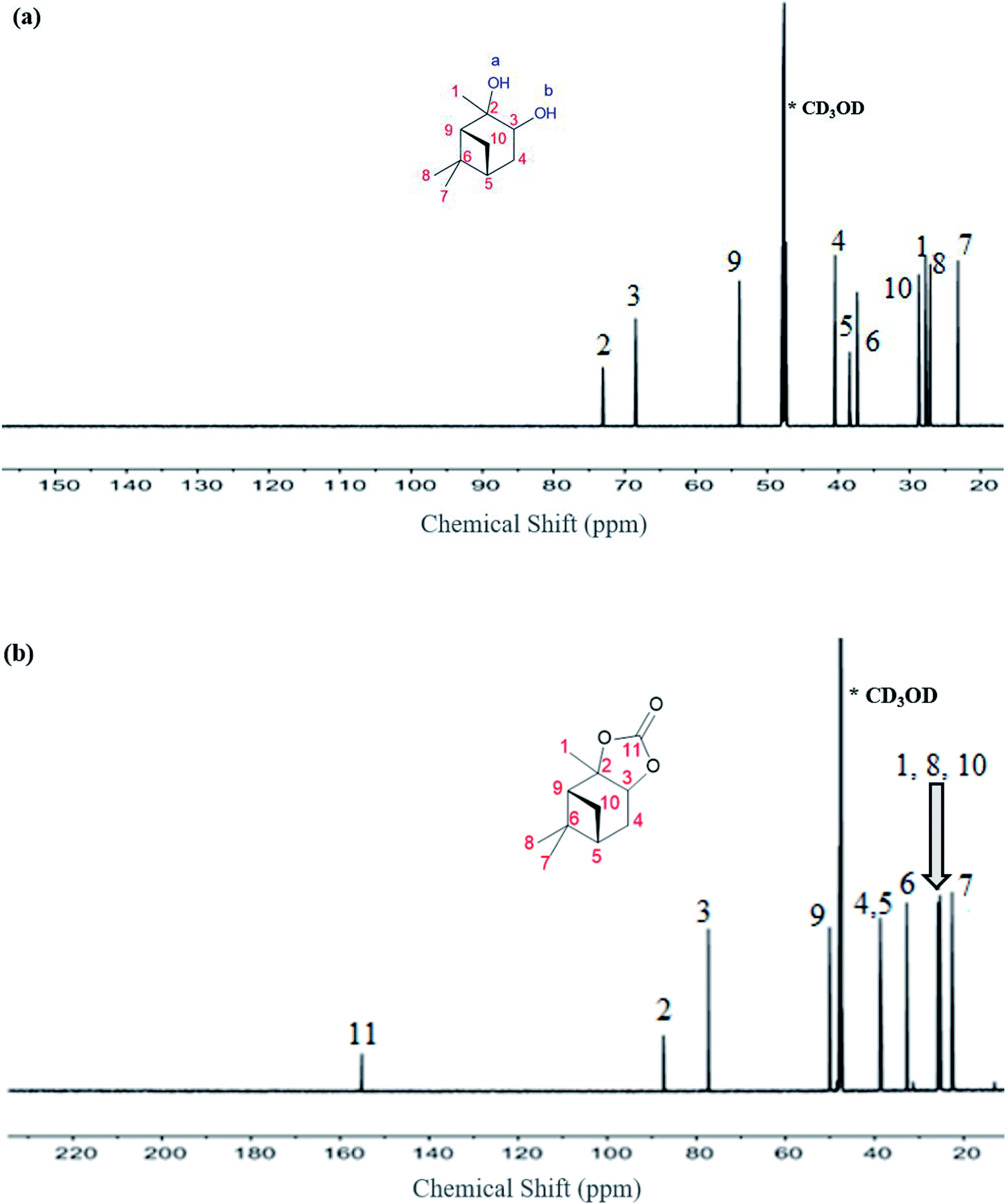 | ||
| Fig. 3 13C-NMR spectroscopy recorded on a Bruker Avance III HD spectrometer with nitrogen-cooled cryoprobe prodigy™, with methanol-d (CD3OD) as a solvent and operating at 700 MHz, (a) α-pinanediol and, (b) α-pinane carbonate. | ||
The chemical shifts observed in α-pinane carbonate differed from pinanediol. An additional carbon peak was observed at δ (ppm) of 155.1, as shown in Fig. 3(b), corresponding to CO of the α-pinane carbonate. The carbon peak at 155.1 ppm chemical shift is consistent with CO carbon for organic carbonates.9,43 Fig. 3(b) shows that the α-pinane carbonate has 13C NMR peaks (700 MHz, CD3OD) at the following chemical shifts (δ/ppm): 22.6 (C7), 25.3 (C8), 25.3 (C1), 25.7 (C10), 32.8 (C6), 38.4 (C5), 38.7 (C4), 50.1 (C9), 77.3 (C3), 87.4 (C2), and 155.1 (C11). These 11 carbon environments correspond to the various carbon atoms on the C11H16O3 for the α-pinane carbonate.
The major m/z mass spectra peaks for the α-pinane carbonate (ESI Fig. 3†) were 55.0, 67.0 (100%), 69.0, 83.0, 109.1, and 137.0. A molecular ion peak for pinane carbonate was also not observed at the expected m/z of about 196.2. Mass spectra for the α-pinane carbonate in ESI Fig. 3† showed that the m/z peaks are consistent with fragmentation patterns for α-pinene epoxide in the National Institute of Standards and Technology (#21620 in NIST98.1 MS database), indicating that loss of CO2 and molecular rearrangement to form α-pinene epoxide was the plausible fragmentation mechanism of α-pinane carbonate. Molecular ions of the α-pinanediol (152.1 g mol−1) and the α-pinane carbonate (196.1 g mol−1) were subsequently determined as shown in ESI Fig. 4† with the QTOF mass spectrometer.
It is expected that α-pinane carbonate can be polymerised to obtain poly(α-pinane carbonate), which can be used as a bio-based thermoplastic, like poly(limonene carbonates).9,15 poly(limonene carbonate) has a substantially high glass-transition temperature (Tg = 130 °C),15 and this is the expected range for a poly(α-pinane carbonate). Therefore, it is envisaged that a poly(α-pinane carbonate) could be a sustainable replacement for petroleum-derived polycarbonates.
3.2 Recovery of α-pinanediol via base hydrolysis of the pinane carbonate
Currently, a major challenge for the plastics industry is the development of efficient strategies for the handling of waste plastic at the end of life.44,45 Proposed routes for waste plastics handling include recycling44 or composting, in the case of biodegradable materials, such as bioplastics used in packaging.45 However, a more acceptable route would be through recovery and recycling of the starting materials for further utilisation. To develop a feasible route for possible recovery and recycling of the α-pinanediol starting material, the reactivity of the α-pinane carbonate towards base hydrolysis was investigated. The results in Fig. 4 show that quantitative recovery of α-pinanediol was achieved from the base hydrolysis of the α-pinane carbonate. A poly(α-pinane carbonate) is expected to undergo similar reactions.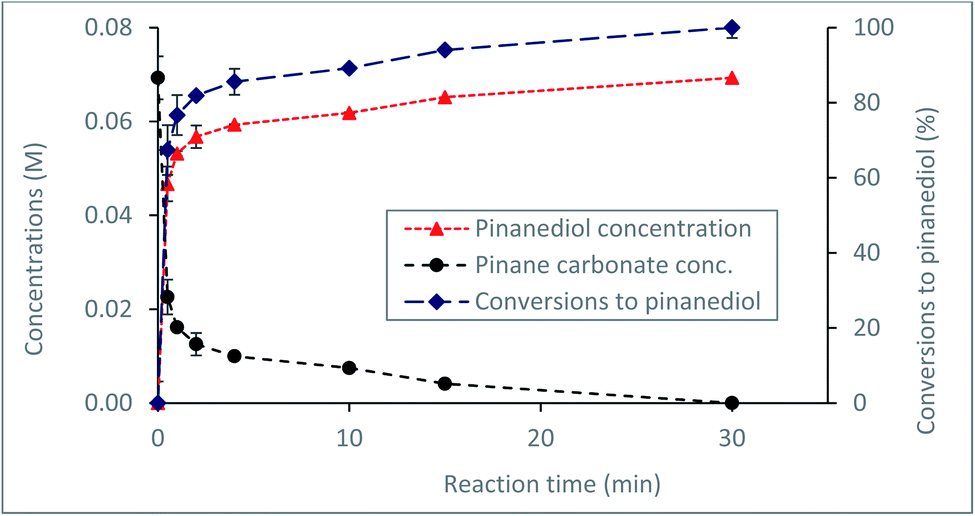 | ||
| Fig. 4 Recovery of α-pinanediol via base hydrolysis of the α-pinane carbonate in a batch reactor at 60 °C using 0.5 M NaOH ethanol/water solution (90:10 v/v)%. | ||
Over 80% of the pinane carbonate was saponified within 2 min using 0.5 M NaOH prepared in ethanol/water solution containing 90:10 (v/v)% and 60 °C. At about 30 min reaction time, all the pinane carbonate had been completely (100%) saponified and converted to pinanediol and sodium carbonate (Scheme 4). Base hydrolysis of the α-pinane carbonate via reactions with hydroxide ions to form carbonic acid salt and α-pinanediol is shown in Scheme 4. This reaction proceeds similarly to typical ester saponification. It should be noted that such reactions can also be performed using different hydroxide ion solutions, such as aqueous hydroxide solutions,46 or hydroxide solutions prepared in a mixture of water and soluble organic solvents, including dimethyl sulphoxide (DMSO),47 ethanol,48,49 dichloromethane50 and other suitable solvents.
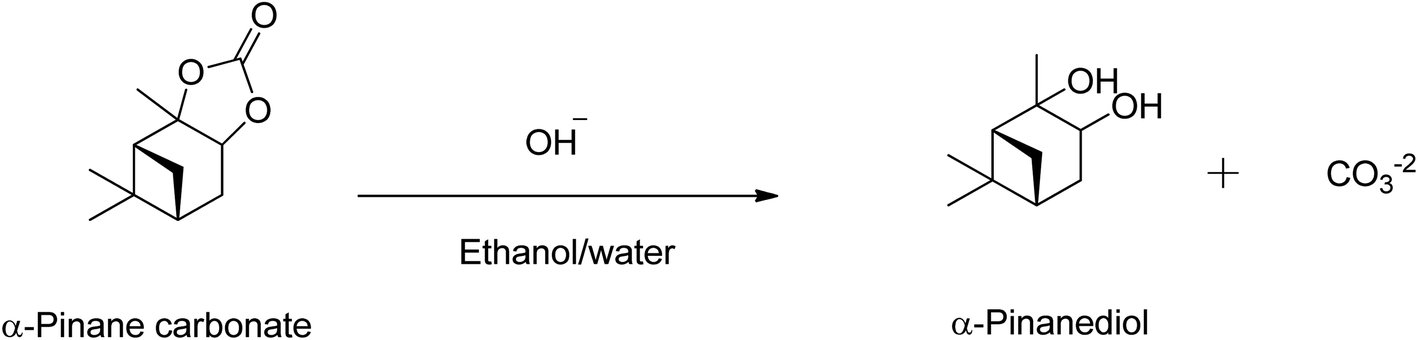 | ||
| Scheme 4 Base hydrolysis of α-pinane carbonate using NaOH in ethanol/water. | ||
The use of ethanol/water systems (i.e., water and soluble organic solvents) allows for a complete dissolution of the NaOH and the α-pinane carbonate, which eliminates the mass transfer limitations that can occur due to poor immiscibility of carbonate esters and water. To ensure complete saponification of the α-pinane carbonate, the NaOH solution was used in a large excess of the stoichiometric amounts.39 A proposed reaction mechanism for base hydrolysis of the α-pinane carbonate is shown in Scheme 5, with a rate-determining step involving the bimolecular collision of the pinane carbonate and hydroxide ion to form a tetrahedral intermediate, which decomposes to give the products, as previously reported for saponification of alkyl esters.51
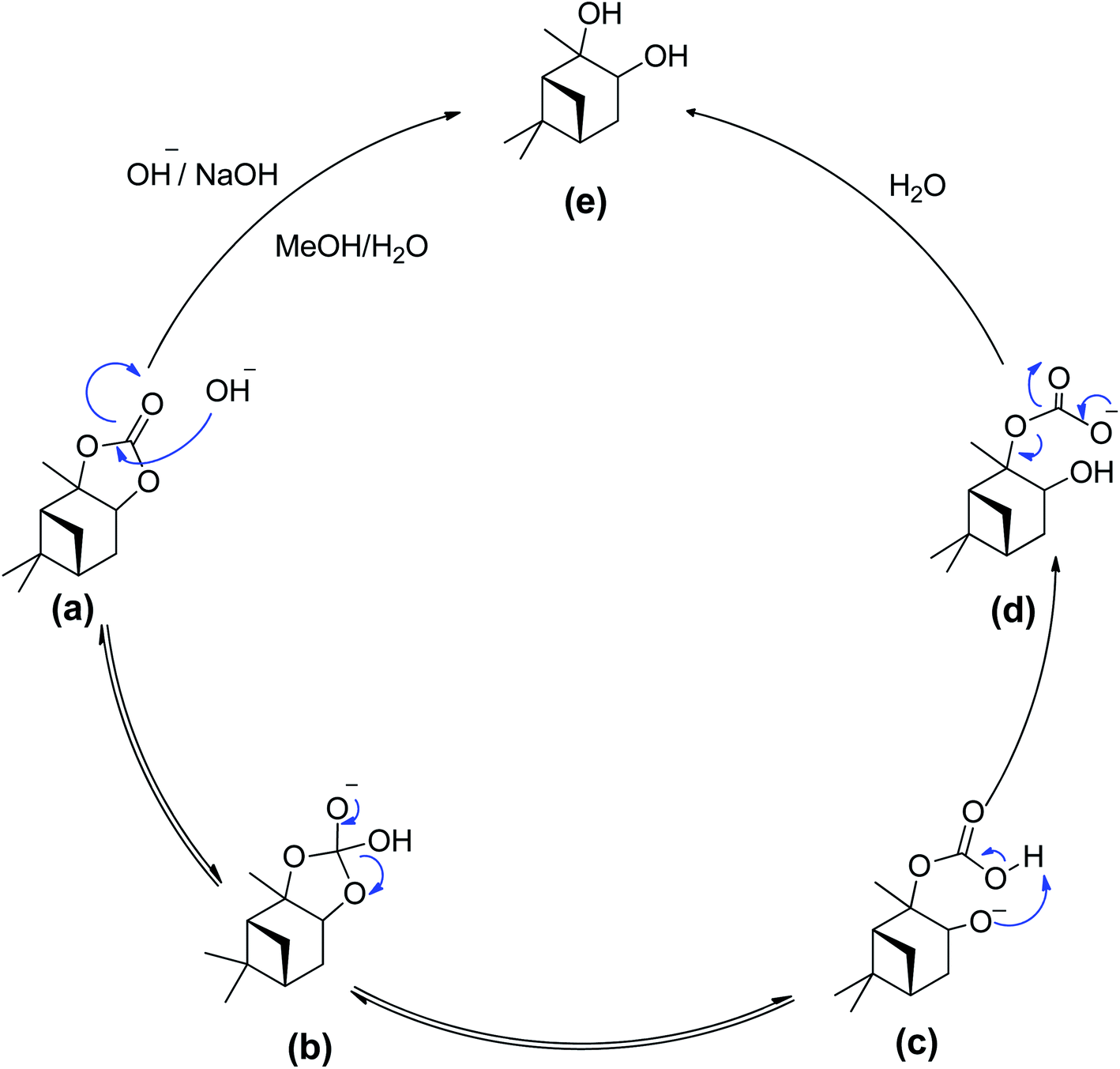 | ||
| Scheme 5 Proposed reaction mechanism – a bimolecular collision of solvated hydroxide ion with the alkyl esters carbonyl carbon. | ||
In this reaction mixture, a molecule of water is required to stabilise the intermediates and the same can also be provided by other protic solvents such as simple alcohols, like base hydrolysis of alkyl esters.50,51 During the reaction as shown in the Scheme 5, the hydroxyl group acted as a nucleophile and reacted with α-pinane carbonate (a) to yield intermediate (b) which decomposes and the carbonyl group reforms to form 2-carboxylate-3-pinacol (d) via intermediate (c) in the basic ethanol/water solution that further rearranges to furnish final product (e). As one side of α-pinane carbonate is more crowded thus it is most likely that the hydroxyl group as a nucleophile attacks on the opposite side on the most electropositive position however work is in progress to determine the exact mechanism of the reaction. Overall, the α-pinane carbonate hydrolysis was rapid, leading to the quantitative recovery of the α-pinanediol within 30 min. Previously, studies have shown that the reactivity of carboxylic acids decreases with the length of alkyl substituent due to a combination of polar and steric influences of the alpha substituent on the carboxylic group.52–54 The carbonic acid moiety on the α-pinane carbonate has no alkyl substituent, hence, its base hydrolysis is rapid. It is envisaged that poly(α-pinane carbonate) would follow a similar reaction pattern at slower reaction rates due to higher molecular weight.
4. Conclusions
This study is the first case of synthesis of α-pinane carbonate from α-pinanediol, a derivative of the most abundant naturally occurring monoterpene, α-pinene. Previous attempts to produce pinane carbonate by reactions of α-pinene epoxide with CO2 have been unsuccessful due to the lack of reactivity of the epoxide in ring-opening polymerisation because of steric hindrance. Here, α-pinane carbonate was synthesised by the reactions of α-pinanediol with DMC in the presence of a TBD guanidine catalyst. The α-pinane carbonate is a white crystalline solid with 86 °C melting point. The maximum α-pinane carbonate yield achieved was 81.1 ± 2.8%, at 98.7% purity, after recrystallization. The α-pinane carbonate produced was characterised using FTIR, NMR (1H and 13C), GC and GCMS and QTOF mass spectrometer. Reactivity of the α-pinane carbonate towards base hydrolysis with NaOH in ethanol/water was investigated to develop a feasible route for possible recovery and recycling of the α-pinanediol starting material. It was shown that α-pinane carbonate could be completely saponified within 30 min to the α-pinanediol and sodium carbonate. The α-pinane carbonate can likely be polymerised to obtain poly(α-pinane carbonate), which would be a bio-based thermoplastic, like poly(limonene carbonates) in an industrial scale process. The used poly(α-pinane carbonate) could be hydrolysed to recycle the α-pinanediol starting feedstock in the same way as the α-pinane carbonate monomer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Dr Rachael Dack of the Newcastle University School of Chemistry for the QTOF mass spectrometry services. This work was supported by the UK Engineering and Physical Sciences Research Council (EPSRC) funding for Sustainable Polymers (Grant reference EP/L017393/1).References
- M. Colonna, C. Berti, M. Fiorini, E. Binassi, M. Mazzacurati, M. Vannini and S. Karanam, Synthesis and radiocarbon evidence of terephthalate polyesters completely prepared from renewable resources, Green Chem., 2011, 13(9), 2543–2548 RSC.
- A. Gandini, The irruption of polymers from renewable resources on the scene of macromolecular science and technology, Green Chem., 2011, 13(5), 1061–1083 RSC.
- H. Sixta, Handbook of pulp, Wiley-WCH, Germany, 2006 Search PubMed.
- P. Knuuttila, Wood sulphate turpentine as a gasoline bio-component, Fuel, 2013, 104, 101–108 CrossRef CAS.
- M. Gscheidmeier and H. Fleig, Turpentines, Ullmann’s Encyclopedia of Industrial Chemistry, Wiley–VCH, Weinheim, 2005 Search PubMed.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Terpene-based renewable monomers and polymers via thiol–ene additions, Macromolecules, 2011, 44(18), 7253–7262 CrossRef CAS.
- R. Ciriminna, M. Lomeli-Rodriguez, P. D. Cara, J. A. Lopez-Sanchez and M. Pagliaro, Limonene: a versatile chemical of the bioeconomy, Chem. Commun., 2014, 50(97), 15288–15296 RSC.
- M. F. M. G. Resul, A. Rehman, A. M. L. Fernández, V. C. Eze and A. P. Harvey, Development of rapid and selective epoxidation of α-pinene using single-step addition of H2O2 in an organic solvent-free process, RSC Adv., 2021, 11(52), 33027–33035 RSC.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Bio-based polycarbonate from limonene oxide and CO2 with high molecular weight, excellent thermal resistance, hardness and transparency, Green Chem., 2016, 18(3), 760–770 RSC.
- M. F. M. Gunam Resul, A. M. López Fernández, A. Rehman and A. P. Harvey, Development of a selective, solvent-free epoxidation of limonene using hydrogen peroxide and a tungsten-based catalyst, React. Chem. Eng., 2018, 3(5), 747–756 RSC.
- A. Rehman, A. M. López Fernández, M. F. M. Gunam Resul and A. Harvey, Highly selective, sustainable synthesis of limonene cyclic carbonate from bio-based limonene oxide and CO2: A kinetic study, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- L. Charbonneau, X. Foster, D. Zhao and S. Kaliaguine, Catalyst-Free Epoxidation of Limonene to Limonene Dioxide, ACS Sustainable Chem. Eng., 2018, 6(4), 5115–5121 CrossRef CAS.
- A. Rehman, E. Russell, F. Saleem, F. Javed, S. Ahmad, V. C. Eze and A. Harvey, Synthesis of trans-limonene bis-epoxide by stereoselective epoxidation of (R)-(+)-limonene, J. Environ. Chem. Eng., 2021, 9(1), 104680 CrossRef CAS.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, Alternating Copolymerization of Limonene Oxide and Carbon Dioxide, J. Am. Chem. Soc., 2004, 126(37), 11404–11405 CrossRef CAS PubMed.
- F. Parrino, A. Fidalgo, L. Palmisano, L. M. Ilharco, M. Pagliaro and R. Ciriminna, Polymers of Limonene Oxide and Carbon Dioxide: Polycarbonates of the Solar Economy, ACS Omega, 2018, 3(5), 4884–4890 CrossRef CAS PubMed.
- N. Kindermann, À. Cristòfol and A. W. Kleij, Access to Biorenewable Polycarbonates with Unusual Glass-Transition Temperature (Tg) Modulation, ACS Catal., 2017, 7(6), 3860–3863 CrossRef CAS.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Monomer design strategies to create natural product-based polymer materials, Nat. Prod. Rep., 2017, 34(4), 433–459 RSC.
- V. Schimpf, B. S. Ritter, P. Weis, K. Parison and R. Mülhaupt, High Purity Limonene Dicarbonate as Versatile Building Block for Sustainable Non-Isocyanate Polyhydroxyurethane Thermosets and Thermoplastics, Macromolecules, 2017, 50(3), 944–955 CrossRef CAS.
- M. Bähr, A. Bitto and R. Mülhaupt, Cyclic limonene dicarbonate as a new monomer for non-isocyanate oligo- and polyurethanes (NIPU) based upon terpenes, Green Chem., 2012, 14(5), 1447–1454 RSC.
- A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, Recent advances in the synthesis of cyclic carbonates via CO2 cycloaddition to epoxides, J. Environ. Chem. Eng., 2021, 105113 CrossRef CAS.
- G. Fráter, J. A. Bajgrowicz and P. Kraft, Fragrance Chem., 1998, 54(27), 7633–7703 Search PubMed.
- M. Stekrova, N. Kumar, A. Aho, I. Sinev, W. Grünert, J. Dahl, J. Roine, S. S. Arzumanov, P. Mäki-Arvela and D. Y. Murzin, Isomerization of α-pinene oxide using Fe-supported catalysts: Selective synthesis of campholenic aldehyde, Appl. Catal., A, 2014, 470, 162–176 CrossRef CAS.
- S. Qiu, G. Li, P. Liu, C. Wang, Z. Feng and C. Li, Chirality transition in the epoxidation of (−)-α-pinene and successive hydrolysis studied by Raman optical activity and DFT, Phys. Chem. Chem. Phys., 2010, 12(12), 3005–3013 RSC.
- L. A. Paquette, Handbook of Reagents for Organic Synthesis, Chiral Reagents for Asymmetric Synthesis, John Wiley & Sons, 1999 Search PubMed.
- P. A. Wender and T. P. Mucciaro, A new and practical approach to the synthesis of taxol and taxol analogs: the pinene path, J. Am. Chem. Soc., 1992, 114(14), 5878–5879 CrossRef CAS.
- D. J. Darensbourg, J. R. Wildeson, S. J. Lewis and J. C. Yarbrough, Solution and Solid-State Structural Studies of Epoxide Adducts of Cadmium Phenoxides. Chemistry Relevant to Epoxide Activation for Ring-Opening Reactions, J. Am. Chem. Soc., 2002, 124(24), 7075–7083 CrossRef CAS PubMed.
- S. J. Poland and D. J. Darensbourg, A quest for polycarbonates provided via sustainable epoxide/CO2 copolymerization processes, Green Chem., 2017, 19(21), 4990–5011 RSC.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, Catalytic Strategies for the Cycloaddition of Pure, Diluted, and Waste CO2 to Epoxides under Ambient Conditions, ACS Catal., 2017, 8(1), 419–450 CrossRef.
- M. Murayama, F. Sanda and T. Endo, Anionic Ring-Opening Polymerization of a Cyclic Carbonate Having a Norbornene Structure with Amine Initiators, Macromolecules, 1998, 31(3), 919–923 CrossRef CAS.
- F. Suriano, O. Coulembier, J. L. Hedrick and P. Dubois, Functionalized cyclic carbonates: from synthesis and metal-free catalyzed ring-opening polymerization to applications, Polym. Chem., 2011, 2(3), 528–533 RSC.
- T. Ariga, T. Takata and T. Endo, Cationic Ring-Opening Polymerization of Cyclic Carbonates with Alkyl Halides To Yield Polycarbonate without the Ether Unit by Suppression of Elimination of Carbon Dioxide, Macromolecules, 1997, 30(4), 737–744 CrossRef CAS.
- M. Selva, A. Caretto, M. Noè and A. Perosa, Carbonate phosphonium salts as catalysts for the transesterification of dialkyl carbonates with diols. The competition between cyclic carbonates and linear dicarbonate products, Org. Biomol. Chem., 2014, 12(24), 4143–4155 RSC.
- J. M. Coxon, E. Dansted, M. P. Hartshorn and K. E. Richards, Some rearrangements of 2,3- and 2,10-oxygenated pinane derivatives, Tetrahedron, 1969, 25(16), 3307–3312 CrossRef CAS.
- M. Aresta and E. Quaranta, Carbon dioxide: a substitute for phosgene, CHEMTECH, 1997, 27(3) CAS.
- E. Erdik and D. S. Matteson, Kinetics of osmium tetraoxide catalyzed trimethylamine N-oxide oxidations of cyclohexene and alpha-pinene to diols, J. Org. Chem., 1989, 54(11), 2742–2748 CrossRef CAS.
- A. Bansode and A. Urakawa, Continuous DMC Synthesis from CO2 and Methanol over a CeO2 Catalyst in a Fixed Bed Reactor in the Presence of a Dehydrating Agent, ACS Catal., 2014, 4(11), 3877–3880 CrossRef CAS.
- H. Huang, R. C. Samsun, R. Peters and D. Stolten, Greener production of dimethyl carbonate by the Power-to-Fuel concept: a comparative techno-economic analysis, Green Chem., 2021, 23(4), 1734–1747 RSC.
- AOCS, Official Methods and Recommended Practices of the American Oil Chemists' Society, Champaign Illinois, USA, 5th edn, 1998 Search PubMed.
- V. C. Eze, A. P. Harvey and A. N. Phan, Determination of the kinetics of biodiesel saponification in alcoholic hydroxide solutions, Fuel, 2015, 140, 724–730 CrossRef CAS.
- V. C. Eze, A. N. Phan and A. P. Harvey, A more robust model of the biodiesel reaction, allowing identification of process conditions for significantly enhanced rate and water tolerance, Bioresour. Technol., 2014, 156, 222–231 CrossRef CAS PubMed.
- A. A. S. Araújo, M. d. S. Bezerra, S. Storpirtis and J. d. R. Matos, Determination of the melting temperature, heat of fusion, and purity analysis of different samples of zidovudine (AZT) using DSC, Braz. J. Pharm. Sci., 2010, 46, 37–43 CrossRef.
- Z. Guo and Q. Lin, Coupling reaction of CO2 and propylene oxide catalyzed by DMC with co-complexing agents incorporated via ball milling, J. Mol. Catal. A: Chem., 2014, 390, 63–68 CrossRef CAS.
- J. Geschwind and H. Frey, Poly(1,2-glycerol carbonate): A Fundamental Polymer Structure Synthesized from CO2 and Glycidyl Ethers, Macromolecules, 2013, 46(9), 3280–3287 CrossRef CAS.
- J. Hopewell, R. Dvorak and E. Kosior, Plastics recycling: challenges and opportunities, Philos. Trans. R. Soc. London, Ser. B, 2009, 364(1526), 2115–2126 CrossRef CAS PubMed.
- G. Kale, T. Kijchavengkul, R. Auras, M. Rubino, S. E. Selke and S. P. Singh, Compostability of Bioplastic Packaging Materials: An Overview, Macromol. Biosci., 2007, 7(3), 255–277 CrossRef CAS PubMed.
- K. Das, P. Sahoo, M. Sai Baba, N. Murali and P. Swaminathan, Kinetic studies on saponification of ethyl acetate using an innovative conductivity-monitoring instrument with a pulsating sensor, Int. J. Chem. Kinet., 2011, 43(11), 648–656 CrossRef CAS.
- J. A. Vinson, J. S. Fritz and C. A. Kingsbury, Quantitative determination of esters by saponification in dimethyl sulphoxide, Talanta, 1966, 13(12), 1673–1677 CrossRef CAS PubMed.
- H. S. Levenson and H. A. Smith, Kinetics of the Saponification of the Ethyl Esters of Several Phenyl Substituted Aliphatic Acids1, J. Am. Chem. Soc., 1940, 62(9), 2324–2327 CrossRef CAS.
- H. A. Smith and H. S. Levenson, Kinetics of the Saponification of the Ethyl Esters of Normal Aliphatic Acids, J. Am. Chem. Soc., 1939, 61(5), 1172–1175 CrossRef CAS.
- V. Theodorou, K. Skobridis, A. G. Tzakos and V. Ragoussis, A simple method for the alkaline hydrolysis of esters, Tetrahedron Lett., 2007, 48(46), 8230–8233 CrossRef CAS.
- J. F. Mata-Segreda, Hydroxide as general base in the saponification of ethyl acetate, J. Am. Chem. Soc., 2002, 124(10), 2259–2262 CrossRef CAS PubMed.
- V. C. Eze, A. N. Phan, C. Pirez, A. P. Harvey, A. F. Lee and K. Wilson, Heterogeneous catalysis in an oscillatory baffled flow reactor, Catal. Sci. Technol., 2013, 3(9), 2373–2379 RSC.
- Y. Liu, E. Lotero and J. G. Goodwin, Effect of carbon chain length on esterification of carboxylic acids with methanol using acid catalysis, J. Catal., 2006, 243(2), 221–228 CrossRef CAS.
- V. C. Eze, J. C. Fisher, A. N. Phan and A. P. Harvey, Intensification of carboxylic acid esterification using a solid catalyst in a mesoscale oscillatory baffled reactor platform, Chem. Eng. J., 2017, 322, 205–214 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1ra07943c |
| This journal is © The Royal Society of Chemistry 2022 |