
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly covalent molecular cage based porous organic polymer: pore size control and pore property enhancement†
Zhen Wang *a,
Yan-Qun Liub,
Yu-Hang Zhaoa,
Qing-Pu Zhanga,
Yu-Ling Suna,
Bin-Bin Yanga,
Jian-Hua Bu*c and
Chun Zhang*a
*a,
Yan-Qun Liub,
Yu-Hang Zhaoa,
Qing-Pu Zhanga,
Yu-Ling Suna,
Bin-Bin Yanga,
Jian-Hua Bu*c and
Chun Zhang*a
aCollege of Life Science and Technology, National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: zhenwang89@hust.edu.cn; chunzhang@hust.edu.cn
bHenan Industry and Trade Vocational College, Zhengzhou, Henan 451191, China
cXi'an Modern Chemistry Research Institute, Xi'an, Shanxi 710065, China. E-mail: bujianhua@gmail.com
First published on 6th June 2022
Abstract
It remains a great challenge to effectively control the pore size in porous organic polymers (POPs) because of the disordered linking modes. Herein, we used organic molecular cages (OMCs), possessing the properties of fixed intrinsic cavities, high numbers of reactive sites and dissolvable processability, as building blocks to construct a molecular cage-based POP (TPP-pOMC) with high valency through covalent cross coupling reaction. In the formed TPP-pOMC, the originating blocking pore channels of TPP-OMC were “turned on” and formed fixed pore channels (5.3 Å) corresponding to the connective intrinsic cavities of cages, and intermolecular pore channels (1.34 and 2.72 nm) between cages. Therefore, TPP-pOMC showed significant enhancement in Brunauer–Emmett–Teller (BET) surface area and CO2 adsorption capacity.
Introduction
Porous organic polymers (POPs),1–4 a new generation porous material with salient advantages including high surface area, low mass density, easy functionalization, and high stability, have been used for tackling the greenhouse effect since Haszeldine5 put forward that carbon capture and storage (CCS) has the potential to reduce CO2 emissions in 2009. Of course, plenty of other porous materials like metal organic frameworks (MOFs)6–8 and covalent organic frameworks (COFs)9–12 have been developed for CCS. With well-ordered and unambiguous linking, although the topological structures MOFs and COFs can be purposefully targeted by a judicious selection of build blocks and exhibit superior performances, their poor chemical stability may give hindrance in practical application. For CCS, the CO2 behaved acidic under humid conditions, which would cause the breakage of their well-defined topological structures, and further may lead to leakage of metal ions in MOFs that could have influence on the environment.POPs, including hypercross-linked polymers (HCPs),13 polymers of intrinsic microporosity (PIMs)14 and conjugated microporous polymers (CMPs),15–17 constructed by irreversible covalent bonds, had proven to possess excellent chemical stability. To date, the majority of reported POPs were constructed by two-dimensional building blocks with no well-defined pore size distribution and modest pore performances. The rigidity of building blocks was considered to be important to form connective pore channels during the formation process of polymer networks. Endeavors had been made by using three-dimensional rigid building blocks, such as tetraphenyl methane,18 tetraphenyl silane,19,20 hexaphenylbenzene21 and triptycene,22 which can effectively enhance the pore performance. But the regulation of pore size distribution in POPs is still to be challenging.
With well-defined intrinsic cavities, shape persistent organic molecular cages (OMCs)23–25 exhibited unique advantages in selective recognition and separation for guests in the field of host-guest chemistry. Although OMCs had been used as porous organic materials for CCS, the pore performances were in stark contrast to MOFs, COFs and POPs.26–28 Recently, in consideration of well-defined intrinsic cavities of OMCs, a few studies have reported that the use of OMCs as building blocks provides opportunities for controlling the size distribution and enhanced molecular diffusivity in POPs.29–34 In addition, compared with two or three-dimensional organic molecular fragments, the three-dimensional OMCs with high numbers of reactive sites are easily to construct POPs with extending spatial topology.35,36 Zhang's30 and Coskun's31 groups developed cage-to-frameworks strategy and had demonstrated that the “cage effects” played a key role in CO2 adsorption and selectivity. Also, using OMCs as building blocks, Cooper's33 and Wang's groups32 paved a way for porous organic cages to construct crystalline organic frameworks.
However, to synthesize chemical stable organic cage remains difficult because of low yielding, especially for complex topological structures with more reactive sites (>6). Ascribed to the good stability and mild synthetic condition, oxacalixarene cages36–38 with tunable intrinsic cavity should be ideal building blocks for constructing POPs with well-defined pore channels. Herein, we used tetraphenylpyrazine as building fragments to react with 2,3,5,6-tetrachloropyridine to form an oxacalixarene cage (TPP-OMC) by nucleophilic aromatic substitution reaction (SNAr). With eight reacting sites around side rims of cage, the TPP-OMC could be used as monomer to construct a cage-based POP (TPP-pOMC) with highly connected topologies by nickel (0)-catalyzed Yamamoto-type Ullmann cross coupling reaction,15 which showed a well-defined pore channel corresponding to the cavity size (5.3 Å) of TPP-OMC and connective intermolecular pore channels with size of 1.34 nm and 2.7 nm, and showed obvious pore performance enhancement.
Results and discussion
The 4OH-TPP was obtained according to literature,39 and then served as building fragments to react with 2,3,5,6-tetrachloropyridine in the presence of Cs2CO3 at 120 °C in dimethyl sulfoxide (DMSO) for 12 h by one-pot condensation nucleophilic aromatic substitution reaction (SNAr) to construct TPP-OMC with the yield of 24% (Fig. 1). The TPP-OMC structure was confirmed by 1H nuclear magnetic resonance (1H-NMR). As shown in Fig. S1,† the 1H NMR spectrum in d8-toluene showed only a single signal peak at chemical shift of 7.25 ppm, which was ascribed to the protons of the linker pyridine moieties; and the two sets of double signal peaks at chemical shifts of 7.57 ppm and 6.72 ppm were ascribed to the protons of phenyl rings in TPP. Further, MALDI-TOF mass spectrometry (MALDI-TOF MS) analysis also confirmed that the TPP-OMC formed exclusively with m/z = 1473.8 (Fig. S2†).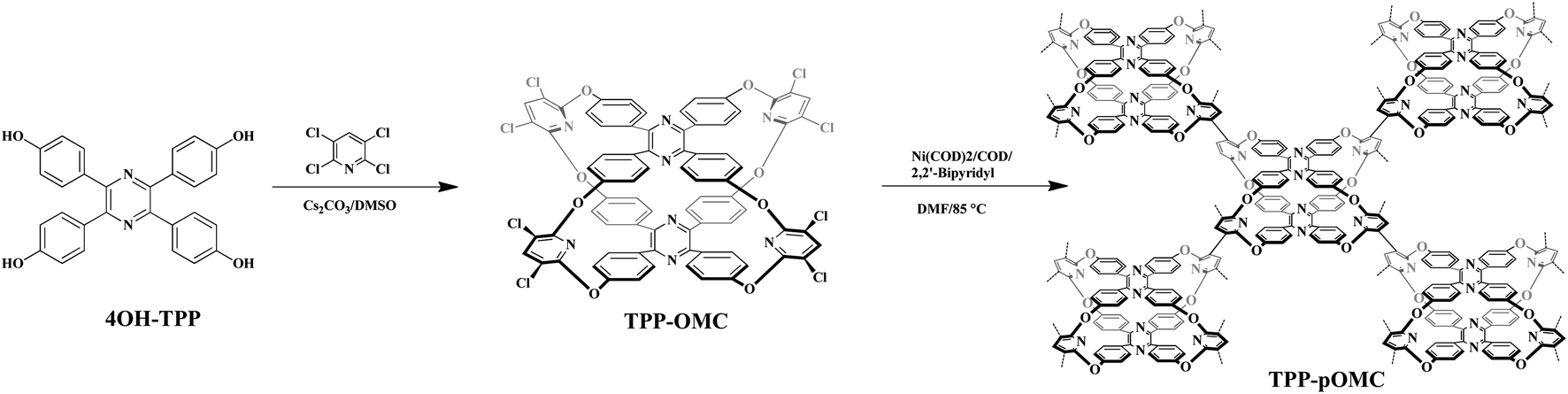 | ||
| Fig. 1 Synthesis of TPP-based oxacalixarene molecular cage TPP-OMC and TPP-pOMC. | ||
Unfortunately, the suitable single crystal of TPP-OMC was never obtained after various solution systems and methods. So, the conformational structure of TPP-OMC was simulated by the Forcite Tools mode in Material Studio 7.0 with geometry optimization. The simulated TPP-OMC adopted minimum energy state with the total energy of 293.85 kcal mol−1, including valence energy of 145.34 kcal mol−1 and non-bond energy of 148.41 kcal mol−1. As shown in Fig. 2, the TPP-OMC formed large intrinsic cavity with size of 11.8 × 8.1 × 5.0 Å3 that was confirmed by the N atom distances on the pyridine rings and pyrazine rings in TPP, which was similar to our reported oxacalixarene cage.15
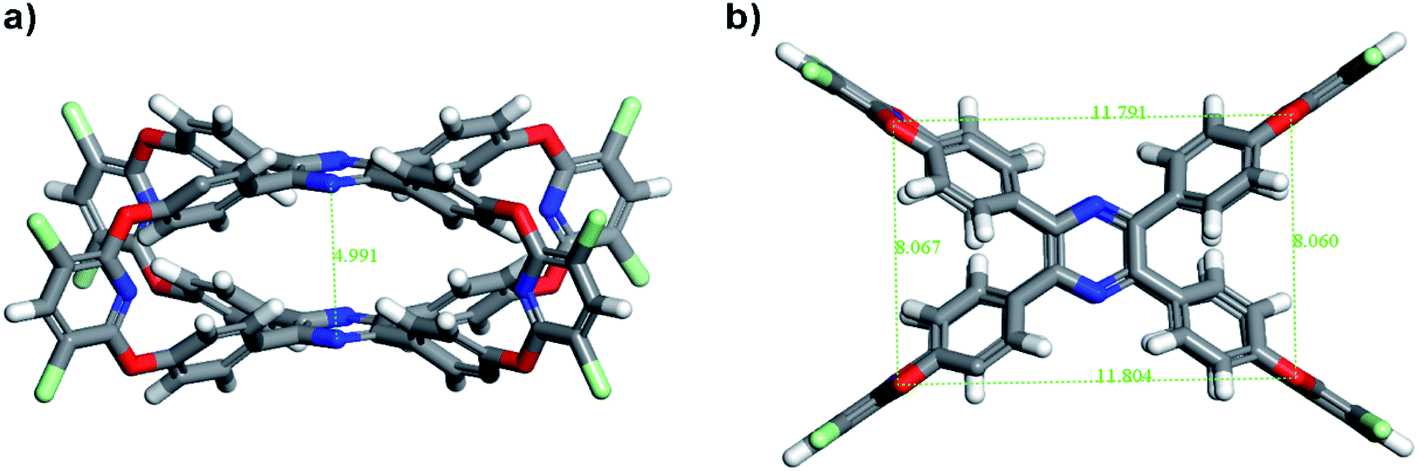 | ||
| Fig. 2 Simulated chemical structures of TPP-OMC, (a) side view and (b) top view. | ||
The phenyl rings of TPP formed a propeller-like structure, and the four blades (phenyl rings) rotated with angle of 40.125° because of the steric hindrance interaction. But, in the simulated structures of 4OH-TPP (minimum energy state), the form blades rotation was distinctly larger with torsion angle of 49.143° (Fig. S3†). In 4OH-TPP, the phenyl rings were only affected by steric hindrance of the themselves, and they can rotate relatively freely to form a minimum energy state with larger blades torsion. While, in TPP-OMC, the rotation of blades was not only affected by the steric hindrance in themselves, but also affected by the steric hindrance between two TPP molecules, which all together restricted the free rotation of phenyl rings after cage structures formed.37
With large intrinsic cavity, the pore performance of TPP-OMC in solid state was estimated by the N2 sorption experiment at 77 K (Fig. S7†). TPP-OMC was heated to 120 °C for 10 h in vacuum for desolvation. The result showed that the TPP-OMC can hardly adsorb N2 with the Brunauer–Emmett–Teller (BET) surface area of 6.62 m2 g−1, Langmuir surface area of 2.62 m2 g−1 and pore volume of 0.0022 cm3 g−1. The absence of interconnective pore channels may ascribe to the formation of interlocking mode, which was similar to the reported OMCs.15 Without the supporting of guest solvents, the intermolecular channels of TPP-OMC would collapse and tend to assemble into tight stacking mode, so that intermolecular pore channels would be “turn off”.40
In order to “turn on” the blocking intermolecular pore channels and non-connective intrinsic cavities, and further to avoid influences of “solvent effect” for intermolecular pore channels supporting. The TPP-OMC was used as monomer to construct covalently linked POP. By the virtue of eight react sites (–Cl), the OMC based POP with high valency was obtained through Yamamoto-type Ullmann cross-coupling reaction. After polymerization, the obtained TPP-pOMC was washed with common organic solvents (dichloromethane, tetrahydrofuran (THF), and acetone) several times and then Soxhlet extraction with methanol for 3 days at 100 °C for eliminating the unreacted TPP-OMCs or oligomers. The successful constructing of TPP-pOMC was characterized through the Fourier transform infrared (FT-IR). As the FI-IR spectra shown in Fig. S4,† the aromatic C–Cl bending vibrations at 1094 cm−1 was attenuated obviously after polymerization. The slight remaining signals of C–Cl bending vibration in TPP-pOMC was duo to the residues of unreacted sites in process of polymerization through nickel (0)-catalyzed Yamamoto-type Ullmann cross-coupling reaction. The aromatic C–O–C bonds vibrations at 1625 cm−1 was preserved well, and the peaks at 1572 and 1500 cm−1 that ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN of phenyl rings and pyridine were also preserved well, indicating the existing of TPP moieties and pyridine moieties. These results can illustrate that the cage structure was not broken in the process of polymerization. Corresponding to the powders X-ray diffraction experiments (PXRD) (Fig. S5†), the 2θ at around 20 degrees shown broaden peaks, which indicates that the TPP-pOMC was non-crystalline structure with poorly ordered linking. The transmission electron microscopy (TEM) (Fig. 3a and b) and scanning electron microscopy (SEM) (Fig. 3c and d) indicated that the TPP-pOMC formed random loose texture structures in the solid state, which were accorded with results of PXRD experiments. The thermal stability of TPP-pOMC was investigated by thermogravimetric analysis (TGA). As shown in Fig. S6,† under nitrogen atmosphere, there is 8% weight lose below 100 °C, originated from loss of the adsorbed guest solvent methanol. By heating to 500 °C, the weight of TPP-pOMC started to further lose. The results illustrated that the high valency of covalent cross-coupling TPP-pOMC possesses excellent thermal stability.
C and CN of phenyl rings and pyridine were also preserved well, indicating the existing of TPP moieties and pyridine moieties. These results can illustrate that the cage structure was not broken in the process of polymerization. Corresponding to the powders X-ray diffraction experiments (PXRD) (Fig. S5†), the 2θ at around 20 degrees shown broaden peaks, which indicates that the TPP-pOMC was non-crystalline structure with poorly ordered linking. The transmission electron microscopy (TEM) (Fig. 3a and b) and scanning electron microscopy (SEM) (Fig. 3c and d) indicated that the TPP-pOMC formed random loose texture structures in the solid state, which were accorded with results of PXRD experiments. The thermal stability of TPP-pOMC was investigated by thermogravimetric analysis (TGA). As shown in Fig. S6,† under nitrogen atmosphere, there is 8% weight lose below 100 °C, originated from loss of the adsorbed guest solvent methanol. By heating to 500 °C, the weight of TPP-pOMC started to further lose. The results illustrated that the high valency of covalent cross-coupling TPP-pOMC possesses excellent thermal stability.
 | ||
| Fig. 3 TEM images (a) and (b) and SEM (c) and (d) images of TPP-pOMC. | ||
To investigate the pore performances of TPP-pOMC, the N2 sorption analysis at 77 K was conducted after heating to 120 °C for 10 h in vacuum for desolvation (Fig. 4a). The TPP-pOMC showed a steep nitrogen adsorption at low pressure indicating the existing of micropores. Low-pressure hysteresis was extending to the lowest attainable pressures, which was associated with the irreversible uptake of gas molecules in the pore channels (or through pore entrances). This phenomenon probably means a swelling of polymer matrix at 77 K by nitrogen. After calculation from the N2 sorption isotherm of TPP-pOMC (Fig. S7†), the Brunauer–Emmett–Teller (BET) surface area was found to be 929.61 m2 g−1 (Langmuir surface area of 1909.56 m2 g−1), and pore volume was found to be 0.15 cm3 g−1, which increased significantly compare to the N2 sorption analysis of monomer TPP-OMC. These results could confirm that the blocking intermolecular pore channels and intrinsic cavities may already “turn on”. Further, using the Nonlocal Density Functional Theory (NLDFT) method, the pore size distribution of TPP-pOMC was confirmed. As shown in Fig. 4b, the main pore size was distributed at 5.3 Å, corresponding to the intrinsic cavity of monomer as calculated in Fig. 2a, and the presence of pores channels at 1.34 nm and 2.72 nm implied the formation of intermolecular pore channel between TPP-OMCs.
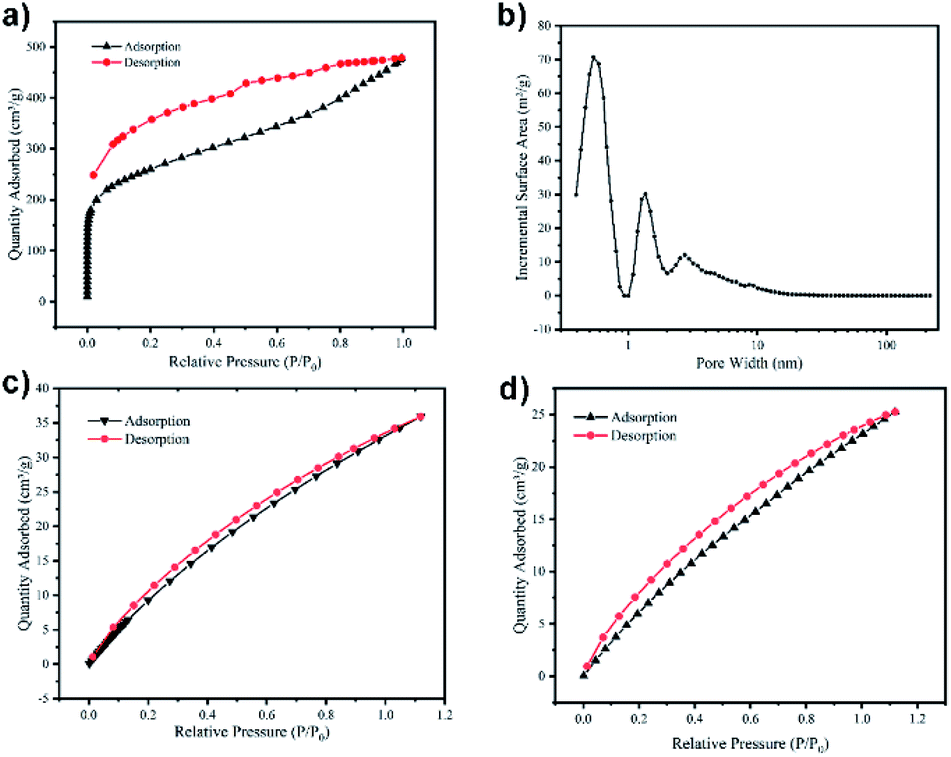 | ||
| Fig. 4 N2 sorption isotherms of TPP-pOMC at 77 K (a). Pore size distributions of TPP-pOMC calculated using the NLDFT method (b). CO2 sorption isotherms of pore size distributions at 273 K (c) and 298 K (d). | ||
The CO2 capture and storage capacity of TPP-pOMC was investigated at 273 K and 298 K. As shown in Fig. 4c and d The CO2 adsorption capacities of TPP-pOMC were 35.92 cm3 g−1 (7.05 wt%) at 273 K/1.0 bar and 25.27 cm3 g−1 (7.05 wt%) at 298 K/1.0 bar, respectively. These CO2 adsorption capacities were much better than those of its building blocks TPP-OMC, which were 8.49 cm3 g−1 (1.67 wt%) at 273 K/1.0 bar and 4.29 cm3 g−1 (0.84 wt%) at 298 K/1.0 bar, respectively. These results showed a full proof of the “turning on” of blocking intermolecular pore channels and intrinsic cavities. Using the Clausius–Clapeyron equation41 for the adsorption isotherms calculating, the isosteric enthalpies (Qst)42 of TPP-pOMC was found to be 25.59 kJ mol−1 (Fig. S8†). All these CO2 sorption experiments imply the TPP-pOMC could use as absorbent for CCS.
Conclusions
In summary, we have designed and synthesized a TPP-based oxacalixarene molecular cage TPP-OMC with a lager intrinsic cavity. By the virtue of eight reacting sites, the blocking intermolecular pore channels and intrinsic cavities in TPP-OMC were “turned on” after polymerized to high valency POPs (TPP-pOMC) through nickel (0)-catalyzed Yamamoto-type Ullmann cross-coupling reaction. The obtained TPP-pOMC showed fixed size pore channels originating from connective pore cavities of cages (5.3 Å) and intermolecular pore channels between cages (1.34 nm and 2.72 nm). Compared to the TPP-OMC, the TPP-pOMC shown obvious enhancement on their porous properties, including BET surface area, pore volume, and CO2 capture capacity that could be used as solid phase absorbents for COS. Our protocol works put forward a method for effective pore size controlling and pore properties enhancement.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (22005110), the China Postdoctoral Science Foundation (2020M672339) and the China Postdoctoral Innovative Talent Support Program (BX20200141). We thank the Analytical and Testing Center of Huazhong University of Science and Technology for related analysis.Notes and references
- S. Das, P. Heasman, T. Ben and S. L. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS.
- S. T. Wang, H. T. Li, H. A. Huang, X. H. Cao, X. D. Chen and D. P. Cao, Chem. Rev., 2022, 51, 2031–2080 CAS.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- L. F. Zou, Y. J. Sun, S. Che, X. Y. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H. C. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
- R. S. Haszeldine, Science, 2009, 325, 1647 CrossRef CAS PubMed.
- M. Younas, M. Rezakazemi, M. Daud, M. B. Wazir, S. Ahmad, N. Ullah, Inamuddin and S. Ramakrishna, Prog. Energy Combust. Sci., 2020, 80, 100849 CrossRef.
- Z. Q. Sun, Y. R. Liao, S. L. Zhao, X. Zhang, Q. Liu and X. Z. Shi, J. Mater. Chem. A, 2022, 10, 5174–5211 RSC.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Sci. Adv., 2022, 4, eaat9180 CrossRef PubMed.
- J. L. Wang and S. T. Zhuang, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- T. Q. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Y. Zhou, J. Niu, L. H. Li, Y. Y. Wang, J. Su, J. Li, X. G. Wang, W. D. Wang, W. Wang, J. L. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS PubMed.
- Y. F. Zeng, R. Q. Zou and Y. L. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- B. Li, Z. Guan, W. Wang, X. Yang, J. Hu and B. Tan, Adv. Mater., 2012, 24, 3390 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163 CrossRef CAS.
- Z. Wang, H. Ma, T. L. Zhai, G. Cheng, Q. Xu, J. M. Liu, J. K. Yang, Q. M. Zhang, Q. P. Zhang, Y. S. Zheng, B. Tan and C. Zhang, Adv. Sci., 2018, 5, 1800141 CrossRef.
- J. Du, H. Ouyang and B. Tan, Chem. – Asian J., 2021, 16, 3833–3850 CrossRef CAS PubMed.
- Y. Y. Tian and G. S. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- X. H. Li, Y. F. Yu, Q. F. Liu and Y. Z. Meng, ACS Appl. Mater. Interfaces, 2012, 4, 3627 CrossRef CAS PubMed.
- T. Ben, C. Y. Pei, D. L. Zhang, J. Xu, F. Deng, X. F. Jing and S. L. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 RSC.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS.
- C. Zhang, L. H. Peng, B. Y. Li, Y. Liu, P. C. Zhu, Z. Wang, D. H. Zhan, B. Tan, X. L. Yang and H. B. Xu, Polym. Chem., 2013, 4, 3663 RSC.
- H. Ma, J. J. Chen, L. X. Tan, J. H. Bu, Y. H. Zhu, B. Tan and C. Zhang, ACS Macro Lett., 2016, 5, 1039–1043 CrossRef CAS PubMed.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- A. I. Cooper, ACS Cent. Sci., 2017, 3, 544–553 CrossRef CAS PubMed.
- B. D. Egleston, K. V. Luzyanin, M. C. Brand, R. Clowes, M. E. Briggs, R. L. Greenaway and A. I. Cooper, Angew. Chem., Int. Ed., 2020, 59, 7362–7366 CrossRef CAS PubMed.
- X. L. Liu, J. Li, B. Gui, G. Q. Lin, Q. Fu, S. Yin, X. F. Liu, J. L. Sun and C. Wang, J. Am. Chem. Soc., 2021, 143, 2123–2129 CrossRef CAS PubMed.
- B. Gui, X. F. Liu, G. Yu, W. X. Zeng, A. Mal, S. L. Gong, C. L. Yang and C. Wang, CCS Chem., 2021, 3, 2054–2062 CrossRef CAS.
- B. Gui, H. M. Ding, Y. P. Cheng, A. Mal and C. Wang, Trends Chem., 2022, 4, 437–450 CrossRef CAS.
- Y. H. Jin, B. A. Voss, R. McCaffrey, C. T. Baggett, R. D. Noble and W. Zhang, Chem. Sci., 2012, 3, 874–877 RSC.
- Y. H. Jin, B. A. Voss, A. Jin, H. Long, R. D. Noble and W. Zhang, J. Am. Chem. Soc., 2011, 133, 6650–6658 CrossRef CAS.
- O. Buyukcakir, Y. Seo and A. Coskun, Chem. Mater., 2015, 27, 4149–4155 CrossRef CAS.
- J. X. Ma, J. Li, Y. F. Chen, R. Ning, Y. F. Ao, J. M. Liu, J. L. Sun, D. X. Wang and Q. Q. Wang, J. Am. Chem. Soc., 2019, 141, 3843–3848 CrossRef CAS PubMed.
- Q. Zhu, X. Wang, R. Clowes, P. Cui, L. J. Chen, M. A. Little and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 16842–16848 CrossRef CAS PubMed.
- K. C. Jie, Y. J. Zhou, Q. Sun, B. Li, R. Zhao, D. E. Jiang, W. Guo, H. Chen, Z. Z. Yang, F. H. Huang and S. Dai, Nat. Commun., 2020, 11, 1086 CrossRef CAS PubMed.
- C. Gropp, T. Q. Ma, N. Hanikel and O. M. Yaghi, Science, 2021, 370, 424 Search PubMed.
- Z. Wang, X. W. He, T. Y. Yong, Y. Miao, C. Zhang and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 512–519 CrossRef CAS PubMed.
- C. Zhang, Z. Wang, L. X. Tan, T. L. Zhai, S. Wang, B. Tan, Y. S. Zheng, X. L. Yang and H. B. Xu, Angew. Chem., Int. Ed., 2015, 54, 9244–9248 CrossRef CAS PubMed.
- Z. Wang, Y. Luo, T. L. Zhai, H. Ma, J. J. Chen, Y. J. Shu and C. Zhang, Org. Lett., 2016, 18, 4574–4577 CrossRef CAS PubMed.
- M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932–1937 RSC.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, J. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- A. Aijaz, N. Fujiwara and Q. Xu, J. Am. Chem. Soc., 2014, 136, 6790–6793 CrossRef CAS PubMed.
- V. Krungleviciute, L. Heroux, A. D. Migone, C. T. Kingston and B. Simard, J. Phys. Chem. B, 2005, 109, 9317–9320 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02343a |
| This journal is © The Royal Society of Chemistry 2022 |