DOI:
10.1039/D2RA01298G
(Paper)
RSC Adv., 2022,
12, 18197-18208
Theoretical study on the mechanism, chemo- and enantioselectivity of the Ag- vs. Rh-catalyzed intramolecular carbene transfer reaction of diazoacetamides†
Received
26th February 2022
, Accepted 13th June 2022
First published on 22nd June 2022
Abstract
To explore the mechanism of silver and rhodium catalysis and reveal the origin of the chemo- and enantioselectivity of the reaction, density functional theory calculations were performed on the first silver-catalyzed highly enantioselective carbene transfer reaction. The calculation results reveal that when silver is used as a catalyst, due to the participation of the phosphate anion in the transition state, the enhanced nucleophilicity of the α-diazoacetamide unit promotes smooth dearomatization before generation of the silver carbene. Because the generated rhodium carbene has stronger electrophilicity, typical carbene reactions (C–H insertion and the Büchner reaction) are favored. In addition, in the process of silver catalyzed dearomatization, the formation of an R-type transition state is determined by the small torsion energy and strong interaction energy.
Introduction
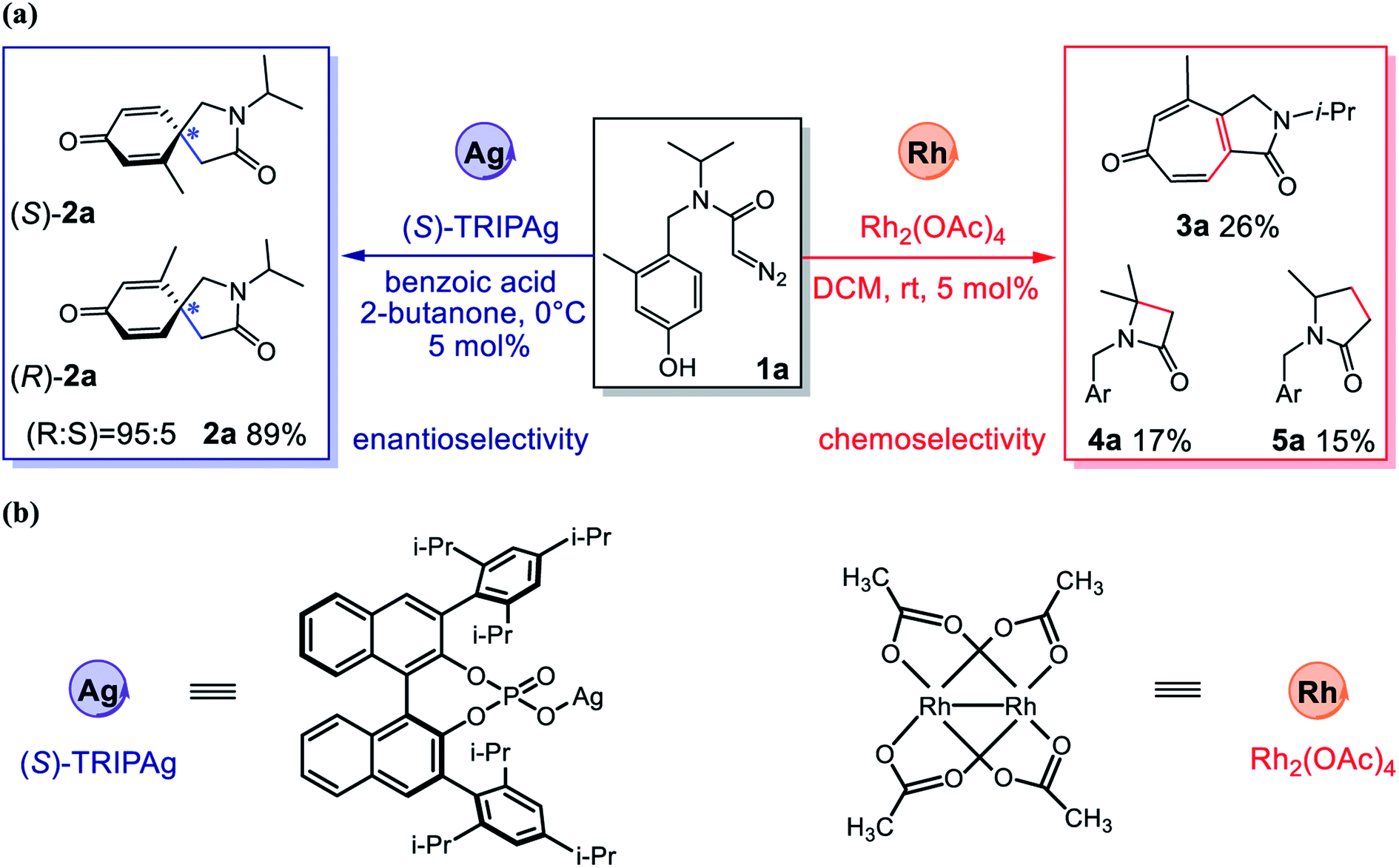
Metal carbenes are a class of highly reactive intermediates, and their catalytic transformations have developed into a very important field in organic synthetic chemistry.1 Compared with widely used catalysts based on metals such as rhodium, copper and gold, silver-based catalysts exhibit unique reactivity and selectivity.2–5 At present, some research groups have successfully realized metal carbene mediated selective addition to arenes.6 However, the field of silver-catalyzed asymmetric carbene transfer reactions has developed very slowly7–11 because of the linear and triangular coordination modes,12 the competitive Lewis acid-catalyzed pathway and the free carbene pathway.13 Since 1996, Burgess,14 Jørgensen13 and others10,11,15,16 have investigated the asymmetric carbene transfer reaction catalyzed by silver salts, but have not achieved satisfactory results. In 2017, Nemoto's research group successfully realized the first silver-catalyzed, highly enantioselective carbene transfer reaction through the intramolecular reaction model.9 They deployed (S)-TRIPAg as a catalyst to achieve the asymmetric dearomatization of phenols with α-diazoamides under mild conditions (Fig. 1). The chiral catalyst can effectively induce the configuration of the product, by circumventing the long distance between the chiral center and the reaction center, which leads to weakening of the induction effect. In this reaction, silver catalysis exhibits high chemoselectivity, effectively preventing the Büchner reaction and the C–H insertion reaction.
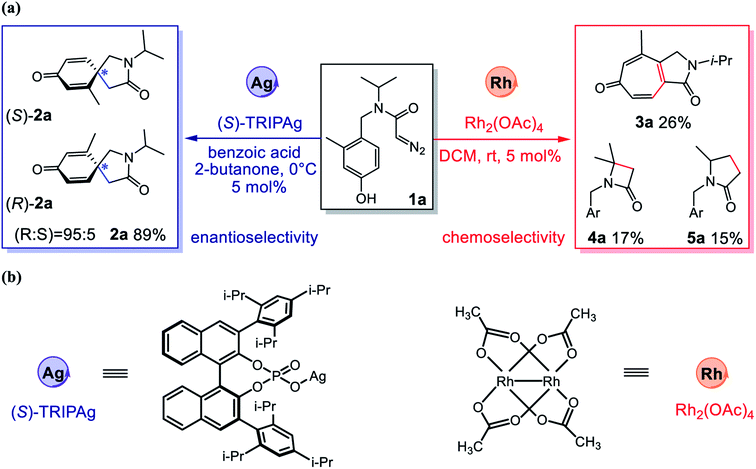 |
| | Fig. 1 (a) Asymmetric intramolecular dearomatization, 1°/3° C–H insertion and Büchner reaction of α-diazoamide 1a catalyzed by Ag/Rh; (b) the structures for Ag and Rh catalysts. | |
Since then, asymmetric carbene transfer reactions based on silver catalysis have been successively developed. In these reaction systems, the use of a silver catalyst was essential for achieving high chemo- and enantioselectivity. However, the reasons for the efficient catalysis of silver salts and the origin of the metal-determined chemo- and enantioselectivity remain unclear. Therefore, this work adopts the work of Nemoto's group as a model, and deeply explores the mechanism of the intramolecular carbene transfer reaction catalyzed by (S)-TRIPAg and Rh2(OAc)4 through density functional theory (DFT) calculations. The optimal path of different, transition metal-catalyzed reactions and the key factors affecting the chemo- and enantioselectivity of the reaction are revealed. A deeper understanding of the mechanism should help provide some insights for the further development of metal-catalyzed carbene transfer reactions and silver chemistry.
Computational details
All DFT calculations were performed using Gaussian16 suite of program.17 All structure geometries were optimized without any symmetry constraints in the gas phase by using B3LYP18 functional with D3BJ dispersion corrections,19 and a mixed basis set of SDD20 for the transition metals and 6-31G(d,p) for other atoms. Frequency calculations were performed at the same theoretical level for all optimized geometries to confirm them as minima (zero imaginary frequencies) or transition-state structures (a single imaginary frequency). Intrinsic reaction coordinate (IRC)21 were traced from the various transition structures to obtain the connected intermediates. Single-point energies were calculated at the ωB97XD/6-311+G(d,p) level of theory with the SMD22 solvation model. Dichloromethane was chosen as solvent for rhodium catalytic system, while butanone was used as solvent for silver catalytic system. Non-covalent interaction (NCI) analysis and energy decomposition analysis (EDA) were calculated by the Multiwfn program.23 Three-dimensional diagrams of the computed species were generated using CYLview visualization software.24
Results and discussion
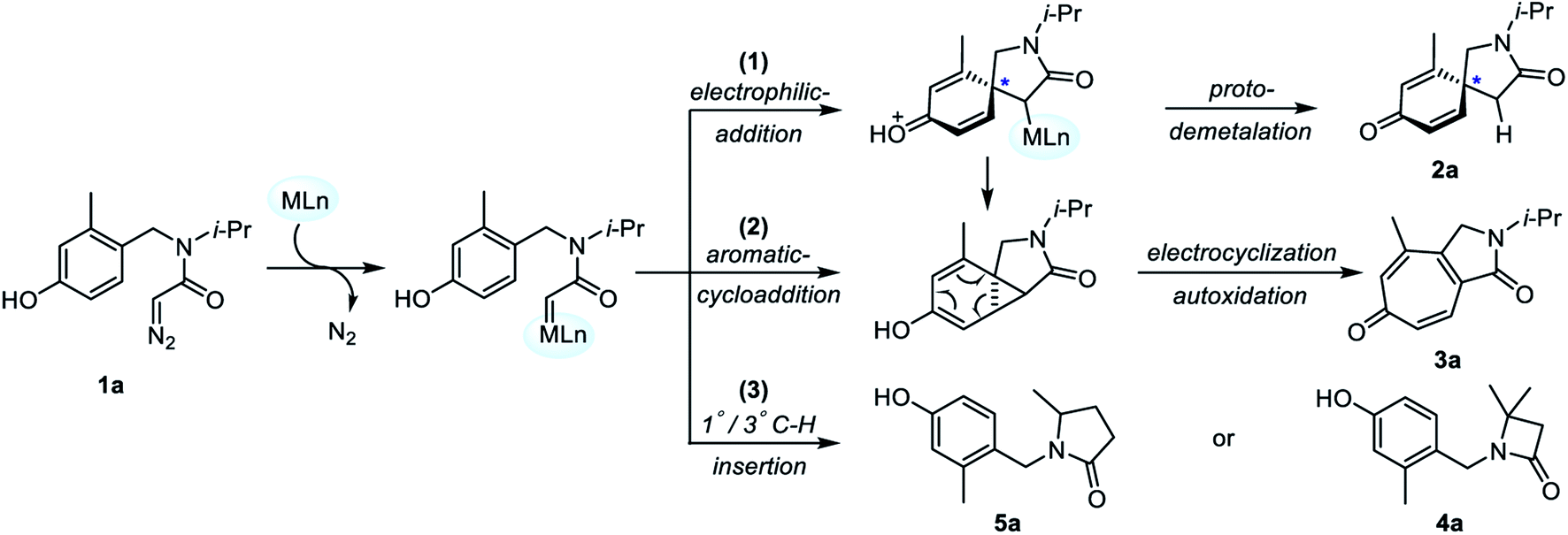
Based on the experimental results, DFT calculations on the asymmetric intramolecular dearomatization of α-diazoamide 1a catalyzed by (S)-TRIPAg, the Büchner reaction and the 1°/3° C–H insertion reaction catalyzed by Rh2(OAc)4 were carried out to further explore the origins of chemical selectivity and enantioselectivity. Firstly, we propose three possible reaction mechanisms catalyzed by metal salts, as shown in Fig. 2.
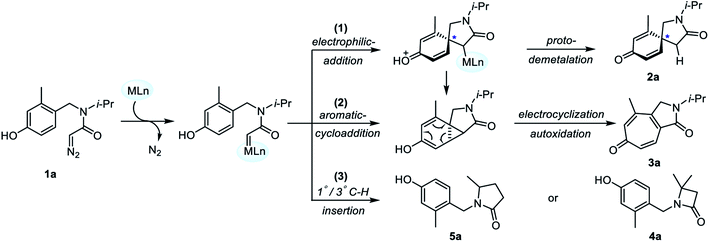 |
| | Fig. 2 Mechanistic pathways of transition metal-catalyzed intramolecular carbene transfer reaction of α-diazoamide 1a. | |
Starting from metal carbene intermediates, there are three reaction pathways: (1) chiral spiro cyclization occurs through intramolecular electrophilic addition, followed by intramolecular protodemetalation to obtain azaspiro[4.5]decane derivative 2a. (2) Through aromatic cycloaddition to form ternary ring intermediates; subsequently, cycloheptatrienone 3a is obtained by ring enlargement and autoxidation. (3) The metal carbene complex leads to γ-lactam 5a or β-lactam 4a by an intramolecular 1°/3° C–H insertion reaction. Considering these three reaction pathways, DFT calculations were performed to reveal the mechanism of Ag and Rh catalyzed intramolecular carbene transfer reactions.
Mechanism of the 1°/3° C–H insertion reaction catalyzed by Rh2(OAc)4
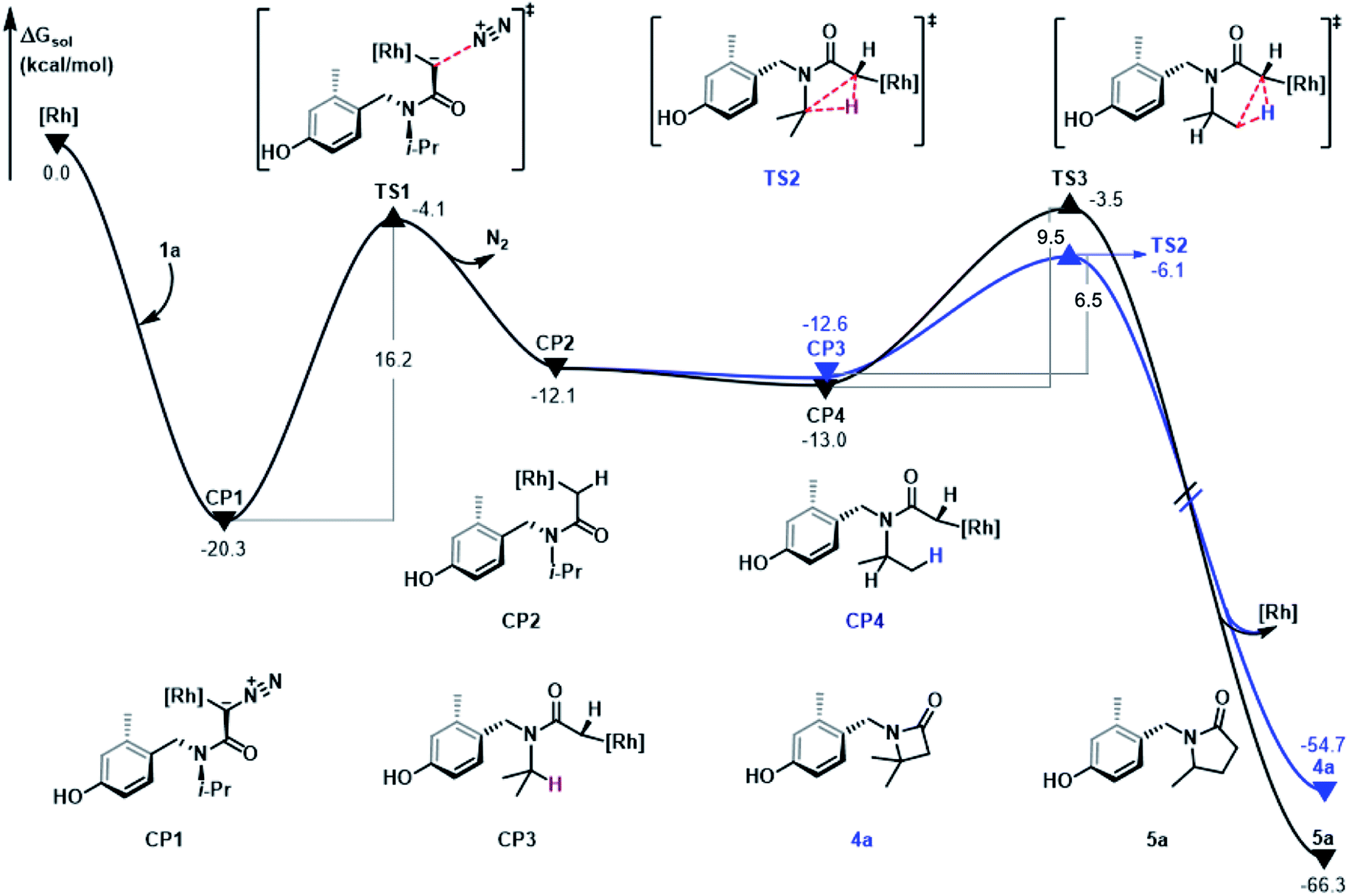
In Fig. 3, the free energy profile of C–H bond insertion is shown based on the mechanism discussed above and the energy profile is discussed using Rh2(OAc)4 as the starting point. The substrate, α-diazoacetamide 1a, contains a formal N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond and a negatively charged α-carbon. The α-carbon site is coordinated with the metal center of the rhodium salt to form the stable complex CP1 and this step is exothermic by an energy difference of 20.3 kcal mol−1. Subsequent denitrogenation of CP1 occurs via transition state TS1 by cleavage of the C–N bond with an energy barrier of 16.2 kcal mol−1, to give rhodium-carbene complex CP2, a process which is endergonic by 8.2 kcal mol−1. CP2 undergoes isomerization following the C–N bond rotation and generates the intermediates CP3 and CP4, steps which are exothermic by 0.5 kcal mol−1 and 0.9 kcal mol−1, respectively. The generation of intermediates CP3 and CP4 facilitate the C–H insertion and then the C–C bond generation. Comparing the two reaction paths, shows that their energy barrier difference is only 3.0 kcal mol−1, which is consistent with the experimental results of obtaining 4a and 5a products with 17% and 15% yields, respectively.
N triple bond and a negatively charged α-carbon. The α-carbon site is coordinated with the metal center of the rhodium salt to form the stable complex CP1 and this step is exothermic by an energy difference of 20.3 kcal mol−1. Subsequent denitrogenation of CP1 occurs via transition state TS1 by cleavage of the C–N bond with an energy barrier of 16.2 kcal mol−1, to give rhodium-carbene complex CP2, a process which is endergonic by 8.2 kcal mol−1. CP2 undergoes isomerization following the C–N bond rotation and generates the intermediates CP3 and CP4, steps which are exothermic by 0.5 kcal mol−1 and 0.9 kcal mol−1, respectively. The generation of intermediates CP3 and CP4 facilitate the C–H insertion and then the C–C bond generation. Comparing the two reaction paths, shows that their energy barrier difference is only 3.0 kcal mol−1, which is consistent with the experimental results of obtaining 4a and 5a products with 17% and 15% yields, respectively.
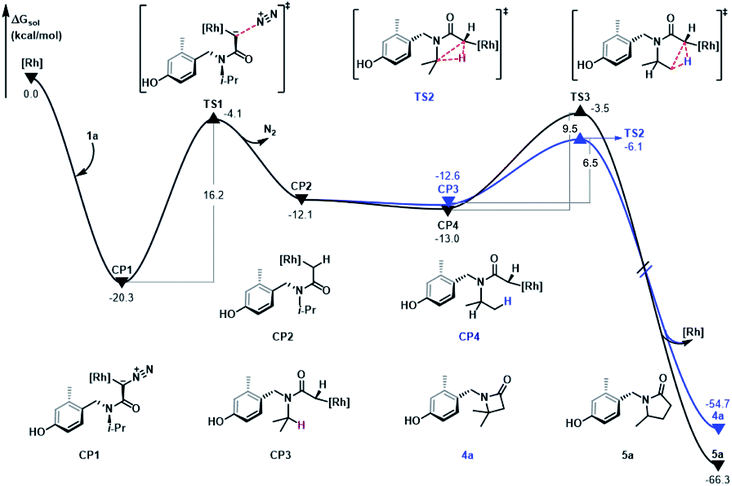 |
| | Fig. 3 Gibbs free energy profile (in kcal mol−1) for the pathways of 1°/3° C–H insertion reaction of α-diazoamide 1a catalyzed by Rh2(OAc)4. | |
The Büchner reaction mechanism
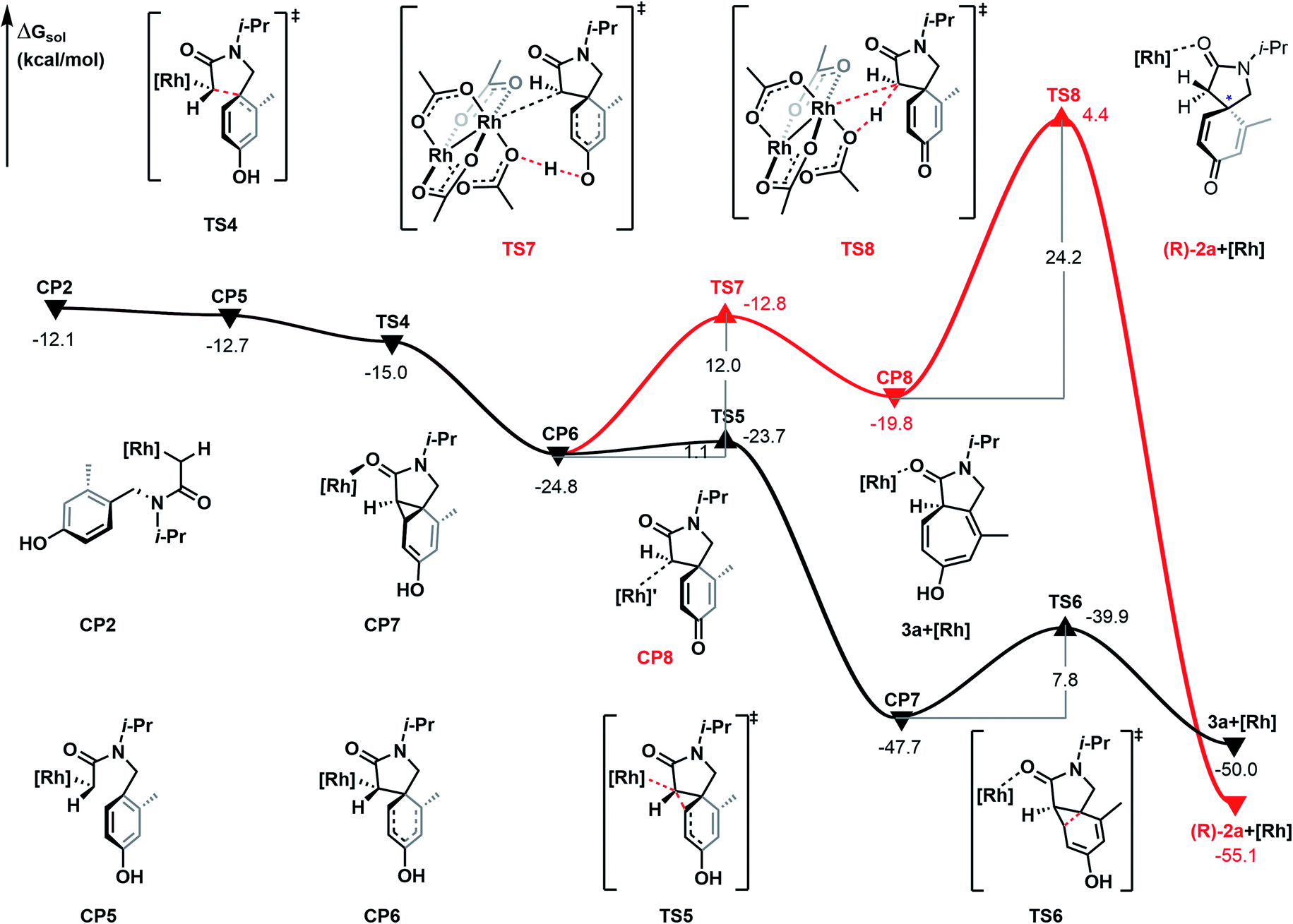
As shown in Fig. 4, the Büchner reaction mechanism has been explored, taking the rhodium-carbene complex CP2 as the starting point of the energy profile. Activation of 4 °C contains two steps, cyclopropanation and ring-expansion reactions. Firstly, CP2 undergoes isomerization following the C–N bond rotation and generates the intermediate CP5, a step which is exothermic by 0.6 kcal mol−1. The intermediate CP5 leads to the electrophilic addition reaction. Intramolecular C–C bond formation between the C-atom of Rh-carbene and the C-atom of the phenyl to generate spirocyclic intermediate CP6, with a free energy release of 12.1 kcal mol−1. Subsequently, the 5 membered ring of CP6 undergoes aromatic-addition via the transition state TS5 to form the cyclopropane CP7, with a free energy release of 22.9 kcal mol−1 and the energy barrier of this step is 1.1 kcal mol−1. In the next step, the Csp2–Csp3 bond of the fused ring of CP6 is cleaved and generates the ring-expansion product 3a, with a relative free energy of −50.0 kcal mol−1. Of note, 3a is the bicyclic [5.3.0] derivative and the main product of the Büchner reaction. It is formed through transition state TS6 with an energy barrier of 7.8 kcal mol−1. Obviously, the energy barrier of each transition state in the Büchner reaction path is lower than in the C–H bond insertion process and on the potential energy surface, the relative free energy of each intermediate and transition state is lower. Therefore, the Büchner reaction process is the most favorable pathway in the intramolecular carbene transfer reaction of α-diazoamide 1a catalyzed by Rh2(OAc)4. This is consistent with the experimental result that cycloheptatriene derivative 3a was obtained with a yield of 26%.
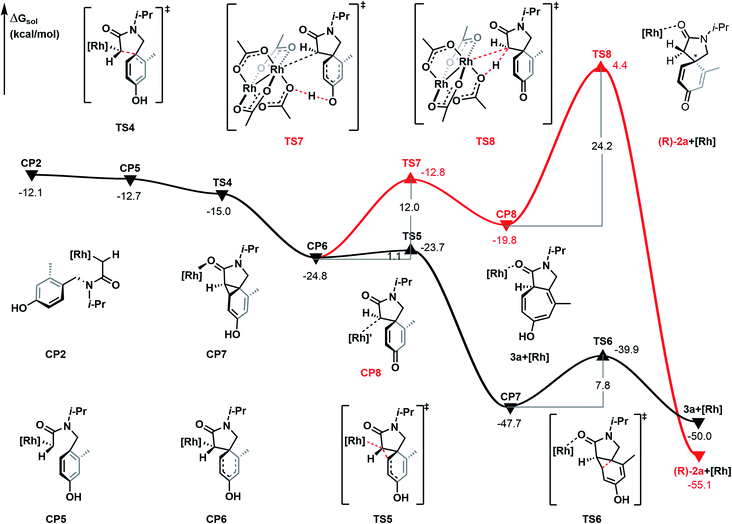 |
| | Fig. 4 Gibbs free energy profile (in kcal mol−1) for the Büchner reaction mechanism catalyzed by Rh2(OAc)4. | |
In addition, we also explored the process of carboxylic acid-assisted proto-demetallation to form azaspiro[4.5]decane derivative 2a, which is shown by the red line in Fig. 4. Firstly, the proton transfer of the phenolic hydroxyl group occurs via transition state TS7 with an energy barrier of 12.0 kcal mol−1 to give complex CP8 and is endergonic by 5.0 kcal mol−1. Subsequently, proto-demetallation occurs via transition state TS8, which transfers the proton from the oxygen atom of the ligand to the α-carbon atom of the α-diazoamide unit. The energy barrier of this step is as high as 24.2 kcal mol−1. Therefore, the dearomatization process catalyzed by Rh2(OAc)4 is kinetically unfavorable.
Mechanism of asymmetric dearomatization catalyzed by chiral silver phosphate
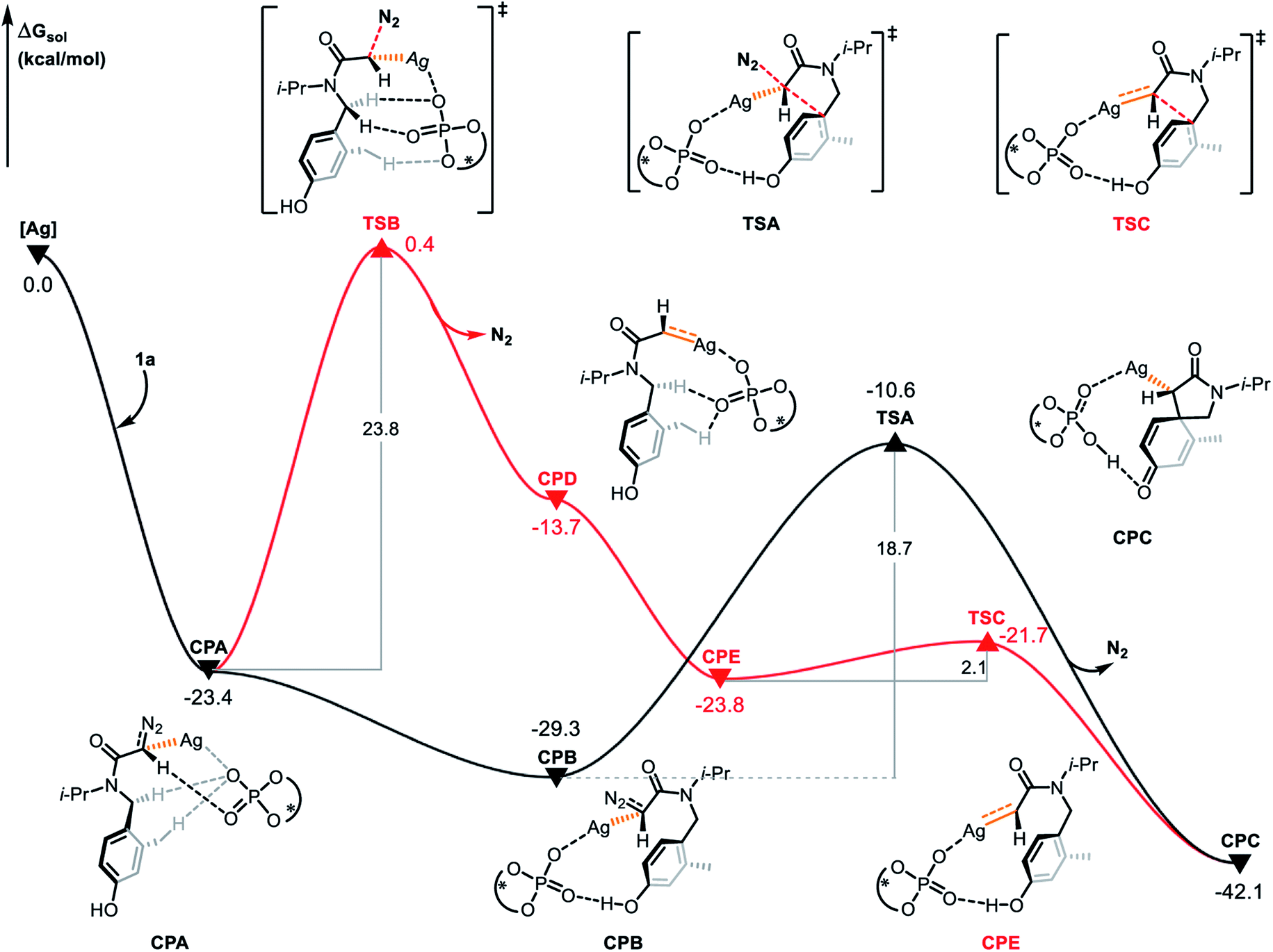
In order to explore the origin of the enantioselectivity, we conducted DFT calculations at the level of ωB97XD/6-311+G(d,p)-SDD(Ag). Based on the mechanism of silver-catalyzed chemo- and enantioselective intramolecular dearomatization of indoles calculated by Nemoto's group,9 we first explored the reaction modes featuring silver-carbene generation and concerted silver-carbene reactivity with diazotization taking place simultaneously with dearomatization. The active catalytic species (S)-TRIPAg was chosen as the starting point for the free energy profiles (Fig. 5). Coordination of diazoamide 1a generates complex CPA and denitrogenation occurs via transition state TSB by cleavage of the C–N bond with an energy barrier of 23.8 kcal mol−1 to give silver-carbene complex CPD. Subsequently, the silver-carbene CPD forms the cyclophane-like intermediate CPE after the rotation of the C–N bond. In the cyclophane-like intermediate CPE, the distance between the hydrogen atom of the hydroxyl group and the oxygen atom of the phosphate is 1.62 Å, indicating the potential of subsequent electrophilic addition. The hydrogen bond interaction aids in achieving the remote stereocontrol. In the next step, the carbene carbon attacks the carbon at the ipso position via transition state TSC with a barrier of 2.1 kcal mol−1. This produces the spirocyclic intermediate CPC and its relative free energy is −42.1 kcal mol−1.
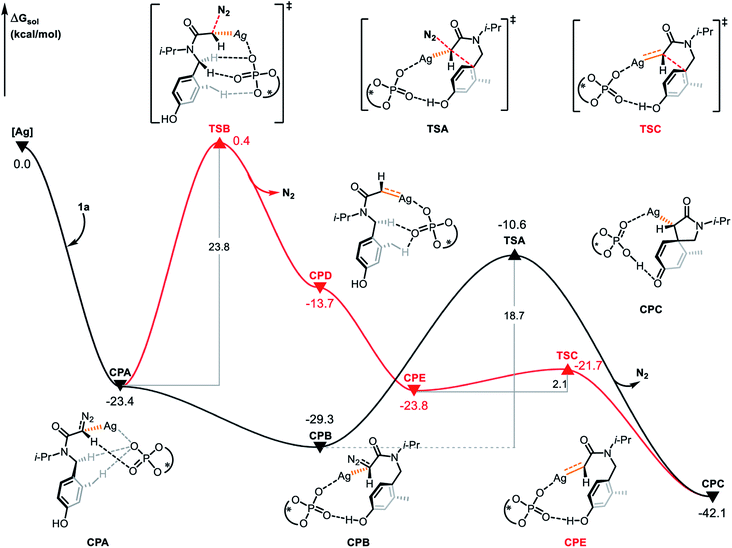 |
| | Fig. 5 Gibbs free energy profile (in kcal mol−1) for the electrophilic addition of R configuration catalyzed by chiral silver phosphate. | |
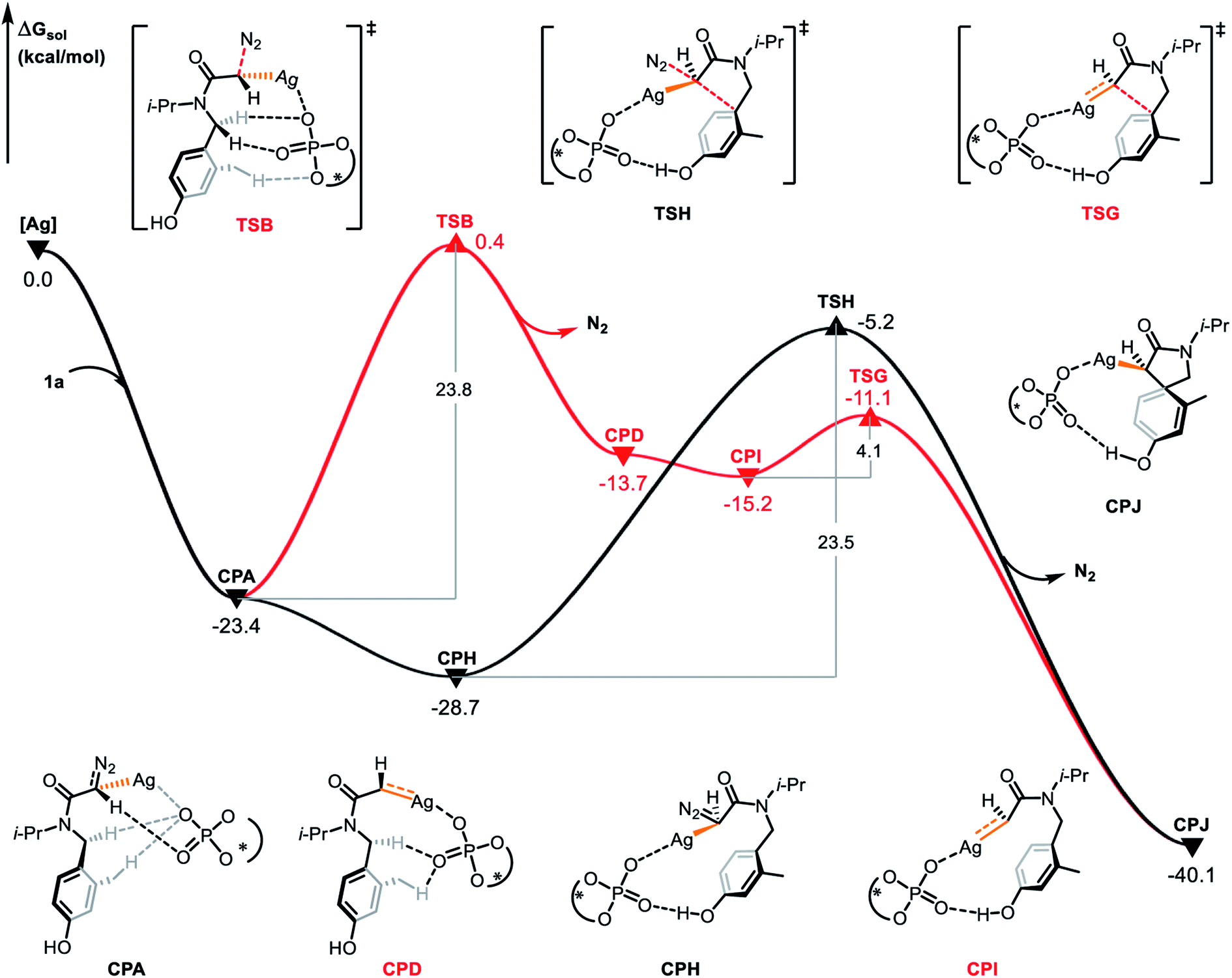
The concerted carbene reaction mode is shown by the black line in Fig. 5. The spirocyclic intermediate CPC is directly formed through the transition state TSA, and the asynchronous C–C bond formation and C–N bond cleavage process occurs with an energy barrier of 18.7 kcal mol−1, which is the rate-determining step. When comparing the two reaction paths, it is found that the energy barrier of the concerted silver-carbene pathway is 5.1 kcal mol−1 lower than that of the stepwise silver-carbene generation mode (TSA vs. TSB). Therefore, the concerted silver-carbene pathway is kinetically more favorable. At the same time, it shows that the concentration of silver-carbene in the reaction is low. The preference for the concerted silver-carbene pathway might be ascribed to the enhanced nucleophilicity of the aryl unit of the α-diazoamide, which has been intramolecularly activated by the phosphate anion, promoting smooth dearomatization before generation of the silver-carbene. For comparision, we have also calculated the pathway to form the (S)-spirocyclic intermediate CPJ through the concerted addition transition state TSH (Fig. 6), the results show that the electrophilic addition of S configuration is both kinetically and thermodynamically unfavorable than that of R configuration.
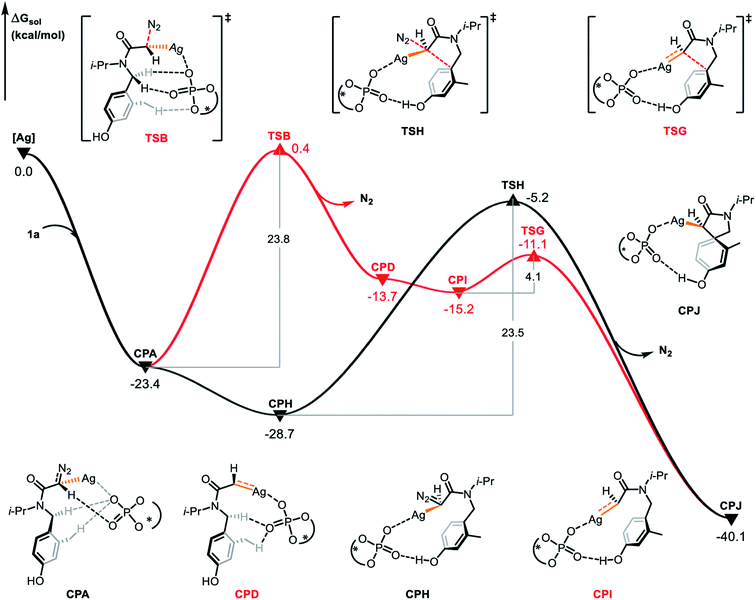 |
| | Fig. 6 Gibbs free energy profile (in kcal mol−1) for the electrophilic addition of S configuration catalyzed by chiral silver phosphate. | |
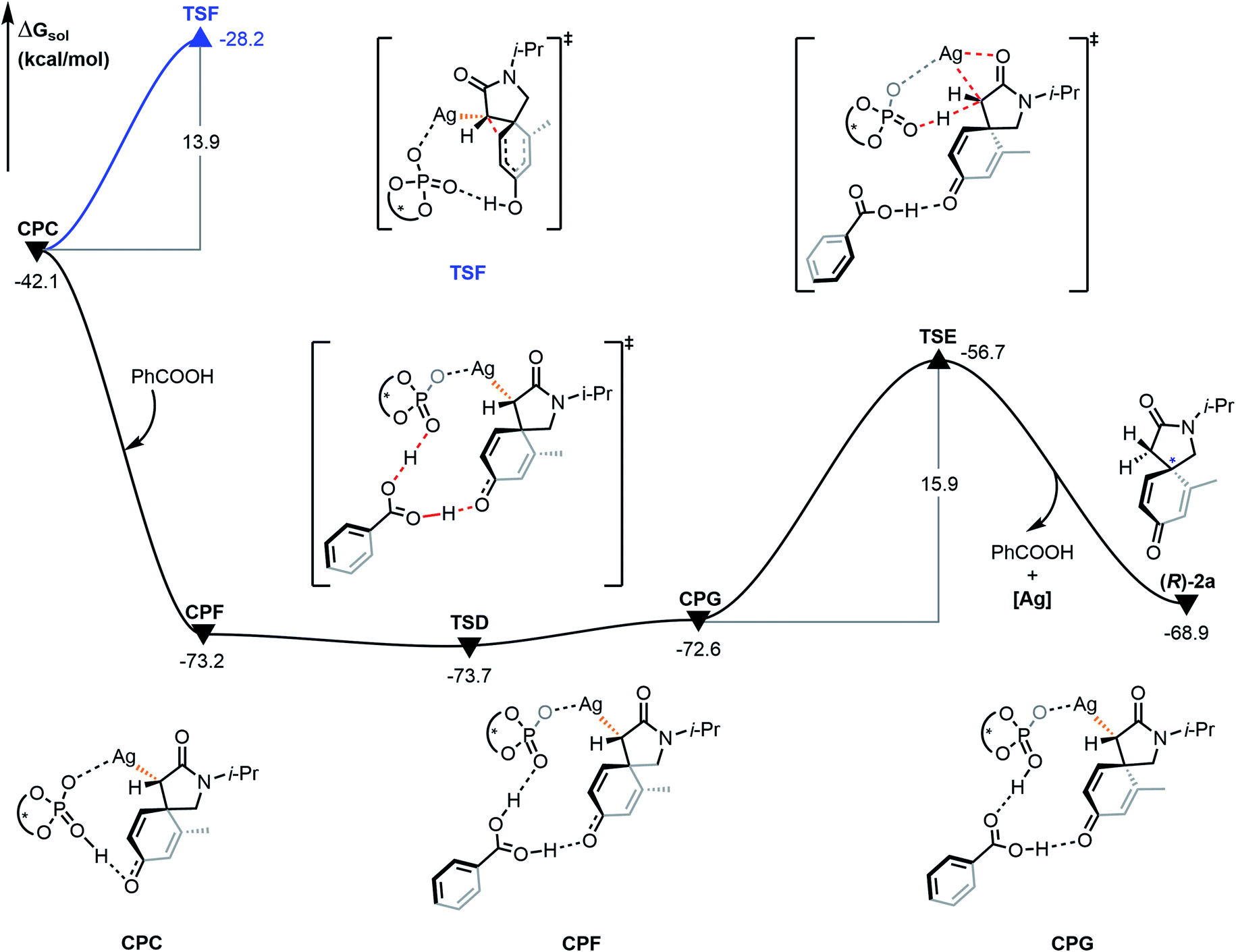
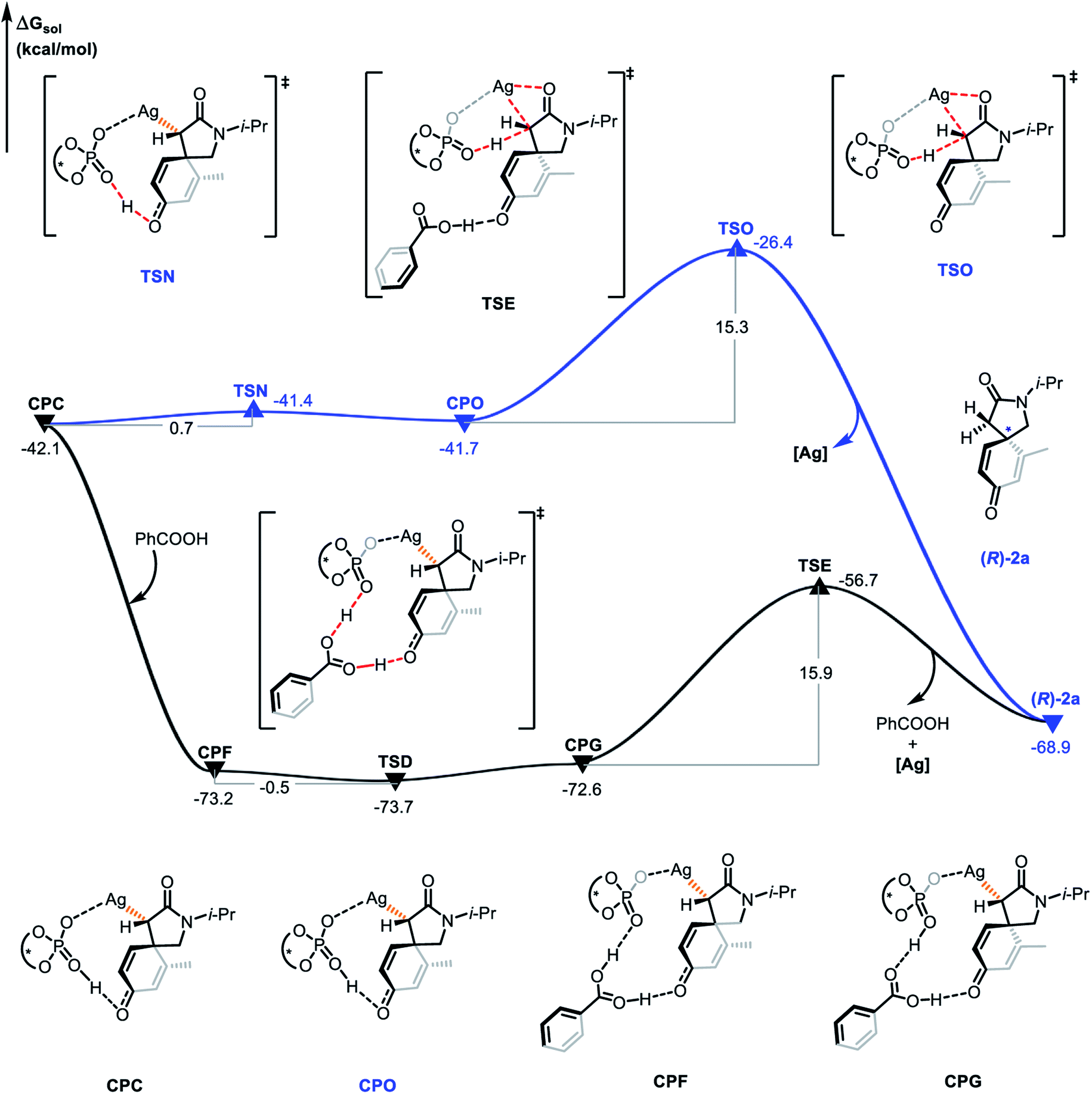
The proto-demetalation process assisted by benzoic acid was also calculated (Fig. 7). Association of benzoic acid generates complex CPF and is exothermic by 31.1 kcal mol−1. The presence of benzoic acid directly promotes the transfer of the hydroxyl proton. Notably, the free energy of complex CPG is 0.6 kcal mol−1 higher than that of complex CPF, which indicates that the proton migration process is an endergonic and reversible process. Due to the similar energy and no energy barrier between the two complexes, it is feasible for them to be in a rapid equilibrium. In the next step, the complex CPG undergoes proto-demetalation through the transition state TSE (ΔΔG≠ = 15.9 kcal mol−1). The hydroxyl proton is transferred from the oxygen atom of the phosphate anion to the α-carbon atom of the α-diazoamide unit. At the same time, the silver atom dissociates from the α-carbon atom and coordinates with the carbonyl oxygen atom. Finally, the (R)-2a product is formed and the catalytic species (S)-TRIPAg is regenerated. For a comprehensive theoretical study, we have also calculated this process without the assistance of benzoic acid (Fig. 8). It can be seen that although the activation barriers are very similar (0.7 kcal mol−1, 15.3 kcal mol−1 vs. −0.5 kcal mol−1, 15.9 kcal mol−1; it is the process with the assistance of acid that is a bit more favorable). The relative free energy for each intermediate and transition state in the presence of benzoic acid is lower than that of the process without the assistance of benzoic acid on the potential energy surface. The role of benzoic acid here is to assist the proto-demetallation process to increase the yield of the desired product without decreasing the enantioselectivity. In 2018, Nemoto's group investigated the role of benzoic acid and pointed out that benzoic acid would promote dissociation of the catalytically inactive homochiral dimer to liberate it for the generation of a monomeric species.25
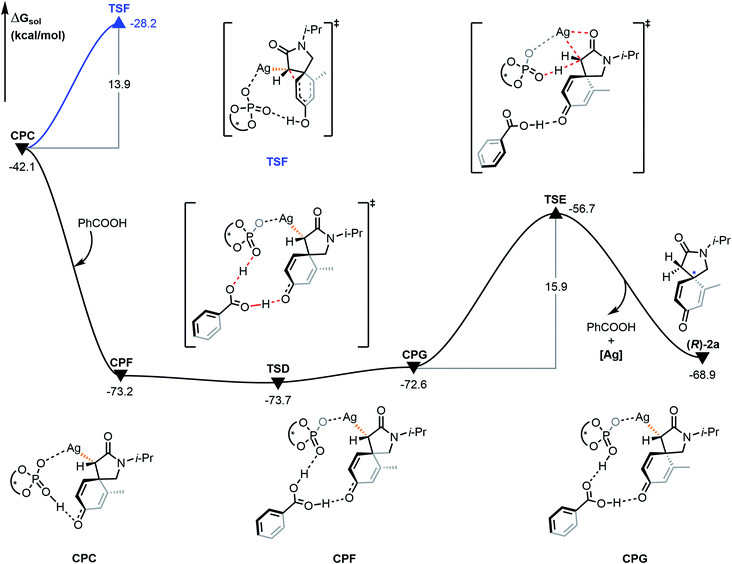 |
| | Fig. 7 Gibbs free energy profile (in kcal mol−1) for the proto-demetalation process and Büchner reaction mechanism of R configuration catalyzed by chiral silver phosphate. | |
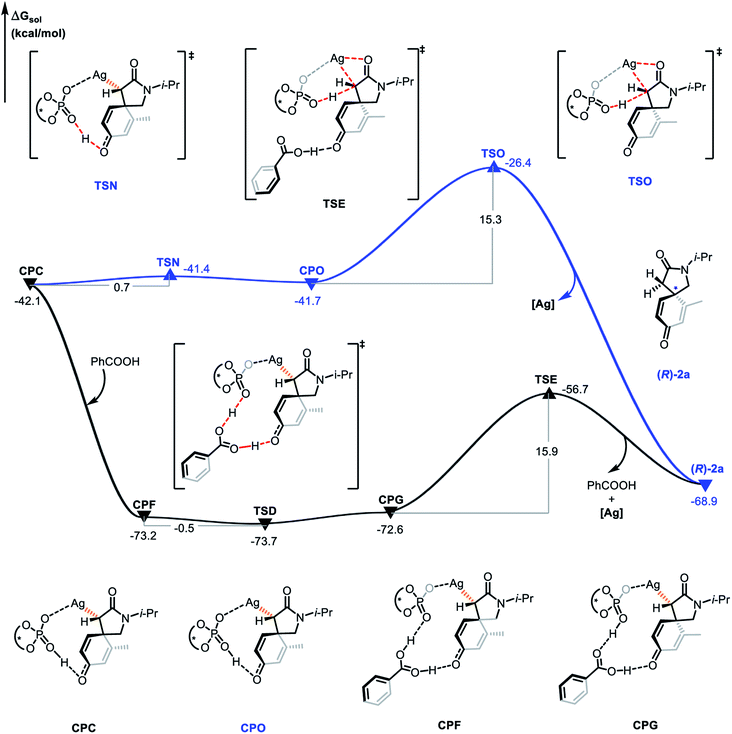 |
| | Fig. 8 Gibbs free energy profile (in kcal mol−1) for the proto-demetalation process with or without the assistance of benzoic acid. | |
The possible Büchner reaction mechanism was also calculated. The energy barrier of the ring expansion of CPC via transition state TSF is 13.9 kcal mol−1, and the free energy of TSF is 45.5 kcal mol−1 higher than that of hydrogen transfer transition state TSD. Therefore, generation of benzoic acid-associated compounds is kinetically and thermodynamically favorable because of the low relative free energy and no energy barrier, which is consistent with the experimental result that the 3a product is not formed. Besides, we also compared the process without benzoic acid with Büchner reaction (ESI, Fig. S6†). The results show that the proto-demetalation is favorable as well, which is also nicely consistent with the experimental outcome that only a small amount of 3a product is found.
In addition to the DFT calculations of above reactions, we have also performed theoretical study for the 1°/3° C–H insertion reaction catalyzed by chiral silver phosphate (ESI, Fig. S1†), the rate-determining step for this process is the carbene formation with an energy barrier of 23.8 kcal mol−1, which is both kinetically and thermodynamically unfavorable compared with the proto-demetalation process, this is also nicely consistent with the experimental results that no C–H insertion products was observed.
Comparison of reactivity between rhodium carbene and silver carbene
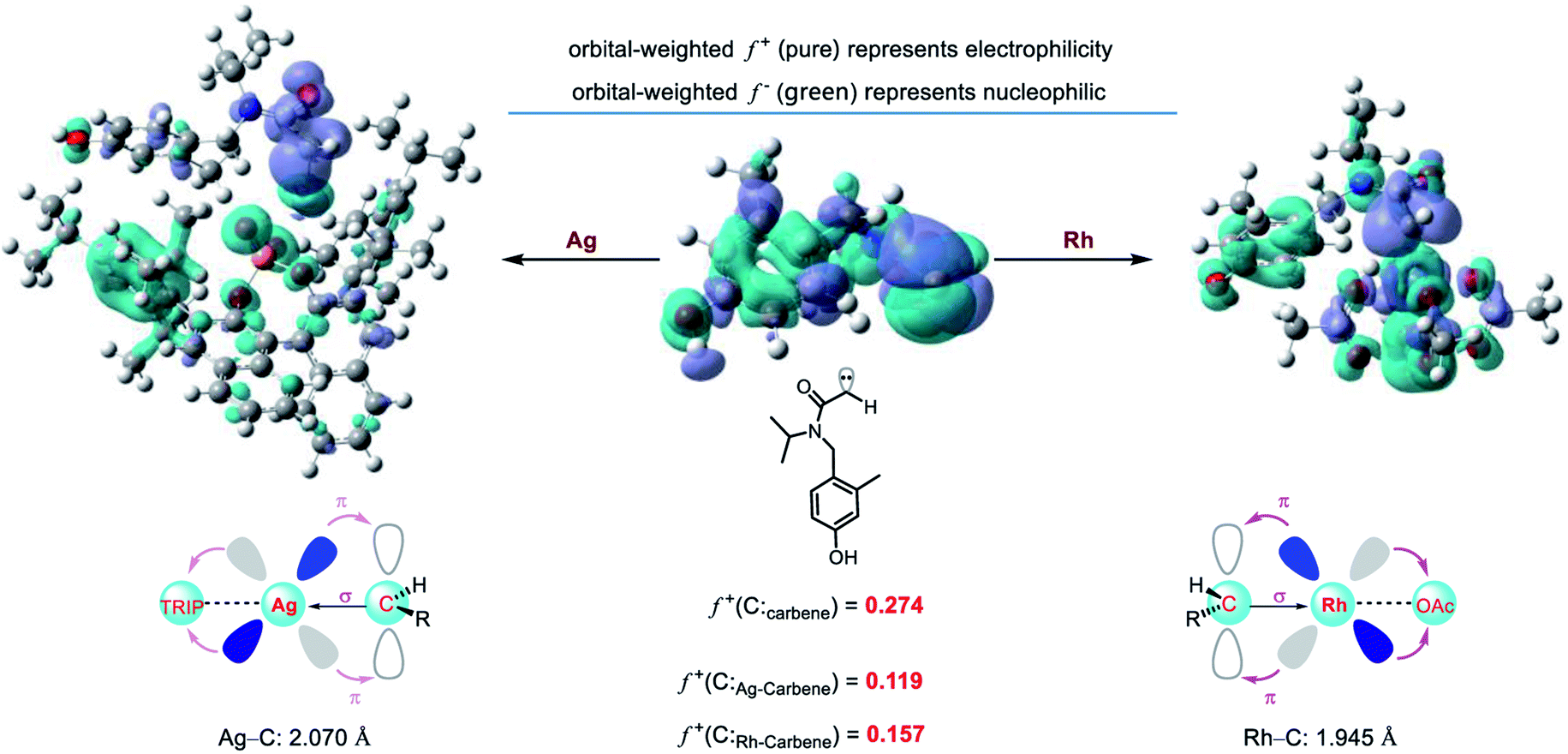
To explore the origin of metal-dependent chemo-selectivity, we performed the orbital weight Fukui function to analyze the different catalytic activities between Rh-carbene and Ag-carbene (Fig. 9). The greater the LUMO orbital weight Fukui function (f+), the stronger its electrophilicity. Thus, the rhodium carbene is more electrophilic than chiral silver phosphate carbene. The chiral phosphate ligand has weak σ-bonding with silver, and the weak π-backdonation effect of the silver 4d10 orbital, resulting in the weak electrophilicity of the chiral silver phosphate carbene. While the 4d orbitals of the rhodium atom are not completely filled with electrons, and the π-backdonation to the acetic acid ligand is strong, then rhodium accepts more electrons from the carbene carbon, resulting in higher electrophilicity. In addition, we also performed charge decomposition analysis (CDA) and orbital interaction diagram (ESI, Fig. S9†) to study their electrophilicity. It is found that the LUMO energy of Rh-carbene is 0.53 eV lower than that of Ag-carbene, which is easier to accept electrons and shows strong electrophilicity and is consistent with the calculation results of the orbital Fukui function. Therefore, the carboxylic acid anion renders the rhodium carbene more electrophilic and promotes the subsequent intramolecular C–H insertion and Büchner reaction, and the theoretical results are consistent with the experimental phenomena.
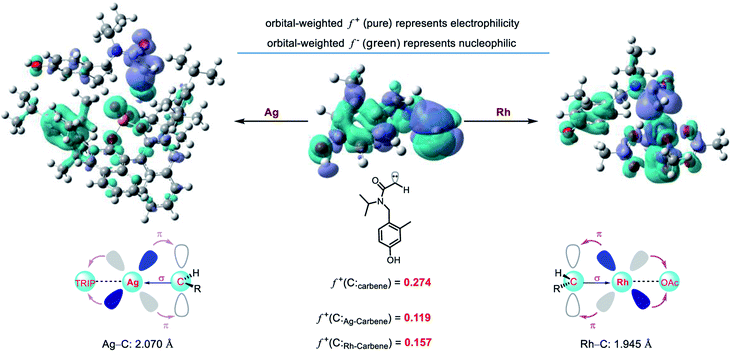 |
| | Fig. 9 The orbital weight Fukui function diagram of Ag-carbene and Rh-carbene. | |
However, it is worth noting that ligands can modify the electrophilicity of a metal carbene. Recently, silver-catalyzed site-selective C(sp3)–H alkylation of alkanes has been developed by our group using donor carbenes.26 The donor rhodium carbene could not achieve the same results as the silver congener. To explore the difference of electrophilicity between the two-donor metal carbenes, orbital weight Fukui function was also performed. The results show that the donor silver carbene is more electrophilic than the donor rhodium carbene. (f+(Ag-carbene) = 0.260 vs. f+(Rh-carbene) = 0.135, Fig. S10†).
The origin of the enantioselectivity for silver catalyzed dearomatization
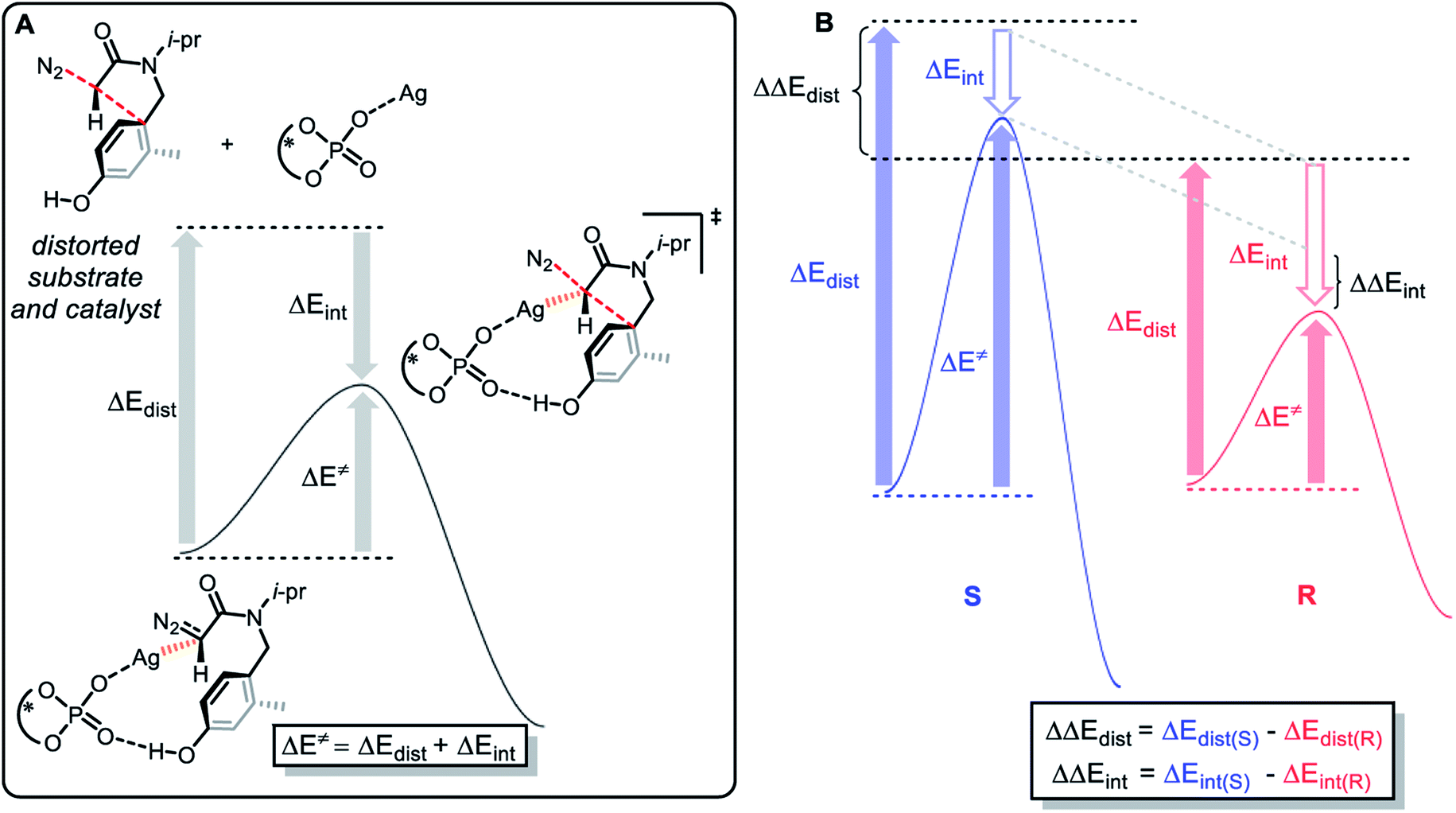
To explore the origin of enantioselectivity, we conducted a distortion–interaction analysis27 on the concerted addition transition state TSA and TSH to gather further evidence for the role of ligand steric effect on selectivity. As shown in Fig. 10, this type of analysis involves the decomposition of activation energy (ΔE≠) into a distortion (ΔEdist) and an interaction term (ΔEint). The distortion term is usually positive (unfavorable), which represents the energy penalty for distorting the reactants into the transition state geometry. The interaction term is usually negative, which represents the favorable interaction between the substrate and the catalyst. The relationship between distortion and interaction is defined as ΔE≠ = ΔEdist + ΔEint.
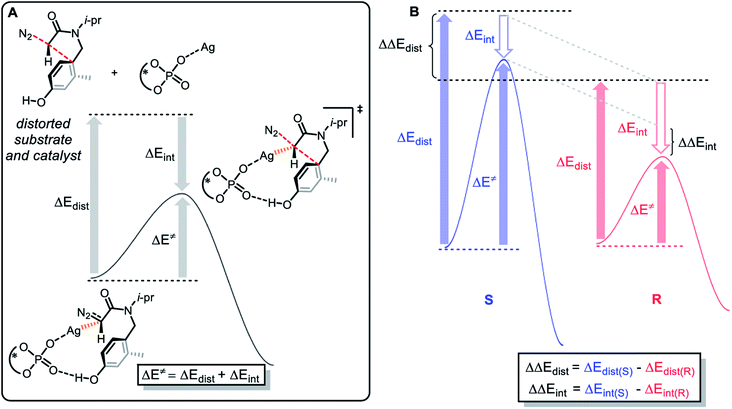 |
| | Fig. 10 (A) Definitions of torsional energy and interaction energy; (B) the determining factors of enantioselectivity. | |
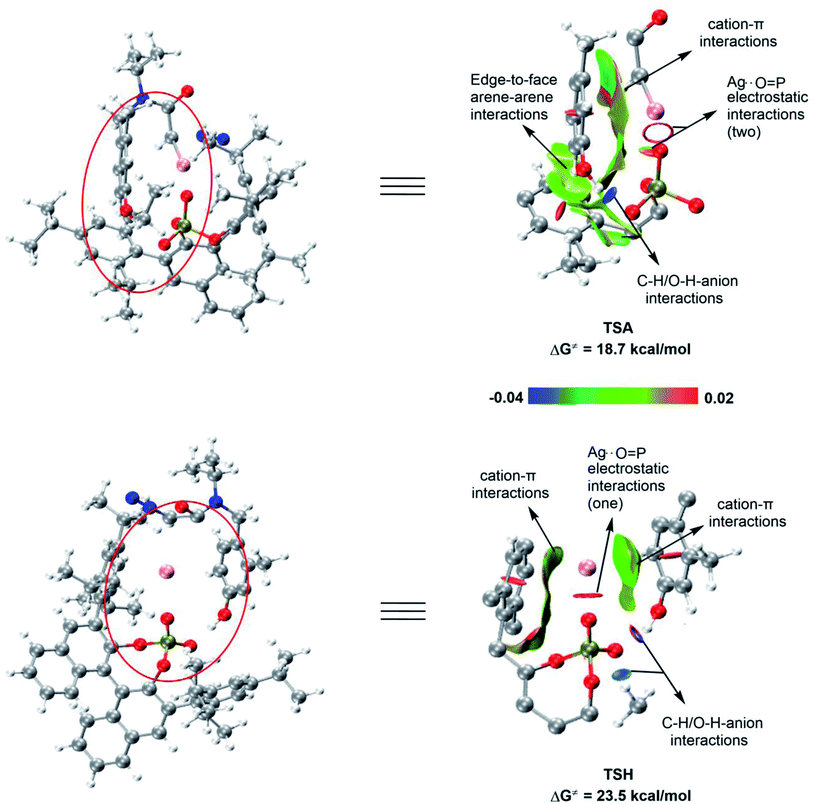
The enantioselectivity is determined by the difference between the distortion and interaction energy of the intramolecular electrophilic addition reaction between the two configurations of R and S (Fig. 10), which is defined as: ΔΔEdist = ΔEdist(S) − ΔEdist(R); ΔΔEint = ΔEint(S) − ΔEint(R). According to the results in Table 1, the larger the value of ΔΔEdist, the stronger the effect of distortion energy, which means that the S configuration reaction is more unfavorable. In addition, interaction energy of R configuration is more negative, which shows that the R configuration reaction is more favorable. This can be rationalized by (1) the stronger coulombic attraction between Ag and the carbene carbon in the R configuration transition state TSA, which can be proven by a relatively short distance (TSH(S), d(Ag⋯C) = 2.10 Å vs. TSA(R), d(Ag⋯C) = 2.05 Å); (2) the ability for another oxygen atom of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O to interact with Ag; (3) there is also a favorable edge-to-face arene–arene interaction between the arene of the substrate and the ligand (Fig. 11).
O to interact with Ag; (3) there is also a favorable edge-to-face arene–arene interaction between the arene of the substrate and the ligand (Fig. 11).
Table 1 Analysis of torsional energy and interaction energy of electrophilic addition transition state structure
| TS |
ΔEdist |
ΔΔEdist |
ΔEint |
ΔΔEint |
ΔE≠ |
ΔΔE≠ |
| Cat. |
Subst. |
Total |
| R |
4.8 |
42.7 |
47.5 |
6.8 |
−59.8 |
4.2 |
−12.3 |
11.0 |
| S |
9.8 |
44.5 |
54.3 |
−55.6 |
−1.3 |
 |
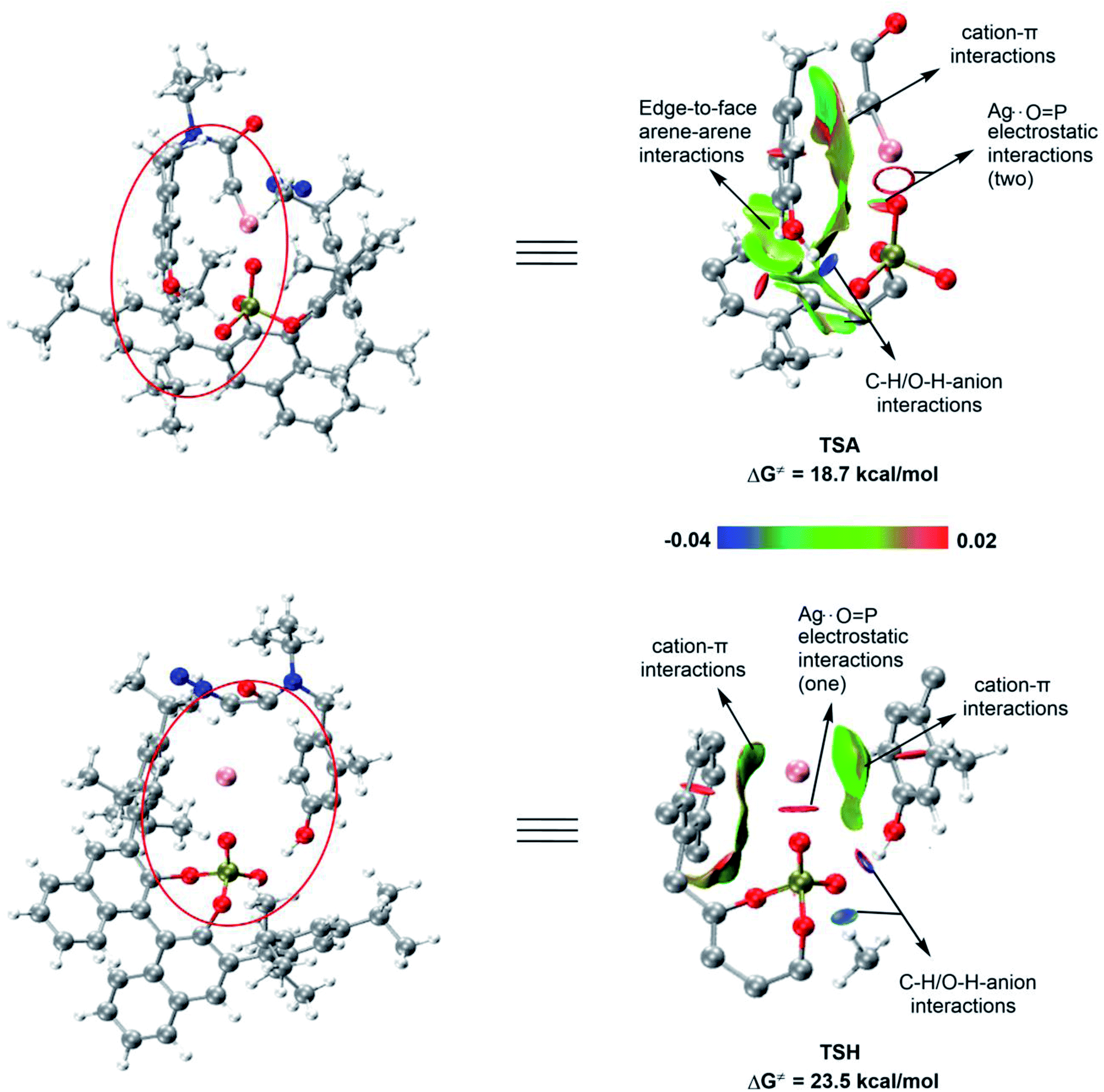
| | Fig. 11 Non-covalent interaction (NCI) of TSA and TSH (in order to enhance the readability, we only keep isosurfaces for the key parts involved the interactions of interest and for the convenience to observe the specific positions of these parts, we also put the whole structures of TSA and TSH). Blue represents strong attraction; green represents van der Waals interaction; red represents strong repulsion or steric effect. | |
To further explore the difference between the key transition states (TSA and TSH) of the two configurations, we conducted a non-covalent interaction (NCI) analysis (Fig. 11). The results showed that the cation−π interaction between the silver atom and the arene of the substrate, the edge-to-face arene–arene interaction between the aromatic rings of the substrate and the ligand, the C–H/O–H anion interaction, and the Coulombic attraction between silver and the two oxygen atoms of the phosphate effectively stabilizes TSA, thereby promoting the smooth progress of the R configuration concerted-addition. However, in the transition state TSH, no edge-to-face arene–arene interaction between the aromatic rings of the substrate and the ligand was found, and the Coulombic attraction only exists between silver and one oxygen atom of the phosphate. In addition, due to the existence of cation−π interactions between the aromatic rings of substrate and ligand with Ag in TSH, the geometry of AgL is more constrained. Therefore, the calculation results show that the reaction of the S configuration is relatively less favorable.
Conclusion
Due to the unique coordination mode and catalytic activity of silver, the asymmetric carbene transfer reactions catalyzed by silver have developed slowly for a long time. Based on the first silver-catalyzed high enantioselective carbene transfer reaction published by Nemoto in 2017, we examined the mechanism of silver versus rhodium catalysis and revealed the origin of the chemo- and enantioselectivity of the reaction. The calculation results reveal that when silver is used as a catalyst, due to the intramolecular activation of the phosphate anion, the enhanced nucleophilicity of α-diazoacetamide unit results in dearomatization before generation of the silver carbene. When rhodium is used as a catalyst, since the rhodium carbene has stronger electrophilicity, typical carbene reactions are prone to occur. In addition, through energy decomposition analysis based on molecular force field of the key concerted-addition transition state, it is found that the preference for the (R)-2a product is due to a much smaller distortion disadvantage and a larger interaction energy advantage. Therefore, the dearomatization reaction of the R configuration is strongly favored.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support was provided by the Fundamental Research Funds for the Central Universities (2412020FZ006), and the Education Department of Jilin Prov. China (JJKH20211283KJ). For work performed in Ghent, the iBOF (C3) and the FWO (fellowship to NVT) are gratefully acknowledged for support.
References
-
(a) A. DeAngelis, R. Panish and J. M. Fox, Rh-catalyzed intermolecular reactions of α-alkyl-α-diazo carbonyl compounds with selectivity over β-hydride migration, Acc. Chem. Res., 2016, 49, 115–127 CrossRef CAS PubMed;
(b) Y. Deng, C. Jing, H. Arman and M. P. Doyle, Reactivity and selectivity in catalytic reactions of enoldiazoacetamides. Assessment of metal carbenes as intermediates, Organometallics, 2016, 35, 3413–3420 CrossRef CAS;
(c) S. Harada, M. Kono, T. Nozaki, Y. Menjo, T. Nemoto and Y. Hamada, General approach to nitrogen-bridged bicyclic frameworks by Rh-catalyzed formal carbene insertion into an amide C–N bond, J. Org. Chem., 2015, 80, 10317–10333 CrossRef CAS PubMed.
-
(a) M. P. Munoz, Silver-catalysed reactions of alkynes: recent advances, Chem. Soc. Rev., 2014, 43, 3164–3183 RSC;
(b) N. T. Patil and Y. Yamamoto, Coinage metal-assisted synthesis of heterocycles, Chem. Rev., 2008, 108, 3395–3442 CrossRef CAS PubMed.
- Y. Cui and C. He, Efficient aziridination of olefins catalyzed by a unique disilver(I) compound, J. Am. Chem. Soc., 2003, 125, 16202–16203 CrossRef CAS PubMed.
- J. M. Alderson, J. R. Corbin and J. M. Schomaker, Tunable, chemo- and site-selective nitrene transfer reactions through the rational design of silver(I) catalysts, Acc. Chem. Res., 2017, 50, 2147–2158 CrossRef CAS PubMed.
- G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, A powerful chiral counterion strategy for asymmetric transition metal catalysis, Science, 2007, 317, 496–499 CrossRef CAS PubMed.
-
(a) R. R. Nani and S. E. Reisman, α-Diazo-β-ketonitriles: uniquely reactive substrates for arene and alkene cyclopropanation, J. Am. Chem. Soc., 2013, 135, 7304–7311 CrossRef CAS PubMed;
(b) V. N. G. Lindsay, D. Fiset, P. J. Gritsch, S. Azzi and A. B. Charette, Stereoselective Rh2(S-IBAZ)4-Catalyzed Cyclopropanation of Alkenes, Alkynes, and Allenes: Asymmetric Synthesis of Diacceptor Cyclopropylphosphonates and Alkylidenecyclopropanes, J. Am. Chem. Soc., 2013, 135, 1463–1470 CrossRef CAS PubMed;
(c) V. K.-Y. Lo, Z. Guo, M. K.-W. Choi, W.-Y. Yu, J.-S. Huang and C.-M. Che, Highly Selective Intramolecular Carbene Insertion into Primary C–H Bond of α-Diazoacetamides Mediated by a (p-Cymene)ruthenium(II) Carboxylate Complex, J. Am. Chem. Soc., 2012, 134, 7588–7591 CrossRef CAS PubMed;
(d) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, Highly Site-Selective Direct C–H Bond Functionalization of Phenols with α-Aryl-α-diazoacetates and Diazooxindoles via Gold Catalysis, J. Am. Chem. Soc., 2014, 136, 6904–6907 CrossRef CAS PubMed.
-
(a) Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Chemoselective carbophilic addition of α-diazoesters through ligand-controlled gold catalysis, Angew. Chem., Int. Ed., 2014, 53, 9817–9821 CrossRef CAS PubMed;
(b) Y. Liu, Z. Yu, J. Z. Zhang, L. Liu, F. Xia and J. Zhang, Origins of unique gold-catalysed chemo- and site-selective C-H functionalization of phenols with diazo compounds, Chem. Sci., 2016, 7, 1988–1995 RSC.
-
(a) M. Alvarez-Corral, M. Munoz-Dorado and I. Rodriguez-Garcia, Enantioselective silver-catalyzed transformations, Chem. Rev., 2008, 108, 3174–3198 CrossRef CAS PubMed;
(b) Q. Z. Zheng and N. Jiao, Ag-catalyzed C–H/C–C bond functionalization, Chem. Soc. Rev., 2016, 45, 4590–4627 RSC;
(c) J. M. Weibel, A. Blanc and P. Pale, Ag-mediated reactions: coupling and heterocyclization reactions, Chem. Rev., 2008, 108, 3149–3173 CrossRef CAS PubMed;
(d) K. Sekine and T. Yamada, Silver-catalyzed carboxylation, Chem. Soc. Rev., 2016, 45, 4524–4532 RSC;
(e) R. Karmakar and D. Lee, Reactions of arynes promoted by silver ions, Chem. Soc. Rev., 2016, 45, 4459–4470 RSC;
(f) U. Halbes-Letinois, J. M. Weibel and P. Pale, The organic chemistry of silver acetylides, Chem. Soc. Rev., 2007, 36, 759–769 RSC;
(g) G. Fang and X. Bi, Silver-catalysed reactions of alkynes: recent advances, Chem. Soc. Rev., 2015, 44, 8124–8173 RSC.
- H. Nakayama, S. Harada, M. Kono and T. Nemoto, Chemoselective Asymmetric Intramolecular Dearomatization of Phenols with α-Diazoacetamides Catalyzed by Silver Phosphate, J. Am. Chem. Soc., 2017, 139, 10188–10191 CrossRef CAS PubMed.
- S. Harada, K. Tanikawa, H. Homma, C. Sakai, T. Ito and T. Nemoto, Silver-catalyzed asymmetric insertion into phenolic O-H bonds using aryl diazoacetates and theoretical mechanistic studies, Chem.–Eur. J., 2019, 25, 12058–12062 CrossRef CAS PubMed.
- T. Shi, S. Teng, A. G. K. Reddy, X. Guo, Y. Zhang, K. H. Moore, T. Buckley, D. J. Mason, W. Wang, E. Chapman and W. Hu, Catalytic asymmetric synthesis of 2,5-dihydrofurans using synergistic bifunctional Ag catalysis, Org. Biomol. Chem., 2019, 17, 8737–8744 Search PubMed.
- G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, A powerful chiral counterion strategy for asymmetric transition metal catalysis, Science, 2007, 317, 496–499 CrossRef CAS PubMed.
- S. Bachmann, D. Fielenbach and K. A. Jørgensen, Cu(I)- carbenoid- and Ag(I)-Lewis acid-catalyzed asymmetric intermolecular insertion of α-diazo compounds into N–H bonds, Org. Biomol. Chem., 2004, 2, 3044–3049 RSC.
- K. Burgess, H.-J. Lim, A. M. Porte and G. A. Sulikowski, New catalysts and conditions for a C–H insertion reaction identified by high throughput catalyst screening, Angew. Chem., Int. Ed., 1996, 35, 220–222 CrossRef CAS.
- J. Ueda, S. Harada, A. Kanda, H. Nakayama and T. Nemoto, Silver-catalyzed, chemo- and enantioselective intramolecular dearomatization of indoles to access sterically congested azaspiro grameworks, J. Org. Chem., 2020, 85, 10934–10950 CrossRef CAS PubMed.
- T. Ito, S. Harada and H. Homma, et al., Asymmetric intramolecular dearomatization of nonactivated arenes with ynamides for rapid assembly of fused ring system under silver catalysis, J. Am. Chem. Soc., 2021, 143, 604–611 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
-
(a) A. D. A. Becke, New Mixing of Hartree-Fock and Local Density-functional Theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS;
(b) A. D. Becke, Density-functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
-
(a) L. von Szentpály, P. Fuentealba, H. Preuss and H. Stoll, Pseudopotential calculations on Rb+2, Cs+2, RbH+, CsH+ and the mixed alkali dimer ions, Chem. Phys. Lett., 1982, 93, 555–559 CrossRef;
(b) P. Schwerdtfeger, M. Dolg, W. H. E. Schwarz, G. A. Bowmaker and P. D. W. Boyd, Relativistic effects in gold chemistry. I. Diatomic gold compounds, J. Chem. Phys., 1989, 91, 1762–1774 CrossRef CAS;
(c) M. Dolg, U. Wedig, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the first row transition elements, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
-
(a) K. Fukui, Formulation of the reaction coordinate, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS;
(b) K. Fukui, The path of chemical reactions - the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
-
(a) T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed , accessed August, 2020, available at: https://www.sobereva.com/multiwfn;
(b) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 Search PubMed;
(c) T. Lu, Z. Liu and Q. Chen, Comment on “18 and 12 - Member carbon rings (cyclo[n]carbons) - A density functional study”, Mater. Sci. Eng., B, 2021, 273, 115425–115428 CrossRef CAS.
- C. Y. Legault, CYLview, version 1.0b, Université de Sherbrooke, Canada, 2009 Search PubMed.
- H. Nakayama, S. Harada, A. Kanda, I. M.-Y. Kwork and T. Nemoto, Binary additive effect of benzoic acid in ipso-Friedel-Crafts-type dearomatization of phenols using a chiral silver phosphate, Tetrahedron, 2018, 74, 2435–2439 CrossRef CAS.
- Z. Liu, S. Cao, W. Yu, J. Wu, F. Yi, E. A. Anderson and X. Bi, Site-Selective C–H Benzylation of Alkanes with N-Triftosylhydrazones Leading to Alkyl Aromatics, Chem, 2020, 6, 2110–2124 CAS.
- F. M. Bickelhaupt and K. N. Houk, Analyzing reaction rates with the distortion/interaction-activation strain model, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Steven P. Nolan
*a and
Steven P. Nolan