
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoupling of acceptor-substituted diazo compounds and tertiary thioamides: synthesis of enamino carbonyl compounds and their pharmacological evaluation†
Jim Seckaa,
Arpan Pala,
Francis A. Acquah bcd,
Blaine H. M. Mooersbcd,
Anand B. Karkie,
Dania Mahjoube,
Mohamed K. Fakhre,
David R. Wallacef,
Takuya Okadag,
Naoki Toyookag,
Adama Kutaa,
Naga Koduria,
Deacon Herndona,
Kenneth P. Robertsa,
Zhiguo Wanga,
Bethany Hilemana,
Nisha Rajagopala and
Syed R. Hussainia
bcd,
Blaine H. M. Mooersbcd,
Anand B. Karkie,
Dania Mahjoube,
Mohamed K. Fakhre,
David R. Wallacef,
Takuya Okadag,
Naoki Toyookag,
Adama Kutaa,
Naga Koduria,
Deacon Herndona,
Kenneth P. Robertsa,
Zhiguo Wanga,
Bethany Hilemana,
Nisha Rajagopala and
Syed R. Hussainia
aDepartment of Chemistry and Biochemistry, The University of Tulsa, 800 S. Tucker Drive, Tulsa, Oklahoma 74104, USA. E-mail: syed-hussaini@utulsa.edu
bDepartment of Biochemistry and Molecular Biology, University of Oklahoma of Health Sciences Center, Oklahoma City, OK 73104, Unites States
cStephenson Cancer Center, University of Oklahoma Health Sciences Center, Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
dLaboratory of Biomolecular Structure and Function, University of Oklahoma of Health Sciences Center, Oklahoma City, OK 73104, USA
eDepartment of Biological Science, The University of Tulsa, 800 S. Tucker Drive, Tulsa, Oklahoma 74104, USA
fDepartment of Pharmacology and Physiology, Oklahoma State University Center for Health Sciences, Tulsa, Oklahoma 74107, USA
gFaculty of Engineering, University of Toyama, 3190 Gofuku, Toyama, 930-8555, Japan
First published on 5th July 2022
Abstract
This paper describes the synthesis of enamino carbonyl compounds by the copper(I)-catalyzed coupling of acceptor-substituted diazo compounds and tertiary thioamides. We plan to use this method to synthesize indolizidine (−)-237D analogs to find α6-selective antismoking agents. Therefore, we also performed in silico α6-nAchRs binding studies of selected products. Compounds with low root-mean-square deviation values showed more favorable binding free energies. We also report preliminary pharmacokinetic data on indolizidine (−)-237D and found it to have weak activity at CYP3A4. In addition, as enamino carbonyl compounds are also known for antimicrobial properties, we screened previously reported and new enamino carbonyl compounds for antibacterial, antimicrobial, and antifungal properties. Eleven compounds showed significant antimicrobial activities.
Introduction
Enamino carbonyl compounds are also called enaminones despite sometimes having carbonyl-containing functional groups that are not ketones.1 Enamino carbonyl compounds are essential synthetic intermediates.2–4 They possess multiple nucleophilic and electrophilic sites that are manipulable to make diverse substrates. Due to their importance in synthesis, several methods exist for preparing enamino carbonyl compounds.2,3,5,6 However, only a few of these methods make enamino carbonyl compounds with an exocyclic double bond (Fig. 1). Enamino carbonyl compounds containing an exocyclic double bond are common structural motifs in bioactive compounds.2,7–9 They also serve as critical intermediates in the synthesis of bioactive heterocycles and pharmaceuticals.2,7,10 Traditionally exocyclic enamino carbonyl synthesis is achieved by the imino ester approach or the Eschenmoser coupling reaction.2,3,11,12 New strategies, such as involving rhodium azavinylcarbenes and isatins, have been reported that complement the imino ester approach and the Eschenmoser coupling reaction.13 However, they all have limitations. For instance, involving rhodium azavinylcarbenes and isatins only provides α-amino enaminones.13 | ||
| Fig. 1 Traditional methods for synthesizing enamino carbonyl compounds with an exocyclic bond. | ||
The Eschenmoser coupling reaction and the imino ester approach both have substrate scope limitations and reactivity issues.2,14–16 For instance, product formation is disfavored in the Eschenmoser coupling reaction if the thioamide is sterically hindered.2 We also observed substrate scope limitation when we tried to make a simple enamino carbonyl compound (1) using literature procedures17 and formed the product in a low yield. Even after optimization, the reaction gave only 50% of the product (Scheme 1).
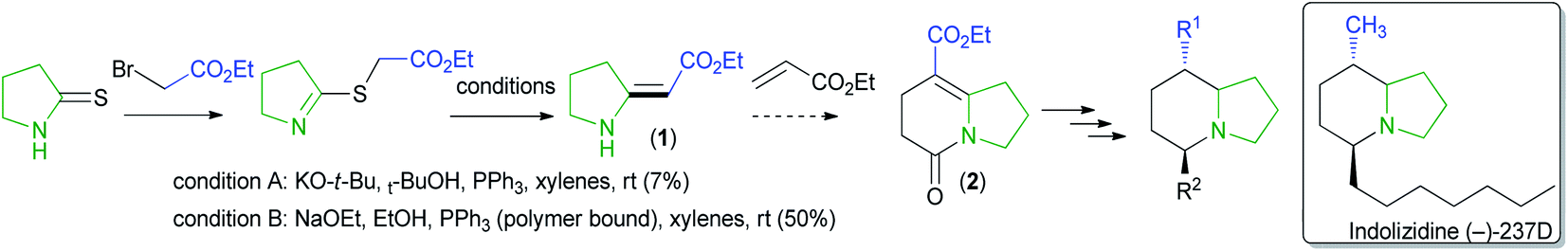 | ||
| Scheme 1 Synthetic plan for preparing indolizidine (−)-237D analog. | ||
We needed compound 1 to be cyclized to 2, which we planned to use for making indolizidine (−)-237D analogs.18 Indolizidine (−)-237D inhibits nicotine-evoked dopamine release from rat brain slices and is thought to inhibit α6β2* nAChRs selectively.19 Although its pharmacological profile (vide infra) is not ideal, it is a reasonable starting point for a subtype-selective antismoking drug-development program. Therefore, we investigated a method for preparing enamino carbonyl compounds that are tri-substituted at the double bond to get to the indolizidine core efficiently.
Our investigation is based on previous contributions to the metal-catalyzed coupling reaction of thioamides and diazo compounds by us and others.20–26 The metal-catalyzed coupling reaction of thioamides and diazo compounds is considered a modified Eschenmoser coupling reaction.2 Although we have reported three reactions between thioamides and diazo-monoketones,25,27 we were unsuccessful in coupling diazomonoesters and thioamides in the past.23,25,27,28 Therefore, this paper's primary emphasis is the Cu(I)-catalyzed coupling of tertiary thioamides and acceptor-substituted diazo compounds. We also report in silico α-6-nAChRs binding studies of selected products, which serve as fragments of indolizidine (−)-237D analogs. Additionally, we communicate preliminary pharmacokinetic data on indolizidine (−)-237D.
As enamino carbonyl compounds are known for therapeutic properties,29,30 we asked ourselves do our enamino carbonyl compounds also possess bioactivity? Therefore, we conducted antibacterial, antimicrobial, and antifungal screening of previously reported and new enamino carbonyl compounds. We share these findings here as well.
Results and discussion
Synthetic methodology
To prepare trisubstituted enamino carbonyl compounds, we initially investigated multiple catalysts for the coupling of thioamides and diazo compounds with a single electron-withdrawing substituent that serves as a precursor to acceptor-substituted carbenoids.31 Then, we attempted the similar reaction conditions we have previously found to be successful in coupling donor/acceptor diazo compounds and thioamides.24 We used these conditions to couple 3a and 4a and compared the results with other catalysts (Table 1) and other reaction conditions. We selected this coupling reaction as debenzylation of 5a could give 1 that we needed for synthesizing indolizidine (−)-237D analogs. We started the screening reaction of 3a with 4a using 5 mol% of CuBr at 40 °C. 1H NMR of crude reaction mixture showed 78% conversion (Table 1, entry 1) after 24 h. When the catalyst amount was increased to 10 mol%, it increased the conversion to 89% (Table 1, entry 2). As diazo compounds can act as a nucleophile rather than a carbene source in the presence of Lewis acids, we tested AgSbF6 in the reaction (Table 1, entry 3).32,33 No reaction was observed in this experiment. Although Rh2(OAc)4 has been used in the coupling of thioamides and diazo compounds,20–22 no product formation was observed with 10 mol% Rh2(OAc)4 at room temperature (Table 1, entry 4). Heating the reaction mixture to 40 °C led to the complete dimerization of 4a (Table 1, entry 5). Ruthenium(II) catalysts effectively coupled acceptor/acceptor substituted diazo compounds with thioamides.23,27 However, the use of tris(triphenylphosphine)ruthenium(II) dichloride in the coupling of thiolactam 3a with ethyl diazo ester 4a led to rapid dimerization of 4a at room temperature within 2 h (Table 1, entry 6). The Grubbs 1st generation catalyst also was ineffective to promote the reaction at room temperature (Table 1, entry 7). When the reaction was heated to 40 °C, the reaction produced 38% of enamino ester 5a, and the rest of the diazo compounds dimerized (Table 1, entry 8). Ru(II)–indenylidene complex was also ineffective for this transformation (Table 1, entries 9 and 10). No product formation was observed in a control reaction without any catalyst (Table 1, entry 11). Although we had a good conversion of 3a and 4a into 5a (Table 1, entry 2), the reaction gave inconsistent results. Finally, the re-purification of 3a and the gradual addition of two equivalents of 4a gave consistent results, and the product could be isolated in high yields (Table 1, entry 12). Re-purification possibly reduces the amount of sulfur present as an impurity in thioamides which could retard the coupling reaction.34 On the other hand, the gradual addition of diazo compounds slows the unwanted dimerization reaction.35 Although we have previously coupled one diazoketone to thioamides,25,27 we could never couple thioamides and diazoesters23 and did not attempt other diazoketones. Therefore, this study solves this challenge and produces acceptor-substituted enamino carbonyl compounds in high yields.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Temp.b (°C) | Time (h) | 5ac (%) |
| a Reaction conditions: 3a (0.20 mmol), 4a (0.26 mmol), catalyst (x mol%) in 2.0 mL dichloroethane.b Reactor temperature.c Percent conversion of 3a into 5a as determined by 1H NMR analysis.d As judged by TLC analysis.e 2.0 eq. of 4a was used.f As judged by TLC analysis. Re-purified 3a was used along with the gradual addition of 4a.g Isolated yield. | ||||
| 1 | CuBr (5 mol%) | 40 | 24 | 78 |
| 2 | CuBr (10 mol%) | 40 | 24 | 89 |
| 3d | AgSbF6 (10 mol%) | 40 | 16 | 0 |
| 4d | Rh2(OAc)4 (10 mol%) | rt | 2 | 0 |
| 5d | Rh2(OAc)4 (10 mol%) | 40 | 2 | 0 |
| 6d | Ru(PPh3)3Cl2 (10 mol%) | rt | 2 | 0 |
| 7d | Grubbs 1st gen (5 mol%) | rt | 14 | 0 |
| 8 | Grubbs 1st gen (5 mol%) | 40 | 2 | 38 |
| 9d | Ru(II)-indenylidene (5 mol%) | rt | 14 | 0 |
| 10d | Ru(II)-indenylidene (5 mol%) | 40 | 2 | 0 |
| 11d | No catalyst | 40 | 18 | 0 |
| 12e,f | CuBr (10 mol%) | 40 | 24 | 100 (86)g |
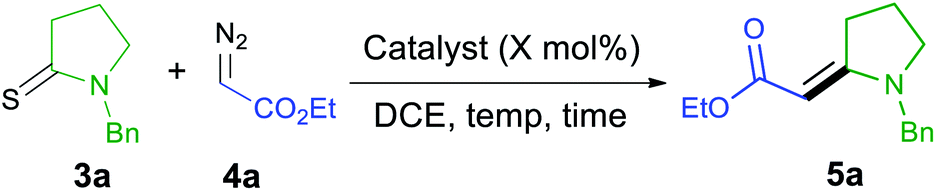
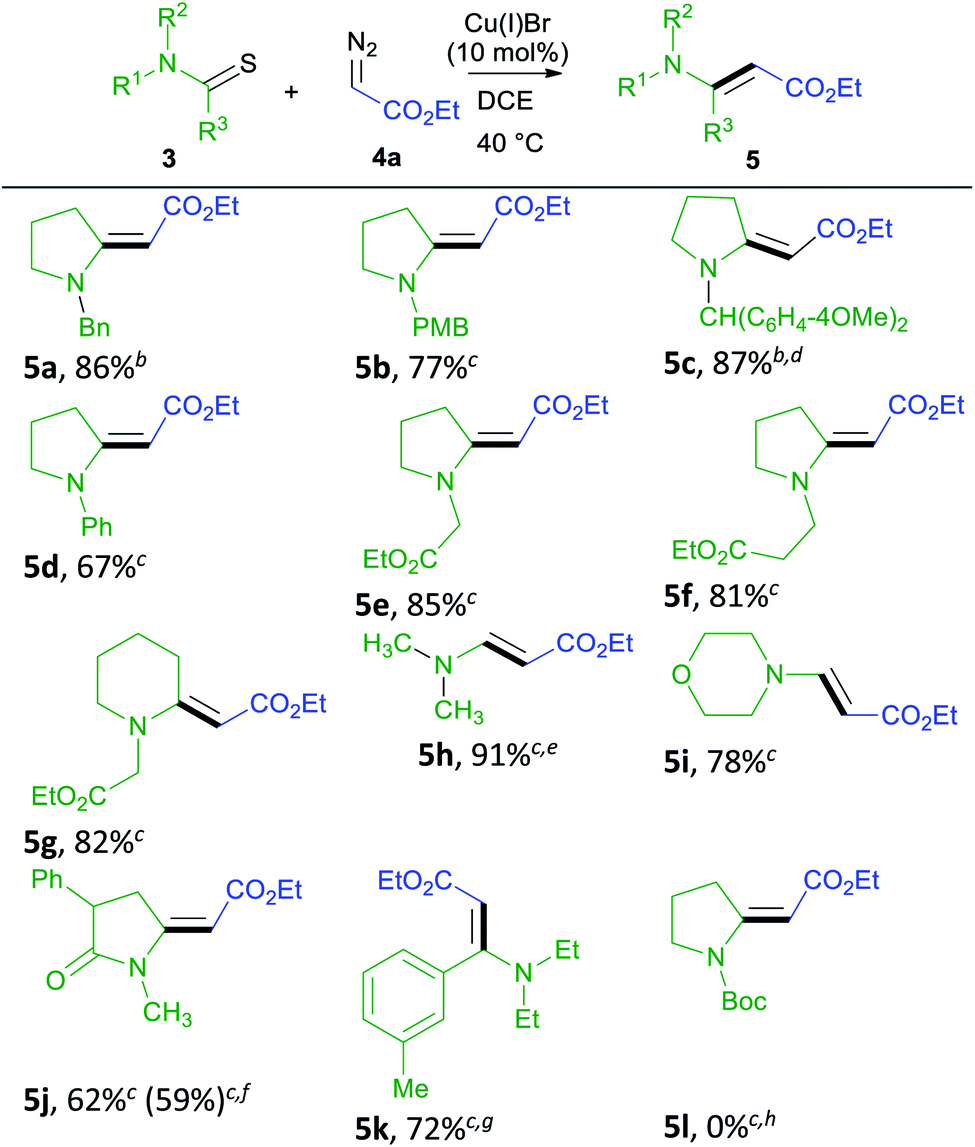
We conducted a rapid assessment of the coupling reaction (see ESI,† rapid assessment of reaction) using Glorius's method.36 It indicated that the reaction tolerates several functional groups and heterocycles (Table S2†). However, terminal alkenes, terminal alkynes, aldehydes, phenols, alkyl chlorides, aryl esters, primary amines, chloropyrimidines, and N-alkyl indoles could retard the formation of enamino carbonyl compounds, and primary amines, chloropyrimidines, and alkyl chlorides may not survive under reaction conditions. Based on these observations, we first screened various thioamides in this reaction (Table 2). Compounds 5a–5c contain nitrogen protecting groups that can be easily removed, and the resulting products could be transformed into indolizidine using the transformation shown in Scheme 1 (1 into 2).37 Compound 5d contains Ph directly attached to the nitrogen atom increasing the steric hindrance close to the reacting centers (thioamide carbon and sulfur). Steric hindrance is known to be detrimental to the metal-catalyzed coupling reaction of thioamides and diazo compounds.23 Not surprisingly, 5d is obtained in a lower yield, and no starting material could be recovered at the end of this reaction. Alkyl esters attached to the nitrogen atom can be useful in the formation of bicyclic structures, including indolizidines. Such groups are also tolerated in the reaction, and we obtained 5e–5g in good yields.3,38,39 Thioformamides are rarely reported in the metal-catalyzed coupling of thioamides and diazo compounds.24,25 However, they gave good yields of coupled products (5h and 5i). Biologically important compounds could also be modified through this coupling. Thus monothiodicarboximide, which is the monothioimide-derivative of the anticonvulsant Phensuximide,40 could be converted into 5j in moderate yield, and the starting material could not be consumed entirely after 24 h. As was for 5d, we suspect that this lower yield is due to the sterically hindered nature of the thioamide (3j). Finally, the thioamide analog of the insecticide DEET (3k) could also be converted into (5k). In this case, no reaction was observed at 40 °C, and the reaction required heating at 110 °C to get a reasonable yield. We attribute the stereochemical preference of products to the equilibrating nature of stereoisomers41 and do not consider this reaction to be stereoselective. Although N-Boc-protected thioamides are known to couple with donor/acceptor-substituted diazo compounds, 5l could not be prepared via the coupling reaction,24 and the attempted coupling resulted in decomposition of the thioamide (3l) under the reaction conditions. Although N-Boc-protected thioamides are known to give trisubstituted enamino carbonyl compounds (similar to 5l),42 no experimental procedures are reported in detail. Our attempts to prepare 5l via the thio-Reformatsky reaction, using experimental procedure43 reported for the thio-Reformatsky reaction of N-Ph-protected thioamides failed.44 Similar failures are reported by others,45 and the conversion of N-Boc thioamides into enamino carbonyl compounds remains a challenge. Examples presented in Table 2 also show that the coupling reaction is successful for cyclic- (5a–5g) and acyclically-positioned (5h, 5i and 5k) thioamides. The stereochemistry of products 5a–5e, 5g–5h, and 5j–5k was assigned by NOESY experiments (ESI†) or by comparison with literature NMR values (5f (ref. 46) and 5i (ref. 47)).
| a Reaction conditions: 3 (0.125 mmol), 4a (0.250 mmol), and CuBr in 2.00 mL DCE and the reaction stirred.b 4a added in three portions over 24 h.c 4a added using a syringe pump over 24 hours.d Reaction was heated at 50 °C (reactor temperature).e NMR yield.f Reaction was performed at 1.48 mmol scale.g Reaction was heated at 110 °C (reactor temperature).h Thioamide decomposed. |
|---|
 |
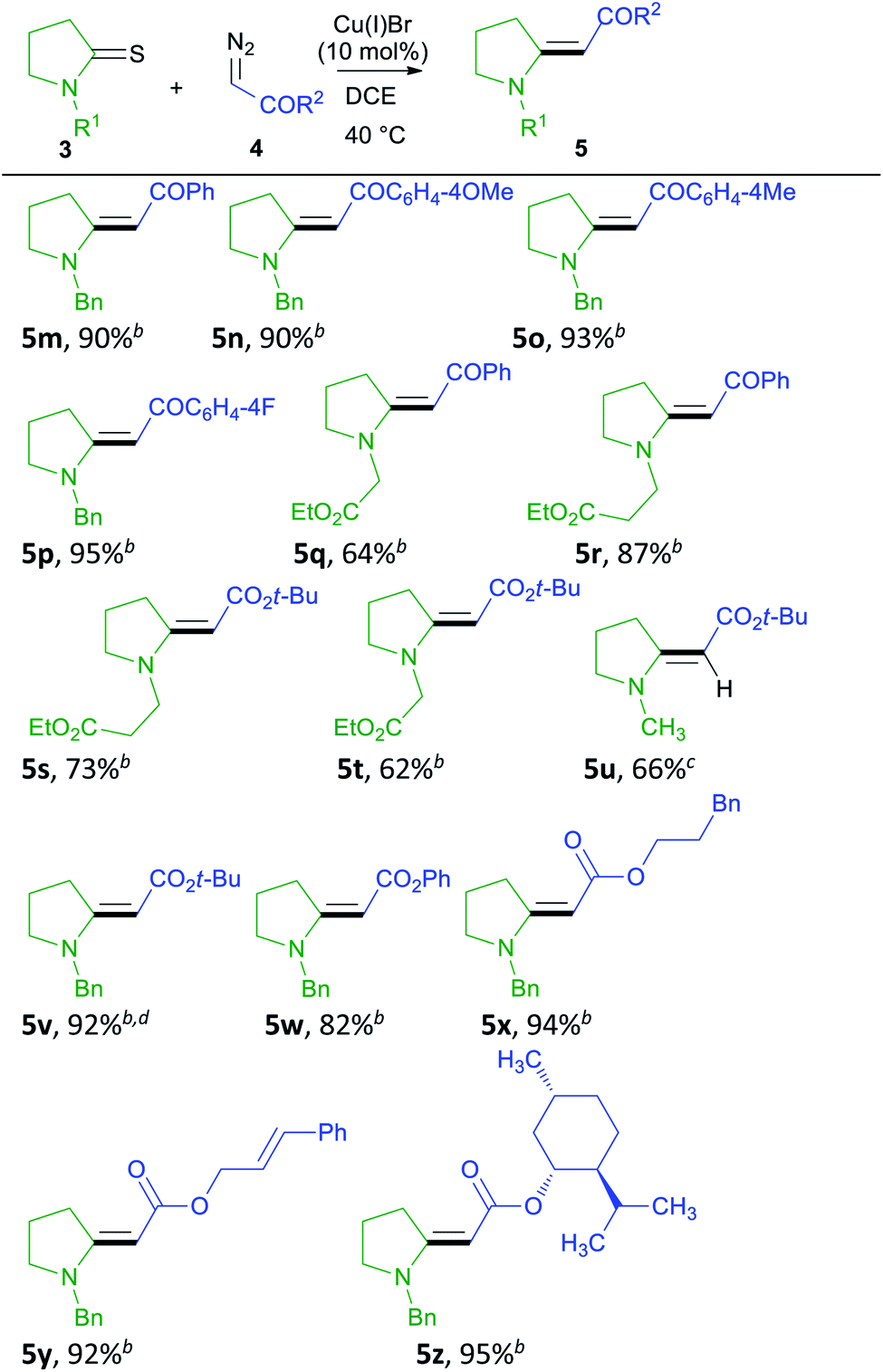
Next, we explored the scope of diazo compounds in this coupling reaction (Table 3). We tested ketones and esters. Ketones gave excellent yields of the coupled products (5m–5r), and both electron-donating and withdrawing groups on diazo compounds gave enamino carbonyl products (compare 5m–5p) in excellent yields. However, in the case of 3q, only a moderate yield of 5q could be obtained as the reaction did not go to completion. With tert-butyl diazoacetate, the yields of products remained modest (5s–5u) as the reaction did not go to completion. With more diazo compound (3 eq.), the yield could be improved (5v). Diazo esters containing aryl groups gave better yields of enamino carbonyl compounds (5w–5y). No cyclopropanation was observed, and the product 5y was obtained chemoselectively. In the end, we also tested the possibility of attaching a chiral ester, and product 5z could be obtained in excellent yield. The stereochemistry of 5m (ref. 27) was assigned by comparing its NMR values with literature, while the stereochemistry of 5n–5z was assigned by NOESY experiments (ESI†). Although diazo esters are known to undergo other reactions with enamino carbonyl compounds in the presence of Cu(I) salts,48 no further reactions were observed in any of the examples listed here. We believe that the reaction mechanism of this reaction is the same as what we proposed for the coupling of thioamides and donor/acceptor-substituted diazo compounds.24
Molecular docking of selected enamino carbonyl compounds against α6β2 nicotinic acetylcholine receptor
Enamino esters presented above can serve as synthetic intermediates for the synthesis of indolizidine (−)-237D (Scheme 1) analogs and are simpler fragments of indolizidine (−)-237D analogs. Therefore, we were interested in conducting molecular docking analysis of selected enamino carbonyl compounds and comparing the results with the parent indolizidine (−)-237D.28 A simpler fragment with a similar binding affinity towards α6β2 nAChRs as that of indolizidine (−)-237D is easier to make and modify to provide a selective inhibitor of α6β2 nAChRs.To rank the indolizidine (−)-237D-related compounds by binding affinity towards α6β2 nAChR for computational studies, we docked the compounds with AutoDock Vina.49 Docking energies ranged from −7.3 to −5.4 kcal mol−1 at one of the binding sites for the α6β2 receptor (Table S4†). We summarize the detailed interactions between the compounds and the binding site residues in Table S5.† While the major forces driving the interactions between the analogs and the receptor are hydrophobic, some compounds made hydrophilic contacts with the active site residues (Table S5,† Fig. 2).
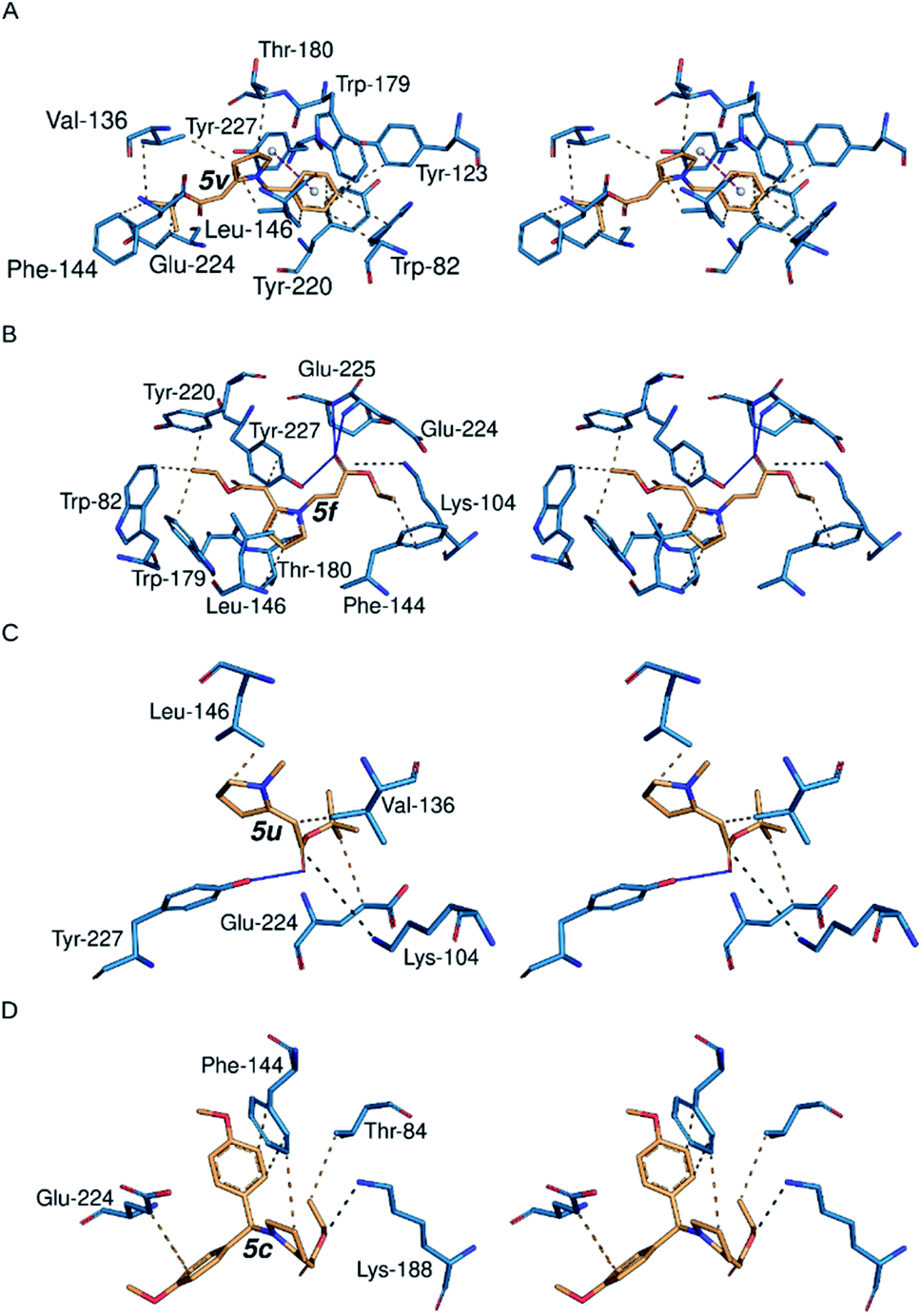 | ||
| Fig. 2 Binding site 2 receptor–ligand interactions using PyMOL and PLIP. (A – D) PyMOL stereo figures of the interactions between representative compounds and the binding site residues of the α6β2 nAChR; (A) 5v, (B) 5f, (C) 5u, and (D) 5c. Broken orange lines represent hydrophobic interactions; magenta broken lines with white spheres = pi-stacking; black broken lines represent salt bridges; straight blue lines represent hydrogen bonds. | ||
Molecular dynamic simulation and analysis
Next, we used MD simulations with each of the nine receptor–analog complexes to check the docking complex's stability during the simulation and verify the binding affinity results from the docking operations by comparing the docking energy to the binding energy from MD simulation. We extracted the α6β2 nAChR backbone root-mean-square deviation (RMSD) from the simulation trajectories to check the convergence of the simulation. We also extracted the RMSD of the compounds in the binding pockets. Analysis of the RMSD of the compounds in the α6β2 binding sites showed stable equilibration in the binding pocket for six out of the nine compounds (Fig. 3). While two out of the three remaining compounds (5e and 5t) show low RMSD values up to 60 ns of the simulation, compound 5c quickly moved out of the pocket with high average RMSD values. As expected, compounds with more favorable docking energies also showed low RMSD values during simulation. Conversely, analogs with less favorable docking energies showed high RMSD values, indicating instability and short residence time at the binding sites. | ||
| Fig. 3 Stability analysis of the compounds during simulations. (A–C) The root-mean-square deviation (RMSD) plot of the compounds in complex with α6β2 nAChR with respect to their starting structures as a function of simulation time. | ||
Predicted binding energies from the MMGBSA calculations were also consistent with previous results. Compounds with low average RMSD values also had the more favorable binding free energies and vice versa (Fig. 4).
 | ||
| Fig. 4 Comparison of docking energy and predicted binding energy using the MMGBSA method. Predicted binding energies are shown as mean ± sd; n = 1000. | ||
Preliminary pharmacokinetic evaluation of indolizidine (−)-237D
As the synthetic methodology described in this paper is planned to be used to synthesize indolizidine (−)-237D analogs, we conducted preliminary PK studies on the indolizidine (−)-237D to provide data that we could compare with the prepared compounds. Although indolizidine (−)-237D has been tested towards a6B2* nAChRs,19 no PK data exists on this compound. Displacement of radioligand binding was accomplished with compounds of known affinity for α6β2* nAChRs,19 nicotine, and anabasine.50–52Radioligand binding
Displacement of [3H] cytisine binding was observed with both the lower and higher concentration of each drug (Fig. 5). Whole-brain homogenates displayed specific binding of about 70 fmol mg−1 protein. To determine whether the variation within the data sets contributed to the overall difference between groups, we used the Brown–Forsythe test (p = 0.9609), which suggests that the differences observed were due to drug effects and not variation within the data sets. One-way ANOVA revealed a significant effect of drugs on the binding of [3H] cytisine across each drug/concentration combination (F6,14 = 16.46; p < 0.0001). Each drug/concentration combination was significantly (p < 0.01) lower than control values. There was a non-significant concentration-dependent reduction (∼10%) in [3H] cytisine binding in each high concentration group compared to the appropriate lower concentration group.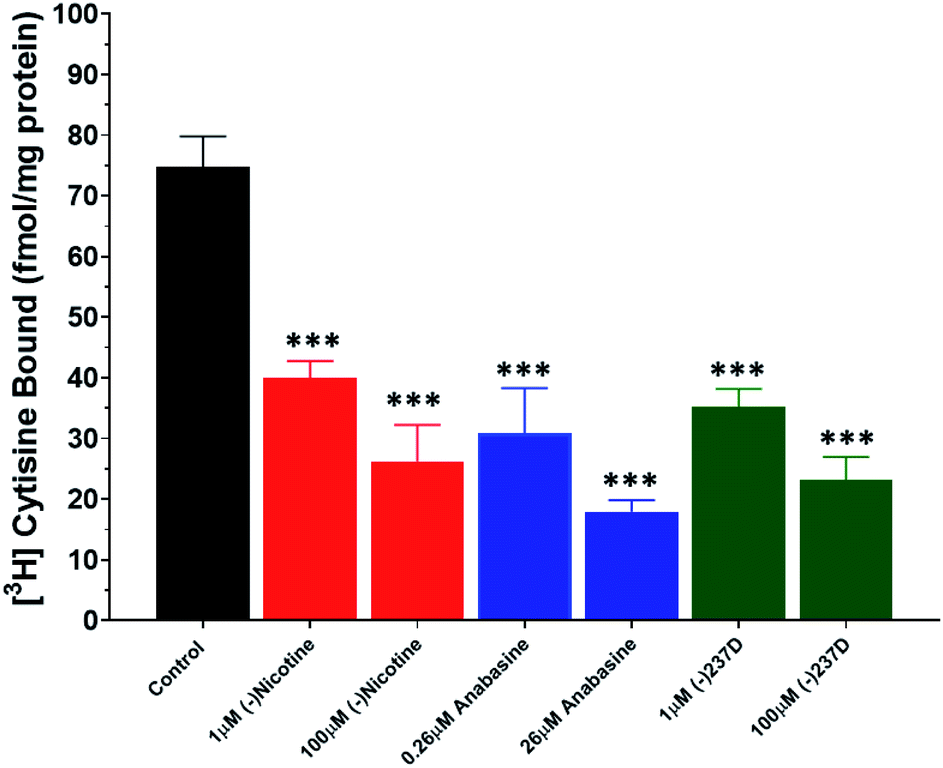 | ||
| Fig. 5 Displacement of [3H] cytisine binding in whole brain homogenates. Homogenates were incubated in the presence of 3 nM [3H] cytisine and a low or high concentration of nicotine, anabasine or (−)-237D for 90 min. Specific binding was calculated by subtracting binding in the presence of 50 μM nicotine from total binding. Data represent the mean ± SEM of 3 brains (n = 3) assayed in duplicate. ***p < 0.001. | ||
Inhibition of CYP3A4
The activity of CYP3A4 and the potential inhibition of CYP3A4 activity was determined using a commercially available kit (Abcam, Cambridge UK). The assay uses the conversion of the weakly fluorescent compound resazurin by CYP3A4 to the strongly fluorescent product resorufin. The fluorescent response was determined with increasing concentrations of resorufin up to 50 pmol per well (Goodness of Fit (Sy.x) = 996.9). To determine if the vehicle (acetonitrile) exhibited any inhibitory effect of CYP3A4 activity, a time course was performed using 10% acetonitrile, 100 μM (−)-237D, or 10 μM ketoconazole. Ketoconazole is a potent inhibitor of CYP3A4 and was included as positive control. Examining the data (Fig. 6A), 10 μM ketoconazole was near 100% effective in inhibiting CYP3A4 activity as indicated by the flat line that doesn't rise much above baseline.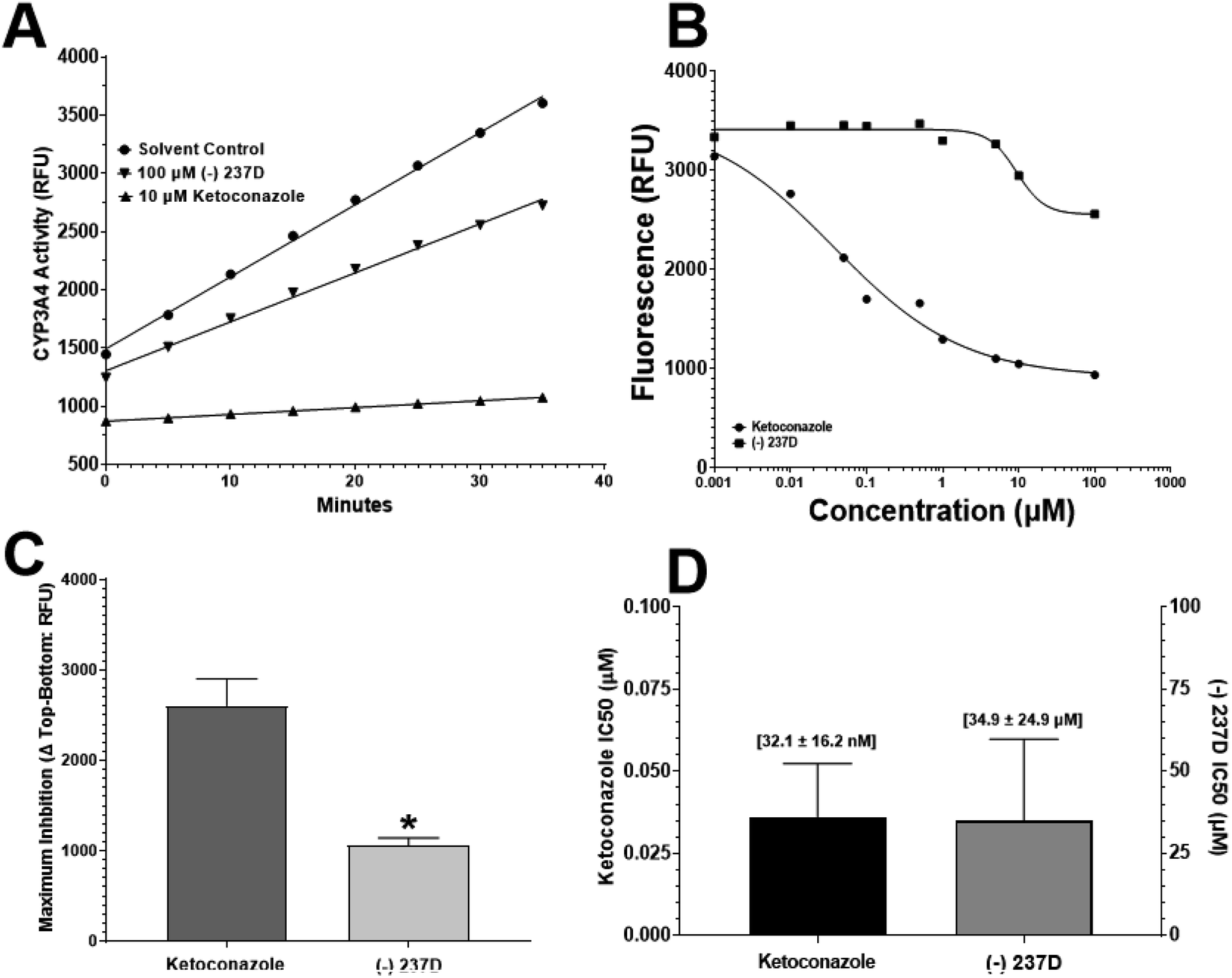 | ||
| Fig. 6 CYP3A4 activity and potential inhibition by (−)-237D. Assay validation was performed using a linear response curve of resorufin fluorescence and determination if the solvent of acetonitrile exerted any inhibition (A). IC50 curves determined the potency of ketoconazole or (−)-237D inhibition of CYP3A4 activity, and maximum enzyme inhibition (B). From the IC50 curves, calculated maximum inhibition (C) and the IC50 values (D) are presented. It is evident that (−)-237D inhibits significantly less CYP3A4 activity and has a much higher IC50 value. Data represent the mean ± SEM of 3 brains (n = 3) assayed in duplicate. *p < 0.05. | ||
Acetonitrile (10%) solvent had minimal effect on CYP3A4 activity. The slope of the (−)-237D response was slightly different from that of control after 35 minutes of exposure. IC50 analyses (Fig. 6B) support the comparison of (−)-237D to the vehicle with only a small percentage of CYP3A4 inhibition compared to ketoconazole. Goodness of fit (Sy.x) values for each set of curves were 506.8 and 587.7 for ketoconazole and (−)-237D, respectively. Comparison of the maximum inhibition exerted by either ketoconazole or (−)-237D is represented in Fig. 6C, with inhibition calculated by using the formula: ΔRFU(Topcalculated − Bottomcalculated). The calculated ‘Top’ and ‘Bottom’ values are determined by the curve fitting of GraphPad. The maximum inhibition of CYP3A4 by (−)-237D is significantly (p < 0.05) lower by 41% compared to the ketoconazole response. Comparing IC50 values (Fig. 6D) ketoconazole is significantly (p < 0.0001) lower than (−)-237D. Ketoconazole is nearly 1000-fold more potent at inhibiting CYP3A4 activity.
From the data presented here, it is concluded that (−)-237D has only very weak activity at CYP3A4. Collectively, there is only a slight reduction in CYP3A4 activity as indicated by the time course and inhibition curve analysis showing that (−)-237D is only marginally more inhibitor alone and that the maximum inhibition is only 25–30% of control baseline values compared to 75–80% inhibition by ketoconazole. Comparing IC50 values, ketoconazole is approximately 1000-fold more potent than (−)-237D.
Antimicrobial properties of enamino carbonyl compounds
Antimicrobial resistance among microbial pathogens has become a major health crisis around the globe.53 Hence, research towards the synthesis and characterization of novel compounds with antimicrobial activities is ongoing, which might provide a better tool (compound) to combat the antimicrobial resistance problem.54 Meanwhile, the community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA), hospital-acquired methicillin-resistant S. aureus (HA-MRSA), Campylobacter jejuni, and Candida albicans are some common human pathogens found in clinical settings and retail food products.55–58 Reports of these microbes with multidrug resistance (antibacterial or antifungal) have indicated the health system's challenges for effective clinical treatment.59–62 So, this study was designed to investigate the biological activity of enamino carbonyl compounds presented in this paper and reported earlier by us elsewhere2,23–25,41,63 (Table S6†) against those common microbial pathogens.Among the screened enamino carbonyl compounds (total of 45, Table S6†), eleven compounds showed antimicrobial activity against one or more tested pathogens (Table S7†). None of the enamino carbonyl compounds synthesized in the current study showed antimicrobial activity. Six compounds could inhibit the growth of all tested pathogens (Fig. 7A and B). Meanwhile, eight compounds showed antimicrobial activity against both MRSA (CA-MRSA and HA-MRSA) strains with similar inhibition zones. However, NK-VIII-169 was found effective against only the CA-MRSA strain but not the HA-MRSA strain. Likewise, organic compounds (NK-III-49, API-II-51) were effective against Gram-positive bacterial strains (CA-MRSA and HA-MRSA) and fungal strain (Candida strain) but could not inhibit the growth of Gram-negative bacterial strain (Campylobacter strain). However, AP-II-61 and AP-II-104 were effective against the Campylobacter strain only. Six of the 11 compounds (AP-II-104, AP-II-61, AP-II-51, AP-II-100, AP-II-142, and AP-II-144) are substituted with a geminal ester and an aryl group at the alkene.
 | ||
| Fig. 7 Screening for antimicrobial activity of organic compounds against common pathogens (CA-MRSA, HA-MRSA, Candida albicans, and Campylobacter jejuni). (A) Number of tested organic compounds (shown inside bracket) which showed antimicrobial activity against used pathogens. (B) Zone of inhibition of all effective organic compounds against all tested pathogens. Table S7† contains structures of enamino carbonyl compounds listed in the figure. | ||
Conclusions
We have successfully shown the coupling reaction between thioamides and acceptor-substituted diazo compounds. With this report, the metal-catalyzed coupling of all major classes of diazo compounds (acceptor/acceptor-substituted diazo compounds, donor/acceptor-substituted diazo compounds, and acceptor-substituted diazo compounds) and thioamides has become possible. The products of this coupling reaction are enamino carbonyl compounds which could be potential intermediates for the synthesis of indolizidine (−)-237D analogs. These enamino carbonyl compounds could also be considered as simpler fragments of indolizidine (−)-237D analogs. Indolizidine (−)-237D is known to inhibit nicotine-evoked dopamine release, likely via selective inhibition of a6B2* nAChRs. Therefore, molecular docking and molecular dynamic simulation of selected enamino carbonyl compounds were compared with indolizidine (−)-237D. Enamino carbonyl compounds with low average RMSD values were found to have more favorable binding free energies. We have also reported preliminary PK studies of indolizidine (−)-237D, which suggests that (−)-237D has moderate affinity α6β2* nAChRs. A potency series from the binding data indicate that Ki values follow this potency: anabasine < nicotine = (−)-237D. Our early studies exploring the metabolizing system for (−)-237D suggest that CYP3A4, a major CYP isozyme responsible for drug metabolism, is not a significant factor in (−)-237D metabolism. We are currently pursuing the coupling reaction presented in this paper for synthesizing indolizidine (−)-237D analogs. Additionally, we have also reported results of antimicrobial screening of selected enamino carbonyl compounds. These compounds belong to three major types of enamino carbonyl compounds (i) compounds where the germinal alkene is substituted with one carbonyl group, (ii) two carbonyl groups, or (iii) one carbonyl one non-carbonyl group. Eleven compounds belonging to each class were found to possess significant antimicrobial activities.Experimental
Synthesis
General procedure A for the synthesis of enamino carbonyl compounds. A solution of a doubly columned thioamide 3 (0.20 mmol) in DCE (1.0 mL) was added to a flamed dried screw-capped reaction vessel containing copper(I) bromide (10 mol%) and a stirring bar. The vial containing the thioamide was rinsed twice with dried DCE (0.50 mL each), and the washings were transferred to the reaction vessel. Diazo carbonyl compound 4 (0.40 mmol) was divided into three portions. The first portion was transferred to the reaction vessel at 0 h (zero h), and the reaction was heated at 40 °C. The second portion of 4 was added after 8 h of stirring, and the third portion after another 8 h (16 h). After 24 h the reaction was allowed to come to rt. The solvent was evaporated, and the reaction mixture was chromatographed to give pure 5.
General procedure B for the synthesis of enamino carbonyl compounds. A solution of a doubly columned thioamide 3 (0.200 mmol) in DCE (1.00 mL) was added to a flamed dried screw-capped reaction vessel containing copper(I) bromide (10 mol%) and a stirring bar. The vial containing the thioamide was rinsed twice with dried DCE (0.500 mL each), and the washings were transferred to the reaction vessel. Liquid diazo compounds 4 (0.400 mmol) were directly taken into a gas-tight syringe, while solid diazo compounds 4 were dissolved in 100 μL of DCE and then taken into a gas-tight syringe. The diazo compounds 4 were dispensed using a syringe pump (syringe diameter 2 mm, rate 0.200 μL min−1) over 24 h. After 24 h, the reaction was allowed to come to rt. The solvent was evaporated, and the reaction mixture was chromatographed to give pure 5.
(E)-Ethyl 2-(1-(4-methoxybenzyl)pyrrolidin-2-ylidene)acetate (5b). Using general procedure B, 0.200 mmol of 3b was converted into crude 5b. Flash column chromatography (5% EtOAc in petroleum ether) gave pure 5b (42.0 mg, 0.153 mmol, 77%) as a yellow viscous oil. The stereochemistry was assigned using 1D-NOESY. Rf = 0.46 (10% ethyl acetate in petroleum ether). 1H NMR (400 MHz, CDCl3): δ: 7.12 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 4.71 (s 1H), 4.29 (s, 2H), 4.09 (q, J = 7.2 Hz, 2H), 3.80 (s, 3H), 3.31 (t, J = 7.2 Hz, 2H) 3.22 (t, J = 7.6 Hz, 2H), 1.95 (tt, J = 7.2, 7.6 Hz, 2H), 1.24 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 169.7, 165.4, 159.1, 128.7, 128.2, 114.2, 78.3, 58.4, 55.4, 52.3, 49.5, 32.8, 21.2, 14.9. HRMS (ESI+) m/z: (M + H)+: calcd for C16H22NO3, 276.1600; measured, 276.1609.
Ethyl 2-(1-(bis(4-methoxyphenyl)methyl)pyrrolidin-2-ylidene)acetate (5c). Using general procedure A, with the exception that the reaction was heated at 50 °C, 0.200 mmol of 3c was converted into crude 5c. Flash column chromatography (20% ethyl acetate in petroleum ether and 1% Et3N) gave pure 5c (42.0 mg, 0.152 mmol, 76%) as a mixture of equilibrating isomers41 (66.0 mg, 0.173 mmol, 87%). The major isomer was identified as the E isomer using 1D-NOESY. The NMR data below is for the E isomer as individual peak assignment of other minor isomers could not be possible due to lower intensity or broad peaks that are typical of rotamers.67 Rf = 0.18 (dichloromethane); 1H NMR (400 MHz, CDCl3): δ: 7.02 (d, J = 8.6 Hz, 4H), 6.86 (d, J = 8.6 Hz, 4H), 5.85 (s, 1H), 4.61 (s, 1H), 4.06 (q, J = 7.2 Hz, 2H), 3.80 (s, 3H), 3.26 (t, J = 7.6 Hz, 2H), 3.07 (t, J = 7.2 Hz, 2H), 1.92 (t, J = 7.2, 7.6 Hz, 2H), 1.22 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 169.7, 165.0, 159.0, 131.1, 129.6, 113.9, 79.2, 61.9, 58.4, 55.4, 49.7, 32.9, 21.3, 14.8; HRMS (ESI+) m/z: (M + H)+ calcd for C23H28NO4, 382.2018; measured, 382.2019.
(E)-Ethyl 2-(1-(2-ethoxy-2-oxoethyl)pyrrolidin-2-ylidene)acetate (5e). Using general procedure B, 0.200 mmol of 3e was converted into crude 5e. Flash column chromatography (10% EtOAc in dichloromethane) gave pure 5e (41.0 mg, 0.169 mmol, 85%) as a yellow solid. The stereochemistry was assigned using 1D-NOESY. Rf = 0.5 (10% ethyl acetate in dichloromethane); mp 63–65 °C; 1H NMR (400 MHz, CDCl3): δ: 4.46 (s, 1H), 4.20 (q, J = 7.6 Hz, 2H), 4.08 (q, J = 7.2 Hz, 2H), 3.91 (s, 2H), 3.47 (t, J = 7.2 Hz, 2H), 3.19 (t, J = 7.6 Hz, 2H), 2.00 (tt, J = 7.2, 7.6 Hz, 2H), 1.28 (t, J = 7.6 Hz, 3H), 1.24 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 169.2, 168.5, 165.2, 79.5, 61.5, 58.6, 53.5, 47.8, 32.6, 21.4, 14.8, 14.3. HRMS (ESI+) m/z: (M + H)+ calcd for C12H20NO4, 242.1392; measured, 242.1399.
(E)-Ethyl 2-(1-(2-ethoxy-2-oxoethyl)piperidin-2-ylidene)acetate (5g). Using general procedure B, 0.200 mmol of 3g was converted into crude 5g. Flash column chromatography (30% EtOAc in petroleum ether) gave pure 5g (42.0 mg, 0.164 mmol, 82%) as a yellow viscous oil. Rf = 0.59 (50% ethyl acetate in petroleum ether). The stereochemistry was determined by 1D-NOESY experiment. 1H NMR (400 MHz, CDCl3): δ: 4.42 (s, 1H), 4.23 (q, J = 7.2 Hz, 2H), 4.05 (q, J = 7.2 Hz, 2H), 3.87 (s, 2H), 3.31 (t, J = 6.0 Hz, 2H), 3.12 (t, J = 6.8 Hz, 2H), 1.85–1.79 (m, 2H), 1.69 (tt, J = 6.0, 6.8 Hz, 2H), 1.30 (t, J = 7.2 Hz, 3H), 1.23 (t, J = 7.2, 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 169.0 × 2, 168.7, 83.8, 61.5, 58.5, 53.8, 51.4, 26.8, 23.8, 19.8, 14.8, 14.3. HRMS (ESI+) m/z: (M + H)+ calcd for C13H22NO4, 256.1549; measured, 256.1559.
(E)-Ethyl 2-(1-methyl-5-oxo-4-phenylpyrrolidin-2-ylidene)acetate (5j). Using general procedure B, 0.200 mmol of 3j was converted into crude 5j. Flash column chromatography (10% EtOAc in petroleum ether) gave pure 5j (32.0 mg, 0.123 mmol, 62%) as a light yellow oil. Rf = 0.21 (100% dichloromethane). The stereochemistry was determined by 1D-NOESY experiment. 1H NMR (400 MHz, CDCl3): δ: 7.36–7.21 (m, 5H), 5.28 (s, 1H), 4.19 (q, J = 7.2 Hz, 2H), 3.87–3.81 (m, 2H), 3.39 (dd, J = 2.4, 2.8 Hz, 1H), 3.08 (s. 3H), 1.30 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 177.0, 167.3, 159.0, 138.5, 129.1, 127.7, 127.6, 92.2, 59.8, 45.6, 34.0, 27.5, 14.8. HRMS (ESI+) m/z: (M + H)+ calcd for C15H18NO3, 260.1287; measured, 260.1317.
(E)-Ethyl 3-(diethylamino)-3-(m-tolyl)acrylate (5k). Using general procedure B, 0.200 mmol of 3k was converted into crude 5k. Flash column chromatography (100% dichloromethane) gave pure 5k as a yellow viscous oil. The yield (72%) was determined by 1H-NMR using benzaldehyde (0.200 mmol, 20.8 μL) as an internal standard. The compound was obtained as a mixture of rotamers. The stereochemistry of the major rotamer was determined by 1D-NOESY experiment. Rf = 0.17 (100% dichloromethane). 1H NMR (400 MHz, CDCl3): δ: 7.37–6.99 (m, 4H), 4.80 (s, 1H (major)), 4.38–3.98 (m, 2H (minor(s))) 3.89 (q, J = 7.2 Hz, 2H (major)), 3.41–3.13 (br m, 4H), 2.29–2.42 (br, m, 3H with 2.37 (s, 3H) (major)), 1.10–1.04 (m, 9H). 13C NMR (100 MHz, CDCl3) δ: 168.3, 162.4, 137.7, 137.0, 129.0, 128.6, 128.1, 125.3, 85.4, 58.4, 43.9 (major), 46.9 (minor), 21.7 (major), 21.5 (minor) 14.6 (major), 14.3 (minor), 13.6 (major), 13.0 (minor). HRMS (ESI+) m/z: (M + H)+ calcd for C16H24NO2, 262.1807; measured 262.1815.
(E)-2-(1-Benzylpyrrolidin-2-ylidene)-1-(p-tolyl)ethanone (5o). Using general procedure B, 0.200 mmol of 3a was converted into crude 5o. Flash column chromatography (20% EtOAc in dichloromethane) gave pure 5o (54.0 mg, 0.185 mmol, 93%) as a yellow solid. The stereochemistry was assigned using 1D-NOESY. Rf = 0.15 (25% ethyl acetate in petroleum ether). mp 59–60 °C; 1H NMR (400 MHz, CDCl3): δ: 7.73 (d, J = 7.6 Hz, 2H), 7.38–7.23 (m, 5H), 7.16 (d, J = 8.0 Hz, 2H), 5.90 (s, 1H) 4.53 (s, 2H), 3.57 (t, J = 7.2 Hz, 2H), 3.44 (t, J = 7.6 Hz, 2H), 2.36 (s, 3H), 2.05 (tt, J = 7.2, 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ: 188.0, 167.4, 140.7, 139.3, 135.8, 129.0, 128.8, 127.9, 127.4, 127.3, 86.9, 52.8, 50.5, 34.0, 21.5, 21.2. HRMS (ESI+) m/z: (M + H)+ calcd for C20H22NO, 292.1701; measured, 292.1709.
(E)-2-(1-Benzylpyrrolidin-2-ylidene)-1-(4-fluorophenyl)ethanone (5p). Using general procedure B, 0.200 mmol of 3a was converted into crude 5p. Flash column chromatography (20% EtOAc in dichloromethane) gave pure 5p (56.0 mg, 0.190 mmol, 95%) as a yellow solid. The stereochemistry was assigned using 1D-NOESY. Rf = 0.15 (25% ethyl acetate in petroleum ether). mp 103–105 °C; 1H NMR (400 MHz, CDCl3): δ: 7.84–7.80 (m, 2H), 7.39–7.30 (m, 3H), 7.26–7.23 (m, 2H), 7.05–7.01 (m, 2H), 5.83 (s, 1H) 4.54 (s, 2H), 3.51–3.45 (m, 4H), 2.07 (tt, J = 7.6 Hz, 7.6 Hz). 13C NMR (100 MHz, CDCl3) δ: 186.6, 166.7 (d, J = 233.6), 163.1, 138.2 (d, J = 3.0 Hz), 135.5, 129.6 (d, J = 8.9 Hz), 129.0, 128.0, 127.3, 115.0 (d, J = 21.6 Hz), 86.6, 52.9, 50.5, 34.1, 21.1. 19F NMR (376 MHz, CDCl3) δ: −110.61. HRMS (ESI+) m/z: (M + H)+ calcd for C19H19FNO, 296.1451; measured, 296.1455.
(E)-Ethyl 3-(2-(2-oxo-2-phenylethylidene)pyrrolidin-1-yl)propanoate (5r). Using general procedure B, 0.200 mmol of 3f was converted into crude 5r. Flash column chromatography (50% EtOAc in petroleum ether) gave pure 5r (50.0 mg, 0.174 mmol, 87%) as a brown viscous oil. The stereochemistry was assigned using 1D-NOESY. Rf = 0.25 (50% ethyl acetate in petroleum ether). 1H NMR (400 MHz, CDCl3): δ: 7.89–7.87 (m, 2H), 7.43–7.38 (m, 3H), 5.75 (s, 1H), 4.16 (q, J = 7.2 Hz, 2H), 3.66 (t, J = 7.0 Hz, 2H), 3.50 (t, J = 7.2 Hz, 2H), 3.39 (t, J = 7.6 Hz, 2H), 2.66 (t, J = 7.0 Hz, 2H), 2.02 (tt, J = 7.2, 7.6 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 188.0, 171.5, 166.9, 142.0, 130.5, 128.2, 127.4, 86.8, 61.2, 53.2, 42.3, 33.9, 31.3, 21.2, 14.3. HRMS (ESI+) m/z: (M + H)+ calcd for C17H22NO3, 288.1600; measured, 288.1606.
(E)-Ethyl 3-(2-(2-(tert-butoxy)-2-oxoethylidene)pyrrolidin-1-yl)propanoate (5s). Using general procedure B, 0.200 mmol of 3f was converted into crude 5s. Flash column chromatography (50% EtOAc in petroleum ether) gave pure 5s (41.0 mg, 0.145 mmol, 73%) as a yellow viscous oil. The stereochemistry was assigned using 1D-NOESY. Rf = 0.29 (30% ethyl acetate in hexane). 1H NMR (400 MHz, CDCl3): δ: 4.47 (s, 1H), 4.15 (q, J = 7.2 Hz, 2H), 3.48 (t, J = 7.2 Hz, 2H), 3.37 (t, J = 7.2 Hz, 2H), 3.10 (t, J = 7.6 Hz, 2H), 2.57 (t, J = 7.2 Hz, 2H), 1.91 (tt, J = 7.2, 7.6 Hz, 2H), 1.47 (s, 9H), 1.27 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 171.8, 169.3, 163.7, 80.3, 77.7, 60.9, 52.8, 42.1, 32.5, 31.2, 28.8, 21.4, 14.3. HRMS (ESI+) m/z: (M + H)+ calcd for C15H26NO4, 284.1862; measured, 284.1886.
(E)-tert-Butyl 2-(1-(2-ethoxy-2-oxoethyl)pyrrolidin-2-ylidene)acetate (5t). Using general procedure B, 0.200 mmol of 3e was converted into crude 5t. Flash column chromatography (30% EtOAc in petroleum ether) gave pure 5t (33.0 mg, 0.123 mmol, 62%) as a yellow viscous oil. The stereochemistry was assigned using 1D-NOESY. Rf = 0.35 (30% ethyl acetate in hexane). 1H NMR (400 MHz, CDCl3): δ: 4.40 (s, 1H), 4.20 (q, J = 7.2 Hz, 2H), 3.89 (s, 2H), 3.43 (t, J = 7.2 Hz, 2H), 3.15 (t, J = 7.2 Hz, 2H), 1.97 (tt, J = 7.2, 7.2 Hz, 2H), 1.45 (s, 9H), 1.28 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 169.0, 168.7, 164.2, 81.3, 77.9, 61.5, 53.3, 47.8, 32.2, 28.8, 21.5, 14.3. HRMS (ESI+) m/z: (M + H)+ calcd for C14H24NO4, 270.1705; measured, 270.1714.
(E)-Phenyl 2-(1-benzylpyrrolidin-2-ylidene)acetate (5w). Using general procedure B, 0.200 mmol of 3a was converted into crude 5w. Flash column chromatography (15% EtOAc in petroleum ether) gave 5w (48.0 mg, 0.164 mmol, 82%) as a yellow solid. The compound was obtained as a mixture of rotamers. The stereochemistry of the major rotamer was determined by the 1D-NOESY experiment. Rf = 0.39 (25% ethyl acetate in petroleum ether). mp 90–92 °C; 1H NMR (400 MHz, CDCl3): δ: 7.39–7.08 (m, 10H), 4.99 (s, 1H (minor)), 4.90 (s, 1H (major)), 4.44 (s, 2H), 3.59–3.55 (m, 2H (minor)), 3.41 (t, J = 6.8 Hz, 2H (major)), 3.27 (t, J = 7.6 Hz, 2H (major)), 3.12–3.08 (m, 2H (minor)), 2.00 (tt, J = 6.8, 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ: 168.0, 167.3, 151.8, 135.8, 129.2, 129.0, 127.8, 127.4, 124.7, 122.4, 77.4, 52.9, 50.3, 33.0, 21.1. HRMS (ESI+) m/z: (M + H)+ calcd for C19H20NO2, 294.1494; measured, 294.1505.
(E)-3-Phenylpropyl 2-(1-benzylpyrrolidin-2-ylidene)acetate (5x). Using general procedure B, 0.200 mmol of 3a was converted into crude 5x. Flash column chromatography (30% EtOAc in petroleum ether) gave 5x (63.0 mg, 0.188 mmol, 94%) as a light-yellow viscous oil. The stereochemistry was determined by the 1D-NOESY experiment. Rf = 0.45 (30% ethyl acetate in petroleum ether) 1H NMR (400 MHz, CDCl3): δ: 7.35–7.14 (m, 10H), 4.71 (s, 1H), 4.37 (s, 2H), 4.06 (t, J = 6.4 Hz, 2H), 3.34 (t, J = 6.8 Hz, 2H), 3.24 (t, J = 8.0 Hz, 2H), 2.69 (t, J = 7.6 Hz, 2H), 2.00–1.90 (m, 4H). 13C NMR (100 MHz, CDCl3) δ: 169.7, 165.4, 141.9, 136.1, 128.8, 128.5, 128.4, 127.6, 127.3, 125.9, 78.4, 62.1, 52.5, 50.1, 32.8, 32.5, 30.9, 21.2. HRMS (ESI+) m/z: (M + H)+ calcd for C22H26NO2, 336.1964.0; measured, 336.1994.
(E)-Cinnamyl 2-(1-benzylpyrrolidin-2-ylidene)acetate (5y). Using general procedure B, 0.200 mmol of 3a was converted into crude 5y. Flash column chromatography (30% EtOAc in petroleum ether) gave 5y in a semi-pure form. Another column in CH2Cl2 gave pure 5x (61.0 mg, 0.183 mmol, 92%) as a yellow viscous oil. The stereochemistry was determined by the 1D-NOESY experiment. Rf = 0.39 (5% ethyl acetate in dichloromethane). 1H NMR (400 MHz, CDCl3): δ: 7.39–7.17 (m, 10H), 6.64 (d, J = 15.6 Hz, 1H), 6.33 (dt, J = 6.0, 15.6 Hz 1H), 4.75 (s, 1H), 4.71 (d, J = 6.0 Hz, 2H), 4.37 (s, 2H), 3.36 (t, J = 7.2 Hz, 2H), 3.26 (t, J = 7.6 Hz, 2H), 1.99 (tt, J = 7.2, 7.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ: 169.2, 165.8, 136.8, 136.0, 132.8, 128.8, 128.5, 127.7, 127.6, 127.3, 126.6, 125.3, 78.0, 63.3, 52.6, 50.1, 32.8, 21.1. HRMS (ESI+) m/z: (M + H)+ calcd for C22H24NO2, 334.1807; measured, 334.1812.
(E)-(1R,2S,5R)-2-Isopropyl-5-methylcyclohexyl 2-(1-benzylpyrrolidin-2-ylidene)acetate (5z). Using general procedure B, 0.200 mmol of 3a was converted into crude 5z. Flash column chromatography (20% EtOAc in petroleum ether) gave 5z (67.0 mg, 0.189 mmol, 95%) as a dark yellow viscous oil. The stereochemistry was determined by the 1D-NOESY experiment. Rf = 0.55 (30% ethyl acetate in petroleum ether). 1H NMR (400 MHz, CDCl3): δ: 7.35–7.18 (m, 5H), 4.69–4.63 (m, 2H), 4.04–4.31 (m, 2H), 3.31 (t, J = 7.2 Hz, 2H), 3.26–3.16 (m, 2H), 2.05–1.92 (m, 4H), 1.75–1.41 (m, 3H) 1.36–1.30 (m, 1H), 1.09–0.84 (m, 9H), 0.78 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 169.4, 165.3, 136.3, 128.9, 127.6, 127.4, 78.9, 71.7, 52.4, 50.2, 47.6, 41.9, 34.7, 32.8, 31.6, 26.4, 23.8, 22.3, 21.3, 21.0, 16.7. HRMS (ESI+) m/z: (M + H)+ calcd for C23H34NO2, 356.2590; measured, 356.2614.
To prepare the candidate compounds for docking with AutoDock Vina,49 we converted the structures of the ligands from the Structure Data File (SDF) format to the pdbqt file format with Open Babel.70 We ran the docking simulations on the Oklahoma Center for Supercomputing in Education and Research (OSCER) at the University of Oklahoma. We used the pose with the lowest binding energy for further analysis with the molecular graphics program PyMOL,71 the Protein–Ligand Interaction Profiler (PLIP),72 and molecular dynamics (MD) simulation with GROMACS.73
PK studies
Animals. Male Sprague-Dawley rats, 4 months old, were purchased from Harlan (now Envigo) Labs (Indianapolis, IN). Animals were maintained on a 12 hours light/dark cycle and had access to food (Rat Chow 5012) ad libitum. Animal care and use were approved by the Oklahoma State University Center for Health Sciences, Institutional Animal Care and Use Committee (IACUC protocol number 2019-03), in accordance with the NIH guidelines for the care and use of laboratory animals in “Guide for The Care and Use of Laboratory Animals.”
Binding assays. Whole brains (minus brainstem and cerebellum) from male Sprague-Dawley rats were weighed and homogenized in 20 v/w of 50 mM Tris (pH 7.4). Binding assays were performed from modification described by others.82,83 Briefly, tissue homogenates were centrifuged for 5 min at 500×g (+4 °C). After centrifugation, the supernatant is decanted and recentrifuged at 20
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g for 15 min (+4 °C). The semi-purified pellet was resuspended in binding buffer (in mM: 20 HEPES; 118 NaCl; 4.8 KCl; 1.2 MgCl2; 2.5 CaCl2; pH 7.4) at approximately 100–150 v/w. All compounds were initially diluted to a concentration of 10 mM in DMSO. The final concentration of DMSO was ≤1% in the binding reaction tube. Based on the reported affinities of each drug, the following concentrations were utilized, 1 and 100 μM (−) nicotine; 0.26 and 26 μM (+) anabasine; or 1 and 100 μM (−)-237D. Nonspecific binding was defined as binding in the presence of 50 μM (−)-nicotine. Tissue homogenate (400 μL), tritiated drug (50 μL), and either buffer or drug (50 μL) were added together in a 12 × 75 mm polypropylene tube and allowed to equilibrate. The binding reaction was initiated by the addition of cytisine HCl [3,5-3H] (31.8 Ci mmol−1, PerkinElmer, Waltham, MA) to a final concentration of 2–3 nM. Cytisine was chosen as the radioligand due to its high binding affinity to brain nicotinic receptors.84 The binding reaction was allowed to continue to equilibrium for 90 min at room temperature (22–23 °C). To terminate the binding reaction, samples were filtered under reduced pressure using a Brandel Tissue harvester onto a GF/B Whatman fiberglass filter that was presoaked for at least 1 h in 0.3% polyethyleneimine (PEI). After filtration, the filters were washed for 15 s with ice-cold Tris–HCl washer buffer (pH 7.4). The amount of radioligand bound was determined using scintillation spectrophotometry.
000×g for 15 min (+4 °C). The semi-purified pellet was resuspended in binding buffer (in mM: 20 HEPES; 118 NaCl; 4.8 KCl; 1.2 MgCl2; 2.5 CaCl2; pH 7.4) at approximately 100–150 v/w. All compounds were initially diluted to a concentration of 10 mM in DMSO. The final concentration of DMSO was ≤1% in the binding reaction tube. Based on the reported affinities of each drug, the following concentrations were utilized, 1 and 100 μM (−) nicotine; 0.26 and 26 μM (+) anabasine; or 1 and 100 μM (−)-237D. Nonspecific binding was defined as binding in the presence of 50 μM (−)-nicotine. Tissue homogenate (400 μL), tritiated drug (50 μL), and either buffer or drug (50 μL) were added together in a 12 × 75 mm polypropylene tube and allowed to equilibrate. The binding reaction was initiated by the addition of cytisine HCl [3,5-3H] (31.8 Ci mmol−1, PerkinElmer, Waltham, MA) to a final concentration of 2–3 nM. Cytisine was chosen as the radioligand due to its high binding affinity to brain nicotinic receptors.84 The binding reaction was allowed to continue to equilibrium for 90 min at room temperature (22–23 °C). To terminate the binding reaction, samples were filtered under reduced pressure using a Brandel Tissue harvester onto a GF/B Whatman fiberglass filter that was presoaked for at least 1 h in 0.3% polyethyleneimine (PEI). After filtration, the filters were washed for 15 s with ice-cold Tris–HCl washer buffer (pH 7.4). The amount of radioligand bound was determined using scintillation spectrophotometry.
All of the enamino carbonyl compounds (Table S6†) included in this study were dissolved in one ml of DMSO (otherwise stated differently). Fifty milliliters of the organic solution were loaded to the wells created in microbes inoculated MHA media plate. For media plates with MRSA and Candida albicans, they were incubated at 37 °C for 72 hours in an aerobic environment. For C. jejuni inoculated media plates, they were incubated at 42 °C in a microaerobic environment for 72 hours. Zones of inhibition for effective enamino carbonyl compounds were measured and recorded as “zone of inhibition” (Table S7†). The organic compound showing a zone of inhibition of more than 9 mm towards the tested microbial pathogen was considered effective, and required analyses were performed.
Author contributions
Jim Secka conducted experimental work for most of the compounds mentioned in Tables 2 and 3. Along with Adama Kuta, he also completed the rapid screening of the reaction (Table S2†). In addition, Jim Secka prepared all compounds listed in Table S6† that start with numbers (except 5a and 5c). Arpan Pal conducted experimental work for Table 1, and prepared 5a. In addition, Arpan Pal prepared all compounds listed in table S6† that start with the letters AP and 5a. Francis A. Acquah performed molecular docking studies under the guidance of Blaine H. M. Mooers. They also wrote the section on molecular docking studies. Anand B. Karki and Dania Mahjoub performed antimicrobial testing of enamino carbonyl compounds in the direction of Mohamed K. Fakhr. David R. Wallace performed all pharmacological experiments and wrote the pharmacological section of the manuscript. Takuya Okada prepared indolizidine (−)-237D under Naoki Toyooka. Naoki Toyooka provided the sample for pharmacology. Adama Kuta prepared 1 and 5c, and contributed to S8 (AK-I-118 and 5c). Naga Koduri made all compounds listed in table S6† that start with the letters N. K. Deacon Herndon is an undergraduate student who prepared 3a, 4b, 3k and made N,N′-ditosylhydrazine to prepare diazo compounds. He also helped Jim Secka and Adama Kuta synthesize and purify multiple starting materials and enamino carbonyl compounds. Kenneth P. Roberts performed multiple HRMS and guided Jim Secka and Adama Kuta during GCMS studies toward rapid assessment of the reaction (ESI†). Bethany Hileman, Nisha Rajagopal, and Zhiguo Wang prepared compounds for Table S6† that start with BNH, NR, and BZW. Syed R. Hussaini guided all organic synthesis (excluding synthesis of indolizidine (−)-237), wrote the manuscript, and prepared compounds starting with the initials srh (Table S6†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded through the award HR-18-049 from the Oklahoma Center for the Advancement of Science and Technology. This material is based upon work supported by the National Science Foundation under grant no. CHE-104874. The molecular simulations were supported by OCAST grant HR20-002 and NIH grants RO1 CA242845, P21 GM103640, P 30 CA225520 and were done using the computing resources of the Laboratory of Biomolecular Structure and Function and the Oklahoma State University High Performance Computing Center, which is supported by NSF OAC-1126330.Notes and references
- J. V. Greenhill, Chem. Soc. Rev., 1977, 6, 277–294 RSC.
- S. R. Hussaini, R. R. Chamala and Z. Wang, Tetrahedron, 2015, 71, 6017–6086 CrossRef CAS.
- R. Klintworth, G. L. Morgans, S. M. Scalzullo, C. B. de Koning, W. A. L. van Otterlo and J. P. Michael, Beilstein J. Org. Chem., 2021, 17, 2543–2552 CrossRef CAS PubMed.
- H. P. A. Khan and T. K. Chakraborty, J. Org. Chem., 2018, 83, 2027–2039 CrossRef CAS PubMed.
- S. Suresh, P. Bhimrao Patil, P.-H. Yu, C.-C. Fang, Y.-Z. Weng, V. Kavala and C.-F. Yao, Adv. Synth. Catal., 2021, 363, 4915–4925 CrossRef CAS.
- M. Saito, Y. Kobayashi and Y. Takemoto, Chem.–Eur. J., 2019, 25, 10314–10318 CrossRef CAS PubMed.
- D. Ma and H. Sun, Tetrahedron Lett., 2000, 41, 1947–1950 CrossRef CAS.
- B. Pettersson, V. Hasimbegovic and J. Bergman, J. Org. Chem., 2011, 76, 1554–1561 CrossRef CAS PubMed.
- M. Butters, C. D. Davies, M. C. Elliott, J. Hill-Cousins, B. M. Kariuki, L.-l. Ooi, J. L. Wood and S. V. Wordingham, Org. Biomol. Chem., 2009, 7, 5001–5009 RSC.
- A. K. Chattopadhyay and S. Hanessian, Chem. Commun., 2015, 51, 16450–16467 RSC.
- L. Marek, L. Kolman, J. Váňa, J. Svoboda and J. Hanusek, Beilstein J. Org. Chem., 2021, 17, 527–539 CrossRef CAS PubMed.
- S. Braverman and M. Cherkinsky, in Comprehensive Organic Synthesis II, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, 2014, ch. 3.18, pp. 887–942 Search PubMed.
- J. Meng, R. Jia, J. Leng, M. Wen, X. Yu and W.-P. Deng, Org. Lett., 2017, 19, 4520–4523 CrossRef CAS PubMed.
- C. Liu, J. Li, H. Chen and R. N. Zare, Chem. Sci., 2019, 10, 9367–9373 RSC.
- A. Eschenmoser, Helv. Chim. Acta, 2015, 98, 1483–1600 CrossRef CAS.
- Y. Yamada, P. Wehrli, D. Miljkovic, H.-J. Wild, N. Bühler, E. Götschi, B. Golding, P. Löliger, J. Gleason, B. Pace, L. Ellis, W. Hunkeler, P. Schneider, W. Fuhrer, R. Nordmann, K. Srinivasachar, R. Keese, K. Müller, R. Neier and A. Eschenmoser, Helv. Chim. Acta, 2015, 98, 1921–2054 CrossRef CAS.
- H. W. Pinnick and Y.-H. Chang, J. Org. Chem., 1978, 43, 4662–4663 CrossRef CAS.
- J.-P. Céaléarier, C. Eskéanazi, G. Lhornmet and P. Maitte, J. Heterocycl. Chem., 1979, 16, 953–955 CrossRef.
- M. Pivavarchyk, A. M. Smith, Z. Zhang, D. Zhou, X. Wang, N. Toyooka, H. Tsuneki, T. Sasaoka, J. M. McIntosh, P. A. Crooks and L. P. Dwoskin, Eur. J. Pharmacol., 2011, 658, 132–139 CrossRef CAS PubMed.
- F. G. Fang, G. B. Feigelson and S. J. Danishefsky, Tetrahedron Lett., 1989, 30, 2743–2746 CrossRef CAS.
- A. Padwa, F. R. Kinder and L. Zhi, Synlett, 1991, 1991, 287–288 CrossRef.
- G. Kim, M. Y. Chu-Moyer, S. J. Danishefsky and G. K. Schulte, J. Am. Chem. Soc., 1993, 115, 30–39 CrossRef CAS.
- N. D. Koduri, H. Scott, B. Hileman, J. D. Cox, M. Coffin, L. Glicksberg and S. R. Hussaini, Org. Lett., 2012, 14, 440–443 CrossRef CAS PubMed.
- A. Pal and S. R. Hussaini, ACS Omega, 2019, 4, 269–280 CrossRef CAS PubMed.
- A. Pal, N. D. Koduri, Z. Wang, E. L. Quiroz, A. Chong, M. Vuong, N. Rajagopal, M. Nguyen, K. P. Roberts and S. R. Hussaini, Tetrahedron Lett., 2017, 58, 586–589 CrossRef CAS.
- L. Mohammadi, M. A. Zolfigol, M. Ebrahiminia, K. P. Roberts, S. Ansari, T. Azadbakht and S. R. Hussaini, Catal. Commun., 2017, 102, 44–47 CrossRef CAS.
- N. D. Koduri, Z. Wang, G. Cannell, K. Cooley, T. M. Lemma, K. Miao, M. Nguyen, B. Frohock, M. Castaneda, H. Scott, D. Albinescu and S. R. Hussaini, J. Org. Chem., 2014, 79, 7405–7414 CrossRef CAS PubMed.
- F. A. Acquah, M. Paramel, A. Kuta, S. R. Hussaini, D. R. Wallace and B. H. M. Mooers, Int. J. Mol. Sci., 2021, 22, 7934 CrossRef CAS PubMed.
- I. O. Edafiogho, S. B. Kombian, K. V. V. Ananthalakshmi, N. N. Salama, N. D. Eddington, T. L. Wilson, M. S. Alexander, P. L. Jackson, C. D. Hanson and K. R. Scott, J. Pharm. Sci., 2007, 96, 2509–2531 CrossRef CAS PubMed.
- G. Negri, C. Kascheres and A. J. Kascheres, J. Heterocycl. Chem., 2004, 41, 461–491 CrossRef CAS.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS PubMed.
- J. Li, B. Qian and H. Huang, Org. Lett., 2018, 20, 7090–7094 CrossRef CAS PubMed.
- Q.-Q. Cheng, J. Yedoyan, H. Arman and M. P. Doyle, J. Am. Chem. Soc., 2016, 138, 44–47 CrossRef CAS PubMed.
- A. M. Beale, E. K. Gibson, M. G. O'Brien, S. D. M. Jacques, R. J. Cernik, M. D. Michiel, P. D. Cobden, Ö. Pirgon-Galin, L. v. d. Water, M. J. Watson and B. M. Weckhuysen, J. Catal., 2014, 314, 94–100 CrossRef CAS.
- G. Maas, Top. Curr. Chem., 1987, 137, 75–253 CrossRef CAS.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
- P. G. M. Wuts and T. W. Greene, in Greene's Protective Groups in Organic Synthesis, 2006, ch. 7, pp. 696–926, DOI:10.1002/9780470053485.
- J. P. Michael, D. Gravestock and S. Afr, J. Chem., 1998, 51, 146–157 CAS.
- J. P. Michael and D. Gravestock, Eur. J. Org. Chem., 1998, 2–3, 865–870, DOI:10.1002/(sici)1099-0690(199805)1998:5<865::aid-ejoc865>3.0.co.
- DRUGBANK Online, Phensumixide, https://go.drugbank.com/drugs/DB00832, (accessed December, 2021) Search PubMed.
- S. R. Hussaini, A. Kuta, A. Pal, Z. Wang, M. A. Eastman and R. Duran, ACS Omega, 2020, 5, 24848–24853 CrossRef CAS PubMed.
- H. K. Lee, J. Kim and C. S. Pak, Tetrahedron Lett., 1999, 40, 2173–2174 CrossRef CAS.
- R. L. Petersen, PhD, University of the Witwatersrand, 2004.
- J. P. Michael, C. B. de Koning, R. L. Petersen and T. V. Stanbury, Tetrahedron Lett., 2001, 42, 7513–7516 CrossRef CAS.
- Personal communication with J. P. Michael – the inventor of thio-Reformatsky reaction of N-Ph protected thioamides January 1 2021.
- J. P. Michael, C. B. d. Koning, C. S. Fat and G. L. Nattrass, ARKIVOC, 2002, 2002, 62–77 Search PubMed.
- C. B. Kanner and U. K. Pandit, Tetrahedron, 1982, 38, 3597–3604 CrossRef CAS.
- A. Müller, A. Maier, R. Neumann and G. Maas, Eur. J. Org. Chem., 1998, 1998, 1177–1187 CrossRef.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- K. Andrud, H. Xing, B. Gabrielsen, L. Bloom, V. Mahnir, S. Lee, B. T. Green, J. Lindstrom and W. Kem, Mar. Drugs, 2019, 17, 614 CrossRef CAS PubMed.
- W. R. Kem, V. M. Mahnir, R. L. Papke and C. J. Lingle, J. Pharmacol. Exp. Ther., 1997, 283, 979–992 CAS.
- M. J. Parker, A. Beck and C. W. Luetje, Mol. Pharmacol., 1998, 54, 1132–1139 CrossRef CAS PubMed.
- WHO, Global action plan on antimicrobial resistance, World Health Organization, 2017 Search PubMed.
- N. Molchanova, P. R. Hansen and H. Franzyk, Molecules, 2017, 22, 1430 CrossRef PubMed.
- Y.-h. Hsieh and I. M. Sulaiman, Campylobacteriosis: An Emerging Infectious Foodborne Disease, Academic Press, 2018 Search PubMed.
- P. Mao, P. Peng, Z. Liu, Z. Xue and C. Yao, Infect. Drug Resist., 2019, 12, 3709–3717 CrossRef CAS PubMed.
- R. L. Skov and K. S. Jensen, J. Hosp. Infect., 2009, 73, 364–370 CrossRef CAS PubMed.
- J. Talapko, M. Juzbašić, T. Matijević, E. Pustijanac, S. Bekić, I. Kotris and I. Škrlec, J. Fungi, 2021, 7, 79 CrossRef CAS PubMed.
- S. Costa-de-Oliveira and A. G. Rodrigues, Microorganisms, 2020, 8, 154 CrossRef CAS PubMed.
- A. Khan, B. Wilson and I. M. Gould, Expert Opin. Pharmacother., 2018, 19, 457–470 CrossRef CAS PubMed.
- A. Noormohamed and M. K. Fakhr, Open Microbiol. J., 2014, 8, 130–137 CrossRef PubMed.
- Y. Wang, M. Zhang, F. Deng, Z. Shen, C. Wu, J. Zhang, Q. Zhang and J. Shen, Antimicrob. Agents Chemother., 2014, 58, 5405–5412 CrossRef PubMed.
- N. D. Koduri, B. Hileman, J. D. Cox, H. Scott, P. Hoang, A. Robbins, K. Bowers, L. Tsebaot, K. Miao, M. Castaneda, M. Coffin, G. Wei, T. D. W. Claridge, K. P. Roberts and S. R. Hussaini, RSC Adv., 2013, 3, 181–188 RSC.
- T. Murai and F. Asai, J. Am. Chem. Soc., 2007, 129, 780–781 CrossRef CAS PubMed.
- D. Dar’in, G. Kantin and M. Krasavin, Synthesis, 2019, 51, 4284–4290 CrossRef.
- T. Toma, J. Shimokawa and T. Fukuyama, Org. Lett., 2007, 9, 3195–3197 CrossRef CAS PubMed.
- L. M. Harwood and T. D. W. Claridge, Introduction to Organic Spectroscopy, Oxford Science Publications, 2004 Search PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- E. Krieger, K. Joo, J. Lee, J. Lee, S. Raman, J. Thompson, M. Tyka, D. Baker and K. Karplus, Proteins: Struct., Funct., Bioinf., 2009, 77, 114–122 CrossRef CAS PubMed.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed.
- L. Schrodinger and W. DeLano, The PyMOL Molecular Graphics System, Version 2.4.2021, 2021, https://pymol.org/pymol Search PubMed.
- M. F. Adasme, K. L. Linnemann, S. N. Bolz, F. Kaiser, S. Salentin, V. J. Haupt and M. Schroeder, Nucleic Acids Res., 2021, 49, W530–W534 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- L. Verlet, Phys. Rev., 1967, 159, 98–103 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. R. Miller, T. D. McGee, J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314–3321 CrossRef CAS PubMed.
- M. S. Valdes-Tresanco, M. E. Valdes-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef CAS PubMed.
- D. C. Perry and K. J. Kellar, J. Pharmacol. Exp. Ther., 1995, 275, 1030–1034 CAS.
- H. K. Happe, J. L. Peters, D. A. Bergman and L. C. Murrin, Neuroscience, 1994, 62, 929–944 CrossRef CAS PubMed.
- L. A. Pabreza, S. Dhawan and K. J. Kellar, Mol. Pharmacol., 1991, 39, 9–12 CAS.
- L. M. M. Vermeer, C. D. Isringhausen, B. W. Ogilvie and D. B. Buckley, Drug Metab. Dispos., 2016, 44, 453–459 CrossRef PubMed.
- M. A. Gibbs, K. E. Thummel, D. D. Shen and K. L. Kunze, Drug Metab. Dispos., 1999, 27, 180–187 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structures of thioamides and diazo compounds, synthesis of 1-(4-methoxybenzyl)pyrrodidin-2-one, synthesis of thioamides 3, 4d and known enamino carbonyl compounds 5, rapid assessment of reaction scope, characterization of compounds, docking energies of compounds, protein–ligand interactions, structures of compounds screened for antimicrobial activity, effective antimicrobial compounds. See https://doi.org/10.1039/d2ra02415b |
| This journal is © The Royal Society of Chemistry 2022 |