
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe NKT cell TCR repertoire can accommodate structural modifications to the lipid and orientation of the terminal carbohydrate of iGb3†
Garth Cameron‡
 a,
Janice M. H. Cheng‡b,
Dale I. Godfreya,
Mattie S. M. Timmer*bc,
Bridget L. Stocker*bc and
Emma M. Dangerfield*bc
a,
Janice M. H. Cheng‡b,
Dale I. Godfreya,
Mattie S. M. Timmer*bc,
Bridget L. Stocker*bc and
Emma M. Dangerfield*bc
aDepartment of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, University of Melbourne, Victoria 3000, Australia
bSchool of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, 6140, Wellington, New Zealand. E-mail: mattie.timmer@vuw.ac.nz; bridget.stocker@vuw.ac.nz; emma.dangerfield@vuw.ac.nz
cCentre for Biodiscovery, Victoria University of Wellington, PO Box 600, 6140, Wellington, New Zealand
First published on 22nd June 2022
Abstract
Isoglobotrihexosylceramide (iGb3) is a known NKT cell agonist, however the specific interactions required to trigger NKT cell TCR activation in response to this mammalian glycolipid are not fully understood. Here we report the synthesis of 1,3-β-Gal-LacCer (βG-iGb3) that displays a β-linked terminal sugar. βG-iGb3 activated NKT cells to a similar extent as iGb3 with a terminal α-linkage, indicating that the conformation of the terminal sugar residue of iGb3 is not essential to facilitate NKT cell TCR recognition. In addition, the immunological activity of four recently described iGb3 analogues with modifications to their terminal sugar or lipid backbone were also investigated. These iGb3 analogues all induced NKT cell proliferation, with IL-13 the predominate cytokine detected. This highlights the ability of the NKT cell TCR to accommodate variations in iGb3-based glycolipids and suggests that undiscovered NKT cell ligands may exist within the lacto-series of mammalian glycosphingolipids.
Introduction
The realisation in the mid 1990s that α-galactosylceramides (α-GalCer, 1, Fig. 1) could be presented by the Cluster of Differentiation 1d (CD1d) protein and induce Natural Killer T (NKT) cell activation1,2 brought together the fields of glycoscience and immunology by demonstrating that certain glycolipids have the potential to activate specific subsets of T cells through T cell receptor (TCR) mediated interactions.3 Since this discovery, there has been much interest in exploring the therapeutic potential of α-GalCer and derivatives thereof.4–9 In addition, the mammalian glycolipid isoglobotrihexosylceramide (iGb3, 2, Fig. 1) was shown to be an NKT cell agonist,10,11 although the critical moieties of this glycolipid are unknown. Initially, iGb3 (2) was proposed to be the endogenous ligand responsible for thymic selection of the NKT cell population,10 however Speak et al. revealed that iGb3 could not be detected in the human or mouse thymus.12 This study, along with others,13–16 challenged the concept that iGb3 is the selecting ligand for intrathymic NKT cell development. Nonetheless, the fact that the structurally distinct glycolipids α-GalCer and iGb3 both activate NKT cells highlights the promiscuity of the NKT cell TCR.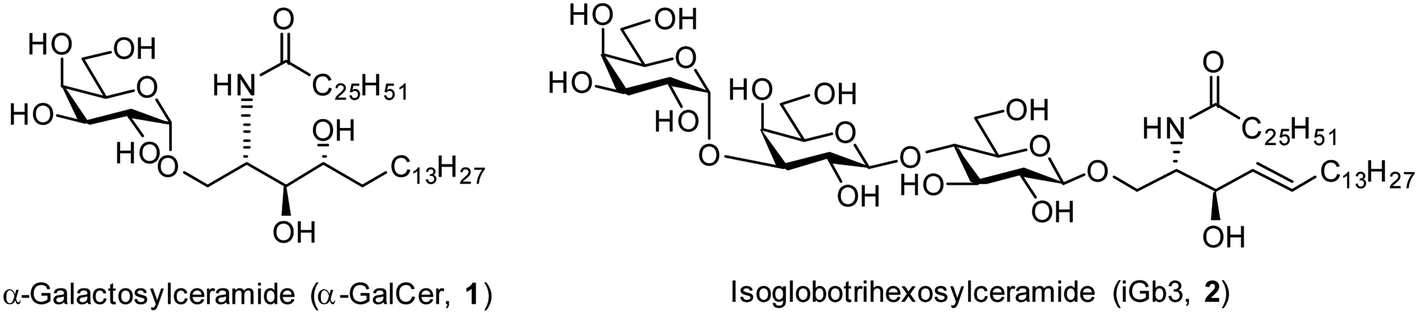 | ||
| Fig. 1 Structurally diverse NKT cell ligands α-GalCer (1) and iGb3 (2). | ||
The ability of NKT cells to accommodate such structurally diverse antigens provides a platform to dissect the requirements for NKT cell agonism. To date, a few groups have synthesised iGb3 analogues including two ceramide modified glycolipids, iGb3-phytosphingosine and iGb3-sphinganine,17 as well as α-iGb3 and α-Gb3, which contain an α-linkage between the proximal sugar and lipid portion, and which lead to enhanced NKT cell activation.18 To probe the effect of the terminal galactose residue on the activity of iGb3, Chen et al. synthesised a series of dehydroxylated analogues of iGb3.19 They observed that the 4′′′- and 6′′′-deoxy iGb3 analogues were able to induce IL-2 production from an NKT cell hybridoma, however to a lesser extent compared to iGb3. Of note, a phytosphingosine backbone was incorporated instead of the sphingosine found on the first form of iGb3 demonstrated to be a NKT cell ligand,10 prompting us to synthesise the 6′′′-dehydro (6′′′-dh) iGb3 derivatives with the sphingosine and sphinganine backbone.20 We also prepared the C20:2 analogue of iGb3, as this acyl chain modification in α-GalCer derivatives has been shown to bias NKT cell responses towards Th2-type cytokine production.21 Furthermore, a C12 acyl chain analogue was prepared, as this variation has previously been shown to enhance the activity of β-GalCer.22
In 2011, Pellicci et al.23 and Yu et al.24 reported the crystal structure of the ternary complex, NKT TCR iGb3-CD1d and suggested that the main reason for β-linked glycolipids activating NKT cells in a manner similar to α-linked glycolipids was because the TCR of the NKT cell is able to “bulldoze” the protruding headgroup so that it sits flat against the α2-helix of CD1d. Furthermore, crystal structure studies revealed that the terminal galactose sugar formed interactions with CD1d. Specifically, the 6′′′-hydroxyl of iGb3 formed a hydrogen bond with Thr159 of CD1d, and both the 4′′′-and 6′′′-hydroxyl moieties partook in van der Waals interactions with Thr159 and Met162.
To further understand the structural requirements of iGb3 to bind CD1d and activate NKT cells, we studied the activity of a modified βG-iGb3 analogue (3, Fig. 2) to probe the importance of the terminal 1,3-α-glycosidic linkage of iGb3. In addition, we assessed the biological activity of iGb3-C12 (4), iGb3-C20:2 (5), 6′′′-dh-iGb3-sphinganine (6) and 6′′′-dh-iGb3-sphingosine (7, Δ4,5)20 to ascertain the importance of changes to the lipid and sugar composition on immunological activity. Such insight would be useful for understanding the key structural requirements of CD1d ligand binding and NKT cell activation, which in turn will allow for a better understanding of how iGb3 analogues might be used in a therapeutic setting.25
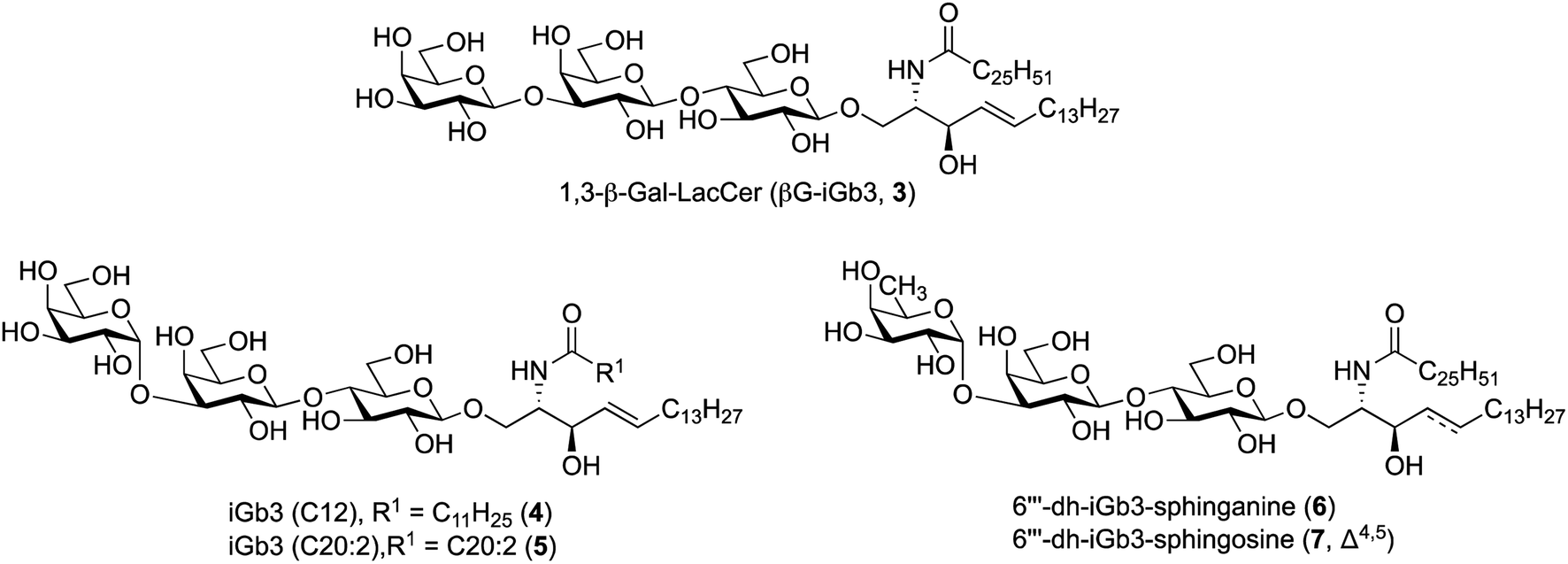 | ||
| Fig. 2 iGb3 analogues 1,3-β-Gal-LacCer (βG-iGb3, 3), iGb3-C12 (4), C20:2 (5), 6′′′-dh-iGb3-sphinganine (6) and 6′′′-dh-iGb3-sphingosine (7). | ||
To prepare βG-iGb3 (3), we envisioned using lactosyl 2-azido-sphingosine intermediate 8, previously identified as a versatile intermediate for the preparation of iGb3 analogues.20 Accordingly, we proposed that the synthesis of βG-iGb3 (3, Scheme 1) would be carried out via the installation of the C26 amide after the reduction of the azide on intermediate 8 and coupling with hexacosanoic acid (9) followed by selective acetylation via orthoester opening26 to give the 4′′-O-acetyl derivative, and subsequent glycosylation with imidate donor 10 containing a β-directing C-2 participating group. Global deprotection would then yield βG-iGb3 (3). Lactosyl 2-azido-sphingosine 8, in turn, would be prepared from the glycosylation reaction between lactose imidate 11 and sphingosine acceptor 12,20 which can be prepared from D-lactose27,28 and D-arabinose,7 respectively. Despite the syntheses of several iGb3 derivatives,11,18,29 there are no published syntheses of βG-iGb3 (3). Moreover, while the trisaccharide has been attached to a sphinganine lipid backbone bearing a C16 acyl chain,30 its biological activity has not been explored. Changes to the lipid can also influence the immunological activity of NKT cell activating glycolipids.21,31–34 Accordingly, our proposed synthesis of βG-iGb3 (3) incorporated the original C26 acyl lipid so that any changes in activity can be attributed to the modification of the terminal glycoside linkage.
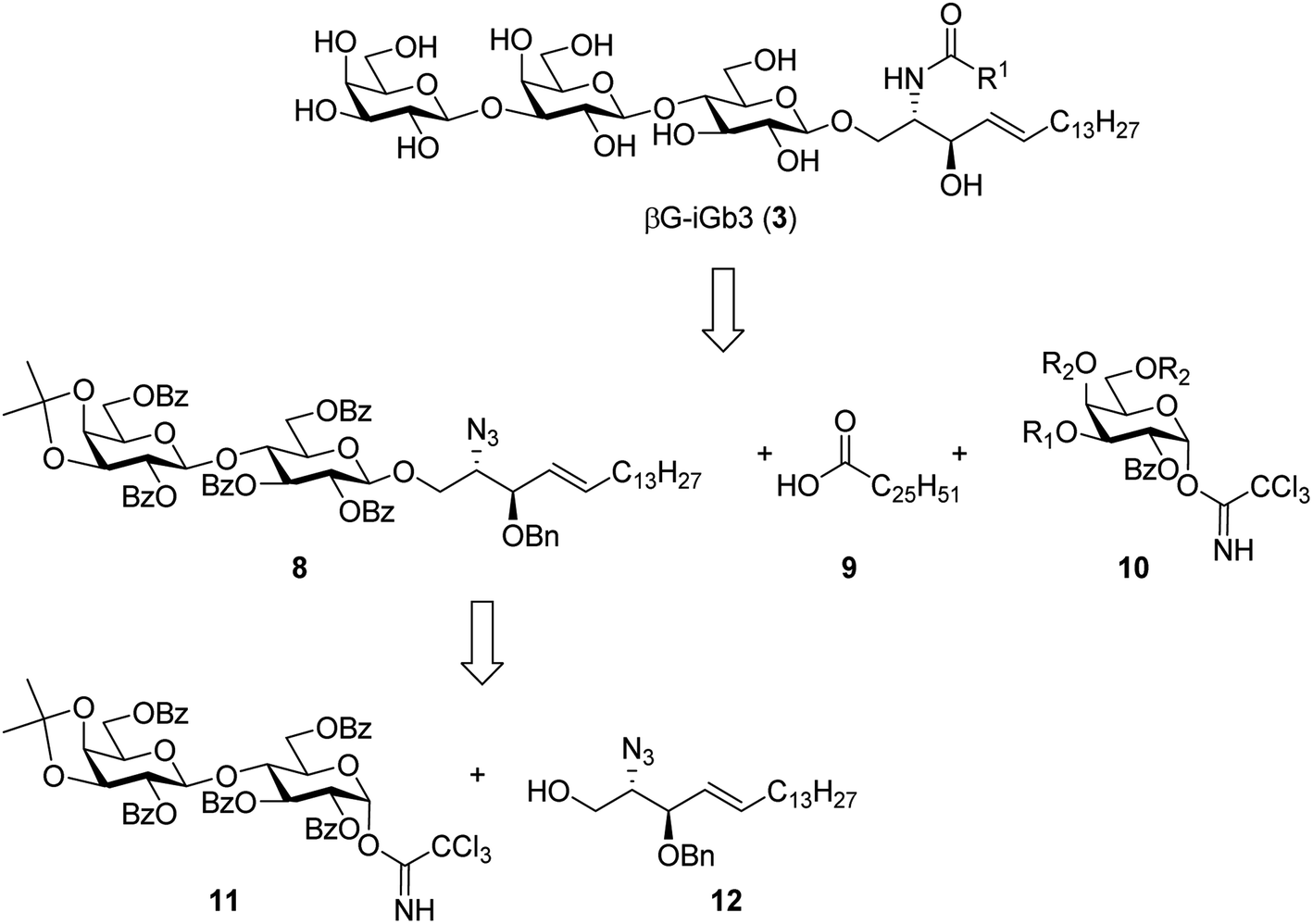 | ||
| Scheme 1 Retrosynthesis of βG-iGb3 (3). | ||
Experimental
See the ESI† for a detailed description of the methods for the biological analysis, and synthetic chemistry including 1H and 13C NMR.Results and discussion
Synthesis of βG-iGb3
The synthesis of βG-iGb3 (3) commenced with the preparation of the key lactosyl 2-azido-spinogosine intermediate 8 from D-lactose (13) according to our previously published procedures (Scheme 2).20 The azide functionality in 8 was then reduced, under Staudinger conditions,35 to give the corresponding amine, which was used without further purification. An EDCI/DMAP-mediated condensation of hexacosanoic acid (9) with the lactosyl amine then resulted in the target lactosyl ceramide (LacCer) 14 in excellent (72%) overall yield for the two steps. The acetonide in LacCer 14 was then removed via treatment with TFA/H2O and an acetate group regioselectively installed at the 4′′-OH, as first reported by Lemieux et al.,26 to give the lactosyl acceptor 15 ready for coupling to an appropriately functionalised galactose donor.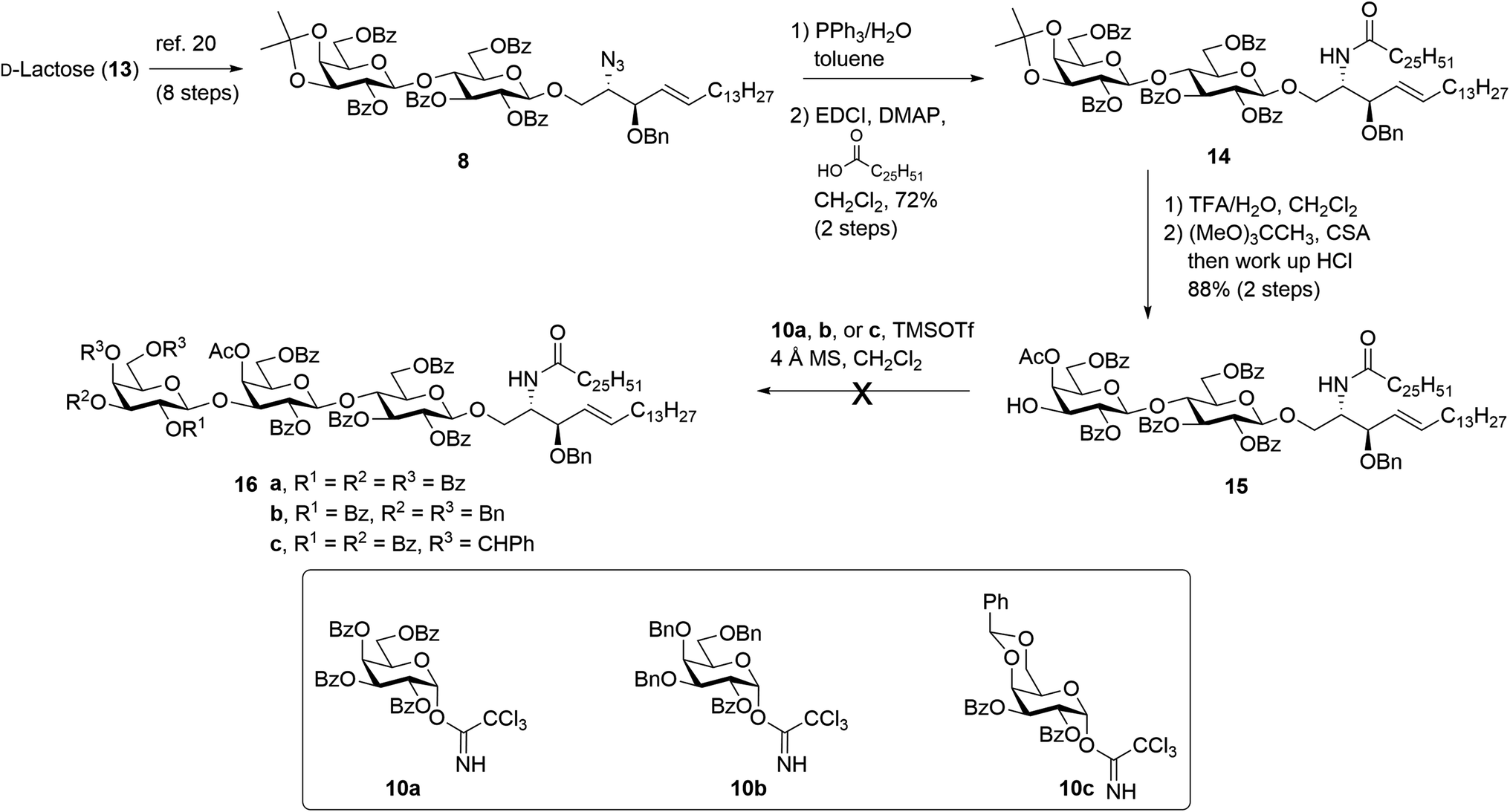 | ||
| Scheme 2 Attempted synthesis of βG-iGb3 (3). | ||
Initially, a glycosylation reaction between lactose acceptor 15 and per-benzoylated imidate donor 10a was attempted (Scheme 2). While it is known that benzoyl groups lead to a disarmed donor and potentially sluggish reactions,36 the ease of synthesis of 1-O-(2,3,4,6-tetra-O-benzoyl-D-galactopyranosyl)trichloroacetimidate37 (10a) saw us first explore the potential of this reagent. Lactose acceptor 15 (1.0 equiv.) and imidate donor 10a (2 equiv.) were reacted in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf). However, despite several attempts altering the number of equivalents of TMSOTf, the reaction length and temperature, no trace of the desired trisaccharide 16a was observed. In view of this, it was proposed that the more armed 1-O-(2-O-benzoyl-3,4,6-tri-O-benzyl-D-galactopyranosyl)trichloroacetimidate (10b) would enhance the glycosylation yield and a strategy for the preparation of this donor was developed utilising an orthoester at the 1- and 2-positions in a manner similar to that developed for mannose donors (Scheme S1†).38,39 The TMSOTf-mediated glycosylation reaction with LacCer acceptor 15 was attempted, yet despite numerous attempts, only trace amounts of the target trisaccharide 16b were observed. We attributed this to the incompatible reactivity between the sluggish acceptor and armed donor.
Next, we attempted the glycosylation reaction using 2,3-di-O-benzoyl-4,6-O-benzylidene-α-D-galactosyl trichloroacetimidate (10c), which is more armed than the per-benzoylated imidate 10a, but less armed than donor 10b. Benzylidine-protected donor 10c was synthesised from phenyl 4,6-O-benzylidene-1-thio-β-D-galactopyranoside (Scheme S2†), itself prepared according to previously published procedures.40 However, a TMSOTf-mediated glycosylation of donor 10c with LacCer 15 only yielded trace amounts of the desired trisaccharide 16c. It was reasoned that the poorly soluble LacCer acceptor 15, which was only moderately soluble at the low temperatures required for the glycosylation reaction, was unsuitable for the formation of the terminal β-galactose linkage, despite our earlier success at using disaccharide 15 for the synthesis of similar α-galactose linked LacCer derivatives.20 Thus, an alternative approach for the synthesis of βG-iGb3 (3) was proposed whereby the required trisaccharide core would be synthesized prior to the incorporation of the C26 N-acyl chain.
Our new strategy began with use of the key lactosyl 2-azido-sphinogosine intermediate 8 (Scheme 3), however this time the acetonide was removed and the 4-O-Ac installed to give lactosyl acceptor 17 in excellent (91%) yield over the two steps. Much to our gratification, the TMSOTf-mediated glycosylation between acceptor 17 and benzylidene imidate donor 10c was successful, with the target trisaccharide 18 being isolated in 51% yield following silica gel column chromatography. The formation of the desired β-linkage was confirmed via 1H NMR (J1′′′,2′′′ = 8.0 Hz) and no trace of the corresponding α-anomer was observed. With trisaccharide 18 in hand, the azide on the sphingosine backbone was then reduced and the resulting amine directly coupled to hexacosanoic acid (9) under the mediation of EDCI and DMAP to give the fully protected βG-iGb3 16c in 70% yield over two-steps. Finally, global deprotection under Birch conditions gave the target βG-iGb3 (3).
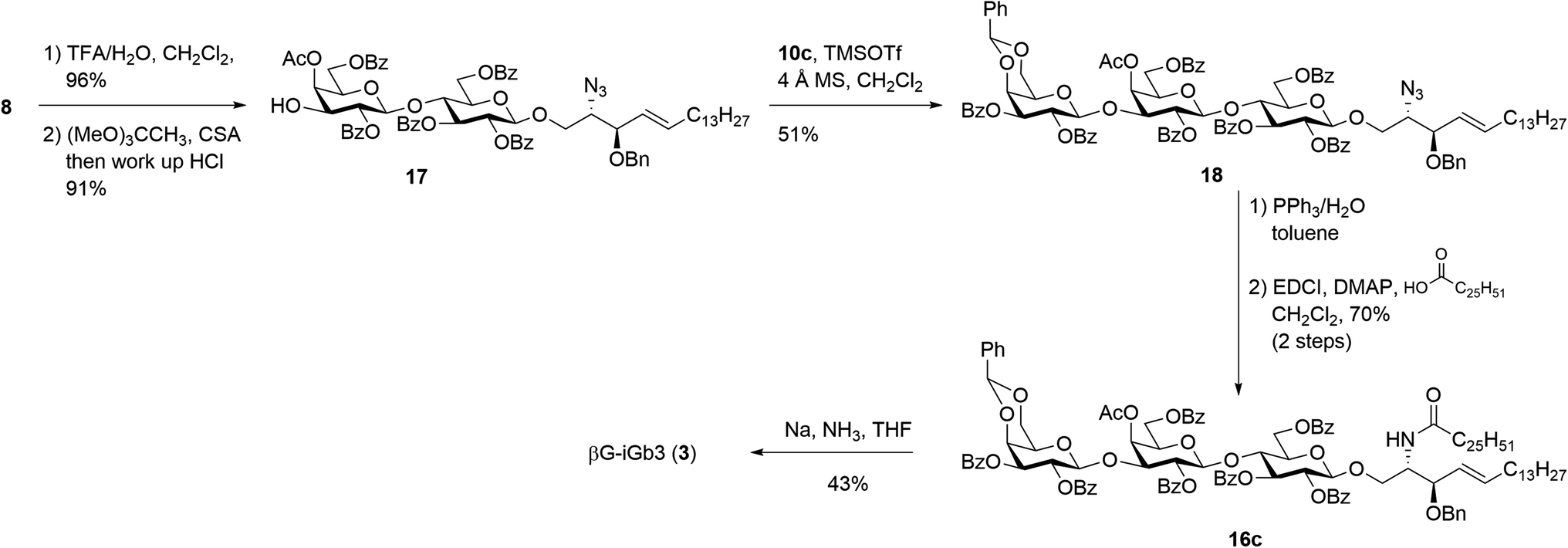 | ||
| Scheme 3 Synthesis of βG-iGb3. | ||
Structure–activity relationship of iGb3 analogues
With βG-iGb3 (3) in hand, we explored the ability of this glycolipid, alongside our other previously generated iGb3 analogues (4 to 7),20 to activate NKT cells. We assessed this by gauging the in vitro proliferation of thymic NKT cells in response to these glycolipids and observed that all iGb3 analogues were able to initiate NKT cell responses (Fig. 3). Notably, variants with shorter lipid acyl chains (C12 4 and C20:2 5) enhanced the extent of NKT cell proliferation compared to the prototypic version of iGb3 (2). Whether this was the result of altered CD1d and/or TCR contacts, or differences in their CD1d-loading efficiencies is unclear. In contrast, the removal of the 6′′′ hydroxyl on the terminal galactose moiety [6′′′-dh-iGb3 sphinganine (6) and sphingosine (7)] did not significantly impact NKT cell proliferation, suggesting that while the terminal galactose 6-OH residue contributes to interactions with CD1d, this is not essential for the TCR-mediated activation of NKT cells. This finding is consistent with previous reports, which demonstrated that 6′′′-dh-iGb3 phytosphingosine could elicit IL-2 production from a NKT cell hybridoma.19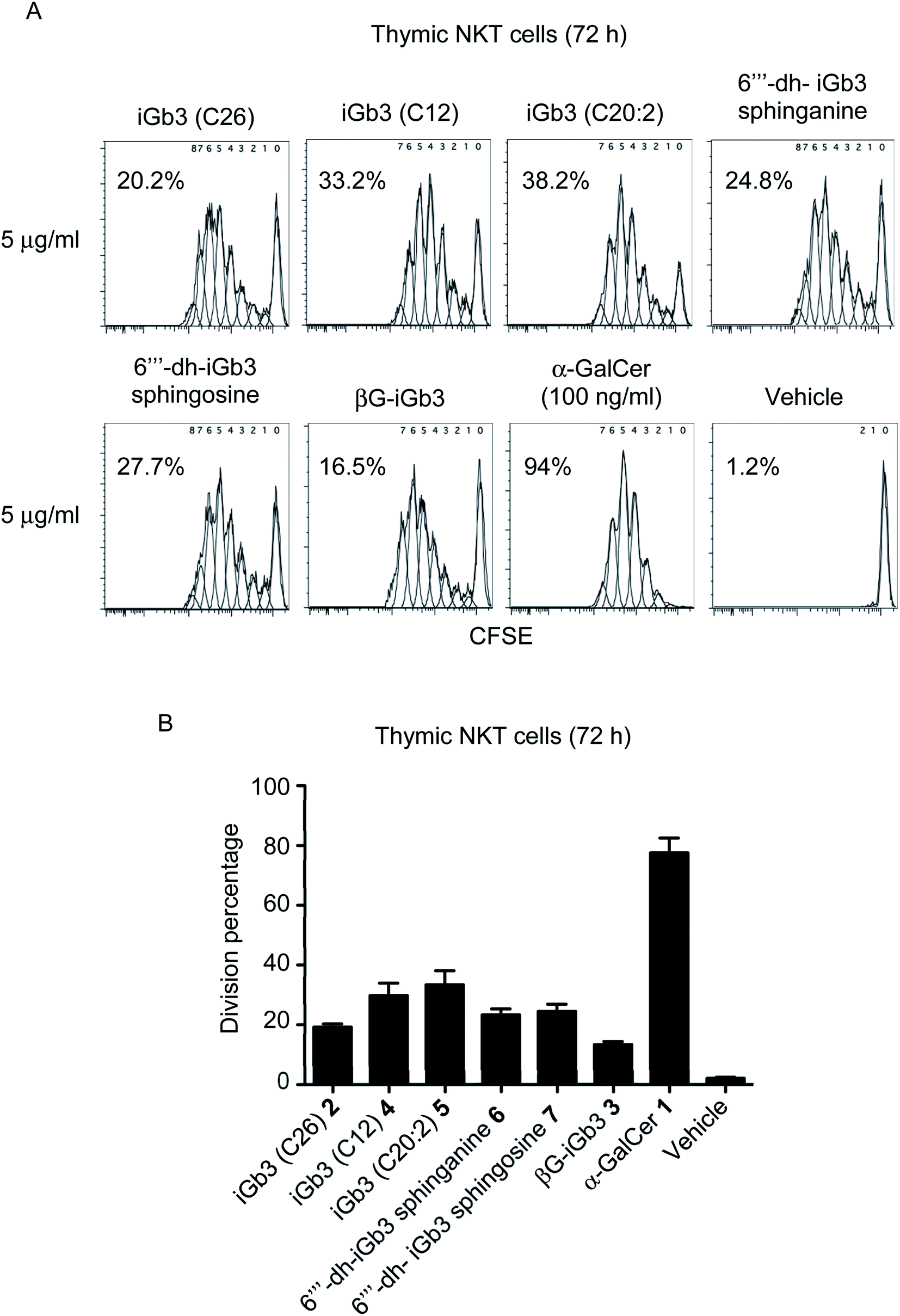 | ||
| Fig. 3 (A) Plots depict the dilution of CFSE amongst thymic (C57BL/6) NKT cells co-cultured for 72 h with Jα18−/− (C57BL/6) splenocytes pulsed overnight with the indicated antigen. Numbers on plots represent the estimated division percentages (FlowJo). (B) Graph depicts the division percentages of thymic NKT cells after 72 h (mean ± SEM). All lipids were used at 1 μg ml−1 except for α-GalCer which was used at 100 ng ml−1 n = 5, except iGb3 (C26), where n = 2. | ||
Notably, βG-iGb3 (3), with a more ‘linear’ sugar head group, was able to induce NKT cell proliferation, albeit at slightly reduced levels in comparison to the commonly used version of iGb3 (2). This suggests that significant structural changes in the composition of endogenous ligands may be accommodated by the inherent diversity within the NKT cell TCR repertoire. This is surprising considering the reportedly crucial role of the terminal sugar conformation for iGb3 activity,10 and given that globotrihexosylceramide (Gb3), a regioisomer of iGb3 which varies only in the α-1,4 linkage of the terminal galactose, is unable to stimulate NKT cells.23 Nonetheless, this supports the hypothesis that the NKT cell TCR can bind disparate antigens by flattening the conformation of the terminal sugar group,23,24 and presumably the TCR pushes the terminal sugar of this lipid into an orientation that accommodates binding. Furthermore, the observed ability of βG-iGb3 (3) to activate NKT cells opens the potential for other classes of glycolipids with 1,3-β linkages, such as the lacto-series glycosphingolipids,41 to contribute to the thymic selection and peripheral activation of NKT cell subsets. Indeed, lactotriaosylceramide (Lc3Cer), a lacto-series glycosphingolipid where the terminal trisaccharide is a 1,3-β-linked GlucNAc, is expressed in murine thymocytes.42
Of those NKT cells that proliferated following in vitro-challenge with each of the iGb3 analogues (Fig. 3), all underwent a similar number of cellular-divisions compared to those stimulated with α-GalCer, which can be observed by the extent of CFSE dilution (Fig. 3A). However, in accordance with previous findings,3,43 the portion of the NKT cell pool that were induced to divide in response to iGb3 (and analogues thereof) was restricted in comparison to the pan-NKT cell antigen α-GalCer. These data support the concept that the NKT cell TCR repertoire can accommodate structural variations among mammalian-derived glycolipids, yet the scope of NKT cell activation elicited by such antigens may remain limited to sub-populations of the broader NKT cell pool.
We and others44–46 have shown that structural modifications in the lipid and/or sugar component of NKT cell agonists can impact the scope and phenotype of cytokine responses following CD1d-restricted activation. To assess this in relation to these novel iGb3 analogues, cell culture supernatants were collected from the in vitro experiments depicted in Fig. 3, with cytokine production being assessed by cytometric bead array. The Th2-type cytokine IL-13 was most prominently detected in response to the iGb3 analogues and α-GalCer (Fig. 4A and B). However, the levels of IL-13 elicited by α-GalCer were at least two-fold greater than what was observed from any iGb3 analogue. The pro-inflammatory Th1-type cytokine IFN-γ was detected at high levels in cultures exposed to α-GalCer, whereas this cytokine was present at background levels in response to the iGb3 analogues, similar to the vehicle control (Fig. 4A). While the Th2-type cytokines IL-4 and IL-10 were relatively low in all conditions, the pro-inflammatory cytokine IL-17A was notably detected at higher levels in response to α-GalCer, demonstrating how glycolipid-antigen composition may influence functional outcomes.
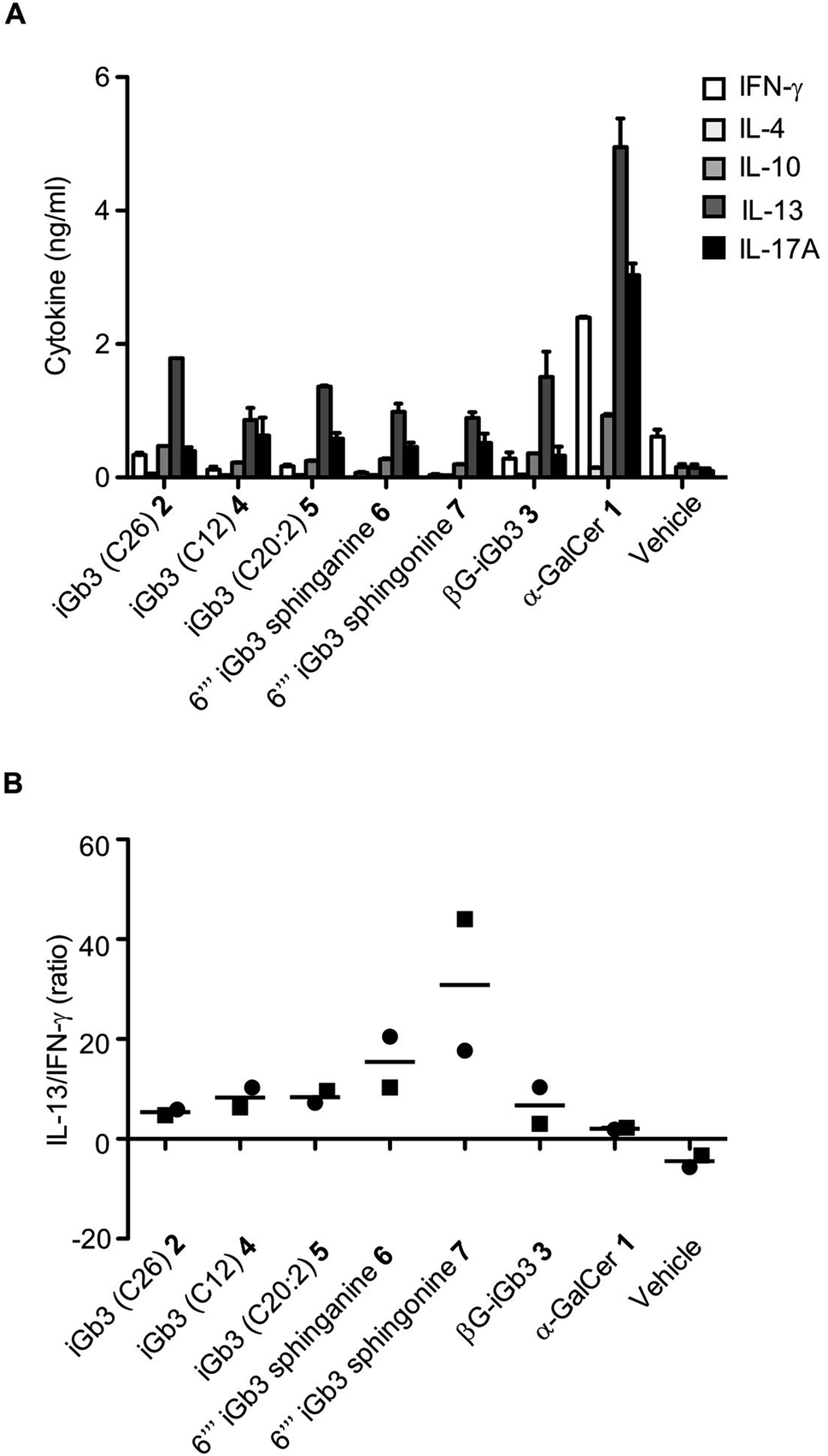 | ||
| Fig. 4 (A) Cell culture supernatants were collected at 72 h following the co-culture of antigen-pulsed Jα18−/− (C57BL/6) splenocytes with NKT cell-enriched thymocytes (C57BL/6). Graph depicts the cytokine concentrations as gauged by CBA. Pooled means (n = 2 replicates) from 2 independent experiments (mean ± SEM). (B) Graph depicts the ratio of IL-13 to IFN-γ from the indicated conditions. Data taken from 2 independent experiments, each represented by square or circular datapoints that depict the mean of two technical replicates for each experiment | ||
Conclusions
To date, the critical moieties involved in NKT cell responses to iGb3 have not been extensively studied. In this work we prepared an iGb3 analogue βG-iGb3 (where the terminal galactose is connected via a β-glycosidic linkage rather than an α-linkage) and assessed the ability of this and four other α-linked iGb3 analogues to activate NKT cells. We observed that NKT cells proliferated in response to each iGb3 analogue, however, in line with a previous report3 this was only observable among a portion of the NKT cell population. Consistent with this, the levels of cytokines detected in response to iGb3 analogues were markedly reduced in comparison to the pan-agonist α-GalCer, with IFN-γ being notably absent. These data indicate the degree of flexibility in glycolipid components that contact both CD1d and the NKT cell TCR. Notably, βG-iGb3 was able to induce NKT cell proliferation, highlighting the promiscuous nature of the NKT cell TCR repertoire for glycolipids with seemingly remarkable differences in the orientation (either an α- or β-linkage) of their terminal sugar residues. This observation is surprising given that NKT cells are not able to recognise Gb3, an analogue of iGb3 where the terminal galactose is connected via a 1,4-α linkage.23 Nonetheless, this suggests that other classes of glycolipids with 1,3-β linkages, such as the lacto-series glycosphingolipids,41 might be involved in the thymic selection and peripheral activation of NKT cells. These data develop our understanding of how the intricacies of glycolipid structure and CD1d/NKT cell interactions may be better manipulated for therapeutic outcomes.Author contributions
J. C. prepared the synthetic glycolipids, G. C. carried out the biological analysis and wrote the related portion of the main manuscript text. D. G. and G. C. directed the biological analysis, and M. T. and B. S. directed the chemical synthesis. E. D. wrote and prepared the manuscript for submission. All authors reviewed the manuscript.Conflicts of interest
D. G. and G. C. have provisional patent applications submitted regarding targeting of unconventional T cells and their ligands for immunotherapy.Acknowledgements
The authors would like to thank The Health Research Council New Zealand (Hercus Fellowship, B. L. S., 2013/33) and the National Health and Medical Research Council of Australia (NHMRC): 1113293, 1140126, 1117766, 2008913 and Australian Research Council (CE140100011) to D. I. G. for funding. The graphical abstract was created with BioRender.com (https://www.BioRender.com).References
- A. Bendelac, O. Lantz, M. E. Quimby, J. W. Yewdell, J. R. Bennink and R. R. Brutkiewicz, Science, 1995, 268, 863–865 CrossRef CAS PubMed.
- T. Kawano, J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, H. Koseki and M. Taniguchi, Science, 1997, 278, 1626–1629 CrossRef CAS PubMed.
- G. Cameron, D. G. Pellicci, A. P. Uldrich, G. S. Besra, P. Illarionov, S. J. Williams, N. L. La Gruta, J. Rossjohn and D. I. Godfrey, J. Immunol., 2015, 195, 4604–4614 CrossRef CAS PubMed.
- R. M. McEwen-Smith, M. Salio and V. Cerundolo, Cancer Immunol. Res., 2015, 3, 425–435 CrossRef CAS PubMed.
- L. J. Carreño, S. S. Kharkwal and S. A. Porcelli, Immunotherapy, 2014, 6, 309–320 CrossRef PubMed.
- S. Inuki, N. Hirata, E. Kashiwabara, J. Kishi, T. Aiba, T. Teratani, W. Nakamura, Y. Kojima, T. Maruyama, T. Kanai and Y. Fujimoto, Sci. Rep., 2020, 10, 15766 CrossRef CAS PubMed.
- E. M. Dangerfield, J. M. H. Cheng, D. A. Knight, R. Weinkove, P. R. Dunbar, I. F. Hermans, M. S. M. Timmer and B. L. Stocker, ChemBioChem, 2012, 13, 1349–1356 CrossRef CAS PubMed.
- J. Guillaume, J. Wang, J. Janssens, S. G. Remesh, M. D. P. Risseeuw, T. Decruy, M. Froeyen, D. Elewaut, D. M. Zajonc and S. Van Calenbergh, Sci. Rep., 2017, 7, 4276 CrossRef PubMed.
- S. S. Kharkwal, P. Arora and S. A. Porcelli, Immunogenetics, 2016, 68, 597–610 CrossRef CAS PubMed.
- D. Zhou, J. Mattner, C. Cantu III, N. Schrantz, N. Yin, Y. Gao, Y. Sagiv, K. Hudspeth, Y.-P. Wu, T. Yamashita, S. Teneberg, D. Wang, R. L. Proia, S. B. Levery, P. B. Savage, L. Teyton and A. Bendelac, Science, 2004, 306, 1786–1789 CrossRef CAS PubMed.
- C. Xia, Q. Yao, J. Schümann, E. Rossy, W. Chen, L. Zhu, W. Zhang, G. De Libero and P. G. Wang, Bioorg. Med. Chem. Lett., 2006, 16, 2195–2199 CrossRef CAS PubMed.
- A. O. Speak, M. Salio, D. C. A. Neville, J. Fontaine, D. A. Priestman, N. Platt, T. Heare, T. D. Butters, R. A. Dwek, F. Trottein, M. A. Exley, V. Cerundolo and F. M. Platt, Proc. Natl. Acad. Sci., 2007, 104, 5971–5976 CrossRef CAS PubMed.
- S. Porubsky, A. O. Speak, B. Luckow, V. Cerundolo, F. M. Platt and H.-J. Gröne, Proc. Natl. Acad. Sci., 2007, 104, 5977–5982 CrossRef CAS PubMed.
- D. Christiansen, J. Milland, E. Mouhtouris, H. Vaughan, D. G. Pellicci, M. J. McConville, D. I. Godfrey and M. S. Sandrin, PLoS Biol., 2008, 6, 1527–1538 CrossRef CAS PubMed.
- S. D. Gadola, J. D. Silk, A. Jeans, P. A. Illarionov, M. Salio, G. S. Besra, R. Dwek, T. D. Butters, F. M. Platt and V. Cerundolo, J. Exp. Med., 2006, 203, 2293–2303 CrossRef CAS PubMed.
- S. Porubsky, A. O. Speak, M. Salio, R. Jennemann, M. Bonrouhi, R. Zafarulla, Y. Singh, J. Dyson, B. Luckow, A. Lehuen, E. Malle, J. Müthing, F. M. Platt, V. Cerundolo and H.-J. Gröne, J. Immunol., 2012, 189, 3007–3017 CrossRef CAS PubMed.
- C. Xia, J. Schümann, R. Emmanuel, Y. Zhang, W. Chen, W. Zhang, G. De Libero and P. G. Wang, J. Med. Chem., 2007, 50, 3489–3496 CrossRef CAS PubMed.
- N. Yin, X. Long, R. D. Goff, D. Zhou, C. Cantu III, J. Mattner, P. Saint Mezard, L. Teyton, A. Bendelac and P. B. Savage, ACS Chem. Biol., 2009, 4, 191–197 CrossRef CAS PubMed.
- W. Chen, C. Xia, J. Wang, P. Thapa, Y. Li, A. Talukdar, J. Nadas, W. Zhang, D. Zhou and P. G. Wang, J. Org. Chem., 2007, 72, 9914–9923 CrossRef CAS PubMed.
- J. M. H. Cheng, E. M. Dangerfield, M. S. M. Timmer and B. L. Stocker, Org. Biomol. Chem., 2014, 12, 2729–2736 RSC.
- J. S. Im, P. Arora, G. Bricard, A. Molano, M. M. Venkataswamy, I. Baine, E. S. Jerud, M. F. Goldberg, A. Baena, K. O. A. Yu, R. M. Ndonye, A. R. Howell, W. Yuan, P. Cresswell, Y. Chang, P. A. Illarionov, G. S. Besra and S. A. Porcelli, Immunity, 2009, 30, 888–898 CrossRef CAS PubMed.
- V. V. Parekh, A. K. Singh, M. T. Wilson, D. Olivares-Villagómez, J. S. Bezbradica, H. Inazawa, H. Ehara, T. Sakai, I. Serizawa, L. Wu, C.-R. Wang, S. Joyce and L. Van Kaer, J. Immunol., 2004, 173, 3693–3706 CrossRef CAS PubMed.
- D. G. Pellicci, A. J. Clarke, O. Patel, T. Mallevaey, T. Beddoe, J. Le Nours, A. P. Uldrich, J. McCluskey, G. S. Besra, S. A. Porcelli, L. Gapin, D. I. Godfrey and J. Rossjohn, Nat. Immunol., 2011, 12, 827–833 CrossRef CAS PubMed.
- E. D. Yu, E. Girardi, J. Wang and D. M. Zajonc, J. Immunol., 2011, 187, 2079–2083 CrossRef CAS PubMed.
- Z. Zhou, C. Zhang, C. Xia, W. Chen, H. Zhu, P. Shang, F. Ma, P. G. Wang, J. Zhang, W. Xu and Z. Tian, Mol. Cancer Ther., 2011, 10, 1375–1384 CrossRef CAS PubMed.
- R. U. Lemieux and H. Driguez, J. Am. Chem. Soc., 1975, 97, 4069–4075 CrossRef CAS PubMed.
- G. Xing, L. Chen and F. Liang, Eur. J. Org. Chem., 2009, 5963–5970 CrossRef CAS.
- L. Lay, R. Windmüller, S. Reinhardt and R. R. Schmidt, Carbohydr. Res., 1997, 303, 39–49 CrossRef PubMed.
- D. Adlercreutz, J. T. Weadge, B. O. Petersen, J. Ø. Duus, N. J. Dovichi and M. M. Palcic, Carbohydr. Res., 2010, 345, 1384–1388 CrossRef CAS PubMed.
- D. Beith-Halahmi and H. M. Flowers, Carbohydr. Res., 1968, 8, 340–343 CrossRef CAS.
- D. M. Zajonc, C. Cantu III, J. Mattner, D. Zhou, P. B. Savage, A. Bendelac, I. A. Wilson and L. Teyton, Nat. Immunol., 2005, 6, 810–818 CrossRef CAS PubMed.
- R. D. Goff, Y. Gao, J. Mattner, D. Zhou, N. Yin, C. Cantu III, L. Teyton, A. Bendelac and P. B. Savage, J. Am. Chem. Soc., 2004, 126, 13602–13603 CrossRef CAS PubMed.
- C. McCarthy, D. Shepherd, S. Fleire, V. S. Stronge, M. Koch, P. A. Illarionov, G. Bossi, M. Salio, G. Denkberg, F. Reddington, A. Tarlton, B. G. Reddy, R. R. Schmidt, Y. Reiter, G. M. Griffiths, P. A. van der Merwe, G. S. Besra, E. Y. Jones, F. D. Batista and V. Cerundolo, J. Exp. Med., 2007, 204, 1131–1144 CrossRef CAS PubMed.
- C. Paget, S. Deng, D. Soulard, D. A. Priestman, S. Speca, J. von Gerichten, A. O. Speak, A. Saroha, Y. Pewzner-Jung, A. H. Futerman, T. Mallevaey, C. Faveeuw, X. Gu, F. M. Platt, R. Sandhoff and F. Trottein, PLoS Biol., 2019, 17, e3000169 CrossRef CAS PubMed.
- H. Staudinger and J. Meyer, Helv. Chim. Acta, 1919, 2, 635–646 CrossRef CAS.
- D. R. Mootoo, P. Konradsson, U. Udodong and B. Fraser-Reid, J. Am. Chem. Soc., 1988, 110, 5583–5584 CrossRef CAS.
- W. Pilgrim and P. V. Murphy, J. Org. Chem., 2010, 75, 6747–6755 CrossRef CAS PubMed.
- A. Hölemann, B. L. Stocker and P. H. Seeberger, J. Org. Chem., 2006, 71, 8071–8088 CrossRef PubMed.
- K. Plé, Carbohydr. Res., 2003, 338, 1441–1454 CrossRef.
- M. S. M. Timmer, B. L. Stocker, P. T. Northcote and B. A. Burkett, Tetrahedron Lett., 2009, 50, 7199–7204 CrossRef CAS.
- A. Togayachi, T. Akashima, R. Ookubo, T. Kudo, S. Nishihara, H. Iwasaki, A. Natsume, H. Mio, J. Inokuchi, T. Irimura, K. Sasaki and H. Narimatsu, J. Biol. Chem., 2001, 276, 22032–22040 CrossRef CAS PubMed.
- Y. Li, P. Thapa, D. Hawke, Y. Kondo, K. Furukawa, K. Furukawa, F.-F. Hsu, D. Adlercreutz, J. Weadge, M. M. Palcic, P. G. Wang, S. B. Levery and D. Zhou, J. Proteome Res., 2009, 8, 2740–2751 CrossRef CAS PubMed.
- W. C. Florence, C. Xia, L. E. Gordy, W. Chen, Y. Zhang, J. Scott-Browne, Y. Kinjo, K. O. A. Yu, S. Keshipeddy, D. G. Pellicci, O. Patel, L. Kjer-Nielsen, J. McCluskey, D. I. Godfrey, J. Rossjohn, S. K. Richardson, S. A. Porcelli, A. R. Howell, K. Hayakawa, L. Gapin, D. M. Zajonc, P. G. Wang and S. Joyce, EMBO J., 2009, 28, 3579–3590 CrossRef CAS PubMed.
- K. S. Wun, G. Cameron, O. Patel, S. S. Pang, D. G. Pellicci, L. C. Sullivan, S. Keshipeddy, M. H. Young, A. P. Uldrich, M. S. Thakur, S. K. Richardson, A. R. Howell, P. A. Illarionov, A. G. Brooks, G. S. Besra, J. McCluskey, L. Gapin, S. A. Porcelli, D. I. Godfrey and J. Rossjohn, Immunity, 2011, 34, 327–339 CrossRef CAS PubMed.
- K. Miyamoto, S. Miyake and T. Yamamura, Nature, 2001, 413, 531–534 CrossRef CAS PubMed.
- K. O. A. Yu, J. S. Im, A. Molano, Y. Dutronc, P. A. Illarionov, C. Forestier, N. Fujiwara, I. Arias, S. Miyake, T. Yamamura, Y.-T. Chang, G. S. Besra and S. A. Porcelli, Proc. Natl. Acad. Sci., 2005, 102, 3383–3388 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02373c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |