
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDIPEA-induced activation of OH− for the synthesis of amides via photocatalysis†
Mei Wu‡
a,
Sheng Huang‡a,
Huiqing Houa,
Jie Linb,
Mei Lina,
Sunying Zhoua,
Zhiqiang Zhengb,
Weiming Sun *a and
Fang Ke*a
*a and
Fang Ke*a
aInstitute of Materia Medica, School of Pharmacy, Fujian Provincial Key Laboratory of Natural Medicine Pharmacology, Fujian Medical University, Fuzhou 350122, China. E-mail: kefang@mail.fjmu.edu.cn; sunwm@fjmu.edu.cn
bDepartment of VIP Dental Service, School and Hospital of Stomatology, Fujian Medical University, Fuzhou 350002, China
First published on 16th May 2022
Abstract
The development of green protocols for photocatalysis where water acts as a nucleophile, induced by a weak organic base, is difficult to achieve in organic chemistry. Herein, an efficient light-mediated strategy for the synthesis of amides in which a weak organic base acts as a reductant to induce the formation of OH– from water under metal-free conditions is reported. A mechanistic study reveals that the generation of an N,N-diisopropylethylamine (DIPEA) radical via single electron transfer (SET), with the assistance of photocatalyst, that increases the nucleophilicity of the water molecules with respect to the cyanides is essential. Moreover, the removal rate of nitrile in wastewater can be as high as 83%, indicating that this strategy has excellent potential for nitrile degradation.
The synthesis of amides is a subject of continuous interest and great importance because of their important applications in detergents, agrochemicals, polymers and pharmaceuticals.1 Traditional methods for their synthesis require the transformation of an acid into the corresponding acyl chloride, facilitated by the use of the Schotten–Baumann reaction.2 Although these methods produce amides in good yields, stoichiometric amounts of an activating reagent are required, making these poorly atom economic processes. Various strategies for carboxamide synthesis, such as oxidative alcohol–amine and aldehyde–amine coupling reactions, amine dehydrogenation or oxidation reactions, and C–N coupling reactions, have been developed in recent years.3 Despite this, one of the most straightforward and atom-economical ways to synthesize amides remains the hydration of organonitriles.4 Conventional strategies mostly use strong inorganic bases to generate strongly nucleophilic hydroxide ions, require tough conditions, and are sensitive, especially when using bioactive molecules,5 to the substrate. Moreover, water molecules are usually used as nucleophiles in the hydration of nitriles. Thus, compared to the use of strong basic conditions, the direct nucleophilic addition of water to the cyano group is kinetically slow due to the high energy of the carbon–nitrogen triple bond.6 To circumvent these problems, transition metal (TM) catalytic procedures, where the metal center of the catalyst acts as a Lewis acid to activate the nitrile and the ligand acts as a Lewis base-activated nucleophile, have been developed in recent years.7 But these protocols are associated with certain debilitating disadvantages that include the presence of toxic transition metal cations within the molecular structure of the reagents and difficulties in preventing over-hydrolysis to the corresponding carboxylic acids.
Recently, there have been some reports that reductants have been used to change the morphology of water to increase its nucleophilicity.8 Organoamine bases, such as N,N-diisopropylethylamine (DIPEA), have acted in the role of both base and nitrogen radical intermediate and are considered to be reductants.9 However, DIPEA does not lose electrons easily and therefore has a low reductive activity, which means the nitrogen center has to cross a higher energetic barrier. Recently, it was shown that DIPEA could reductively quench many excited photocatalysts by single electron transfer (SET) to generate nitrogen-centered radicals.10 For example, Xu and coworkers11 proposed a new approach using DIPEA to construct difluoroalkylated diarylmethane compounds via visible light photocatalytic radical–radical cross-coupling reactions, in which DIPEA can carry out electron transfer due to the induction of the photocatalyst. It indicates that photooxidation–reduction and organic amine reduction are, when exposed to sufficient light intensity, co-catalytic processes and can generate nitrogen-centered radicals so that the downstream reaction process can continue.12 Herein, an efficient light-mediated strategy for the synthesis of amides in which a weak organic base acts as a reductant to induce the formation of OH– from water under metal-free conditions is reported.
Initially, the reaction of the benzonitrile (1a) was selected for the screening of the reaction conditions (Table 1). Eosin Y (0.05 mmol) was used as photocatalyst and DIPEA as the organoamine base (1.0 mmol) in H2O (3 mL) to give the desired product 2a in a yield of 89% (Table 1, entry 1). Next, several photocatalysts were added to the reaction instead of eosin Y (Table 1, entries 2–5). It is noteworthy that changing the photocatalyst from eosin Y to eosin B, Rose bengal, rhodamine B or erythrosin B led to a decrease in the product yield or even a failure to produce the target product. Further experiments indicated that the product yield also decreased when other bases such as triethylamine (TEA) and triethylene diamine (DABCO) were utilized instead of DIPEA (Table 1, entries 6 and 7). Moreover, different solvents were also tested, but the resulting reaction efficiency was not satisfactory (Table 1, entries 8 and 9). This result demonstrates that the participation of water, which is an inexpensive, atom economical oxygen donor, should be conducive to the hydration of nitriles to amides in this reaction system. In addition, when the time of reaction was halved, the yield of 2a was reduced to 47% (Table 1, entry 10), while an increase in the reaction time did not have much effect on the yield (Table 1, entry 11). It was noted that a reduction in light intensity or the use of white light also led to a reduction in yield (Table 1, entries 12 and 13). Finally, we screened the amount of base required for the reaction and demonstrated that decreasing the amount of DIPEA led to a significantly lower yield (Table 1, entry 14). In order to further demonstrate the practicality of the catalytic system, a gram–scale reaction of benzonitrile (10 mmol) was implemented (Table 1, entry 15) and the target product benzamide was obtained in a 72% yield Fig. 1, 2 and 3.
|
||
|---|---|---|
| Entry | Variations from the standard conditions | Yieldb (%) |
| a Standard conditions: 1a (0.5 mmol), DIPEA (2.0 equiv.), eosin Y (0.1 equiv.), blue light, 12 W, H2O 3 mL, 24 h, rt.b Isolated yield.c 10 mmol 1a, 15 equiv. DIPEA and 0.1 equiv. eosin Y, blue light 12 W, H2O 25 mL 36 h. | ||
| 1 | None | 89 |
| 2 | Eosin B instead of eosin Y | 41 |
| 3 | Rose bengal instead of eosin Y | 73 |
| 4 | Rhodamine B instead of eosin Y | Trace |
| 5 | Erythrosin B instead of eosin Y | 78 |
| 6 | TEA instead of DIPEA | 64 |
| 7 | DABCO instead of DIPEA | 13 |
| 8 | DMSO/H2O (1/2) instead of H2O 3 mL | 54 |
| 9 | DMF/H2O (1/2) instead of H2O 3 mL | 27 |
| 10 | 12 h instead of 24 h | 47 |
| 11 | 28 h instead of 24 h | 90 |
| 12 | Blue light 5 W instead of blue light 12 W | 63 |
| 13 | White light instead of blue light | 43 |
| 14 | DIPEA 1.0 equiv. instead of 2.0 equiv. | 59 |
| 15c | Gram-scale experiment | 72 |

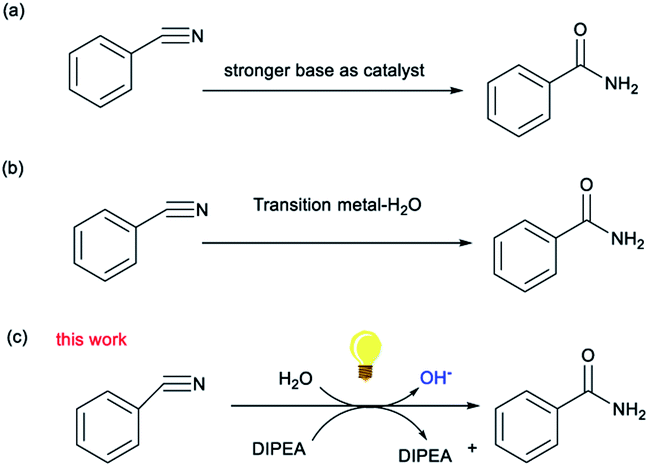 | ||
| Fig. 1 General methods for the hydration of organonitriles. | ||
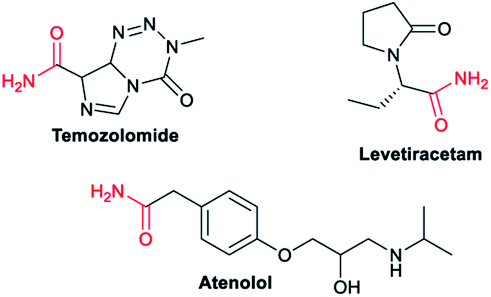 | ||
| Fig. 2 Pharmaceuticals and biomolecules containing a primary amide functional group. | ||
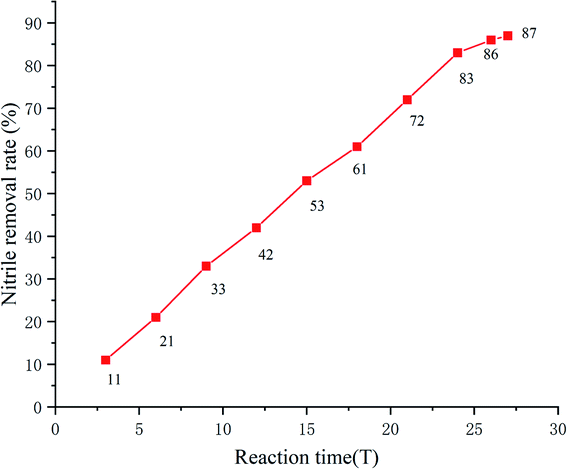 | ||
| Fig. 3 Variation of removal rate of nitrile at different reaction times. | ||
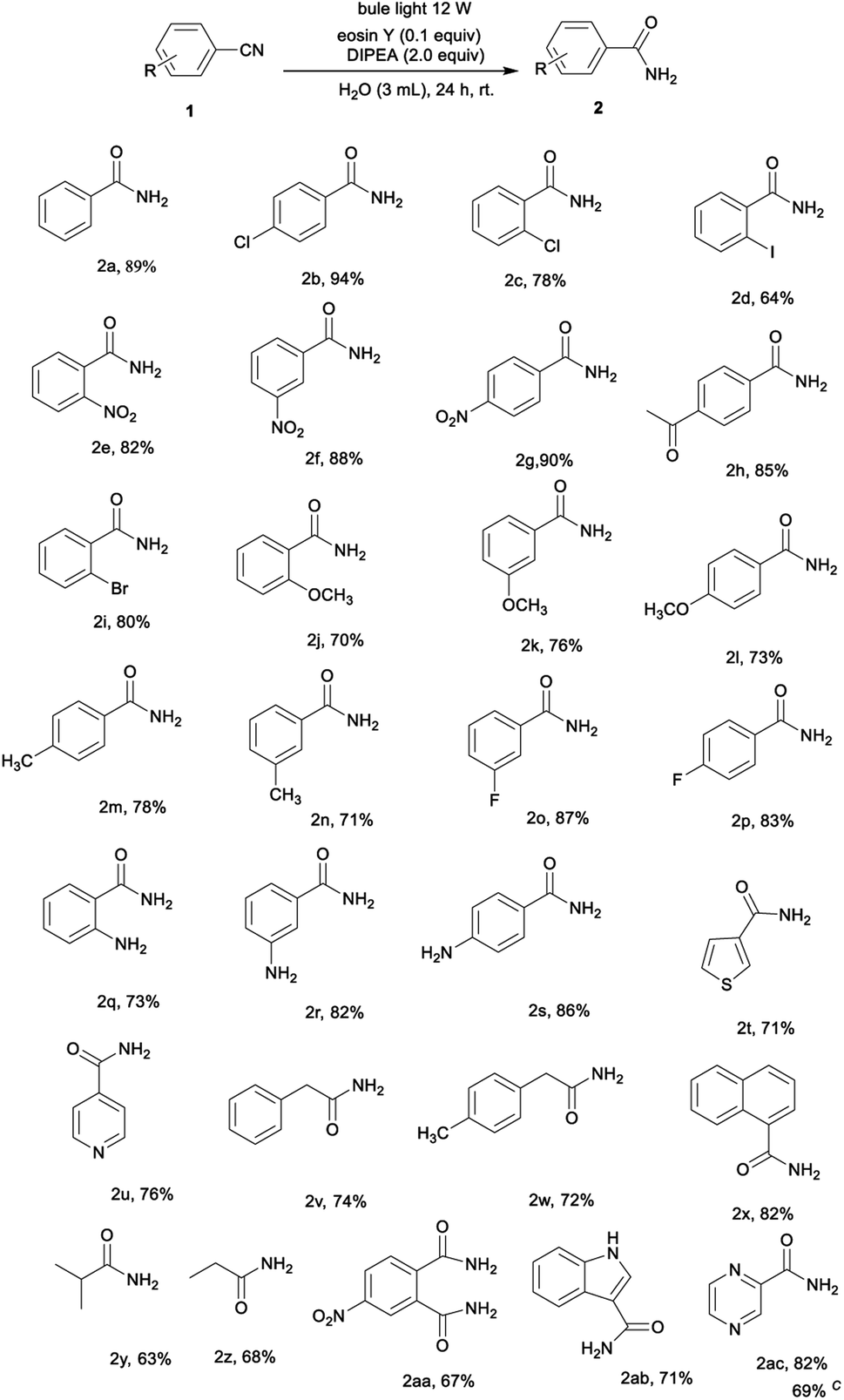
With the optimized conditions in hand, the substrate scope was investigated (Table 2). The method was found to be applicable to a wide range of substituted nitriles. Nitriles bearing electron-donating groups (–Me, –OMe, –NH2) or electron-withdrawing groups (–F, –Cl, –Br, –NO2, –Ac) are very compatible, furnishing the corresponding products in 70–94% yields (Table 2, 2b–2s). Generally, electron-withdrawing nitriles gave better product yields than electron-donating nitriles. For example, the hydration of p-halobenzonitriles offered p-halobenzamides yields of 83–94% (Table 2, 2b and 2p) while p-methylbenzonitrile gave a 78% yield of the corresponding amide (Tables 2 and 2m). This might be due to the electron-withdrawing group making the nitrile carbon more susceptible to nucleophilic attack. Surprisingly, heterocycles such as thiophene-3-carbonitrile and isonicotinonitrile gave amides 2t and 2u in 71% and 76% yields, respectively. The aliphatic nitriles containing phenyl, such as p-methylphenylacetonitrile and phenylacetonitrile, were also found to be suitable reactants under the reaction conditions and produced the corresponding amides in 74% and 72% yields, respectively (Table 2, 2v and 2w). Moreover, the hydration of isobutyronitrile and propionitrile afforded the corresponding amide in good yields (Table 2, 2y and 2z). The protocol was successfully applied for the hydration of a dicyano-substituted arene to give the desired amide in a 67% yield (Table 2, 2aa). Pleasingly, the reaction of 3-cyanoindole gave the carboxamide product 2ab as a single product in a 71% yield. In addition, a pyrazinamide, which has a strong antibacterial effect on tuberculosis bacillus, can also be synthesized using a similar reaction system, and the target product was reached in a 69% yield following a gram–scale reaction (Table 2, 2ac).
Nitrile wastewater is a big threat to the environment, especially to aquatic organisms. With the optimal reaction conditions established, the effect of the reaction time on the removal rate of nitrile in wastewater was investigated (the nitrile concentration of wastewater was 200 mg L−1 as determined using HPLC). It was shown that the removal rate of nitrile increased up to 83% as the reaction time increased to 24 h and remained stable.
To explore the reaction mechanism, a series of control experiments were performed (Scheme 1). These reactions were essentially carried out under conditions in which only one reaction parameter was changed. The control experiments revealed that no reaction occurred in the absence of either the visible light or the photocatalyst, indicating that these two components are essential to the reaction (Scheme 1a, 1b). In addition, no products are formed in the absence of DIPEA, which indicates that the organic base is key to this reaction system (Scheme 1c). Upon conducting the nitrile hydration under an H218O atmosphere (Scheme 1d), we obtained an 18O-labeled product, demonstrating that H2O rather than molecule oxygen serves as the oxygen source.
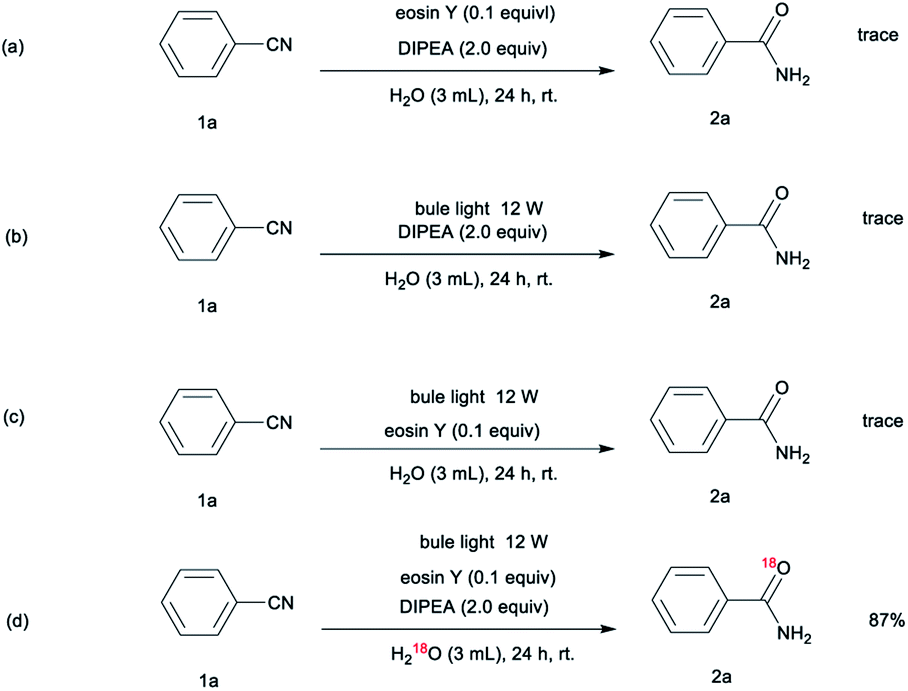 | ||
| Scheme 1 Control reactions. | ||
On the basis of the mechanistic studies above and the literature, a plausible mechanism is outlined in Scheme 2. Initially, the photocatalyst eosin Y is irradiated to give an activated species eosin Y* from which oxygen abstracts an electron to form an O2˙− radical. Then, the oxidation state of the photocatalyst is reduced by the reductive quencher A.10,11,13a Subsequently, the O2˙− radical acquires an electron and H+ from radical B to form HO2−. Next, a water molecule and HO2− instantaneously form OH− and H2O2 (as determined using HPLC).13b,c Then, a nucleophilic addition of OH− to the electrophilic carbon atom of the nitrile generates intermediate D, which is further hydrated to form the product 2a.13d
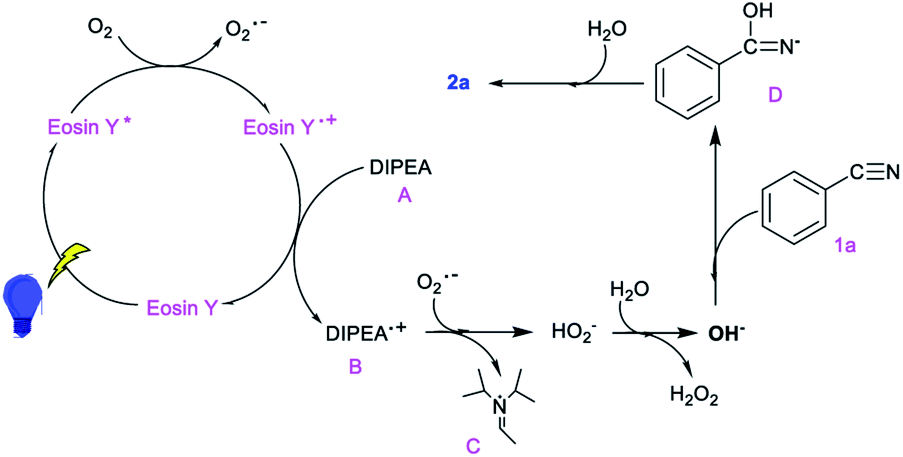 | ||
| Scheme 2 Proposed mechanism for this transformation. | ||
To verify the above proposed mechanism, density functional theory (DFT) calculations were performed and are shown in Fig. 4. First, the generated eosin Y free radical can easily attack DIPEA to generate radical B along with the release of 27.70 kcal mol−1 of energy. Afterwards, the resulting radical B will spontaneously react with the radical O2˙− and an H2O molecule to generate OH−. Then, the obtained OH− further reacts with nitrile 1a to form intermediate D, a process with a very small energy barrier of 11.27 kcal mol−1. Finally, intermediate D is rapidly oxidized to the target product 2a.
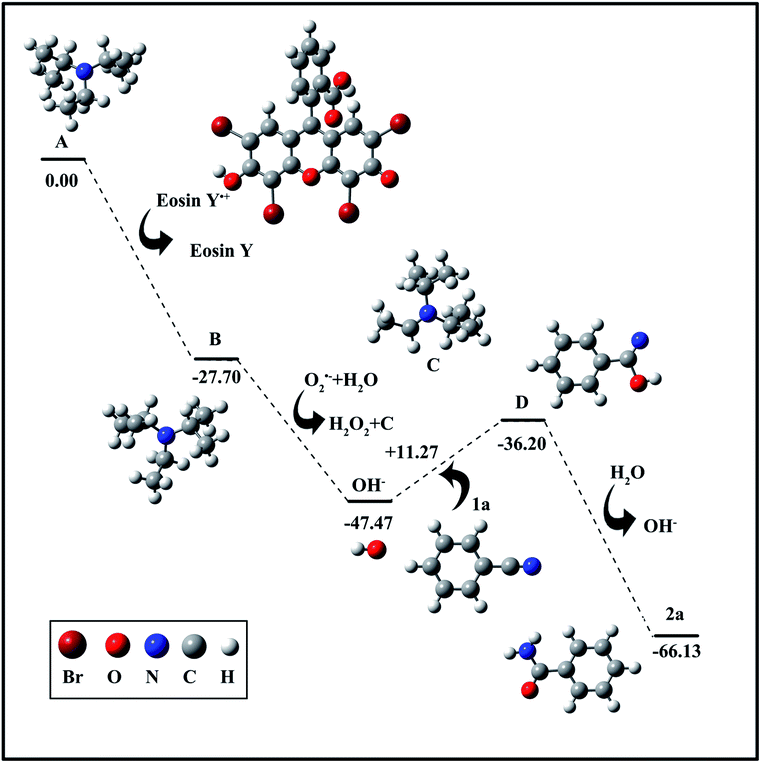 | ||
| Fig. 4 DFT study of the hydration of nitrile. | ||
Conclusions
An efficient photocatalytic transformation of nitriles into amides was developed where a weak organic base acts as reductant to induce the formation of OH− from water. The protocol has excellent functional group tolerance and a broad substrate scope. The key to the strategy is a green photoredox process and organoamine base reductant that cocatalyze nitrile hydration under metal-free conditions. Moreover, photocatalytic nitrile degradation using this system has excellent efficiency and the results showed that the removal rate of nitrile can reach 83%. This indicates that the reaction system effectively degrades nitrile compounds in wastewater.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Fujian Provincial Foundation (No. 2016Y9051, 2016Y9052, 2016Y9053, 2017J01820, 2021kq02), the Research Fund of Fujian Medical University (No. 2015MP008, CA18142), and the Natural Science Foundation of Fujian Province (No. 2021J01692, 2020J01620).Notes and references
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (b) M. Antoszczak, E. Maj, A. Napiórkowska, J. Stefańska, E. Augustynowicz-Kopeć, J. Wietrzyk, J. Janczak, B. Brzezinski and A. Huczyński, Molecules, 2014, 19, 19435–19459 CrossRef; (c) A. Mehta and S. Basu, J. Photochem. Photobiol., A, 2017, 343, 1–6 CrossRef CAS; (d) J.-B. Denis, G. Masson, P. Retailleau and J. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5356–5360 CrossRef CAS.
- (a) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed; (b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (c) C. A. G. N. Montalbetti and V. Falque, Tetrahedron, 2005, 61, 10827–10852 CrossRef CAS; (d) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS.
- (a) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS; (c) C. Chen and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20–26 RSC; (d) K. Ekoue-Kovi and C. Wolf, Chem.–Eur. J., 2008, 14, 6302–6315 CrossRef CAS PubMed; (e) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed; (f) P. Crochet and V. Cadierno, Chem. Commun., 2015, 51, 2495–2505 RSC; (g) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- (a) S. Davulcu, C. L. Allen, K. Milne and J. M. J. Williams, ChemCatChem, 2013, 5, 435–438 CrossRef CAS; (b) E. L. Downs and D. R. Tyler, Coord. Chem. Rev., 2014, 280, 28–37 CrossRef CAS; (c) P. Marcé, J. Lynch, A. J. Blacker and J. M. J. Williams, Chem. Commun., 2016, 52, 1436–1438 RSC; (d) T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949–974 CrossRef CAS; (e) C. L. Allen, A. A. Lapkin and J. M. J. Williams, Tetrahedron Lett., 2009, 50, 4262–4264 CrossRef CAS; (f) S. Murahashi, T. Naota and E. Saito, J. Am. Chem. Soc., 1986, 108, 7846–7847 CrossRef CAS; (g) Y.-M. Liu, L. He, M.-M. Wang, Y. Cao, H.-Y. He and K.-N. Fan, ChemSusChem, 2012, 5, 1392–1396 CrossRef CAS PubMed; (h) A. Goto, K. Endo and S. Saito, Angew. Chem., Int. Ed., 2008, 47, 3607–3609 CrossRef CAS; (i) M. Tamura, H. Wakasugi, K. Shimizu and A. Satsuma, Chem.–Eur. J., 2011, 17, 11428–11431 CrossRef CAS PubMed.
- L. Yang, H. Chen, J. Liu, X. Wan and Q. Xu, Green Chem., 2016, 18, 4865–4870 RSC.
- B. Guo, J. G. de Vries and E. Otten, Chem. Sci., 2019, 10, 10647–10652 RSC.
- (a) R. García-Álvarez, P. Crochet and V. Cadierno, Green Chem., 2013, 15, 46–66 RSC; (b) W. K. Fung, X. Huang, M. L. Man, S. M. Ng, M. Y. Hung, Z. Lin and C. P. Lau, J. Am. Chem. Soc., 2003, 125, 11539–11544 CrossRef CAS; (c) E. Tomás-Mendivil, R. García-Álvarez, C. Vidal, P. Crochet and V. Ca-dierno, ACS Catal., 2014, 4, 1901–1910 CrossRef; (d) X.-Y. Xing, C. Xu, B. Chen, C.-C. Li, S. C. Virgil and R. H. Grubbs, J. Am. Chem. Soc., 2018, 140, 17782–17789 CrossRef CAS.
- H. Li and M. B. Hall, J. Am. Chem. Soc., 2015, 137, 12330–12342 CrossRef CAS PubMed.
- (a) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed; (b) D. B. Freeman, L. Furst, A. G. Condie and C. R. J. Stephenson, Org. Lett., 2012, 14, 94–97 CrossRef CAS PubMed.
- (a) W.-J. Zhou, Z.-H. Wang, L.-L. Liao, Y.-X. Jiang, K.-G. Cao, T. Ju, Y. Li, G.-M. Cao and D.-G. Yu, Nat. Commun., 2020, 11, 1–9 CrossRef; (b) K. Wu, Y. Du and Z. Wei, Chem. Commun., 2018, 54, 7443–7446 RSC.
- Y.-N. Zhao, Y.-C. Luo, Z.-Y. Wang and P.-F. Xu, Chem. Commun., 2018, 54, 3993–3996 RSC.
- (a) Y. Jian, M. Chen, B. Huang, W. Jia, C. Yang and W. Xia, Org. Lett., 2018, 20, 5370–5374 CrossRef CAS PubMed; (b) M. Teders, L. Pitzer, S. Buss and F. Glorius, ACS Catal., 2017, 7, 4053–4056 CrossRef CAS.
- (a) Z. Wei, S. Qi, Y. Xu, H. Liu, J. Wu, H. Li, C. Xia and G. Duan, Adv. Synth. Catal., 2019, 361, 5490–5498 CrossRef CAS; (b) Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed; (c) H. Hao, L. Zhang, W. Wang and S. Zeng, Catal. Sci. Technol., 2018, 8, 1229–1250 RSC; (d) T. Tu, Z.-X. Wang, Z. L. Liu, X.-K. Feng and Q.-Y. Wang, Green Chem., 2012, 14, 921–924 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra02107b |
| ‡ Mei Wu and Sheng Huang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |