
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of diluent gases on sooting transition process in ethylene counterflow diffusion flames
Zhiwei Su†
ab,
Yaoyao Ying† *ab,
Chen Chenab,
Rui Zhaoab,
Xuan Zhaoab and
Dong Liu*ab
*ab,
Chen Chenab,
Rui Zhaoab,
Xuan Zhaoab and
Dong Liu*ab
aMIIT Key Laboratory of Thermal Control of Electronic Equipment, School of Energy and Power Engineering, Nanjing University of Science and Technology, Nanjing 210094, P. R. China. E-mail: yingyaoyao@njust.edu.cn; dongliu@njust.edu.cn
bAdvanced Combustion Laboratory, School of Energy and Power Engineering, Nanjing University of Science and Technology, Nanjing 210094, P. R. China
First published on 21st June 2022
Abstract
The impacts of adding diluent gases (nitrogen, carbon dioxide, and helium) either to the fuel side or the oxidizer side on the sooting transition process in ethylene counterflow diffusion flames are investigated experimentally and numerically. A series of ethylene flames ranging from non-sooting to heavy-sooting are studied by gradually increasing the oxygen concentration on the oxidizer side. The optical method is used to analyze flame images, determining the sooting transition process. It is found that whether CO2 is added to the fuel side or the oxidizer side, the sooting transition process is delayed significantly. This process is slightly delayed when He is added to the fuel side, however, it is promoted when He is introduced to the oxidizer side. The numerical results show that in CO2-diluted flames, the mole fraction of the main soot precursors C2H2, C3H3, and C6H6 are reduced, which leads to the delay of soot formation. In addition, the H radical decreases while the OH radical increases, both of them are important for soot formation. In He-diluted flames, the concentration of C2H2, C3H3, and C6H6 decreased, as well as H and OH radicals. Moreover, adding He obviously changes the distribution area of products.
1. Introduction
The development and progress of human society are inseparable from the consumption of energy. At present, the consumption of fossil energy accounts for about 80% of the total global consumption.1 The energy of fossil fuels is mainly produced through combustion, which is the main source of air pollutants. The nanoparticles produced by combustion are called soot,2,3 and are mainly emitted by the incomplete combustion of fossil fuels.4 Incomplete combustion of fossil fuels in actual equipment such as gas turbines, boilers, and internal combustion engines will produce soot.5–7 Soot particles will harm the human respiratory system.8 The soot particles can also change the climate, such as changing the absorption of solar radiation, and influencing cloud formation and deposition on snow and ice.9,10 Therefore, reducing the emission of soot is an important subject. Several researchers have studied soot generation characteristics in actual equipment, providing a strong basis for reducing soot formation.11,12To reduce the emission of soot particles, the effects of various diluent gas additives on soot emission in hydrocarbon flames have attracted more and more attention. Helium (He), nitrogen (N2), and carbon dioxide (CO2) are currently commonly used diluents. It is well-known that the addition of CO2 to hydrocarbon diffusion flames can effectively suppress the emission of soot particles. Du et al.13 studied the effect of CO2 and oxygen as additives on soot formation in the diffusion flame, finding that CO2, whether added to the fuel or oxidizer side, could suppress soot formation chemically. Oh et al.,14 Liu et al.,15 and Gülder et al.16 combined experiment and numerical simulation in laminar diffusion flames and found that the chemical effect of CO2 would increase the production of O and OH radicals, which were believed to be responsible for soot precursor oxidation. Guo et al.17 conducted a numerical study on the effect of CO2 addition on soot formation in an ethylene/air diffusion flame. The result showed that the chemical effect of CO2 addition was primarily introduced by the reduction of radical H concentration, which suppressed the soot inception and surface growth rate. Some researchers investigated the effect of various diluents on soot production in laminar diffusion flames,18–20 and they all found that when CO2 was adopted as the diluent, there was a strong inhibition on soot formation, but there was no virtually noticeable effect on soot suppression when He was used. Gülder et al.21 investigated the influence of N2 dilution and flame temperature on soot formation in diffusion flames and found that the reduction in soot formation is due to both lowered temperature and fuel concentration in dilution experiments. Yen et al.22 focused on modeling the effects of hydrogen and N2 addition on soot formation in laminar ethylene jet diffusion flames. They found the model was able to qualitatively predict the measured trends of reducing soot concentration as hydrogen or N2 is added to ethylene or substituted.
The incipient stage of soot formation is of great significance for understanding the nucleation of soot. Ergut et al.23,24 investigated the effect of temperature and equivalence ratio on the soot onset chemistry in ethylbenzene flames. They determined the onset of soot with observation, measured the concentrations of major species by GC/MS system, and validated with computations. Therrien25 studied the soot onset threshold and flame properties of ethylbenzene–ethanol blends, and found that the concentration of benzyl groups dropped significantly with ethanol addition, resulting in the reduction of key soot precursors. Gleason et al.26 followed quantitatively the transition from parent fuel molecule to polycyclic aromatic hydrocarbons (PAHs) and eventually to soot. They found that only one- and two-ring aromatic compounds could account for soot nucleation. Zhao et al.27 systematically investigated the sooting transition process in iso-octane flame and performed combustion chemistry during the transition process in details.
From the literature above, it can be concluded that the current detailed study on the sooting transition process under different diluents is still lacking. It is of great significance for deeply understanding the influence of different diluents on the sooting transition process of hydrocarbon flames to understand the nucleation of soot and control soot emission. Therefore, in this study, the diluent gas (N2, CO2, and He) was added to the oxidizer side or the fuel side, and the changes in the sooting transition process were analyzed by experiments and chemical kinetic simulations, aming to investigate the effects of diluent gases on sooting transition process in ethylene counterflow diffusion flames.
2. Experiments and simulations
2.1. Experimental procedure
The experimental setup consisted of a counterflow diffusion flame platform, a fuel/oxidizer flow-control system and a flame image acquisition system, the architecture of this experimental apparatus was illustrated in Fig. 1. | ||
| Fig. 1 Schematic diagram of experimental system. | ||
The counterflow diffusion flame platform was designed by Advanced Combustion Laboratory in NJUST, which had two vertically placed nozzles with a diameter of 6.77 mm for the inner nozzle and a diameter of 14.88 mm for the outer surrounding nozzle. The separation distance between the two nozzles was 8.0 mm. The oxidizer and fuel stream were connected to the lower and upper nozzle, respectively. The shield N2 could protect the flame from indoor air flow and ensure that the fuel burned in a controlled atmosphere. N2, He, and CO2 were chosen as the diluents. The diluent gas was mixed with oxygen and fuel before entering the combustion chamber. The gas flow rates of nitrogen, fuel gas, diluent gas and oxygen were controlled by high precision digital mass flow controllers (Sevenstar, CS200A). The flow rate of the two nozzles was 18.52 cm s−1. The image acquisition system, consisted of a digital single lens reflex camera (Nikon D7100) in manual mode, was used to deal with flame images for distinguishing the sooting transition process. The lens was AF-S NIKKOR 24–85 mm f/3.5–4.5G ED VR without any filter.
The detailed conditions of the flames in the experiment were shown in Table 1. There were five groups of flames were investigated. In Flame A, N2 was added on both the fuel side and the oxidizer side, which could be regarded as the standard flames for comparison. In Flame B1, CO2 was added on the fuel side and N2 was added on the oxidizer side. Flame C1 had a He addition on the fuel side and N2 addition on the oxidizer side. The compositions of Flames B2 and C2 corresponded closely to Flames B1 and C1, respectively, but in Flames B2 and C2, the diluent gases were added on the opposite side. Here XO was defined as the oxygen concentration.
| Flame | Fuel side | Mole fraction | Oxidizer side | XO | ||||
|---|---|---|---|---|---|---|---|---|
| C2H4 | Diluent gas | Min | Increment | Max | ||||
| Flame A | C2H4 and N2 | 0.3 | 0.7 | O2 and N2 | 0.20 | 0.01 | 0.28 | |
| Flame B1 | C2H4 and CO2 | 0.3 | 0.7 | O2 and N2 | 0.32 | 0.01 | 0.40 | |
| Flame C1 | C2H4 and He | 0.3 | 0.7 | O2 and N2 | 0.23 | 0.01 | 0.30 | |
| Flame B2 | C2H4 and N2 | 0.3 | 0.7 | O2 and CO2 | 0.32 | 0.01 | 0.40 | |
| Flame C2 | C2H4 and N2 | 0.3 | 0.7 | O2 and He | 0.17 | 0.01 | 0.25 | |
2.2. Kinetic modeling
The Chemkin/OPPDIF code28 was used to simulate the investigated flames. The reaction mechanism used in this study was developed by the Ranzi et al.29–31 (CRECK_2003_TOT_HT_SOOT), which contained 452 species and 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 041 reactions.
041 reactions.
The boundary conditions were the same as the experiments. Four working conditions in vicinity of the sooting transition process in each group of flames were selected, which covered by the experimental conditions. The detailed conditions of the investigated flames were summarized in Table 2.
| Flames | Fuel side | Mole fraction | Oxidizer side | XO | ||||
|---|---|---|---|---|---|---|---|---|
| C2H4 | Diluent gas | |||||||
| Flame A | C2H4 and N2 | 0.3 | 0.7 | O2 and N2 | 0.21 | 0.23 | 0.24 | 0.27 |
| Flame B1 | C2H4 and CO2 | 0.3 | 0.7 | O2 and N2 | 0.32 | 0.34 | 0.35 | 0.38 |
| Flame C1 | C2H4 and He | 0.3 | 0.7 | O2 and N2 | 0.23 | 0.25 | 0.26 | 0.29 |
| Flame B2 | C2H4 and N2 | 0.3 | 0.7 | O2 and CO2 | 0.32 | 0.34 | 0.35 | 0.38 |
| Flame C2 | C2H4 and N2 | 0.3 | 0.7 | O2 and He | 0.18 | 0.20 | 0.21 | 0.24 |
3. Results and discussion
3.1. Sooting transition point identification
Fig. 2 showed Flame A images with the oxygen concentration (XO) changing from 20% to 28%. Soot particles produced blackbody radiation at high temperatures, causing the flame to glow yellow.32 Figura et al.33 reported that the flame without soot was blue, and the visual observation of a faint yellow luminosity revealed incipient sooting. It can be seen from Fig. 2, the flame color was blue in low oxygen concentrations, which indicated the flame was non-sooting one. As the oxygen concentration increased, a yellow flame gradually appeared above the blue flame, suggesting that the flame was transferring to an incipient sooting state. As XO continued to increase, a significant yellow luminous sooting region could be found at the flame fuel side. The soot formation just occurred when XO increased to a critical value, XO,cr. The XO,cr was defined as sooting critical transition point here. We might determine the XO,cr as XO = 24% where a faint yellow region visible to the naked eye. However, the real XO,cr was actually smaller because human eyes cannot respond to the flame yellow luminosity sensitively under the condition of stronger blue luminosity. To accurately determine the sooting transition process and critical transition point, this article used the method following ref. 27. | ||
| Fig. 2 Flame A images at different oxygen concentration combustion conditions. | ||
All the pixel points in the central axial line throughout the flame were taken, as shown with the yellow dotted line in Fig. 2. The luminosities of tricolor for the target pixel points were obtained, as shown in Fig. 3.
 | ||
| Fig. 3 Profiles of the tricolor luminosity proportion for pixel points in the flame at different O2 concentrations for Flame A, (a) before transition, (b) in transition, (c) after transition. | ||
In Fig. 3, the three corners of the triangle represented one color, respectively. Defining the lower left corner as blue luminous region, the lower right corner as blue luminous region, and the top corner as green luminous region. From Fig. 3(a), it could be found that most pixels were distributed in the blue luminous region, which meant the flames were blue and few soot particles were formed under these three conditions. Therefore, these conditions could be considered as ones before transition, which was consistent with the flame observation from Fig. 2. In Fig. 3(b), there was a clear transition. The pixels shifted from blue luminous region to red region, indicating that there were blue and yellow layers in the flames at the same time. Thus, the two conditions could be considered as ones in the sooting transition process which corresponded to the gradual appearance of the yellow layer of flame in Fig. 2. More soot particles appeared in the process of formation from XO = 23% to XO = 24%. From the analyses above, we could define the critical sooting transition condition as XO,cr = 22%. Fig. 3(c) showed the post-transition with XO larger than 24%. In these four conditions, the distribution of pixels was highly centralized from left to right, and less pixels were distributed in the blue region. It could also be seen from Fig. 2, a bright yellow layer appeared in the flame at 25–28% O2 concentrations. The sooting transition process have been completed and the flame was heavily sooting.
Fig. 4 showed flames at different oxygen concentrations conditions with different diluent gases. Compared to Flame A, the colors of Flames B1 in Fig. 4(a) and B2 in Fig. 4(c) had changed significantly. The blue layers were thicker and more obvious, and the yellow layers were quite weak, especially for Flame B1. As a result, the critical sooting transition condition was very difficult and almost impossible to determine by naked eyes. It can be concluded that no matter CO2 was added to the fuel side or the oxidizer side, the sooting transition process was delayed significantly, which indicated that the CO2 could effectively inhibit soot formation. The weaker yellow layer of Flame B1 meant a stronger inhibitory effect when CO2 was added to the fuel side. Flames C1 in Fig. 4(b) and C2 in Fig. 4(d) were very similar to Flame A visually. It was interesting to note that the sooting transition process of Flame C1 was delayed compared to Flame A, but it was advanced in Flame C2. This phenomenon meant adding He to the fuel side could inhibit the soot formation, but it had a promoting effect when He was added to the oxidizer side.
 | ||
| Fig. 4 Flame images at different oxygen concentration and different diluent gases combustion conditions, (a) Flame B1, (b) Flame C1, (c) Flame B2, (d) Flame C2. | ||
In addition, it was interesting to see that with low O2 concentration, there was a different in the color of the flames. Although the flames are all blue, the color in Fig. 4(a) and (c) was more towards pure blue. This was because the blue color of the flame was mainly due to CH* and C2* radicals.34 The addition of CO2 and He changed the concentration and distribution of the above-mentioned free radicals. In addition, the temperature of the flames was also different. These factors led to the difference in the energy radiated by the free radicals, resulting in a different color in the flames.
Taking the pixel points of the above four flames and drew the tricolor luminosity ratio profiles, as shown in Fig. 5. Similarly, the analysis method described above was used to determine the sooting transition process. The sooting transition process of the four flames could be concluded as follows: Flame B1: XO = 34% to XO = 36%, Flame C1: XO = 25% to XO = 26%, Flame B2: XO = 34% to XO = 35%, Flame C2: XO = 20% to XO = 21%. The critical sooting transition condition could be determined as XO,cr = 33%, 24%, 33%, and 19%, respectively. In order to deeply study the sooting transition process, four representative conditions (one condition before transition, two conditions in the transition process, and one condition after transition) of these flames were selected to perform the kinetic study.
 | ||
| Fig. 5 Different flame profiles of the tricolor luminosity proportion for pixel points in the flame at different O2 concentrations, (a) Flame B1, (b) Flame C1, (c) Flame B2, (d) Flame C2. | ||
3.2. Simulation results
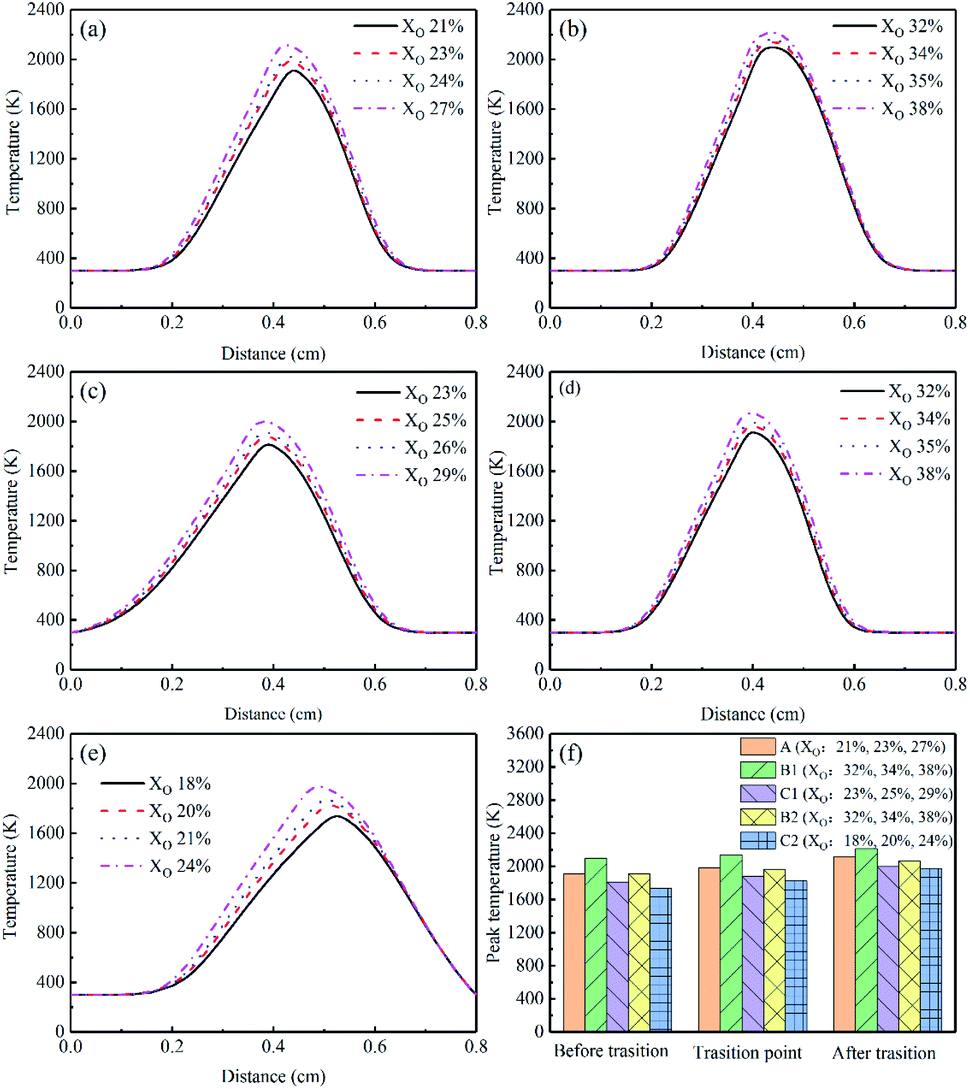 | ||
| Fig. 6 Temperature profiles and peak temperature of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak temperature. | ||
 | ||
| Fig. 7 Ethylene mole fraction profiles of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2. | ||
The reaction pathway analysis of ethylene for different flames were carried out, as shown in Fig. 8. The results showed that the main consumption pathways of ethylene in counterflow diffusion flame was the decomposition of fuel molecules into small alkyl radicals, and secondly was the polymerization into large alkyl radicals. With the increase of oxygen concentration in the sooting transition process, the decomposition of fuel molecules increased. The reason for this phenomenon was that the increasing temperature would promote the scission of C–C bonds. Furthermore, it was worth noting that the important soot precursor C2H2 had a significant increase in the transition process. Comparing the five flames, the addition of diluents had little effect on the main consumption paths of ethylene. When CO2 was added, the conversion of CH3 to C2H6 was inhibited.
 | ||
| Fig. 8 The reaction paths of ethylene, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2 (percentage of each path's contribution to the consumption of the species at the beginning of the corresponding arrow was marked). | ||
The concentration profiles of CH3 radical were exhibited in Fig. 9(a)–(e). In the sooting transition process, the mole fraction of CH3 showed an increasing trend. The addition of diluents changed the distribution of CH3 in the flame, which was more obvious in Flames C1 and C2. Fig. 9(f) showed the corresponding CH3 peak concentrations of different Flames. The CH3 peaks of Flames B1 and B2 were similar to Flame A. However, the results shown in Flames C1 and C2 indicated that the addition of He had an inhibitory effect on CH3 formation. This phenomenon was possibly due to the reaction C2H3 + CH3 + M → C3H6 + M.20
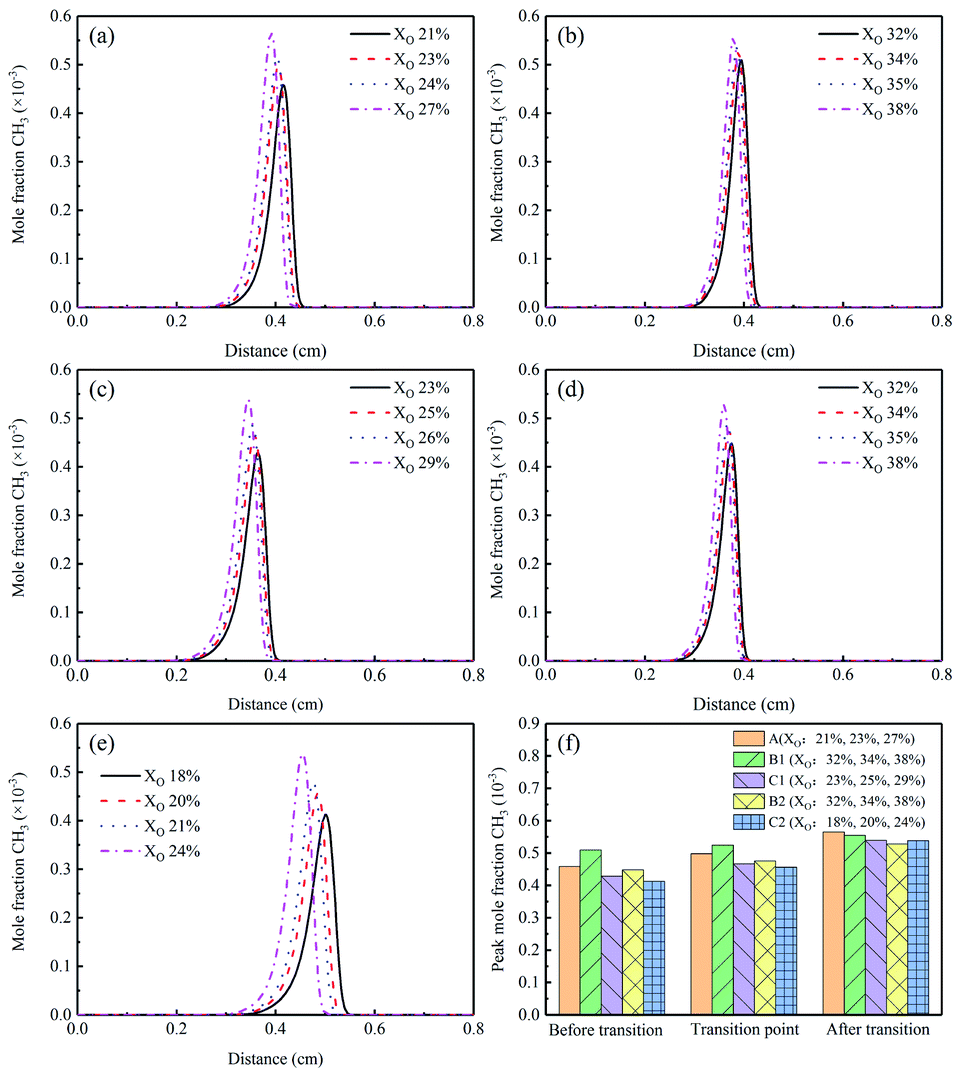 | ||
| Fig. 9 CH3 mole fraction profiles and peak mole fractions of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
The HACA mechanism proposed by Frenklach et al.37 reported that H free radicals could control the growth of polycyclic aromatic hydrocarbons and were related to the surface growth of soot. Therefore, it was necessary to study the formation characteristics of H.
Fig. 10(a)–(e) demonstrated the H mole fraction of different flames, from which we could see that H mole fraction increased with the increasing XO in the sooting transition process. The corresponding peak concentrations were shown in Fig. 10(f). Compared to Flame A, the lower peaks of Flames B1, C1, B2, C2 suggested there was a suppression effect on the H radical formation when adding He or CO2. In Flame B2, this effect was the most obvious. For CO2-diluted flames, some researchers attributed the inhibitory effect to the important reaction CO2 + H ⇌ CO + OH.14,17,38,39 As a result, they all thought that adding CO2 could reduce the production of H, then effectively inhibit the soot formation.
 | ||
| Fig. 10 H mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
The mole fraction of OH profiles of the flames examined in this study were presented in Fig. 11(a)–(e). The mole fraction of OH showed a significant increase in the sooting transition process. The corresponding peak concentrations were implied in Fig. 11(f). Flames B1 and B2 showed that the addition of CO2 had a promoting effect on OH formation, and the effect became stronger when adding to the fuel side. This was due to the important reaction CO2 + H ⇌ CO + OH mentioned above. Some researchers reported that the chemical mechanism of CO2 addition could promote the concentrations of O and OH, thereby increased the soot precursor oxidation.16,40 Therefore, the increase of the hydroxyl in Flames B1 and B2 was one of the factors that CO2 addition could inhibit the soot formation. More hydroxyl in Flame B1 might explain the stronger soot suppression in the experimental results as shown in Fig. 4. In addition, there was a decrease of OH in Flames C1 and C2.
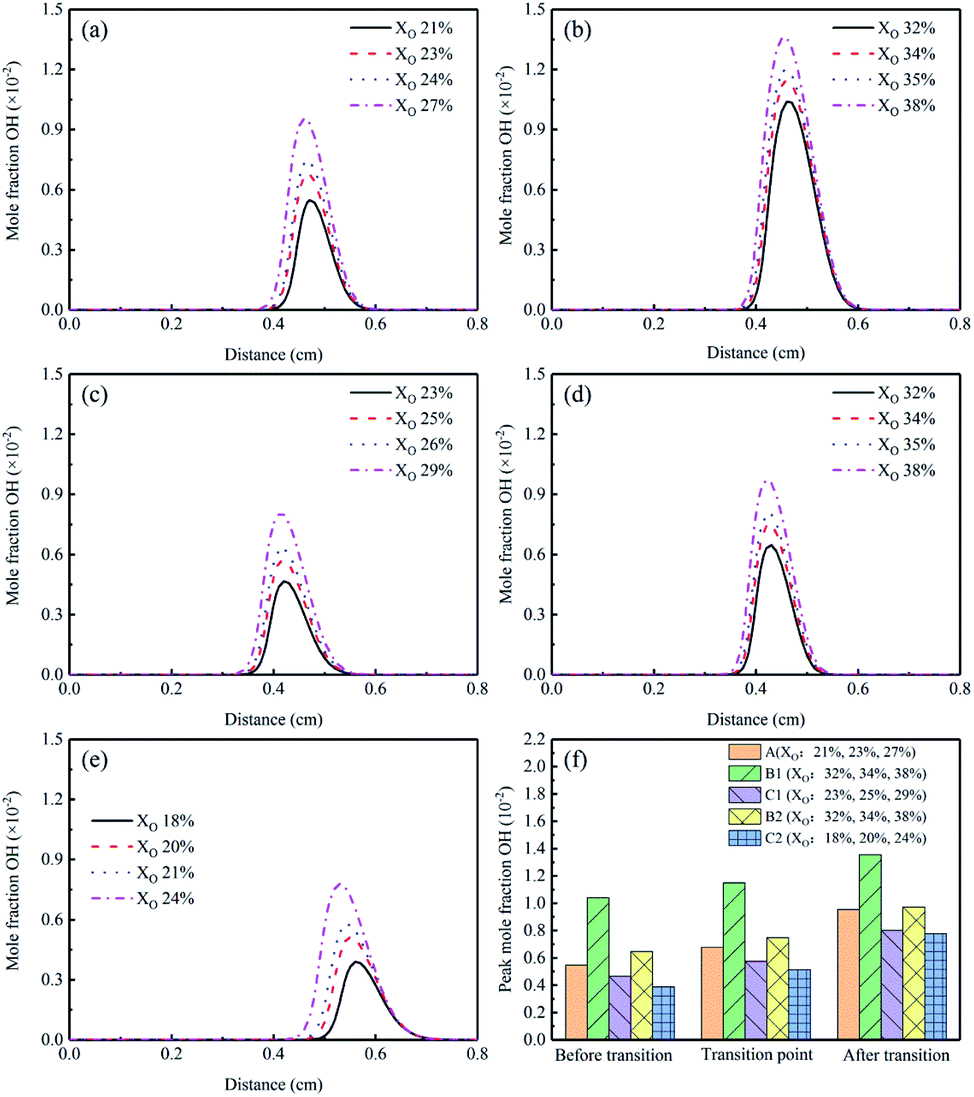 | ||
| Fig. 11 OH mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
The concentration profiles of O were shown in Fig. 12(a)–(e). In the sooting transition process, more oxygen atoms were generated due to the increase in oxygen concentration. The corresponding O peak concentrations were exhibited in Fig. 12(f). Oxygen atoms had an oxidative effect on soot. It was worth to note the Flame B1, the mole fraction of O was the highest. This phenomenon also explained the experimental results that why there was less soot in Flame B1. Moreover, the O was slightly reduced in the Flames C1 and C2.
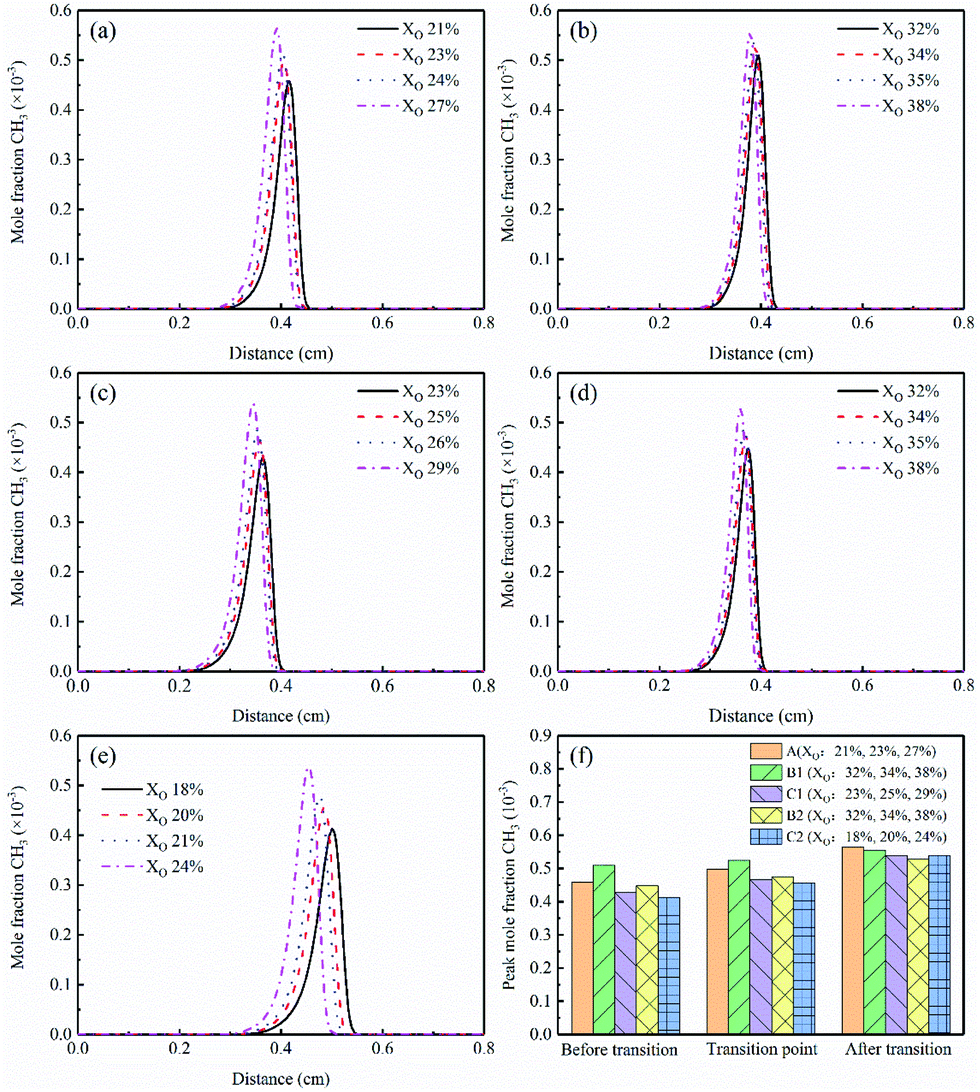 | ||
| Fig. 12 O mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
 | ||
| Fig. 13 CO mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
CO2 was the main product of combustion, and its mole fraction was shown in Fig. 14(a)–(e). CO2 was added to the Flames B1 and B2, so the two Flames were not discussed here. In Flames A, C1, and C2, the mole fraction of CO2 increased in the sooting transition process. This was because the increase of temperature with the increase of XO would intensify the oxidative reactions. The corresponding peak concentrations of CO2 were displayed in Fig. 14(f). The Flame C1 had a slight decrease trend of CO2 while the Flame C2 had a small increase trend.
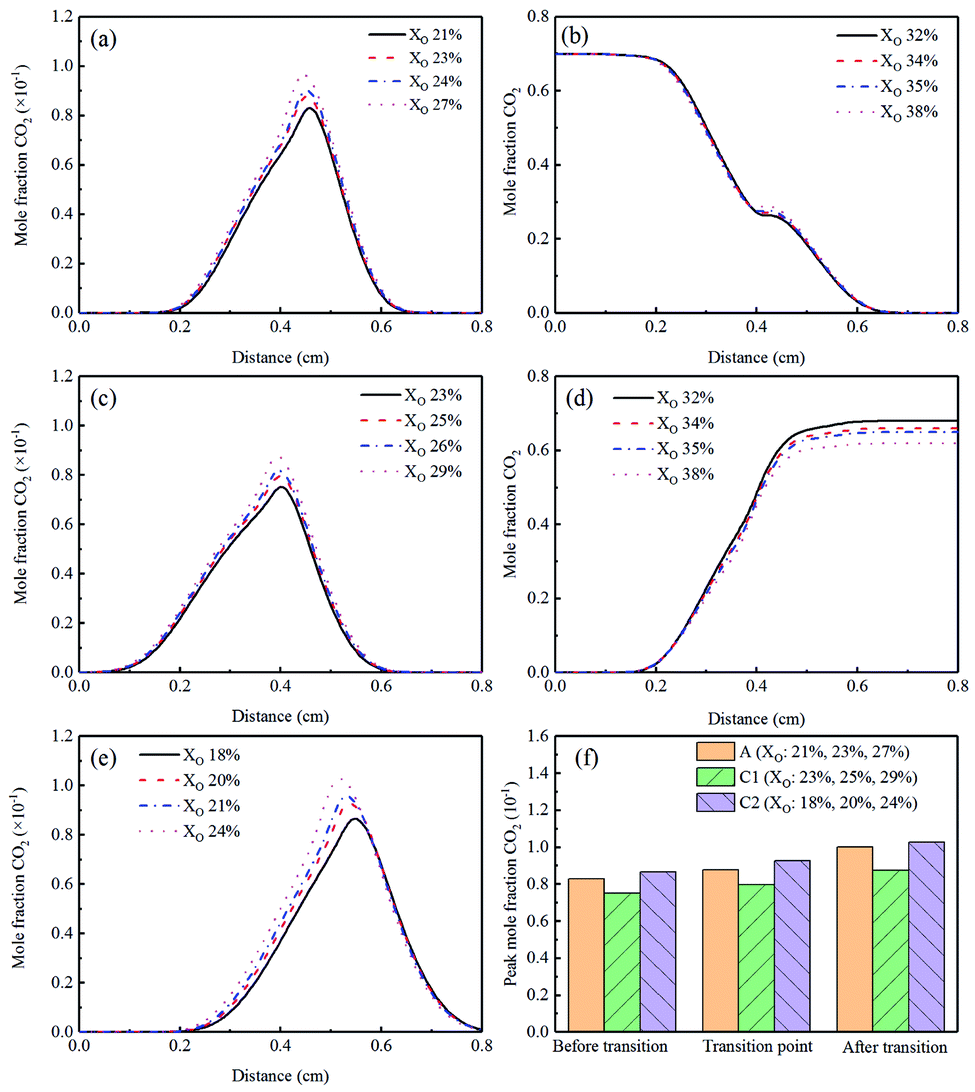 | ||
| Fig. 14 CO2 mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
The study of Gleason et al.26 reported that small aromatic hydrocarbons controlled the onset of soot nucleation, and the growth basis of polycyclic aromatic hydrocarbons was benzene or phenyl.6,41 Therefore, analyzing the formation characteristics of C6H6 could promote the understanding of soot nucleation. Furthermore, according to Naseri et al.,42 C2H2 and C3H3 played an important role in the formation of benzene. Consequently, in this section, the rate of production and reaction path analysis of C2H2, C3H3, and C6H6 were carried out.
Methane (CH4) was an important single-carbon hydrocarbon, and its concentration increased with the increasing XO in the sooting transition process, as shown in Fig. 15(a)–(e). Corresponding peak concentrations could be found in Fig. 15(f). Compared to Flame A, the changes of CH4 in Flames B1 and B2 were very small. While a significant decrease could be seen in Flame C1 and C2, indicating that adding He inhibited the formation of CH4.
 | ||
| Fig. 15 CH4 mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
The nucleation rate of soot was essentially a first-order reaction of acetylene (C2H2) concentration.43,44 Based on the reason, studying the formation characteristics of C2H2 was paramount. The mole fraction of C2H2 was illustrated in Fig. 16(a)–(e), and the corresponding peak concentrations could be obtained in Fig. 16(f). Its mole fraction increased with the increase of XO. From the Flames B1 and B2, it could be concluded that the addition of CO2 had no obvious effect on C2H2. Instead, Flames C1 and C2 suggested that there was an inhibitory effect on the formation of C2H2 when adding He.
 | ||
| Fig. 16 C2H2 mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
Fig. 17 showed the rate of production of C2H2. The main formation reaction of C2H2 was C2H3 (+M) = H + C2H2 (+M), and the primary consumption reaction of C2H2 was O + C2H2 = H + HCCO. The rate of other reactions was small and would vary with the diluents. The rate of all reactions became faster with increasing oxygen concentration in the sooting transition process, and the reaction region shifted to the fuel side.
 | ||
| Fig. 17 C2H2 rate of production diagrams, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2. | ||
Fig. 18 exhibited the reaction paths of C2H2 under different conditions. In these diagrams, the positive value represented the percentage of each path's contribution to the formation of the species at the end of the corresponding arrow (solid line), and the negative value represented the percentage of each path's contribution to the consumption of the species at the beginning of the corresponding arrow (dot line). With different diluent gases, the main products from C2H2 were the same, but their ratios were different. C2H3 and C2H4 were the main sources of C2H2. In CO2-diluted flames, the contribution of C2H2 to C4H2 was significantly reduced.
 | ||
| Fig. 18 Reaction paths diagrams of C2H2 in different Flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2. | ||
In the sooting transition process, with the increasing oxygen concentration, the change of C2H6 formation was not sensitive in Flames A and B1, but it showed a decrease trend in Flames C1, B2, and C2, as shown in Fig. 19(a)–(e). The peak concentrations of C2H6 in Fig. 19(f) showed that the addition of CO2 had a promoting effect on its formation. The C2H6 was a combustion intermediate produced by the direct reaction of ethylene with hydrogen. For Flame C1, the reason why the concentration of C2H6 was higher than Flame A was that the diffusion of helium from the centerline to the periphery of the flame, resulting in an increase of C2H4.20
 | ||
| Fig. 19 C2H6 mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
Based on the fact that the propargyl (C3H3) played a vital role in the formation of the first aromatic ring45 and polycyclic aromatic hydrocarbons (PAHs), studying the characteristics of C3H3 was of great significance. Fig. 20(a)–(e) showed the concentration of C3H3 in each diluted flame. With the increase of XO in the sooting transition process, the concentration of C3H3 had an obvious increasing trend. The corresponding peak concentrations were displayed in Fig. 20(f), from which could be found that adding CO2 and He both inhibited the C3H3 formation, and the later were more effective.
 | ||
| Fig. 20 C3H3 mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
The rate of production of C3H3 was displayed in Fig. 21. In these figures, C3H4-A and C3H4-P were isomers of each other. The main formation reactions of C3H3 were CH2 + C2H2 = H + C3H3, and H + C3H4 = H2 + C3H3. The main reactions responsible for C3H3 consumption were OH + C3H3 = C2H3CHO, and H + C3H3 = C3H4. In Flames A and C2, a formation reaction occurred, CH2(S) + C2H2 = H + C3H3. With the increase of oxygen concentration, the reaction rates of the above reactions were obviously faster. The addition of different diluent gases changed the reaction region of these reactions, which was more significant when He was added.
 | ||
| Fig. 21 C3H3 rate of production diagrams, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame C1, (e) Flame C2. | ||
The reaction paths of C3H3 under different conditions was shown in Fig. 22. In these diagrams, the value represented the percentage of each path's contribution to the formation of the species at the end of the corresponding arrow. The formation of C3H3 was mainly due to the unimolecular C–H scission reaction of propyne (C3H4). The main source of C3H3 is the two isomers of C3H4 (C3H4-A and C3H4-P). And there was an isomerization reaction between two isomers. In the case of high oxygen concentration, it would promote the conversion of C4H6 (butadiene and butyne) to C3H3.
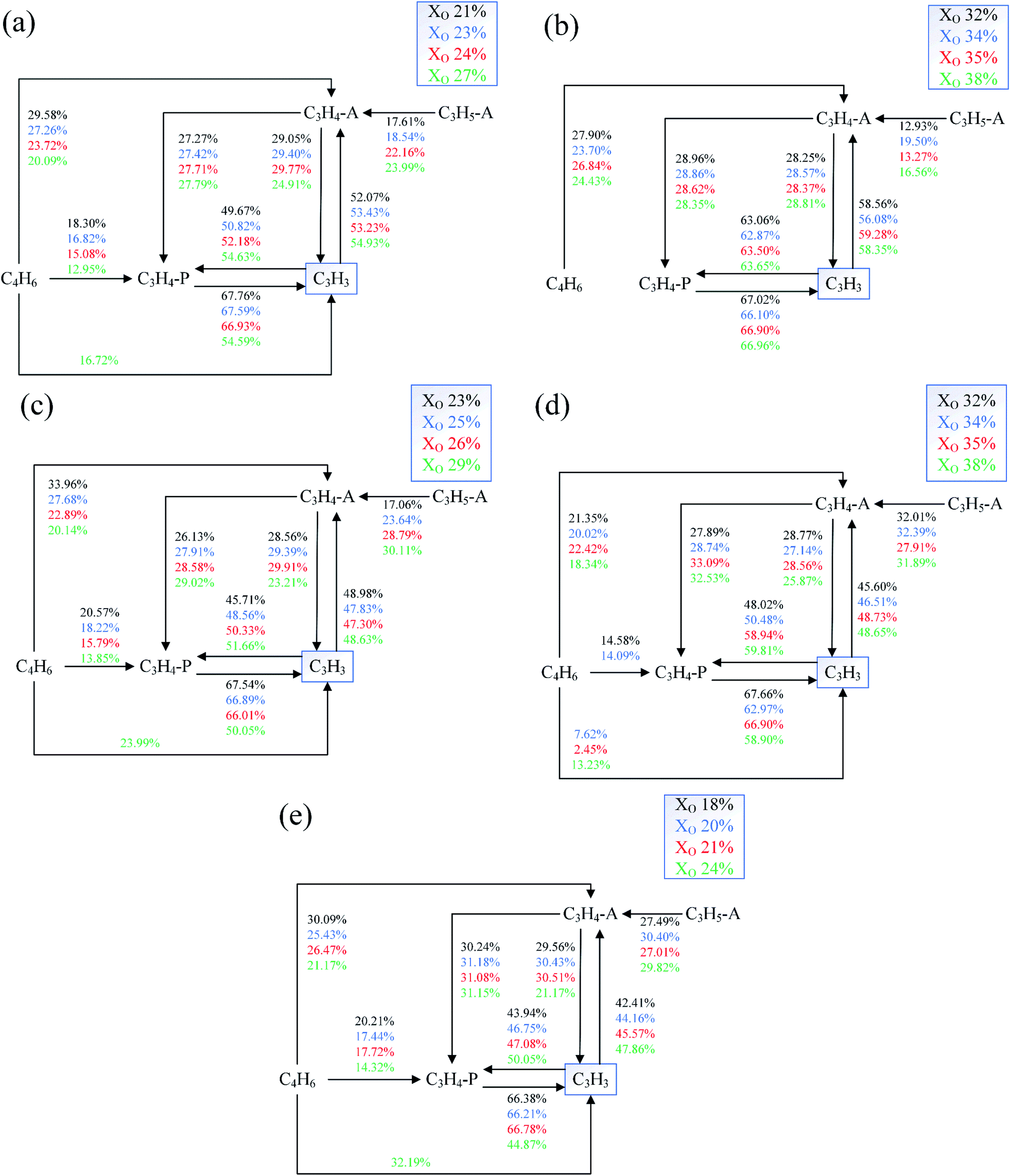 | ||
| Fig. 22 Reaction paths diagrams of C3H3 in different Flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame C1, (e) Flame C2. | ||
Fig. 23(a)–(e) exhibited the profiles of the C3H6 mole fraction, and Fig. 23(f) was a graph of the corresponding peak concentrations. In He-diluted flames, the concentration of C3H6 decreased with increasing XO. However, it had no significant change in N2- and CO2-diluted flames. The peak mole fraction of C3H6 was shown in Fig. 23(f). The results showed that adding CO2 promoted the formation of C3H6, but introducing He to the fuel side and the oxidizer side respectively had different effect. The concentration of C3H6 increased when He was added to the fuel side, while it decreased when He was added to the oxidizer side.
 | ||
| Fig. 23 C3H6 mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
The concentrations of benzene (C6H6) shown in Fig. 24(a)–(e) was of great interest as it was a crucial step in the formation of PAHs, which directly related to soot formation. The C6H6 showed an increasing trend in sooting transition process. In CO2-diluted flames, the change of the curves was not obvious. Fig. 24(f) exhibited corresponding peak concentrations. The concentration of C6H6 in both the CO2-diluted flames and the He-diluted flames was reduced, which suggested CO2 and He both had an inhibitory effect on C6H6. In CO2-diluted flames, the CO2 consumed 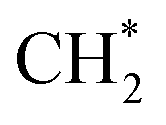 through reaction
through reaction 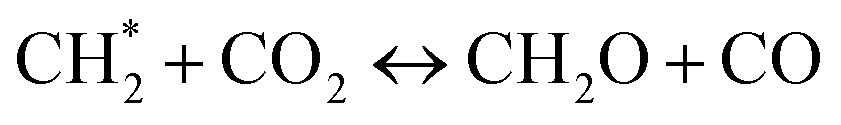 , and then inhibited the formation of C6H6. The
, and then inhibited the formation of C6H6. The  was activated methylene, playing an important role in the generation of C6H6.42
was activated methylene, playing an important role in the generation of C6H6.42
 | ||
| Fig. 24 C6H6 mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
Fig. 25 gave the profiles of C6H6 rate of production in different conditions. H + C6H5 (+M) = C6H6 (+M) was the most important formation reaction. In Flames A, B1, and B2, a formation reaction occurred, H2 + C6H5 = H + C6H6. In addition, the reaction C2H2 + C4H5 = H + C6H6 also influenced the formation of C6H6 in Flames A and B2. The main consumption reaction was H + C6H6 = H2 + C6H5. In sooting transition process, as the oxygen concentration increased, the reaction area shifted to the fuel side.
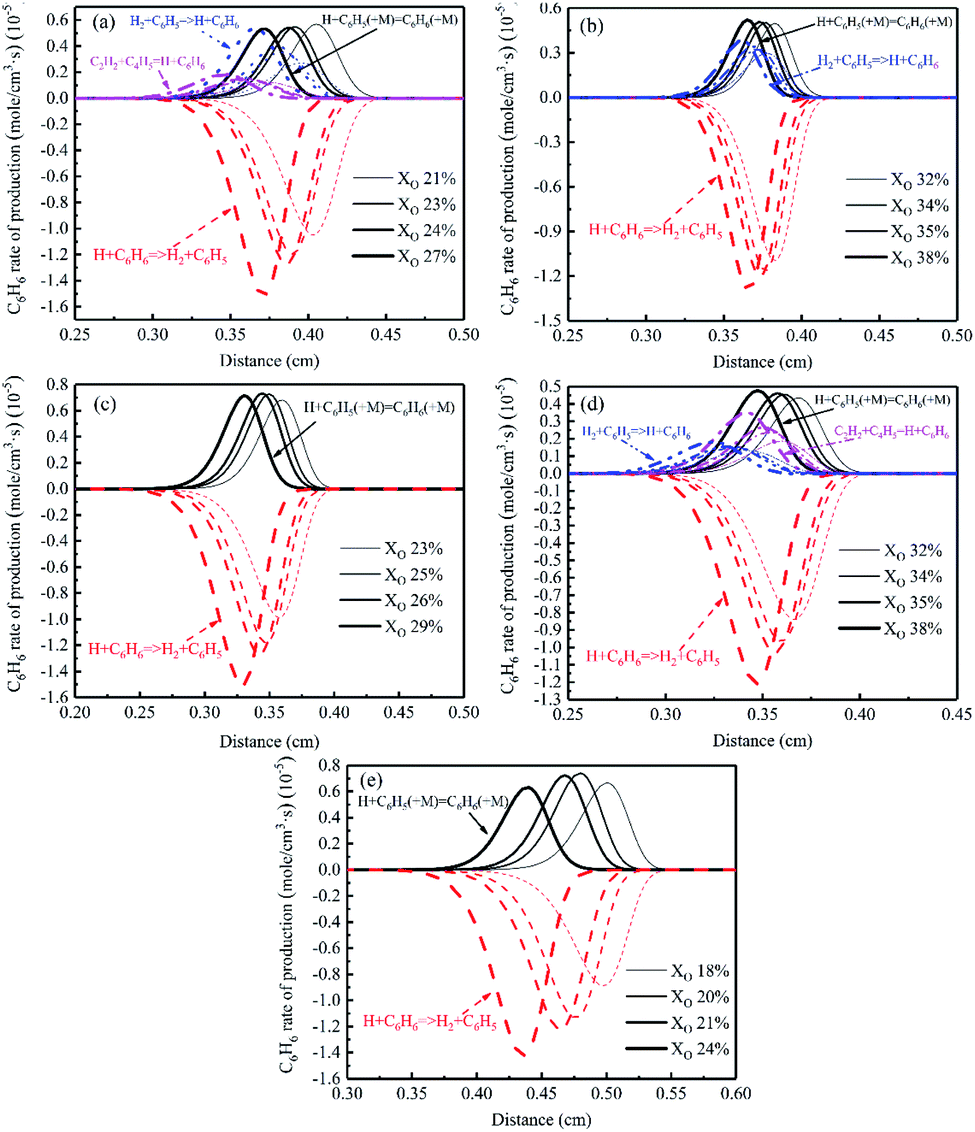 | ||
| Fig. 25 C6H6 rate of production diagrams, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2. | ||
Fig. 26 showed the reaction paths of C6H6 under different conditions. The main source of C6H6 was C6H5, CYC6H8, C3H3, fulvene, and the reaction between C2H2 and C4H5. The consumption reaction of C6H6 was mainly the conversion of C6H6 to C6H5. As the oxygen concentration increased, the contribution of C6H5 and C3H3 to C6H6 increased, and the CYC6H8, C4H5 and C2H2 contributed less to C6H6.
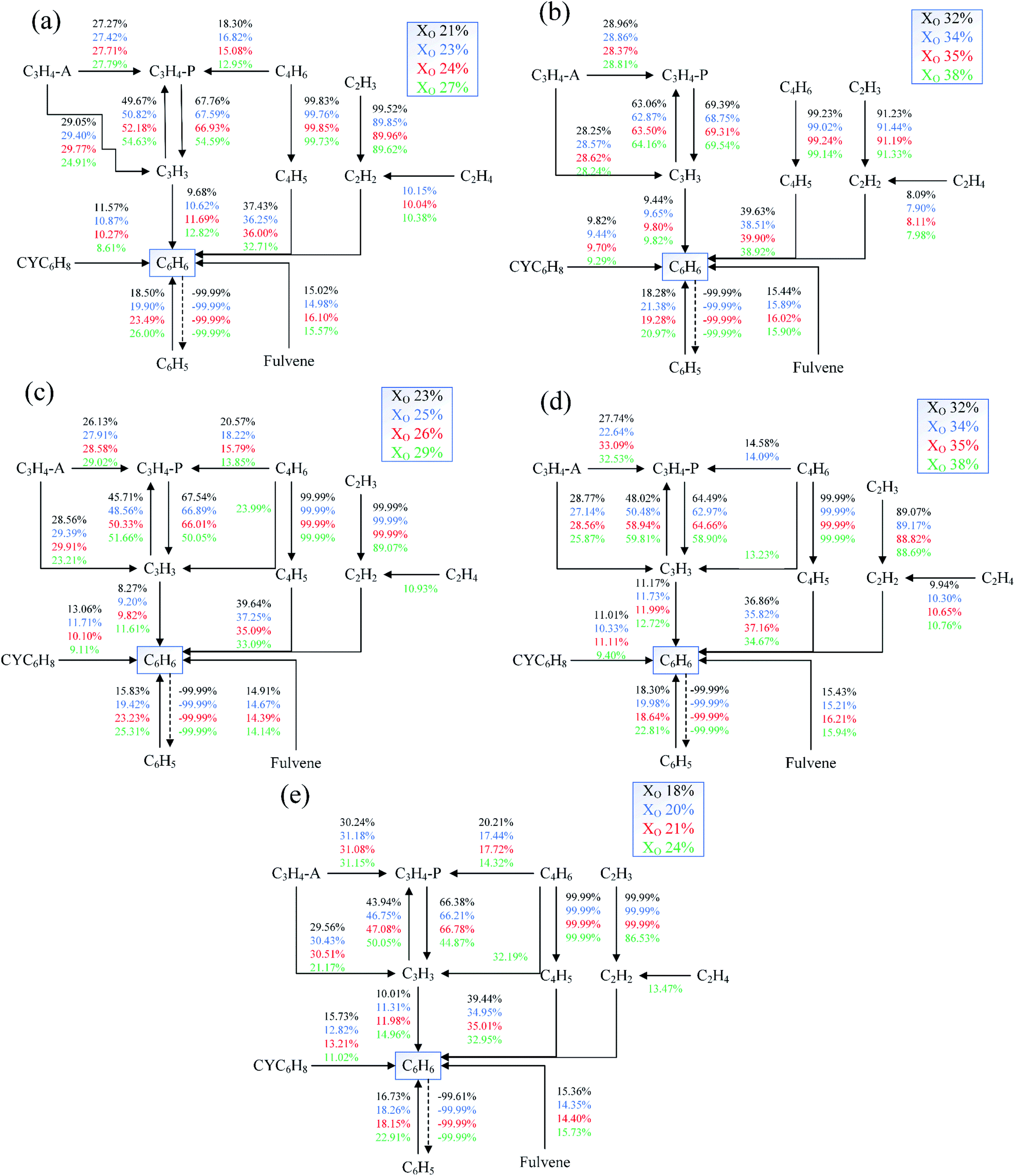 | ||
| Fig. 26 Reaction paths diagrams of C6H6 in different Flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2. | ||
Gleason et al.26 reported that one-ring and two-ring aromatic played an important part in soot nucleation. Therefore, investigating the formation of A2 (naphthalene) could advance the understanding of the sooting transition process. The simulation results of A2 were exhibited in Fig. 1. It could be seen from Fig. 27(a)–(e) that with the increase of XO, except for Flame B1, the concentration of A2 had an obvious upward trend. This result indicated that in sooting transition process, the increase of A2 promoted the formation of soot. From the distribution area of A2, it could be inferred that the generation of soot is closer to the fuel side. The peak mole fraction of A2 was exhibited in Fig. 27(f). Both He and CO2 additions reduced the mole fraction of naphthalene, which was more obvious in CO2-diluted flames. This phenomenon also proved that the addition of CO2 can inhibit the formation of soot.
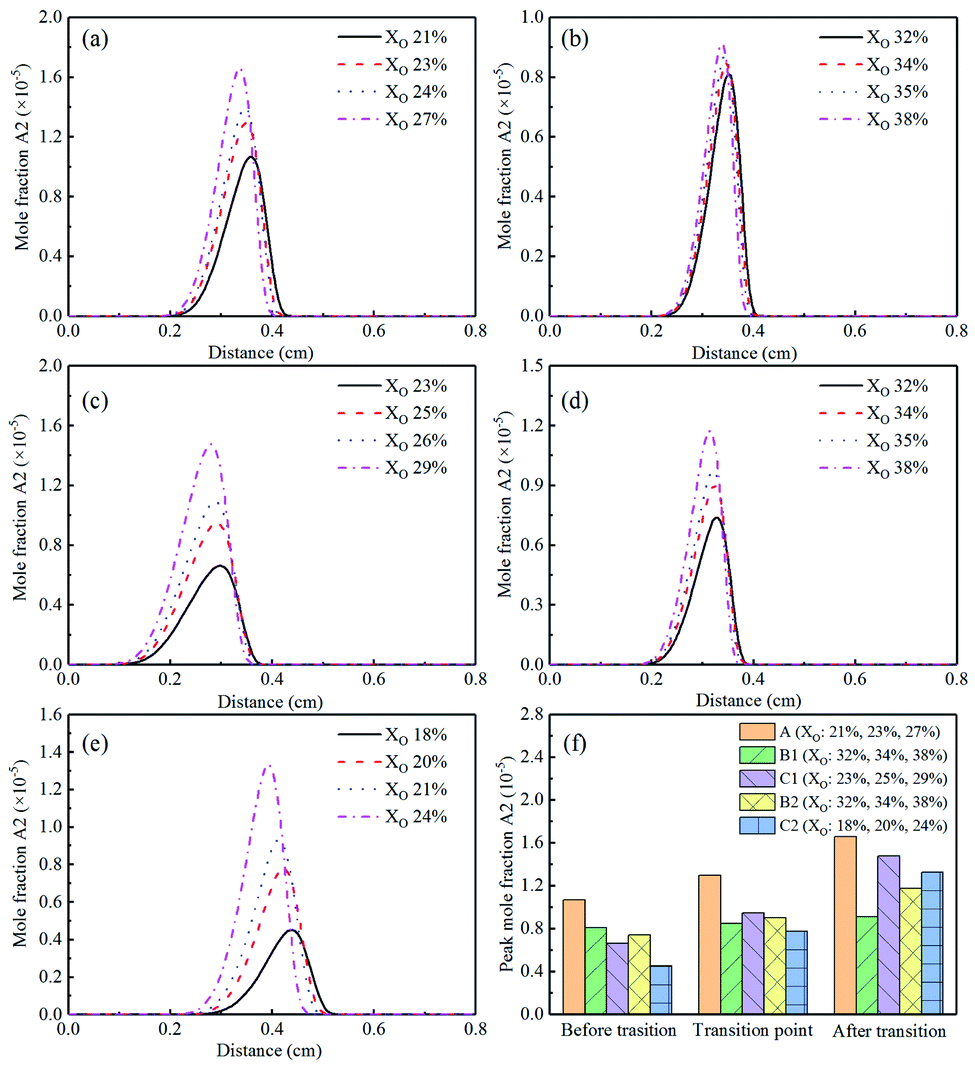 | ||
| Fig. 27 A2 (naphthalene) mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
Fig. 28(a)–(e) showed the concentrations of A3 (anthracene and phenanthrene). Similar to A2, the mole fraction of A3 had an obvious upward trend with the increase of oxygen concentration during the transition process. It could be concluded from Fig. 2(f) that adding CO2 and He could inhibit the formation of A3, and the effect was more significant when adding CO2. A3 was also mainly formed in the region biased towards the fuel side.
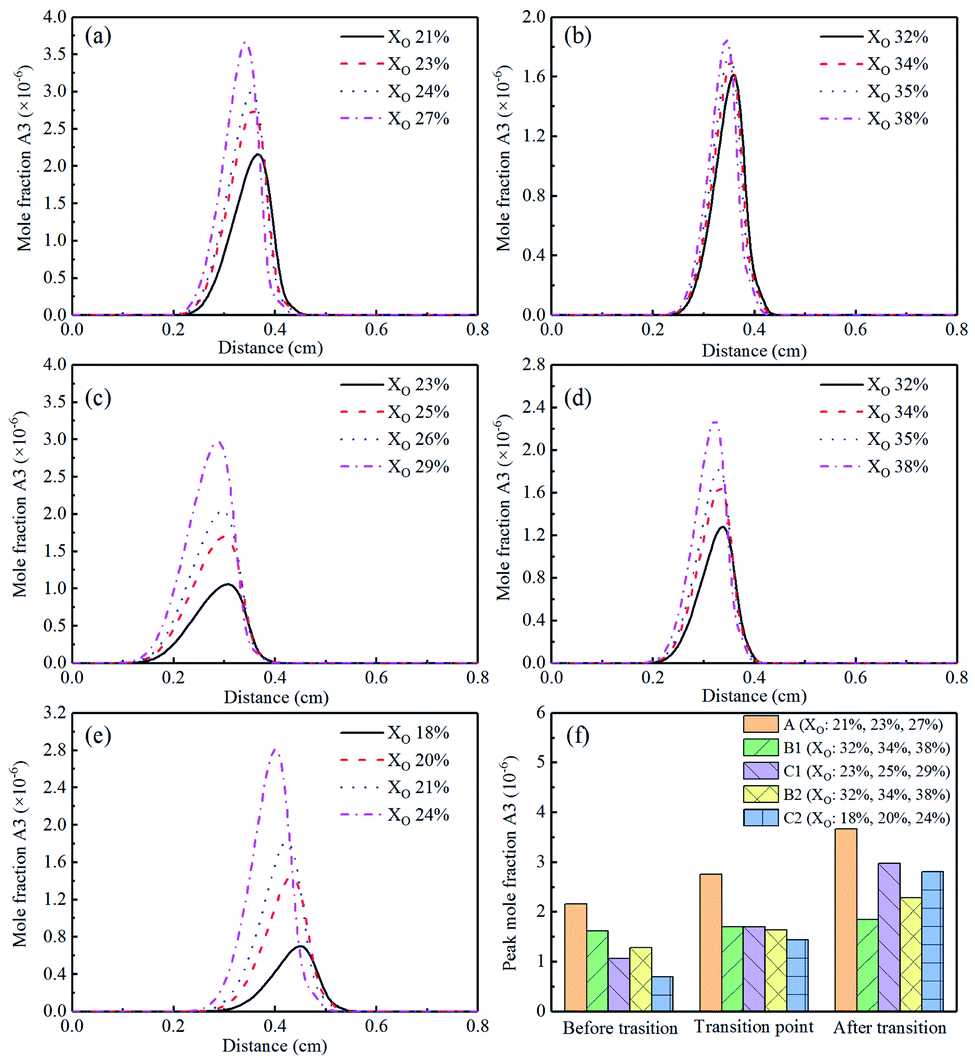 | ||
| Fig. 28 A3 (anthracene and phenanthrene) mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
Fig. 29(a)–(f) showed the simulation results of A4 (pyrene and isomers). In the sooting transition process, the concentration of A4 has an obvious increasing trend, which was similar to A2 and A3. Combining the curves of A2, A3 and A4, it could be concluded that the changes in the sooting transition process were mainly reflected in the promotion of PAHs growth. Both CO2 and He could inhibit the formation of PAHs, and the inhibition effect of CO2 was more significant. Flame B1 was the most interesting because in Flame B1 there was a very slow increase in PAHs as XO increases. Zhang et al.46 reported that gaseous PAHs may also be responsible for the yellow color emissions. This phenomenon was consistent with the experimental results described above: (1) the yellow light of Flame B1 is the weakest (2) the transition process of Flame B1 is the longest.
 | ||
| Fig. 29 A4 (pyrene and isomers) mole fraction profiles and peak mole fraction of different flames, (a) Flame A, (b) Flame B1, (c) Flame C1, (d) Flame B2, (e) Flame C2, (f) peak mole fraction. | ||
4. Conclusions
The sooting transition process was studied in the ethylene counterflow diffusion flames with different diluents (N2, He, and CO2). In each diluted flame, the sooting transition process was determined by optical method. The concentrations of major hydrocarbon species and important radicals were investigated by kinetical modeling. The N2-diluted flames were regarded as the standard flames for comparison. The results were summarized as follows:(1) The CO2-diluted flames significantly delayed the sooting transition process, indicating that the CO2 had an inhibitory effect on soot formation. When CO2 was added to the fuel side, the weaker yellow luminous sooting region meant a stronger soot suppression effect.
(2) He-diluted flames were the most interesting. When He was added to the fuel side, the sooting transition process was delayed, while to the oxidizer side, the transition process was advanced.
(3) The simulation results showed that adding CO2 inhibited the formation of H radical and promoted the OH radical formation, which contributed to suppress the soot formation. When CO2 was added to the fuel side, the concentration of O atom and OH radical increased more significantly, which may explain why less soot was generated in this case.
(4) The concentration of H, OH, and O radicals reduced in He-diluted flames, as well as the important soot precursors C2H2, C3H3, and C6H6. In addition, adding helium significantly changed the reaction region.
(5) In the sooting transition process, the small molecular PAHs (A2, A3, and A4) increased significantly with the increase of oxygen concentration. The addition of carbon dioxide could effectively inhibit the formation of PAHs.
This research could be expected to provide a deep understanding on the effect of different diluent gases on the soot formation, which would be beneficial to the future study on the control of soot emissions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province [BK20200490], the National Natural Science Foundation of China [52106160, 52076110], State Key Laboratory of Engines, Tianjin University and the Fundamental Research Funds for the Central Universities [30920031103].References
- K. Orhan, R. Mayerle and W. W. Pandoe, Energy Proc., 2015, 76, 7–16 CrossRef.
- J. Jeon, J. T. Lee, S. Kwon and S. Park, Energy Convers. Manage., 2016, 116, 174–183 CrossRef CAS.
- C. Shi, C. Ji, S. Wang and J. Yang, Energy Convers., 2020, 205, 112443 CrossRef.
- L. Ying, D. K. Henze, J. Darby, B. H. Henderson and P. L. Kinney, Sci. Total Environ., 2016, 539, 515–525 CrossRef PubMed.
- J. Zhou, X. Sheng and L. He, J. Therm. Sci., 2019, 28, 669–681 CrossRef CAS.
- H. Wang, Proc. Combust. Inst., 2011, 33, 41–67 CrossRef CAS.
- P. Roth, Proc. Combust. Inst., 2007, 31, 1773–1788 CrossRef.
- H. Jung, B. Guo, C. Anastasio and I. Kennedy, Atmos. Environ., 2006, 40, 1043–1052 CrossRef CAS.
- M. T. Lund, T. K. Berntsen, C. Heyes, Z. Klimont and B. H. Samset, Atmos. Environ., 2014, 98, 50–58 CrossRef CAS.
- V. Ramanathan and G. Carmichael, Nat. Geosci., 2008, 1, 221–227 CrossRef CAS.
- S. S. Gill, J. M. Herreros, A. Tsolakis, D. M. Turner, E. Miller and A. P. E. York, RSC Adv., 2012, 2, 10400–10408 RSC.
- O. A. Shromova, N. M. Kinnunen, T. A. Pakkanen and M. Suvanto, RSC Adv., 2017, 7, 46051–46059 RSC.
- D. X. Du, R. L. Axelbaum and C. K. Law, Symp. (Int.) Combust., 1991, 23, 1501–1507 CrossRef.
- K. C. Oh and H. D. Shin, Fuel, 2006, 85, 615–624 CrossRef CAS.
- F. Liu, H. Guo, G. J. Smallwood and Ö. L. Gülder, Combust. Flame, 2001, 125, 778–787 CrossRef CAS.
- Ö. L. Gülder and M. F. Baksh, Angew. Chem., Int. Ed., 1993, 45, 5337–5340 Search PubMed.
- H. Guo and G. J. Smallwood, Combust. Sci. Technol., 2008, 180, 1695–1708 CrossRef CAS.
- I. S. Mclintock, Combust. Flame, 1968, 12, 217–225 CrossRef CAS.
- I. Glassman, Symp. (Int.) Combust., 1998, 27, 1589–1596 CrossRef.
- R. K. A. Kailasanathan, T. L. B. Yelverton, T. Fang and W. J. Roberts, Combust. Flame, 2013, 160, 656–670 CrossRef.
- Ö. L. Gülder and D. Snelling, Combust. Flame, 1993, 92, 115–124 CrossRef.
- M. Yen, V. Magi and J. Abraham, Chem. Eng. Sci., 2019, 196, 116–129 CrossRef CAS.
- A. Ergut, R. J. Therrien, Y. A. Levendis, H. Richter, J. B. Howard and J. B. Garlson, Combust. Flame, 2008, 155, 232–246 CrossRef CAS.
- A. Ergut, R. J. Therrien, Y. A. Levendis, H. Richter, J. B. Howard and J. B. Garlson, Combust. Flame, 2009, 156, 1014–1022 CrossRef CAS.
- R. J. Therrien, Master thesis, Northeastern University, 2008.
- K. Gleason, F. Carbone, A. J. Sumner and B. D. Drollette, Combust. Flame, 2021, 223, 398–406 CrossRef CAS.
- X. Zhao, L. Xu, C. Chen, M. Chen, Y. Ying and D. Liu, J. Energy Inst., 2021, 98, 282–293 CrossRef CAS.
- A. Lutz, R. J. Kee, J. F. Grcar and F. M. Rupley, Office of Scientific and Technical Information Technical Reports, 1997 Search PubMed.
- W. Pejpichestakul, E. Ranzi, M. Pelucchi, A. Frassoldati, A. Cuoci, A. Parente and T. Farvelli, Proc. Combust. Inst., 2019, 37, 1013–1021 CrossRef CAS.
- E. Ranzi, A. Frassoldati, A. Stagni, M. Pelucchi, A. Cuoci and T. Faravelli, Int. J. Chem. Kinet., 2014, 469, 512–542 CrossRef.
- E. Ranzi, C. Cavallotti, A. Cuoci, A. Frassoldati, M. Pelucchi and T. Faravelli, Combust. Flame, 2015, 162, 1679–1691 CrossRef CAS.
- S. A. Tashkun, V. I. Perevalov and J. L. Teffo, Remote Sensing of the Atmosphere for Environmental Security, 2006, pp. 161–169 Search PubMed.
- L. Figura and A. Gomez, Combust. Flame, 2014, 161, 1587–1603 CrossRef CAS.
- Á. Kvaran, Á. H. Haraldsson and T. I. Sigfusson, J. Chem. Educ., 2000, 77, 1345 CrossRef.
- W. Wang, Master thesis, Wuhan University of Technology, 2020.
- F. Takahashi and I. Glassman, Combust. Sci. Technol., 1984, 37, 1–19 CrossRef CAS.
- M. Frenklach, Phys. Chem. Chem. Phys., 2002, 4, 2028–2037 RSC.
- M. Abián, A. Millera, R. Bilbao and M. U. Alzueta, Fuel, 2012, 91, 307–312 CrossRef.
- T. L. Cong and P. Dagaut, Combust. Sci. Technol., 2008, 180, 2046–2091 CrossRef CAS.
- R. D. Kern, K. Xie, H. Chen and J. H. Kiefer, Twenty-Third Symposium (International) on Combustion, 1991, vol. 23, pp. 1–1945 Search PubMed.
- R. A. Dobbins, Aerosol Sci. Technol., 2007, 41, 485–496 CrossRef CAS.
- A. Naseri, A. Veshkini and M. J. Combust, Flame, 2017, 183, 75–87 CrossRef CAS.
- P. B. Sunderland and G. M. Faeth, Combust. Flame, 1996, 105, 132–146 CrossRef CAS.
- K. C. Lin, P. B. Sunderland and G. M. Faeth, Combust. Flame, 1996, 104, 369–375 CrossRef CAS.
- Y. Georgievskii, J. A. Miller and S. J. Klippenstein, Phys. Chem. Chem. Phys., 2007, 9, 4259 RSC.
- C. Zhang, A. Atreya and k. Lee, Proc. Combust. Inst., 1992, 24, 1049–1057 CrossRef.
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2022 |