
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemically stable piperidinium cations for anion exchange membranes†
Jinyuan Liabc,
Congrong Yangab,
Suli Wang*ab,
Zhangxun Xiaab and
Gongquan Sun *ab
*ab
aDivision of Fuel Cells and Battery, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: suliwang@dicp.ac.cn; gqsun@dicp.ac.cn
bKey Laboratory of Fuel Cells & Hybrid Power Sources, Chinese Academy of Sciences, Dalian 116023, China
cUniversity of Chinese Academy of Sciences, Beijing 100039, China
First published on 20th September 2022
Abstract
The chemical stability of the anion exchange membranes (AEMs) is determinative towards the engineering applications of anion exchange membrane fuel cells (AEMFCs) and other AEM-based electrochemical devices, yet remains a challenge due to deficiencies in the structural design of cations. In this work, an effective design strategy for ultra-stable piperidinium cations is presented based on the systematic investigation of the chemical stability of piperidinium in harsh alkaline media. Firstly, benzyl-substituted piperidinium was degraded by about 23% in a 7 M KOH solution at 100 °C after 1436 h, which was much more stable than pyrrolidinium due to its lower ring strain. The introduction of substituent effects at the α-C position was proved to be an effective strategy for enhancing the chemical stability of the piperidinium functional group. As a result, the butyl-substituted piperidinium cation showed no obvious structural changes after being treated in the 7 M KOH solution at 100 °C for 1050 h. Afterwards, GC-MS and NMR analysis indicated that the α-C atoms in the substituents of piperidinium are fragile to the nucleophilic attack of OH−. Based on the above results, the electronic and steric effects of different alkyl substitutions were analyzed. This work provides critical insights into the structural design of chemically stable piperidinium functional groups for the AEM and boosts its application in electrochemical devices, such as fuel cells and alkaline water electrolysis.
1 Introduction
The technology of alkaline exchange membrane fuel cells (AEMFCs) has attracted worldwide interest due to the potential to significantly reduce the cost of the device.1–3 Over the past decade, substantial progress has been made in improving the performance of AEMFCs, which has reached that of the most advanced proton exchange membrane fuel cells (PEMFCs).4–6 However, the lifetime of AEMFCs is still at least an order of magnitude lower than that of the PEMFCs, which is the most significant challenge of its development.7,8 The previous U.S. DOE milestone of <10% voltage degradation over a 2000 h hold test at 0.6 A cm−2 at >60 °C has not yet been achieved.3 The chemical degradation of an alkaline exchange membrane (AEM) under alkaline conditions is identified as one of the main factors for the unrecoverable performance loss of AEMFCs.8–11To meet the requirements of long-lasting AEMFCs, numerous experiments and theoretical studies have been conducted concerning the chemical stabilities of AEMs in alkaline media.10–13 For polymer backbones, aryl ether-free polymers have been designed, prepared and demonstrated to possess superior chemical stability, which is not influenced by the cationic functional group.14–16 Additionally, cations other than common benzyl quaternary ammonium (QA) have been introduced into AEMs as functional groups and developed to reduce their degradation and guarantee conductivity.10–12,17–20 One option is introducing alkyl spacers to isolate quaternary nitrogen from electron-withdrawing groups in the polymer backbone or attaching long alkyl groups to the quaternary nitrogen. It has been indicated that QA groups with carbon chain lengths between 3 and 6 would be less susceptible to degradation.18 However, the stability of these AEMs still could not fully meet the requirement of the fuel cells. Another solution is adopting novel cationic groups, such as imidazolium,21,22 phosphonium,23 sulfonium,24 organometallic25 and nitrogen-containing lipid heterocyclic cations (pyrrolidinium,26–28 piperidinium10,29–32 and spirocyclic QA cations33–35).
Remarkably, piperidinium cations possessing outstanding alkaline stability were reported by Marino and Kreuer in 2015 and attracted wide public attention.10 Since then, many research groups have designed and investigated the alkaline stable piperidinium-based AEMs.29–31,33,36–38 Jannasch et al. synthesized a series of AEMs with piperidinium cations and ether-free backbones, showing excellent conductivity (up to 124 mS cm−1 at 80 °C) and alkaline stability (>91% cations remaining after immersion for 2400 h in 2 M NaOH at 90 °C).31 In addition, Yan et al. reported a series of piperidinium-based AEMs with ether-free aromatic backbones, showing no significant changes in the chemical structure after being treated in 1 M KOH aqueous solution at 100 °C for 2000 h.29 In contrast, we found that piperidinium cations grafted on a benzyl-containing polymer (SEBS) degraded by 46% in 2 M KOH solution at 100 °C, which is comparable to benzyl quaternary ammonium functionalized SEBS (as shown in Table S1†). Coincidentally, Coates et al. also reported that piperidinium-based AEM with a polyethylene backbone degraded up to 20% while being treated in 1 M KOH aqueous solution at just 80 °C for 30 days.39 Therefore, a big controversy has arisen over the stability of piperidinium functional groups, which are greatly dependent on the surrounding chemical environment. Thus, systematically investigating the chemical stability of piperidinium cations is very important for designing and optimizing the molecular structure to enhance the chemical stability of this kind of functional group.
Herein, we have systematically explored the stability of piperidinium cations in harsh alkaline conditions. The chemical stability of benzyl-substituted piperidinium was first explored and compared with QA and pyrrolidinium. Then, the detailed degradation routes of piperidinium cations were revealed by a combination of 1H nuclear magnetic resonance (1H NMR) spectroscopy and gas chromatography-mass spectrometry (GC-MS). Based on the above research, electron-donating substituents were introduced into the piperidinium and their effects on the chemical stability of the cation were carefully examined by experimental measurements. This simple strategy of structural design is very effective for significantly improving the chemical stability of the piperidinium cation. This work elaborates on the structure–stability relationships of piperidinium and will provide further insight into the molecular design of the stable piperidinium cation functional group for long-lifetime AEMs in fuel cell environments.
2 Experimental section
2.1 Materials
The following chemicals were used without further treatment: Benzyl chloride (Sinopharm Chemical Reagent Co., Ltd), trimethylamine solution (Aladdin, 30wt%), N-methylpiperidine (Macklin, >97%), N-methylpyrrolidine (Macklin, 98%), ethyl acetate (Tianjin Damao Chemical, ≥99%), 1-bromobutane (Macklin, >99%), 2-iodopropane (Aladdin, 99%), 1-bromohexane (Macklin, 99%), chloroform (Tianjin Damao Chemical, ≥99%), potassium hydroxide (Tianjin Damao Chemical, ≥85%), anhydrous methanol (Tianjin FUYU Fine Chemical), N,N-dimethylbenzylamine (Macklin, 99%), benzyl alcohol (Macklin, ≥99%), N-benzylpiperidine (Macklin, >98%), deuterium oxide (Qingdao Tenglong Weibo Technology, 99.9%), chloroform-d (Qingdao Tenglong Weibo Technology, 99.8%), dimethyl sulfoxide-d6 (Macklin, 99%).2.2 Synthesis of model compounds
Benzyltrimethylammonium chloride ([BTMA][Cl]) was synthesized by stirring a mixture of benzyl chloride (4 ml) and trimethylamine (6 ml) at room temperature. Afterwards, the mixture was added to an excess of ethyl acetate to precipitate the target solid. The product was washed twice with ethyl acetate. The powder was finally collected and dried under vacuum and at 90 °C to obtain [BTMA][Cl]. White solid. 1H NMR (400 MHz, D2O, ppm, Fig. S2†): δ 7.73–7.23 (Ha–c), δ 4.39 (Hd), δ 3.01 (He).N-Benzyl-N-methylpyrrolidinium chloride ([BzPyr][Cl]) was synthesized by stirring a mixture of benzyl chloride (12 ml) and N-methylpyrrolidine (12 ml) at 30 °C. The mixture reacted rapidly and was added to an excess of ethyl acetate to precipitate the target solid. The product was washed twice with ethyl acetate. The powder was finally collected and dried under vacuum and at 90 °C to obtain [BzPyr][Cl]. White solid. 1H NMR (400 MHz, D2O, ppm, Fig. S3†): δ 7.56 (Ha-c), δ 4.48 (Hd), δ 3.59, 3.40 (Hf), δ 2.92 (He), δ 2.23 (Hg).
Piperidinium cations with different substituents were synthesized by the SN2 reaction between N-methylpiperidine and the respective halohydrocarbon. Here, the synthesis route for N-benzyl-N-methylpiperidinium chloride ([BzPip][Cl]) is provided as an example. The syntheses of other piperidinium cations followed a similar procedure and are provided in the ESI.† [BzPip][Cl] was synthesized by stirring a mixture of benzyl chloride (6 ml) and N-methylpiperidine (6 ml) at 40 °C. Afterwards, the mixture was added to an excess of ethyl acetate to precipitate the target solid. The powder was washed twice with ethyl acetate, collected and dried under vacuum and at 90 °C to obtain [BzPip][Cl]. White solid. 1H NMR (400 MHz, D2O, ppm, Fig. S1†): δ 7.56–7.41 (Ha-c), δ 4.41 (Hd), δ 3.37–3.18 (Hf), δ 2.87 (He), δ 1.85 (Hg), δ 1.66, 1.53 (Hh).
2.3 Structural characterization
1H and 13C NMR spectra were obtained on a Bruker AVANCE III 400 MHz instrument using D2O, CDCl3 or DMSO-d6 as a reference or an internal deuterium lock. The chemical shift data of each signal are given in units of δ (ppm) relative to the solvent signal or tetramethyl silane (TMS) where δ (TMS) = 0.MS analysis was conducted using an Agilent 6540 Q-TOF. A dual electrospray ionization (ESI) source was applied and operated in the positive mode.
A GC-MS analysis was conducted using an Agilent 8890 GC equipped with a DB-35MS column (60 m × 250 μm × 0.25 μm) and a 7250 Q-TOF MS. The temperature of each run was ramped from 50 to 310 °C at 10 °C min−1, and then maintained at 310 °C for 10 min.
2.4 Alkaline stability of small model compounds
The alkaline stability of model compounds was confirmed by analyzing the chemical structural changes before and after they were treated in 7 M KOH solution at 100 °C in a sealed Teflon container. 1H NMR spectroscopy, MS or GC-MS were adopted to determine the chemical structures.2.5 DFT calculation for the degradation pathways of [Bzpip]+ and [Bzpyr]+
Relative solvated Gibbs free energies are shown in Italic bold, unit: kcal mol−1. Calculated at the B3LYP/6-311++G(2d,p) level with the PCM solvation model (solvent = water). All geometry optimizations were performed at the B3LYP/6-311++G(2d,p) level with the Gaussian 16 (ref. 40) packages. The PCM solvation model (solvent = water) was used. To save computational costs, the “g09defaults” keyword was used in all calculations. Frequency calculations were performed with the optimized geometries to ensure only one imaginary frequency for transition state structures and zero imaginary frequencies for reactant or product intermediate structures. In general, different conformations for a certain transformation were found. However, only the conformation with the lowest free energy was adopted for this transformation.3 Results and discussion
To clarify the chemical stability of the piperidinium cation functional group in alkaline environments, three series of small molecule compounds were designed as shown in Fig. 1. First, the effects of different N-containing functional groups on alkaline stability were investigated by comparing the degradation behavior of N-benzyl-N-methylpiperidinium ([BzPip]+), benzyltrimethylammonium ([BTMA]+) and N-benzyl-N-methylpyrrolidinium ([BzPyr]+). [BTMA]+ was typically used as a benchmark to judge the alkaline stability of novel functional groups.10,32,41,42 Then, several factors were taken into account for designing the cation functional groups. One is the selection of alkyl groups as electron-donating substituents, which could decrease the charge density of the nitrogen atom and thus decrease the reactivity. Besides, varying the alkyl chain length would alter the extent of steric effects and change the energy barrier of the degradation pathway. Thus, methyl, butyl and hexyl-substituted piperidinium cations were designed (as shown in Fig. 1b) and investigated in terms of alkaline stability. Besides the electron and steric effects, varying the number of α-C and β-H atoms by tuning the structures of substituents (Fig. 1c) would influence the sensitivity to nucleophilic attack. Thus, we also respectively introduced isopropyl and isobutyl groups into the piperidinium group.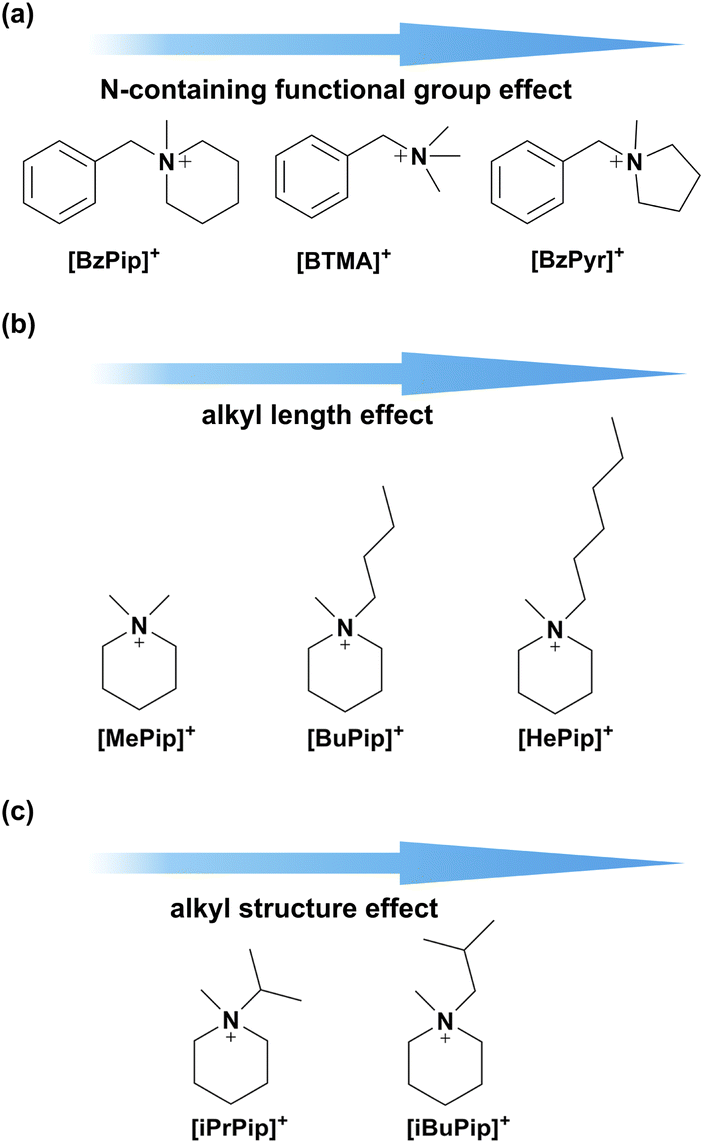 | ||
| Fig. 1 Molecular designs for the optimization of alkaline stable cations for AEMs: (a) modulation of the N-containing functional group; (b) and (c) modulation of the alkyl substituent. | ||
3.1 The N-containing functional group effect on alkaline stability
The chemical structures and the purity of model compounds were confirmed by 1H and 13C NMR spectra (Fig. S1–S3†). To efficiently and precisely judge the alkaline stability, the model compounds were treated in 7 M KOH solution at 100 °C, where the molar ratio of cation and OH− was controlled at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35. Afterwards, the structures of samples were analyzed by 1H and 13C NMR spectroscopy.
:35. Afterwards, the structures of samples were analyzed by 1H and 13C NMR spectroscopy.
During the testing of [BzPip]+, [BTMA]+ and [BzPyr]+, insoluble liquid substances appeared on the surfaces of the piperidinium and pyrrolidinium solutions, while the quaternary ammonium solution was always transparent. To avoid the phase separation and better determine the degradation degree of piperidinium and pyrrolidinium, to the samples were added DMSO-d6 and a slight excess of trifluoroacetic acid (TFA), forming homogeneous solutions. Subsequently, 1H NMR spectroscopy (Fig. 2 and S4–S6†) was used to further determine the chemical structure changes of these cations. After immersion in 7 M KOH solution at 100 °C, several new signals appeared in the 1H NMR spectra of the cations. This indicates that degradation happened during the tests. In order to clarify the degradation degree, we calculated the decrease in the integral area of the N-methyl proton peak (d) relative to that of phenyl proton peaks (a–c) in Fig. 2. [BzPip]+ and [BTMA]+ cations degraded about 9.5% after being treated for 883.5 h and about 23% for the extended period of 1436 h, but [BzPyr]+ suffered a severe degradation up to 25.7% only for 795.5 h. [BzPip]+ showed comparable chemical stability to [BTMA]+, while [BzPyr]+ is much more fragile than [BzPip]+ and [BTMA]+. The alkaline stability results here correlate well with the experimental results obtained by Marino and Kreuer10 and could be explained by the high ring strain of the pyrrolidinium cation.43–45 The lower ring strain of the 6-membered ring was proposed to render the piperidinium with greater tolerance to nucleophilic attack.
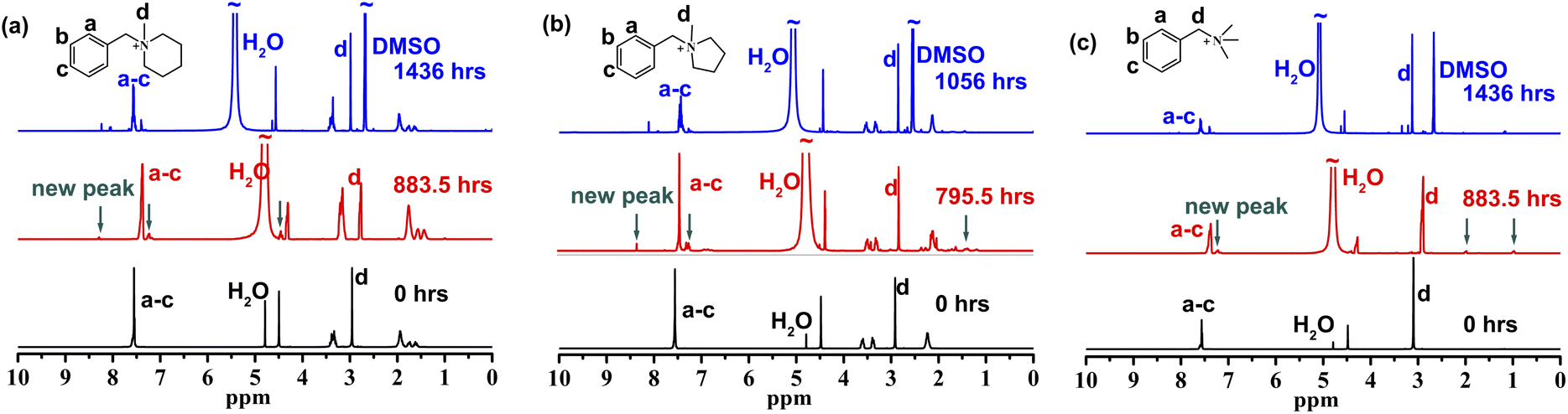 | ||
| Fig. 2 1H NMR of [BzPip]+ (a), [BzPyr]+ (b) and [BTMA]+ (c) before and after immersion in 7 M KOH solution at 100 °C. | ||
The results from the test indicate that piperidinium is a potential alkali-stable functional group for AEMs. Predictably, clarifying the degradation mechanism should be a prerequisite for elucidating the reason for degradation and thus designing a better chemical structure and enhancing the stability of piperidinium-based AEM materials. To elucidate the clear cause of the degradation of benzyl-substituted piperidinium, the degradation mechanism of [BzPip]+ was investigated as follows. First, we used chloroform to successfully extract the insoluble liquid substances from the mixture of the aqueous solutions after being treated. Second, the compositions of organic and aqueous phases were analyzed by GC-MS (Fig. 3a and S7†), MS (Fig. S8†) and 1H NMR spectroscopy (Fig. S9†). The GC-MS analysis suggested the presence of N-methylpiperidine, benzyl alcohol and N-benzylpiperidine. These substances were derived from the SN2 substitution of OH− on [BzPip]+, in which hydroxides attacked the α-C atoms in the methyl and benzyl groups with partial positive charges. Fig. 3b illustrates the degradation routes of [BzPip]+. The piperidinium ring remained intact during the whole test, indicating that it is chemically stable in harsh alkaline conditions. Particularly, the calculation of the integral values of the corresponding byproduct peaks in GC-MS revealed that the benzyl group is more susceptible to nucleophilic attack, which is consistent with an earlier report.10 Since the phenyl group in [BzPip]+ is electron-withdrawing and the benzyl carbon atom possesses more partial positive charges than the methyl carbon atom, hydroxides are more likely to attack the benzyl carbon atom. Therefore, eliminating the adverse effect of benzyl groups on the chemical stability of the piperidinium cation is necessary.
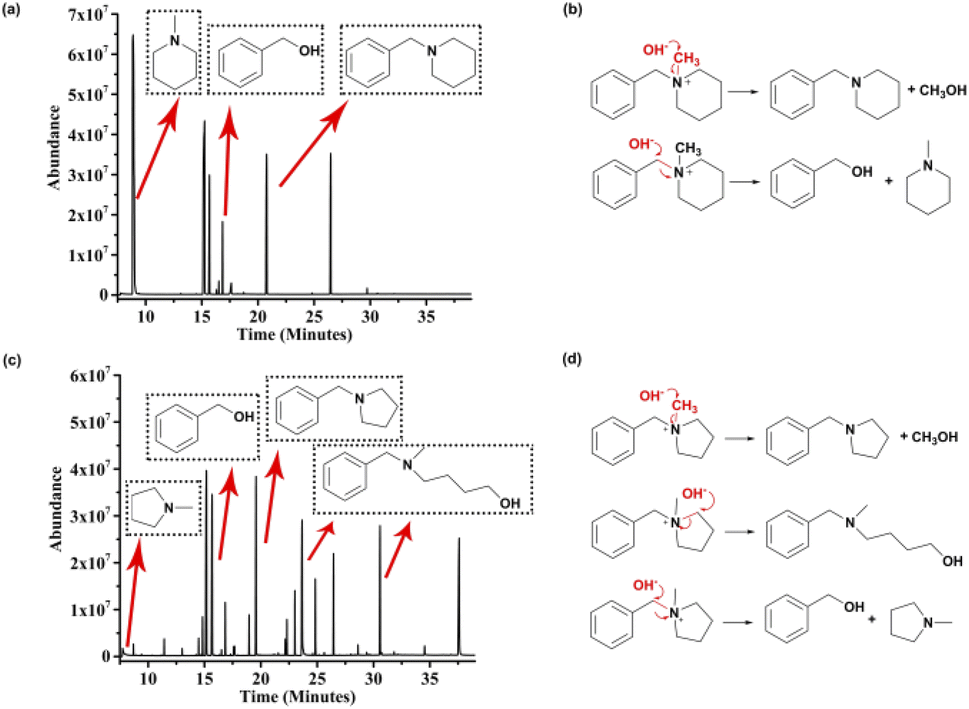 | ||
| Fig. 3 GC-MS analysis of possible byproducts and potential degradation routes for [BzPip]+ (a and b) and [BzPyr]+ in 7 M KOH at 100 °C (c and d). | ||
To evaluate the contributing factors to the alkaline stability of piperidinium cations, the degradation pathways of [BzPyr]+ and [BTMA]+ were also studied as follows. As illustrated in Fig. S13–S15,† [BTMA]+ degraded through SN2 reactions at benzyl and methyl sites. For [BzPyr]+, signals for N-methylpyrrolidine, benzyl alcohol and N-benzylpyrrolidine were present in the GC-MS of the [BzPyr]+ organic phase (Fig. 3c). In addition, signals in GC with longer retention times (23.658 and 30.570 min) correspond to 4-hydroxybutyl-methyl-benzylamine. This suggests that the α-C atoms in the pyrrolidinium ring were also attacked by hydroxides besides those in the benzyl and methyl groups (Fig. 3d), which resulted in the opening of the pyrrolidinium ring and distinguished [BzPyr]+ from [BzPip]+. Herein, the difference in the degradation routes between [BzPip]+ and [BzPyr]+ can provide detailed information on the relationship between the stability and molecular structure of nitrogen-containing lipid heterocyclic rings. According to previous reports, the rate constants for the SN2 reaction at methyl carbon between piperidinium/pyrrolidinium and strong nucleophiles (such as CH3O−) are similar, while the SN2 reaction at the heterocyclic ring of the piperidinium cation is much slower than that of the pyrrolidinium cation.43–45 This is ascribed to the lower strain energy and steric hindrance posed by the C–H equatorial bond of the carbon atom at position 2 of the piperidinium cation.43,45 Additionally, the free energy barrier of the SN2 reaction at the benzyl carbon and methyl carbon was reported to be comparable.11 In this work, the SN2 reaction at the heterocyclic ring was only observed in the degradation of [BzPyr]+. Thus, different extents of ring strain and conformation resulted in the difference in reactivity for different N-containing heterocycles, which is responsible for the difference in the alkaline stability of piperidinium and pyrrolidinium cations.
More importantly, DFT calculations were introduced in our work to further illustrate our view on the stability and degradation pathways of different cations and give a much better insight into chemically stable functional groups for AEM.
Proposed reaction pathways of [Bzpip]+ and [Bzpyr]+ (Fig. S16 and S17†) were studied by DFT calculations (Results are shown in Fig. 4 and 5).
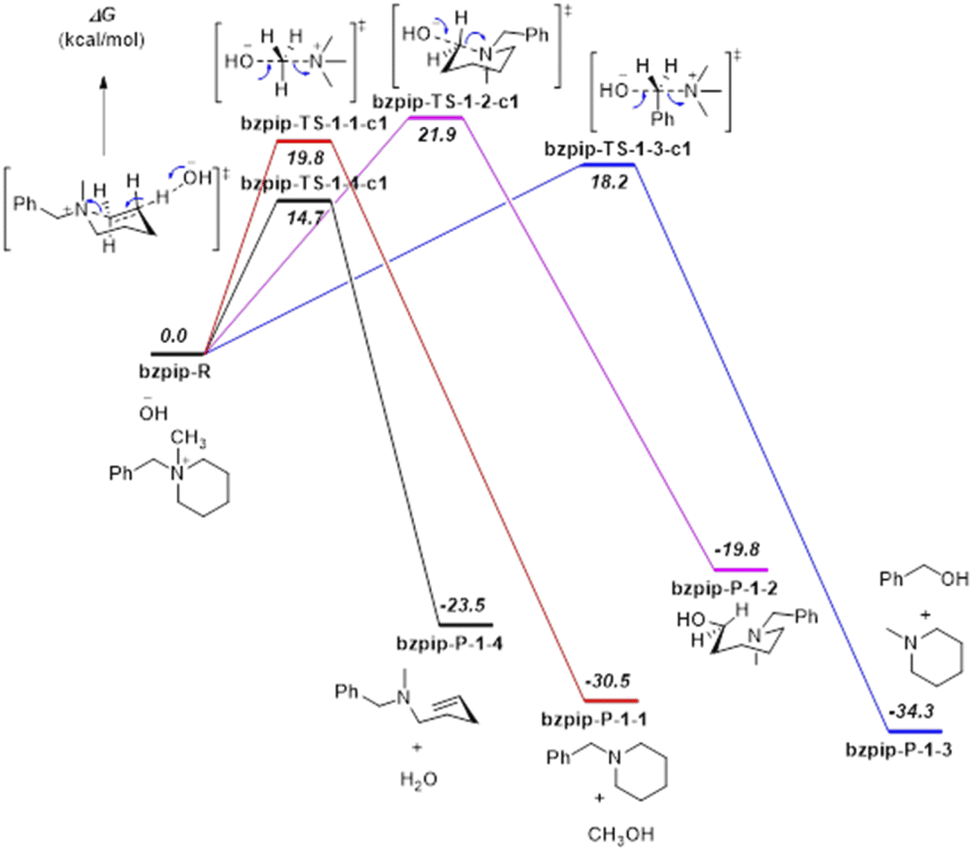 | ||
| Fig. 4 DFT calculation results for the proposed reaction pathways of [Bzpip]+. Relative solvated Gibbs free energies are shown in italic bold, unit: kcal mol−1. Calculated at the B3LYP/6 6-311++G(2d,p) level with the PCM solvation model (solvent = water). | ||
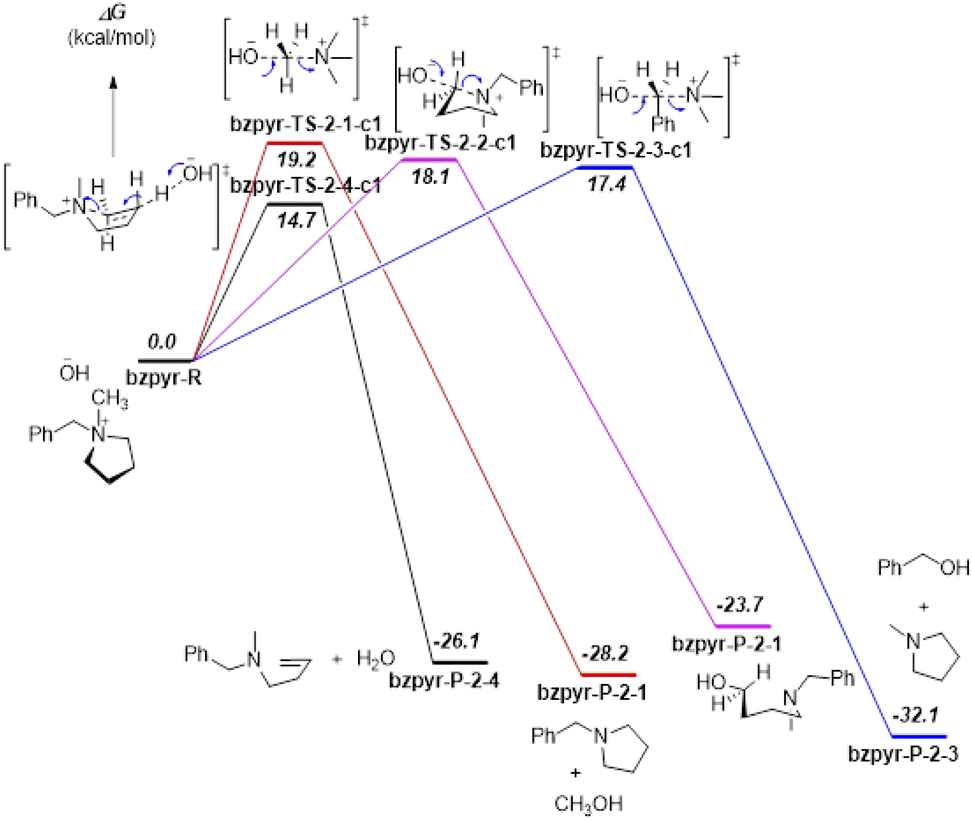 | ||
| Fig. 5 DFT calculation results for the proposed reaction pathways of [Bzpyr]+. Relative solvated Gibbs free energies are shown in italic bold, unit: kcal mol−1. Calculated at the B3LYP/6 6-311++G(2d,p) level with the PCM solvation model (solvent = water). | ||
From the DFT calculation results, all the nucleophilic substitution reaction free energies of [Bzpyr]+ were lower than that of [Bzpip]+, indicating that [Bzpyr]+ would degrade through nucleophilic substitution more easily than [Bzpip]+ when attacked by OH−, which is in full agreement with our experimental results. Moreover, the order of the nucleophilic substitution reaction free energies of [Bzpyr]+ was 2–3 (17.4 kcal mol−1) < 2–2 (18.1 kcal mol−1) < 2–1 (19.2 kcal mol−1), suggesting that the stability order is α-C atoms in the methyl group > α-C atoms in the pyrrolidinium ring > α-C atoms in the benzyl group. However, in [Bzpip]+, the stability order of the [Bzpip]+ is α-C atoms in the piperidinium ring > α-C atoms in the methyl group > α-C atoms in the benzyl group. In the above experimental section, the degradation ratios in different positions of [Bzpyr]+ are α-C atoms in the methyl group (∼33.0%) < α-C atoms in the pyrrolidinium ring (∼34.5%) < α-C atoms in the benzyl group (∼38.5%). From the results, it can be concluded that the experimental results were entirely consistent with DFT calculations. The degradation ratio of α-C atoms in the [Bzpip]+ benzyl group is nearly 80%, which is much greater than that of α-C atoms in methyl groups (free energies and proportion of each substitution reaction is shown in Fig. S18†). The free energy barrier (21.9 kcal mol−1) of α-C atoms in piperidinium by DFT calculation is the most significant in all the reaction pathways, so it can be used to explain that the [Bzpip]+ cation attacked by OH− would produce benzyl alcohol and methylpiperidine and there is no product of the SN2 reaction at the piperidinium cation ring in our experiment.
DFT results have shown that the elimination reaction free energies of both [Bzpyr]+ or [Bzpip]+ are the lowest among all the reaction pathways, which is in accord with the theoretical predictions shown by other reports.18,46 Surprisingly, the product of the elimination reaction was not found in the NMR analysis (Fig. 2 in the manuscript and Fig. S9 and S12 in ESI†), which left us to speculate that there was no elimination reaction in our experiment. The possible reason for this phenomenon is as follows: Hofmann elimination through the E2 mechanism requires the formation of the anti-periplanar conformation, which is not favorable for the 5-membered pyrrolidinium ring.10,45 However, β-protons in the strain-free 6-membered piperidinium ring are expected to move into an anti-periplanar position more easily.10,45 Marino and Kreuer pointed out the geometric constraint of piperidinium on the elimination transition state, in which bond lengths and angles were unfavorable.10 Thus, the inhibition of E2 elimination in the piperidinium cation could be explained by the adverse geometry in the transition state.
In this section, we have combined experimental and DFT calculation methods to explain our view in detail. The results illustrate the stability and degradation pathways of different cation functional groups and could give a better insight into a chemically stable piperidinium cation functional group for the AEM.
3.2 Substituent effect on the alkaline stability of piperidinium
From the above results, it can be concluded that the piperidinium ring would remain relatively stable under harsh alkaline conditions, while the benzyl and methyl substituents would be more liable to nucleophilic attack. Thus, [BzPip]+ is not fully suitable for the stability requirements of AEMs. In addition, N-alkyl substitution has an electron-donating effect and can promote micro-phase separation47,48 and help in constructing a cross-linking structure49,50 to modulate AEM properties and morphology. Thus, we adopted this method for piperidinium and studied its effect on the alkaline stability of the cation. The alkyl-substituted piperidinium cations investigated in this part are denoted as [MePip]+, [BuPip]+, [HePip]+, [iPrPip]+. [iBuPip]+ (Fig. 1b and c).During the test, insoluble liquid substances appeared in [HePip]+, [iPrPip]+ and [iBuPip]+ solutions. However, the appearance of new signals is not significant in 1H NMR spectrum apart from the one appearing at 8.0–8.5 ppm (Fig. 6c–e and S22–S24†), indicating that the extents of degradation were indeed low compared to [BzPyr]+. In contrast, only a small amount of new substance was observed in the solutions of [MePip]+ and [BuPip]+. The new signals in their 1H NMR spectra were also very weak, suggesting their excellent alkaline stability (Fig. 6a, b, S20 and S21†). The percentage of remaining cations was then calculated based on the ratio of the N-methyl peak (peak b) relative to the terminal methyl of the alkyl group (for [HePip]+) or to position 4 of the piperidinium (peak a, for other cations).
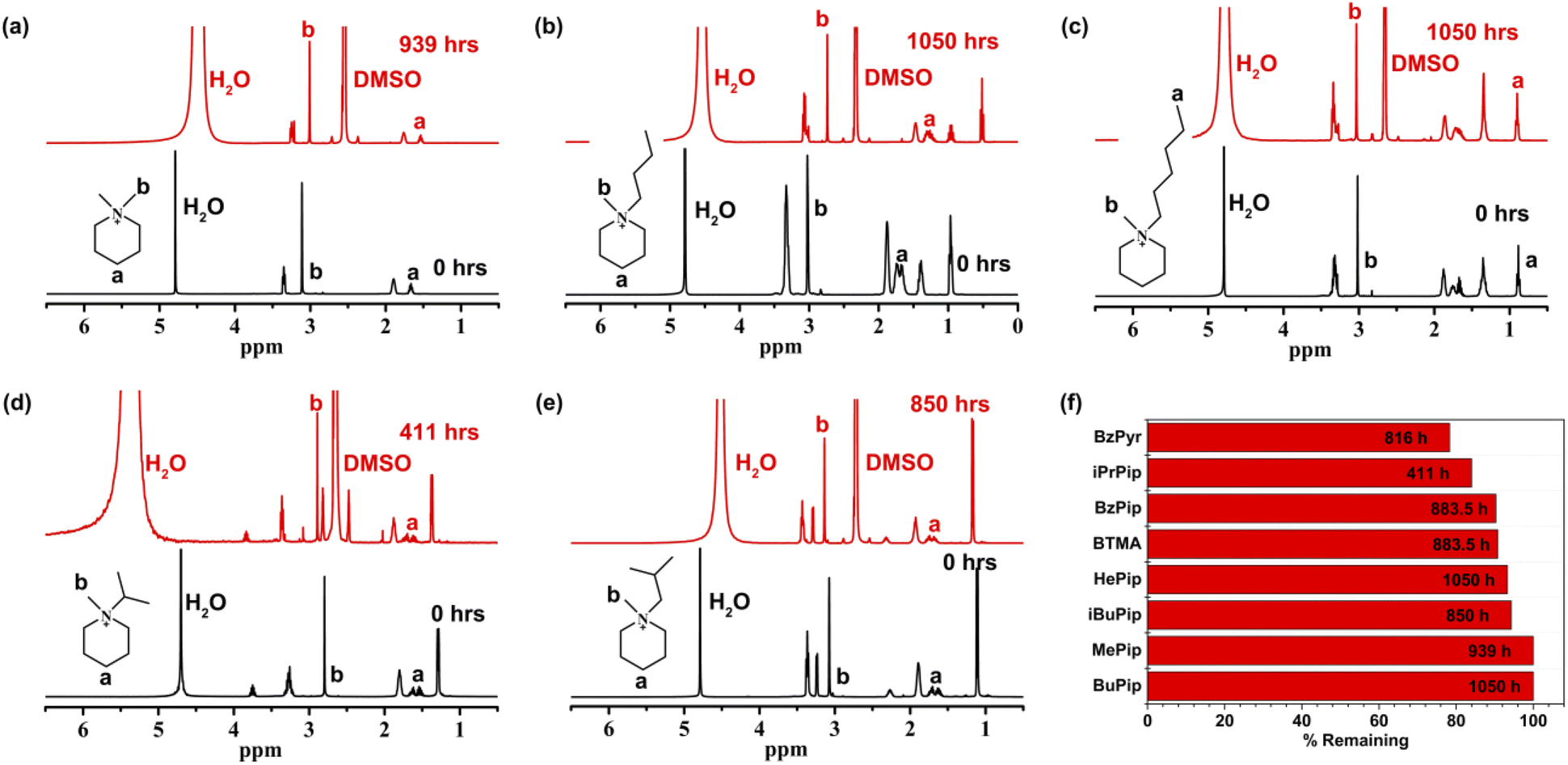 | ||
| Fig. 6 1H NMR spectra of [MePip]+ (a), [BuPip]+ (b), [HePip]+ (c), [iPrPip]+ (d) and [iBuPip]+ (e) before and after immersion in 7 M KOH solution at 100 °C and the remnants of these cations after the test (f). | ||
As shown in Fig. 6f, three n-alkyl-substituted piperidinium cations, [MePip]+, [BuPip]+ and [HePip]+ showed enhanced alkaline stability compared to [BzPip]+. [MePip]+ and [BuPip]+ were particularly stable, with no obvious degradation after the 939 h and 1050 h tests, respectively. [HePip]+ possessed good stability, with 93.3% remaining after the 1050 h test. The results indicated that the introduction of electron-donating n-alkyl groups could protect piperidinium from attack by hydroxides with strong nucleophilicity, in which the charge density on the α-C atoms may decrease. Besides the electronic effect on SN2 reactions, the possibility of the E2 elimination reaction was also investigated. GC-MS analysis of the [HePip]+ solution after the test (Fig. S25†) indicated the presence of N-methylpiperidine and N-hexylpiperidine. This suggests that [HePip]+ degrades through SN2 reactions on the α-C atoms in N-methyl and hexyl groups (Fig. 7). No evidence of E2 elimination products was observed from both NMR spectroscopy and GC-MS (Fig. 6c and S25†). This is in accord with the report by Pivovar et al., which supposed that QAs with an n-alkyl chain length of 4–6 possessed relatively higher E2 elimination barriers than QAs with shorter n-alkyl chains.18 This higher E2 barrier is derived from the enhanced steric hindrance when the chain is longer than four atoms. However, the steric hindrance of the hexyl group seems ineffective in protecting the cation from SN2 degradation in this study, which is in agreement with the slightly lower computational SN2 barrier of hexyl-substituted trimethylammonium than that of butyl-substituted trimethylammonium. One possible explanation is that the hexyl group not only has no additional steric hindrance to the nucleophilic attack on the hexyl group, but it is more flexible than the butyl group, which creates the space for the SN2 reaction between hydroxides and the N-methyl group. GC-MS results showed that the integral area of the signal for N-hexylpiperidine is six times larger than that of the signal for N-methylpiperidine, indicating that the SN2 reaction between N-methyl and hydroxides has a great competitive advantage.
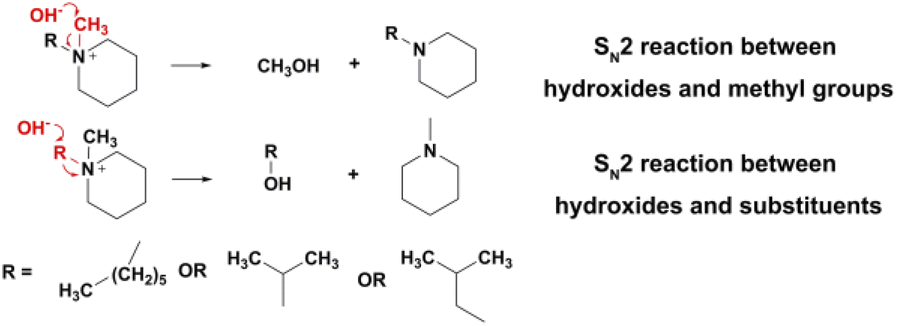 | ||
| Fig. 7 Potential degradation routes for alkyl-substituted piperidinium cations in 7 M KOH at 100 °C. | ||
Among the cations in this study, [iPrPip]+ degraded about 16% only after 411 h of immersion in a harsh alkaline solution, which is the most unstable cation in this part. [iPrPip]+ suffered severe degradation up to 12% for only 96 h, which is even greater than [BzPyr]+ with 9.6% degradation for 716.5 h. Since the isopropyl group has six β-H atoms, [iPrPip]+ is expected to be extremely susceptible to Hofmann elimination. However, GC-MS analysis only revealed the presence of N-methylpiperidine and N-isopropylpiperidine, corresponding to the SN2 reaction between hydroxides and α-C atoms in N-isopropyl and methyl groups (Fig. 7 and S27†). Despite this, Hofmann elimination cannot be completely excluded from the degradation process of [iPrPip]+, as the elimination product propene is only slightly soluble in water and easily evaporates into the ambient environment during the chemical stability test. In contrast to [iPrPip]+, [iBuPip]+ is much more stable, with 94.3% remaining after the 850 h test. Its long-term stability is comparable to [HePip]+ (as shown in Table S3†). GC-MS analysis revealed that [iBuPip]+ also underwent SN2 degradation. However, the contents of N-methylpiperidine and N-isobutylpiperidine are comparable, suggesting that the regioselectivity of the SN2 reaction between hydroxide and [HePip]+ is not apparent. This indicates that steric hindrance posed by the isobutyl group has a similar effect on both α-C atoms in the N-butyl and methyl groups.
Thus, after carefully examining the effects of substitution on the chemical stability of piperidinium, the ultra-high chemical stabilities of [MePip]+ and [BuPip]+ were probably derived from both the electron-donating effects of the alkyl groups and high degradation energy barriers. Additionally, the steric interference exerted by bulky alkyl groups could also reduce the SN2 reactivity of the N-methyl group in [BuPip]+. The stabilities of [MePip]+ and [BuPip]+ were also compared quantitatively with other kinds of cations reported in the studies (Table S3†).10,28,30,39,51 Note that [MePip]+ and [BuPip]+ showed superior alkaline stability even in harsh conditions. The results here shed light on the effectiveness of enhancing chemical stability by introducing and tuning alkyl substituents in piperidinium cations.
4 Conclusions
In this work, we have demonstrated that benzyl-substituted piperidinium ([BzPip]+) possesses comparable chemical stability to benzyl-substituted QA ([BTMA]+) and much higher stability than benzyl-substituted pyrrolidinium ([BzPyr]+). [BzPip]+ followed SN2 degradation routes, but the N-containing heterocycle remained stable because of its strain-free structure, steric hindrance and unfavored geometry in the transition state (possessing the highest energy barrier of 21.9 kcal mol−1). Based on this, we demonstrated the design strategy of chemically stable piperidinium cations by introducing alkyl substituents. This strategy exploits not only the electron-donating inductive and steric effects of alkyl groups, but also high degradation energy barriers rendered by long alkyl chains. Particularly, this simple and effective strategy enables us to find two ultra-stable cations ([MePip]+ and [BuPip]+), which showed no obvious degradation in harsh alkaline conditions. These findings can provide a solid experimental foundation for the molecular design of prevailing piperidinium-based AEMs, which would be excellent candidates for long-term AEMFCs and other AEM-based electrochemical energy systems. Further study should focus on the demonstration of this kind of alkali-stable piperidinium functional group in AEMs.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully thank the financial support from The National Key Research and Development Program of China (2021YFB4001204).References
- H. A. Firouzjaie and W. E. Mustain, ACS Catal., 2019, 10, 225–234 CrossRef.
- Y. Yang, H. Peng, Y. Xiong, Q. Li, J. Lu, L. Xiao, F. J. DiSalvo, L. Zhuang and H. D. Abruña, ACS Energy Lett., 2019, 4, 1251–1257 CrossRef CAS.
- L. Shi, B. P. Setzler, K. Hu, C. M. Weiss, S. Matz, Y. Xue, Z. Xu, Z. Zhuang, S. Gottesfeld and Y. Yan, J. Electrochem. Soc., 2020, 167, 144506 CrossRef CAS.
- T. J. Omasta, A. M. Park, J. M. LaManna, Y. Zhang, X. Peng, L. Wang, D. L. Jacobson, J. R. Varcoe, D. S. Hussey, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2018, 11, 551–558 RSC.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 166, F637–F644 CrossRef CAS.
- S. Maurya, S. Noh, I. Matanovic, E. J. Park, C. N. Villarrubia, U. Martinez, J. Han, C. Bae and Y. S. Kim, Energy Environ. Sci., 2018, 11, 3283–3291 RSC.
- C. G. Arges and L. Zhang, ACS Appl. Energy Mater., 2018, 1, 2991–3012 CrossRef CAS.
- W. E. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805–2838 RSC.
- J. Muller, A. Zhegur, U. Krewer, J. R. Varcoe and D. R. Dekel, ACS Mater. Lett., 2020, 2, 168–173 CrossRef CAS PubMed.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- S. Chempath, J. M. Boncella, L. R. Pratt, N. Henson and B. S. Pivovar, J. Phys. Chem. C, 2010, 114, 11977–11983 CrossRef CAS.
- S. Pusara, S. Srebnik and D. R. Dekel, J. Phys. Chem. C, 2018, 122, 11204–11213 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y.-K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS PubMed.
- C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438–449 CrossRef CAS.
- A. Amel, L. Zhu, M. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2014, 161, F615–F621 CrossRef CAS.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CrossRef CAS.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CrossRef CAS.
- H. Long and B. S. Pivovar, ECS Electrochem. Lett., 2015, 4, F13–F16 CrossRef CAS.
- A. Zhegur, N. Gjineci, S. Willdorf-Cohen, A. N. Mondal, C. E. Diesendruck, N. Gavish and D. R. Dekel, ACS Appl. Polym. Mater., 2020, 2, 360–367 CrossRef CAS.
- K. M. Hugar, H. A. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed.
- Y. Z. Zhuo, A. L. Lai, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Mater. Chem. A, 2015, 3, 18105–18114 RSC.
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik, E. B. Lobkovsky, H. D. Abruna and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161–18164 CrossRef CAS.
- B. Zhang, S. Gu, J. Wang, Y. Liu, A. M. Herring and Y. Yan, RSC Adv., 2012, 2, 12683–12685 RSC.
- Y. Zha, M. L. Disabb-Miller, Z. D. Johnson, M. A. Hickner and G. N. Tew, J. Am. Chem. Soc., 2012, 134, 4493–4496 CrossRef CAS PubMed.
- H. Li, M. R. Kraglund, A. K. Reumert, X. Ren, D. Aili and J. Yang, J. Mater. Chem. A, 2019, 7, 17914–17922 RSC.
- X. Dong, D. Lv, J. Zheng, B. Xue, W. Bi, S. Li and S. Zhang, J. Membr. Sci., 2017, 535, 301–311 CrossRef CAS.
- F. Gu, H. Dong, Y. Li, Z. Sun and F. Yan, Macromolecules, 2014, 47, 6740–6747 CrossRef CAS.
- J. H. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. J. Xu and Y. S. Ya, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- T. H. Pham, A. Allushi, J. S. Olsson and P. Jannasch, Polym. Chem., 2020, 11, 6953–6963 RSC.
- N. Chen, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, C. Hu, J. Y. Bae, E. S. Sim, Y. C. Chung, J. H. Jang, S. J. Yoo, Y. Zhuang and Y. M. Lee, Nat. Commun., 2021, 12, 2367 CrossRef CAS PubMed.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2017, 50, 2784–2793 CrossRef CAS.
- D. J. Strasser, B. J. Graziano and D. M. Knauss, J. Mater. Chem. A, 2017, 5, 9627–9640 RSC.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 11924–11938 RSC.
- H. G. Peng, Q. H. Li, M. X. Hu, L. Xiao, J. T. Lu and L. Zhuang, J. Power Sources, 2018, 390, 165–167 CrossRef CAS.
- F. Wang, B. Xue, S. Zhou, J. Zheng, S. Li, S. Zhang and T. A. Sherazi, J. Membr. Sci., 2019, 591, 117334 CrossRef.
- W. You, J. M. Ganley, B. G. Ernst, C. R. Peltier, H. Y. Ko, R. A. DiStasio Jr, R. R. Knowles and G. W. Coates, Chem. Sci., 2021, 12, 3898–3910 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev B.01. , Wallingford, CT, 2016 Search PubMed.
- Y. Liu, T. P. Pandey, H. N. Sarode, M.-C. Kuo, W. Zhang, R. Gupta, S. Galioto, A. G. Ozioko, S. Seifert, M. W. Liberatore, E. B. Coughlin and A. M. Herring, Solid State Ionics, 2018, 316, 135–142 CrossRef CAS.
- J. Xue, X. Liu, J. Zhang, Y. Yin and M. D. Guiver, J. Membr. Sci., 2020, 595, 117507 CrossRef.
- G. Cerichelli and L. Luchetti, Tetrahedron, 1993, 49, 10733–10738 CrossRef CAS.
- G. Cospito, G. Illuminati, C. Lillocci and H. Petride, J. Org. Chem., 1981, 46, 2944–2947 CrossRef CAS.
- G. Illuminati and C. Lillocci, J. Org. Chem., 1977, 42, 2201–2203 CrossRef CAS.
- R. Espiritu, J. L. Tan, L. H. Lim and S. Arco, J. Phys. Org. Chem., 2020, 33, e4049 CrossRef CAS.
- N. Li, T. Yan, Z. Li, T. Thurn-Albrecht and W. H. Binder, Energy Environ. Sci., 2012, 5, 7888–7892 RSC.
- J. J. Si, H. N. Wang, S. F. Lu, X. Xu, S. K. Peng and Y. Xiang, J. Mater. Chem. A, 2017, 5, 4003–4010 RSC.
- N. Chen, C. Lu, Y. Li, C. Long, Z. Li and H. Zhu, J. Membr. Sci., 2019, 588, 117120 CrossRef CAS.
- J. J. Han, L. Zhu, J. Pan, T. J. Zimudzi, Y. Wang, Y. Q. Peng, M. A. Hickner and L. Zhuang, Macromolecules, 2017, 50, 3323–3332 CrossRef CAS.
- B. Wang, W. Sun, F. Bu, X. Li, H. Na and C. Zhao, Int. J. Hydrogen Energy, 2016, 41, 3102–3112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02286a |
| This journal is © The Royal Society of Chemistry 2022 |