
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond GalNAc! Drug delivery systems comprising complex oligosaccharides for targeted use of nucleic acid therapeutics
Joseph O'Sullivan a,
Jose Muñoz-Muñoza,
Graeme Turnbulla,
Neil Simb,
Stuart Pennyb and
Sterghios Moschos*a
a,
Jose Muñoz-Muñoza,
Graeme Turnbulla,
Neil Simb,
Stuart Pennyb and
Sterghios Moschos*a
aDepartment of Applied Sciences, Northumbria University, Newcastle upon Tyne, UK NE1 8ST. E-mail: sterghios.moschos@northumbria.ac.uk
bHigh Force Research Ltd, Bowburn North Industrial Estate, Durham, UK DH6 5PF
First published on 14th July 2022
Abstract
Nucleic Acid Therapeutics (NATs) are establishing a leading role for the management and treatment of genetic diseases following FDA approval of nusinersen, patisiran, and givosiran in the last 5 years, the breakthrough of milasen, with more approvals undoubtedly on the way. Givosiran takes advantage of the known interaction between the hepatocyte specific asialoglycoprotein receptor (ASGPR) and N-acetyl galactosamine (GalNAc) ligands to deliver a therapeutic effect, underscoring the value of targeting moieties. In this review, we explore the history of GalNAc as a ligand, and the paradigm it has set for the delivery of NATs through precise targeting to the liver, overcoming common hindrances faced with this type of therapy. We describe various complex oligosaccharides (OSs) and ask what others could be used to target receptors for NAT delivery and the opportunities awaiting exploration of this chemical space.
Introduction
There is undeniably frustration for those with untreatable inherited genetic diseases with a known origin, with current options for some only being that of management, until progression of the disease leads to the patient's death.1,2 Where well-characterised genetic changes have been causally linked to heritable disease, treatments can be specifically formulated. For decades the allure of selective gene silencing, quenching, or interference with NATs, and most recently even genome editing, has promised to be the revolutionary jump to a future of precise and personalised therapy for human diseases.3–5 There is an attractive quality in the high specificity of these therapies that can limit the materialisation of unwanted and toxic side effects through careful design and meticulous off-target screening.6 The routine use of NATs has, however, been impeded. Originally by their susceptibility to degradation from endogenous nucleases, later by the unrecognised finesse of the innate immune response, and consistently with the difficulty these generally negatively-charged macromolecular species experience in crossing cell membranes to reach their target.7,8 Persistent research in the field and use of the established interaction of GalNAc with the ASGPR has recently resolved this challenge and enabled enhanced and targeted delivery of functional NATs to the liver, with the ligand–receptor interaction able to facilitate the GalNAc–NAT complex to undergo endocytosis.9 This yielded an upsurge in successful clinical trials leading to FDA approvals such as givosiran, a short interfering RNA (siRNA) bound to a triantennary GalNAc ligand (three GalNAc moieties on three precisely spaced branching arms of defined lengths) that assists in the targeted delivery to hepatocytes.10 GalNAc-therapeutic conjugates demonstrate the effectiveness NATs can have given a suitable delivery system with specific-targeting glycosides, or glycotargeting, and give hope that similar improvements could be achieved for alternative target cell types or tissues given an appropriately constructed glycosylated delivery system.Nucleic acid therapeutics (NATs)
NATs are nucleic acid-based compounds, commonly DNA or RNA in nature but recently oligomers featuring non-natural backbones such as peptide or morpholino chemistries have emerged. These are diverse in therapeutic action and are generally engineered to work by inhibiting, altering the expression, or processing of a gene responsible for a particular disease. Mechanisms of action include endonucleatically active or inactive antisense oligonucleotides (ASOs), antibody-like aptamers, and RNA interference (RNAi) mediators to name a few, which cells can use to tune the outputs of faulty endogenous genes and invasive exogenous genes.11–13 Most NATs work through Watson–Crick base pairing to a target transcript or chromosomal locus to produce an effect,14–16 opening the possibility for bespoke treatments for specific targets. Sizes range from the shorter locked nucleic acids (LNAs) of approximately 10 nucleotides (nts) (∼3300 Da), containing a synthetic bridge linking the 2′-oxygen with the 4′-carbon of the ribose ring with a minimally modified α-phosphorothioate backbone,17 to the larger CRISPR-CAS9 ribonucleoprotein complex involving a bacterially-derived CAS9 protein (∼160 kDa) and a 100 nt guide RNA molecule (∼33 kDa).In 1978, the concept of the therapeutic use of a nucleic acid, in this case an ASO, was first theorised by Zamecnik and Stephenson whilst working on inhibiting virus replication with oligonucleotides.18,19 They suggested that if the target sequence of RNA or DNA was known, a potential future option would be specifically synthesising an oligonucleotide to hybridise to it acting as a virus inhibitor; they also proposed the possibility of hindering protein translation if desired. They then demonstrated how an oligonucleotide 13-mer could prevent translation of certain mRNAs in Rous sarcoma virus. Being aware that nucleic acids are susceptible to degradation, Putney et al. found that the use of an α-phosphorothioate nt restricted digestion from exonuclease III.20 Additional research led to strategies for further modifications that would improve biostability, boost cellular uptake, and increase potency,21–23 including the use of a carrier or drug-delivery system with popular choices being the use of viruses, polymeric nanoparticles, liposomes, and more recently lipid nanoparticle complexes.24–26 Some of these currently used modifications are described further by Moschos et al.27 and Dowdy.28 In 1998, success was achieved with the first approval of a NAT for clinical use in fomivirsen, an ASO to treat cytomegalovirus retinitis for patients with AIDS29 through an aptamer mechanism of action. In the same year, Fire et al. showed that gene silencing could be achieved through use of double-stranded RNA, specifically RNA interference (RNAi), in the nematode worm Caenorhabditis elegans (C. elegans).30 The proceeding years gave the first RNAi-mediated gene silencing in mammalian cells31 and the potential of what could be achieved using NATs became apparent. With the field subsequently exploding in popularity over the following twenty years, the goal of reaching the previously thought ‘undruggable space’ was becoming a reality and an expansive range of NATs have made it to market for a variety of ailments with more in clinical trials, demonstrating their versatility in offering more tailored treatments.32–35
ASOs were the first class of NATs to hit the market and are generally modified short (20–25 nts) oligonucleotides complimentary to a faulty gene's messenger RNA (mRNA). Nusinersen (a chemically modified phosphorothioate DNA ASO) and eteplirsen (a morpholino phosphorodiamidate ASO) have both recently been approved for treatment of spinal muscular atrophy and Duchenne muscular dystrophy, respectively (see Table 1). Both of these ASOs, and indeed the nusinersen-like n of 1 ASO milasen,36 function by controlling the splicing of their target genes to overcome gene changes deleterious to the conversion of mRNA into protein.37 An alternative format of an ASO is a gapmer, a chemistry involving two runs of modified nucleic acids flanking a DNA oligonucleotide segment; this structure can direct gene silencing through RNase H-cleavage of the mRNA.37 In 2018, the FDA approved use of the gapmer inotersen for the treatment of polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.38 Another commonly researched NAT class involves the endogenous gene silencing mechanism of RNAi; a naturally occurring process that consists of RNA, usually 20–25 nts in length, used to silence genes. Therapeutic variations on this theme include double stranded RNA (dsRNA) in the form of custom-designed siRNAs, mimics to the endogenously expressed dsRNA called microRNAs (miRNAs), or single stranded ASOs that are designed to inhibit miRNAs.39 A recent approval by the FDA includes patisiran, a siRNA with a lipid-based delivery system40 for the treatment of transthyretin-mediated amyloidosis,41 with similar systems now also in use in SARS-CoV-2 mRNA vaccines. Aptamers, in part named from the Latin ‘aptus’ meaning ‘to fit’, are single stranded oligonucleotides that can be manipulated to bind to specific molecular targets including proteins in a fashion not dissimilar to antibodies, and with comparable KD's.42 Pegaptanib was the first aptamer approved by the FDA in 2004 for treatment of macular degeneration.43 A final example of the diverse range of NATs is clustered regularly interspaced short palindromic repeats (CRISPR). The principle of CRISPR is that of selective gene editing on the genome with an ability to target a malfunctioning gene with a known error and alter that piece of DNA using either a randomly destructive mechanism (non-homologous end joining), or oligonucleotide template-directed repair (homology directed repair). Commonly used alongside CRISPR Associated (CAS) proteins,16 the CRISPR-CAS9 system has quickly become the dominant method in the field of gene editing. Unlike other systems, CRISPR-CAS9 can use a single or double stranded oligonucleotide, in addition to the 100 nt guide RNA, to drive homology-directed repair; both oligonucleotides can be modified substantially along the same principles of classical ASO modification to improve stability and prevent innate immunity activation. A notable recent success (2021) has been the use of a CRISPR-CAS9 system for treating sickle cell disease and β-thalassemia, in which patients on the trial responded positively to treatment and no longer endured vaso-occulative episodes.44 For additional information, comprehensive reviews on the functions, mechanisms, and delivery of various NATs are available.7,43,45,46
| Name (market name) | Developer | FDA approval | Modality | Administration | Medical indication | Key references |
|---|---|---|---|---|---|---|
| Fomivirsen (Vitravene) | Ionis | 1998 | ASO | Intravitreal | Cytomegalovirus retinitis in immunocompromised individuals | Vitravene study Group52 |
| Pegaptanib (Macugen) | Valeant | 2004 | Aptamer | Intravitreal | Macular degeneration | Gragoudas et al.53 |
| Eteplirsen (Exondys 51) | Sarepta | 2016 | ASO | Intravenous | Duchenne muscular dystrophy | Mendell et al.54 |
| Nusinersen (Spinraza) | Ionis | 2016 | ASO | Intrathecal | Spinal muscular atrophy | Haché et al.55 |
| Inotersen (Tegsedi) | Akcea | 2018 | ASO | Subcutaneous | Hereditary transthyretin-mediated amyloidosis | Benson et al.56 |
| Patisiran (Onpattro) | Alnylam | 2018 | siRNA | Intravenous | Hereditary transthyretin-mediated amyloidosis | Adams et al.57 |
| Milasen (N/A) | Boston Children's Hospital | Allowed use 2018 | ASO | Intrathecal | Batten disease | Kim et al.36 |
| Givosiran (Givlaari) | Alnylam | 2019 | siRNA | Subcutaneous | Acute hepatic porphyria | Balwani et al.58 |
| Lumasiran (Oxlumo) | Alnylam | 2020 | siRNA | Subcutaneous | Primary hyperoxaluria type 1 | Garrelfs et al.59 |
| Casimersen (Amondys 45) | Sarepta | 2021 | ASO | Subcutaneous | Duchenne muscular dystrophy | Shirley60 |
NATs for gene quenching, silencing, or editing have continually been cited as having great potential to herald a new age of personalised therapeutics to treat genetic conditions as well as cancer.5,47 Key to the future of NATs is possessing the ability to treat diseases with known genetic origin irrespective of molecular target location in the body, and offer customisable therapies for patients; in some instances, this is already being carried out.36 Success to date, however, is driven by first pass metabolism-driven liver loading, leaky cell membranes (Duchenne's muscular dystrophy), and cell uptake after intrathecal injection (spinal muscular atrophy). Crucially, an outstanding and persistent issue that remains is the delivery of the NATs to other target tissues, which must overcome a naturally evolved defence designed to prevent exogenous nucleic acids, such as those being administered, from entering cells.43,47–49 ‘Naked’ nucleic acid delivery is therefore challenging due to the susceptibility of these compounds to degradation under physiological conditions from nucleases, non-specific binding, negative charges preventing therapies crossing cell membranes, and inefficient intracellular trafficking and access to cell cytosols.50,51 As a result, the development of precise and potent NATs that work exceptionally well in cells has been relatively straight forward in comparison to the problems endured by the challenge posed in delivering them to cells in animals or humans.47 With NATs beginning to gain market share, their use is becoming a valid therapeutic option, but there is still a need to improve their stability and delivery in vivo, with a hope of perfecting delivery systems to fulfil the promise of these therapies of expanding the druggable space.61–63
GalNAc and the liver
Lectins are carbohydrate-binding proteins with high specificity for particular glycans that can agglutinate cells and aid in the precipitation and removal glycoconjugates.64,65 Studies on hepatic lectins, in both animals and humans, presented common findings in their affinity for desilylated molecules possessing a terminal galactose or GalNAc.66 Once bound, a process influenced by Ca2+ concentration and pH,67 the lectin drives endocytosis of the glycosides by liver parenchymal cells and, particularly in the case of glycoproteins, are removed from circulation.68,69 In 1980, Baenziger and Maynard suggested there could be a greater binding affinity for terminal GalNAc over galactose when comparing inhibition constants (ki) calculated through binding assays (up to 27-fold reduction in ki). A similar reduction in ki (∼30-fold) was also observed comparing three galactose residues to two, and four GalNAc residues to two, suggesting binding at multiple sites simultaneously.70 This work was furthered by Lee et al. showing that branching the glycosides, as well as altering their spacing and flexibility, contributed significantly to the binding (up to 1000-fold).71 The preference for these multivalent moieties brought about the term ‘glycoside cluster effect’, which is dependent on two factors: it must contain a lectin with clustered glycan binding sites, and have a multi-valent ligand that can offer the specific glycans to these sites.72The ASGPR, a hepatic C-type lectin, presents up to 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 copies per cell73–75 and is part of a natural trafficking mechanism into liver cells for molecules bearing a terminal galactose or GalNAc.76 It thus became an obvious target for enhancing the intracellular delivery of therapeutics aimed at cytosolic hepatocyte targets.77 Successful delivery of DNA using a soluble receptor-specific vector through ASGPR was first achieved by Wu and Wu in 1987,78 but there was a need to a develop this technology into a safe and reliable system. Promising work was carried out using delivery systems bearing terminal galactose residues,79,80 but GalNAc-based systems were favoured along with multivalent systems over singular, previously showing a 3000 and 200000-fold greater affinity for the ASGPR in competitive binding assays respectively.70,81–83 The use of triantennary GalNAc ligands, three GalNAc moieties on three precisely spaced branching arms of defined lengths, noted to be key for the efficacy,83,84 became a popular choice of targeting ligand for the ASGPR. Using a ligand of this nature had historically shown promising cellular uptake of oligonucleotide derivative payloads,85 with Rensen et al. showing a 50-fold increase in affinity for ASGPR in comparison to the triantennary galactose equivalent when delivering glycolipids.86 Molecules are attached to the glycoside-based delivery system through the use of a spacer, which itself contributes to the binding affinity of the ligands to the ASGPR; Biessen et al. noted that the affinity of a cluster galactoside increased with the spacer length between 4 and 20 Å, with a 20 Å spacer having a 2000-fold higher affinity than a 4 Å one.87 Working on many of these principals, Khorev et al.88 developed a triantennary GalNAc ligand targeting the ASGPR capable of internalising into human parenchymal liver cells carrying fluorescent cargo, visualising endosomes with fluorescence microscopy, and confirming the ASGPR-mediated uptake with asialofetuin in competitive binding assays. They proposed these GalNAc-based ligands held high potential for delivery of therapeutic agents to the liver as, elsewhere, optimisation of the oligonucleotides used in therapeutic delivery were being undertaken by altering the length, backbone charge and stability through phosphorothioate or 2′-O-methoxyethyl (MOE) modifications to increase their efficiency.89,90 Notable success using therapeutics was limited until 2014 when Prakash et al.75 reported a breakthrough targeting the ASGPR using a triantennary GalNAc ligand linked through amide bonds to an ASO. This delivery system facilitated endocytosis of the GalNAc–ASO complex improving potency by up to 60-fold in mouse livers. Upon internalisation into the endosome, a drop in pH enables its dissociation from the ASGPR which is recycled to the cell surface.91 The therapeutic is lysed from the GalNAc-ligand in the lumen before undergoing endosomal escape, as illustrated in Fig. 1, the mechanism of which is still undetermined.92 In the same year, Nair et al.93 demonstrated dose-dependent gene silencing (ED50 1 mg kg−1) in mice using trivalent GalNAc-conjugated siRNA; the novel RNAi offered a 5-fold enhancement compared to the parent design with no adverse effects after sustained dosing. These achievements of robust gene silencing led to the first in vivo characterization of the drug disposition, metabolism and toxicokinetics of GalNAc-conjugated therapeutics, showing favourable results, with a 20-fold increase in potency in the knockdown of the hepatic apolipoprotein (a) mRNA when given weekly doses of a GalNAc-conjugated therapeutic (ED50 0.32 mg kg−1), in comparison to an unconjugated therapeutic (ED50 6.38 mg kg−1), supporting further development in humans.94 Clinical trials proceeded with givosiran; a siRNA linked to a trivalent GalNAc ligand that targets delta aminolevulinic acid synthase 1 (ALAS1) mRNA in hepatocytes.95 In the phase 1 study, patients with acute intermittent porphyria, inherited disorders in heme biosynthesis, were given monthly or quarterly doses of givosiran (0.035–0.5 mg kg−1) or a placebo.96 Those provided with monthly doses of givosiran showed consistent and dose dependent reduction in ALAS1 mRNA and the neurotoxic intermediates aminolevulinic acid (ALA) and porphobilinogen (PBG) in urine when compared to the placebo; these reductions correlated with the reduction in neurovisceral attacks. A six-month phase 3 trial similarly showed sustained reductions in ALA and PBG along with a 74% reduction in mean annualized attack rate for patients receiving monthly doses (2.5 mg kg−1) of givosiran (P < 0.001) in comparison to the placebo. These results led to the first GalNAc-conjugated oligonucleotide therapy to be approved by the FDA in 2019.58 Givosiran is marketed for treatment of acute hepatic porphyria and this accomplishment inspired use of GalNAc-based drug delivery systems (DDSs) leading to an array of GalNAc-conjugates currently in varying stages of clinical trials, excellently summarised by Debacker et al.10 With the approval and commercial success of givosiran and its GalNAc-based DDS, the question arose as to what other glycosides could be used, based on this model, in delivery systems for targeting non-hepatic tissues and cell types.
000 copies per cell73–75 and is part of a natural trafficking mechanism into liver cells for molecules bearing a terminal galactose or GalNAc.76 It thus became an obvious target for enhancing the intracellular delivery of therapeutics aimed at cytosolic hepatocyte targets.77 Successful delivery of DNA using a soluble receptor-specific vector through ASGPR was first achieved by Wu and Wu in 1987,78 but there was a need to a develop this technology into a safe and reliable system. Promising work was carried out using delivery systems bearing terminal galactose residues,79,80 but GalNAc-based systems were favoured along with multivalent systems over singular, previously showing a 3000 and 200000-fold greater affinity for the ASGPR in competitive binding assays respectively.70,81–83 The use of triantennary GalNAc ligands, three GalNAc moieties on three precisely spaced branching arms of defined lengths, noted to be key for the efficacy,83,84 became a popular choice of targeting ligand for the ASGPR. Using a ligand of this nature had historically shown promising cellular uptake of oligonucleotide derivative payloads,85 with Rensen et al. showing a 50-fold increase in affinity for ASGPR in comparison to the triantennary galactose equivalent when delivering glycolipids.86 Molecules are attached to the glycoside-based delivery system through the use of a spacer, which itself contributes to the binding affinity of the ligands to the ASGPR; Biessen et al. noted that the affinity of a cluster galactoside increased with the spacer length between 4 and 20 Å, with a 20 Å spacer having a 2000-fold higher affinity than a 4 Å one.87 Working on many of these principals, Khorev et al.88 developed a triantennary GalNAc ligand targeting the ASGPR capable of internalising into human parenchymal liver cells carrying fluorescent cargo, visualising endosomes with fluorescence microscopy, and confirming the ASGPR-mediated uptake with asialofetuin in competitive binding assays. They proposed these GalNAc-based ligands held high potential for delivery of therapeutic agents to the liver as, elsewhere, optimisation of the oligonucleotides used in therapeutic delivery were being undertaken by altering the length, backbone charge and stability through phosphorothioate or 2′-O-methoxyethyl (MOE) modifications to increase their efficiency.89,90 Notable success using therapeutics was limited until 2014 when Prakash et al.75 reported a breakthrough targeting the ASGPR using a triantennary GalNAc ligand linked through amide bonds to an ASO. This delivery system facilitated endocytosis of the GalNAc–ASO complex improving potency by up to 60-fold in mouse livers. Upon internalisation into the endosome, a drop in pH enables its dissociation from the ASGPR which is recycled to the cell surface.91 The therapeutic is lysed from the GalNAc-ligand in the lumen before undergoing endosomal escape, as illustrated in Fig. 1, the mechanism of which is still undetermined.92 In the same year, Nair et al.93 demonstrated dose-dependent gene silencing (ED50 1 mg kg−1) in mice using trivalent GalNAc-conjugated siRNA; the novel RNAi offered a 5-fold enhancement compared to the parent design with no adverse effects after sustained dosing. These achievements of robust gene silencing led to the first in vivo characterization of the drug disposition, metabolism and toxicokinetics of GalNAc-conjugated therapeutics, showing favourable results, with a 20-fold increase in potency in the knockdown of the hepatic apolipoprotein (a) mRNA when given weekly doses of a GalNAc-conjugated therapeutic (ED50 0.32 mg kg−1), in comparison to an unconjugated therapeutic (ED50 6.38 mg kg−1), supporting further development in humans.94 Clinical trials proceeded with givosiran; a siRNA linked to a trivalent GalNAc ligand that targets delta aminolevulinic acid synthase 1 (ALAS1) mRNA in hepatocytes.95 In the phase 1 study, patients with acute intermittent porphyria, inherited disorders in heme biosynthesis, were given monthly or quarterly doses of givosiran (0.035–0.5 mg kg−1) or a placebo.96 Those provided with monthly doses of givosiran showed consistent and dose dependent reduction in ALAS1 mRNA and the neurotoxic intermediates aminolevulinic acid (ALA) and porphobilinogen (PBG) in urine when compared to the placebo; these reductions correlated with the reduction in neurovisceral attacks. A six-month phase 3 trial similarly showed sustained reductions in ALA and PBG along with a 74% reduction in mean annualized attack rate for patients receiving monthly doses (2.5 mg kg−1) of givosiran (P < 0.001) in comparison to the placebo. These results led to the first GalNAc-conjugated oligonucleotide therapy to be approved by the FDA in 2019.58 Givosiran is marketed for treatment of acute hepatic porphyria and this accomplishment inspired use of GalNAc-based drug delivery systems (DDSs) leading to an array of GalNAc-conjugates currently in varying stages of clinical trials, excellently summarised by Debacker et al.10 With the approval and commercial success of givosiran and its GalNAc-based DDS, the question arose as to what other glycosides could be used, based on this model, in delivery systems for targeting non-hepatic tissues and cell types.
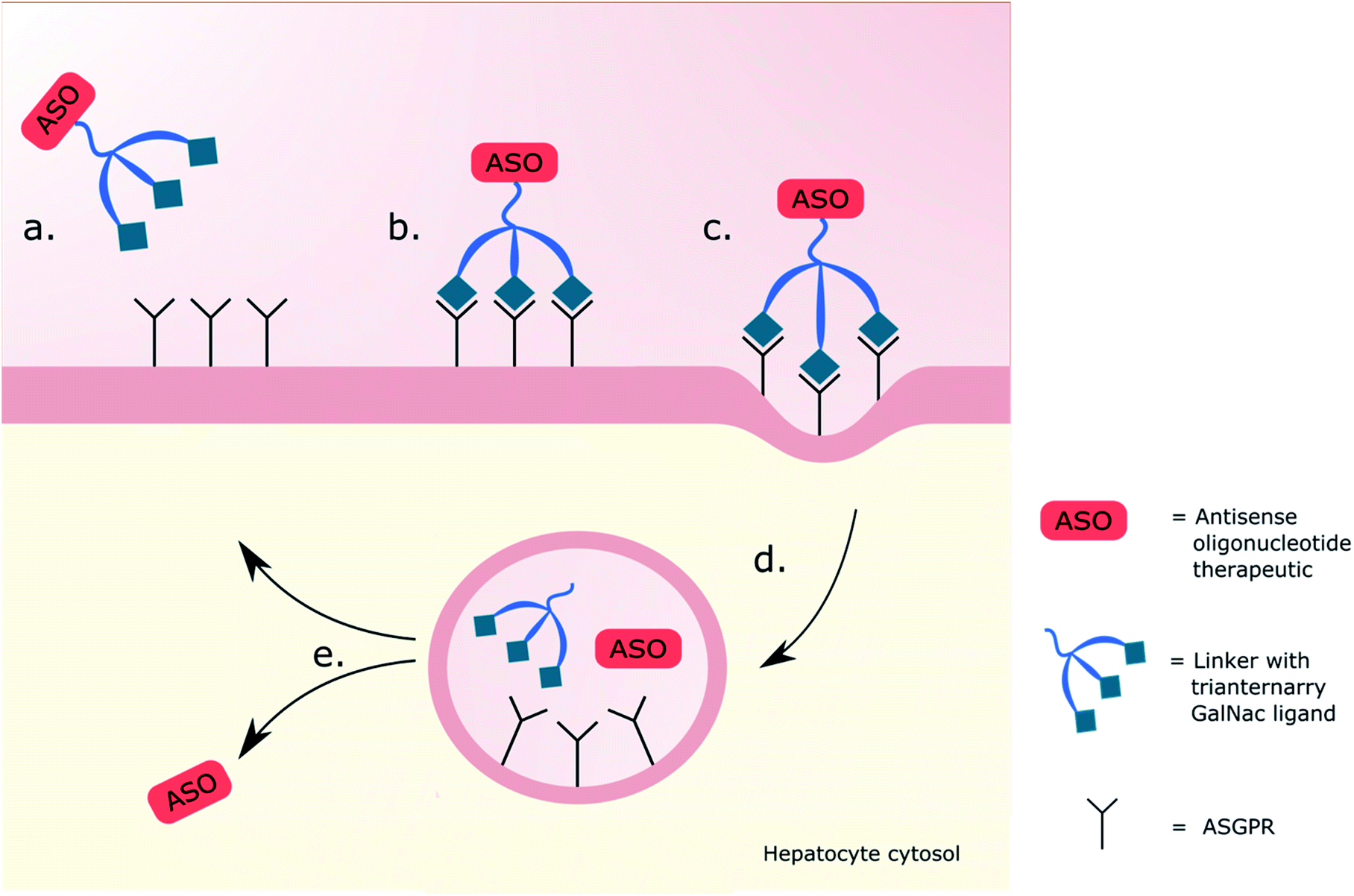 | ||
| Fig. 1 Schematic representation of triantennary GalNAc interaction with ASGPR. (a) A therapy is administered with the intention of targeting hepatocytes. (b) The triantennary GalNAc ligands bind onto three ASGPRs. (c) The GalNAc–receptor interaction initiates clathrin coated pit-mediated endocytosis. (d) Dissociation of ASO from the GalNAc delivery system occurs in the endosome. (e) Endosomal escape of the ASO then allows for release into the cytosol, whilst the ASGPR is recycled to the cell surface. | ||
Alternative drug delivery
There are several alternative DDSs to the glycan-based receptor-mediated endocytic delivery used in the liver that have been formulated and researched over the previous decades, one of which is viral vectors. Viruses often infect human cells, and the use of virus-based vectors to transport nucleic acids as treatments is exemplified by the 1990 work of Rosenberg et al. when they succeeded in gene transfer with the use of retroviruses.97 There was initially unease in the use of viral vectors due to issues regarding their toxicity, with caution encouraged, but their safety and efficacy have dramatically improved recently.98 Today, viral vectors are indeed popular choices for the treatment of monogenic diseases, the most abundantly used being adeno-associated viruses (AAVs)99,100 with notable successes in onasemnogene abeparvovec-xioi.63 On the other hand, many in the field still see viral carriers posing risks such as immunogenicity,16 and more information on AAV gene therapies may be obtained from other reviews.101,102 Nanoparticles (NPs) are typically polymeric structures in the 100 nm diameter range, with variability in shape and size affecting properties including their biodistribution, uptake, and reactivity.103 Because of the size and uniform distribution of synthetically engineered NPs (ENPs), they have been seen as a promising solution to achieving NAT delivery.104,105 Attempts have been made to address site-specific delivery using targeting ligands on drug loaded NPs for cancer treatments. NPs with monoclonal antibody ligands have been used to target the HER2 in breast cancer cells and glycosylated NPs to target a number of receptors in the liver, breast and lung.106,107 Elsewhere, chitosan-modified NPs carrying siRNA showed greater gene silencing in comparison to a lipofectamine control,108 demonstrating the power of effective targeting. Another delivery method involves synthetic lipids for DNA transfection (lipofection), but this is an ageing method. Originally thought to be a promising non-viral alternative for the delivery of nucleic acids,109 this technique has developed into a modern alternative format through the use of lipid nanoparticles (LNPs) of size distributions similar to polymeric nanoparticles, achieving success in the form of patisiran. LNPs are now more commonplace as they have been specifically developed as a vector for gene therapy,110 often targeting immune cells,111,112 and use with siRNAs has been successful in silencing particular genes.113 Moreover, Moderna Therapeutics and Pfizer/BioNTech recently succeeded in using LNPs as the delivery vector for the approved and globally used mRNA-based vaccines for SARS-CoV-2,114,115 and could potentially revolutionise the future of vaccines for many diseases, including therapeutic mRNA delivery for diseases such as cystic fibrosis, where fractional amounts of correctly assembled proteins are considered sufficient for achieving therapy. There remain concerns though, around our continually increasing exposure to NPs such as those from combustion (CNPs) present in diesel exhaust fumes, and ENPs, including TiO2 and carbon nanotubes.116 It is widely acknowledged that something so small and unpredictable could produce detrimental side effects in vivo,117 with studies showing NPs frequently producing toxic side effects (nanotoxicity) including inflammation, production of reactive oxygen species (ROS), and apoptosis,117–119 particularly when these NPs are inhaled, causing damage to lung cells. Investigations of NP nanotoxicity frequently involve experiments in C. elegans, which are easy to cultivate and with a short enough life span to provide generational analysis when exposed to NPs; the advantages and disadvantages of which are well defined by Gonzalez-Moragas et al.120 Using C. elegans, Wu et al. demonstrated a correlation between increase in NP dose and ROS production, with the size of these NPs (4–90 nm) behaving differently when treated with antioxidants.121 Different exposures to gold and silver NPs exhibited multigenerational nanotoxicity, compounding the unpredictability of this technology.122,123 NPs can also induce oxidative stress,116 leading to inflammation where they are deposited causing further problems downstream. Because of their small size, NPs can cross the blood–brain barrier118,124 and could provide an invaluable method of assisted delivery to the brain, but also run a huge risk of neurotoxicity.118 With the lung being another tissue recalcitrant to NAT treatment,125 the recent success of LNPs has garnered substantial interest in evaluating their utility for pulmonary drug delivery. However, exposure of rats to In2O3 ENPs induced problems in the lungs, including pulmonary alveolar proteinosis (PAP), granulomas, and foamy macrophages; it has been therefore advised that the workplace exposure to such NPs is revisited to help protect workers.126 More recently, LNP exposure in the context of pre-existing bacterial lipopolysaccharide-induced acute inflammation was demonstrated to exacerbate inflammation in a macrophage- and toll-like immune receptor-dependent means.127 Although this response was corticosteroid-responsive, it is unclear how chronic NP dosing, especially in the lung where NP exposure drives foamy macrophage phenotypes, will affect tissue homeostasis beyond the anticipated therapeutic effect. Another common concern is the exposure of expectant parents and their unborn children to conditions with the potential to hinder or alter the foetus during the critical stages of development; it is accordingly proposed that CNPs from air pollution may reach the human placenta.128 Overall, the field of NPs needs to be explored further to gain a better understanding of their safety to humans before long term effects of their repeated dosing become unwantedly apparent. It must be emphasised, however, that limited LNP dosage, such as, for example, in the intramuscular administration of a handful of doses of prophylactic vaccines, has not been associated to date with any short or long-term complications in adults, expectant mothers, or foetuses.Drug delivery systems have made huge strides in the last 10 years, but there are still concerns regarding the safety and ethics of viral vectors including insertional mutagenesis leading to cancer, and the possibility of causing viral treatments to be less efficient.129,130 LNPs, after SARS-CoV-2, could conceivably occupy and flourish in the field of vaccinations where infrequent dosing is required. The preference of NPs for dendritic cells and macrophages questions their usefulness for specific alternative targeting and facts around the safety of chronic dosing of NPs still need to be explored, reinforcing the overall need for better and safer targeting systems.
Complex oligosaccharide-containing delivery systems
Carbohydrates are of biological importance and naturally occurring examples are used in an array of cellular functions in which specific carbohydrate moieties are required.131 The ongoing success of the GalNAc-based DDSs for oligonucleotides is fuelling research into the exploration of other glycoconjugates as a potential DDS (or likewise) for numerous targets.107,132 The field of glycan science is vast, primarily due to their diversity and structural complexities in architecture: choosing, building, and isolating a chosen OS for one target can be challenging. On the other hand, complex natural OSs are limited in number and there are difficulties in isolating pure products from their sources, particularly where these might be derived from endangered or pathogenic species, and with issues such as homogeneity decreasing their reliability.133 If obtaining from a natural source is not a viable option, the assembly of an OS by chemical synthesis, glycoenzymatic biocatalysis, or a combination of the two are popular methods of achieving sufficient, scalable quantities.134Carbohydrate chemistry in general is an established field, with the first glycosylations occurring in the late 19th century by Michael and Fischer, whose initial work influenced Koenigs and Knorr in the development of glycosylating agents; combined, their work provides the foundations for our current day knowledge.135,136 Our understanding of carbohydrate synthesis has vastly increased hence, it is the mutual goal of many to form isomerically-pure glycosides and glycoconjugates to explore biological processes.137–140 An attractive feature of using carbohydrates is the unique ability to theoretically assemble a complex of any size and orientation, using only the necessary monosaccharides to build up the desired molecule, giving an array of possible variations for a range of applications. Due to the complexity of OS synthesis, many possible side products may also be obtained, and stereoselective glycosylation in the production of complex OSs has been, and still is, an obstacle for glycomics.136,141–143 The glycosidic linkage is commonly formed through use of an electrophilic glycosyl donor with a suitable leaving group, a nucleophilic glycosyl acceptor, and assisted with help of a promotor. Ample research has been carried out in these areas, particularly on the glycosyl donors, from the earlier years with Fischer's work on thioglycosides, Koenigs and Knorr with glycosyl halides,135,136 and to the more recent trichloroacetimidates.144,145 A multitude of glycosyl donors have also been and are currently being investigated.141,146 With the potential to assemble any glycoside in any conformation, complexities arise in controlling the position of the glycosidic bond and the stereochemistry of the resulting product. Generally, each step that increases the size of an OS increases the difficulty in making it due to the possibility of further stereoisomers being formed. Isolating one particular form of an OS to suit the intended target often involves arduous and complex sugar protection strategies (SPSs) with difficulties in separating isomers.147 With a substantial body of literature available on how glycosyl donors, promotors, and catalysts can affect the stereospecificity of glycosylation reactions,148–150 there are now suggestions to consider how the molar equivalents, reaction temperature, and reaction solvent could also influence this,151 which will ultimately be a useful tool in achieving stereospecific glycosylations. The protecting group strategy for building a chosen OS also plays a vital role in concise synthesis, with the priority of hydroxyl groups and orthogonality of the protecting groups key. A popular option is to direct the centre of reactivity through the anomeric carbon with specific activating groups and the protection of all reactive groups before the selective deprotection of the acceptor prior to glycosylation, as shown in Scheme 1. In this partial synthesis, taken from Wang et al.,152 a protected glycoside is selectively deprotected before it is coupled to another selectively protected monosaccharide with a suitable leaving group. This scheme uses only protected glucosides but shows the varying complexities that must be endured; it is inevitable that using different monosaccharides will affect the yield of isomerically pure complex OSs. An example of a selective deprotection strategy using glucosides is through the use of benzylidene acetal chemistry, explored by Johnsson et al.,153 shown in Scheme 2. This work shows that the same protected glucoside (12) can undergo selective ring opening to yield the preferred 6′-OH (13) or 4′-OH (14), with the other remaining protected by a benzyl group, and that the regioselectivity is not dependent on the Lewis acid used, but on the type of borane ligand.153,154 The influence on selectivity using specific protecting groups must also be considered, one aspect of this being participating protecting groups, and in particular neighbouring-group participation. A recognised example from Guo and Ye involves the formation of 1,2-trans glycosides through use acyl groups in the C2 position.142,155 As shown in Scheme 3, the C2 acyl group promotes departure of the leaving group (X), forming an intermediate dioxolenium ion (16), leaving only the back side open to attack from a glycosyl acceptor to form only the stereospecific b-glycoside.146 Influences such as this must be considered when developing individual synthetic protection strategies to yield isomerically pure OSs and has been the pursuit of many researchers.
 | ||
| Scheme 1 Synthesis of OS 11. | ||
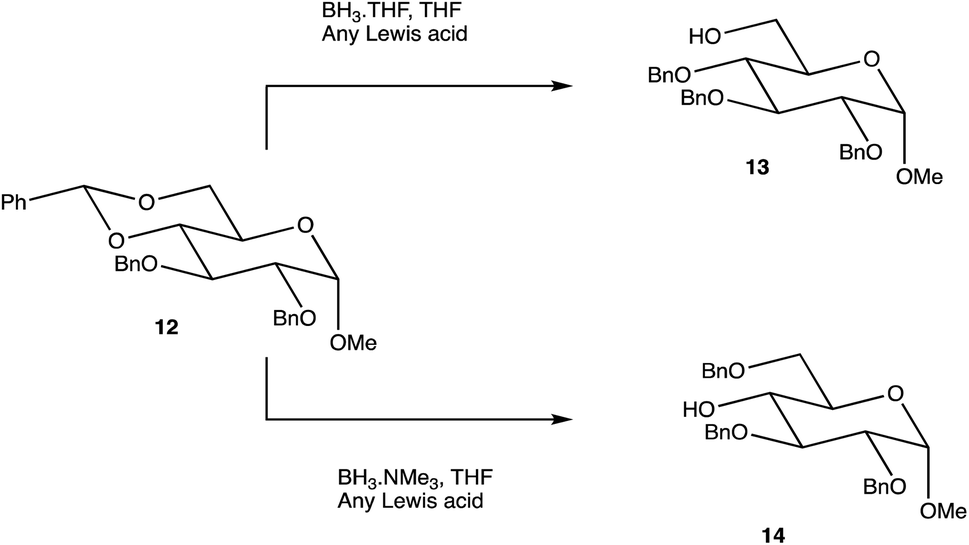 | ||
| Scheme 2 Selective benzylidene ring opening dependent on the borane ligand. | ||
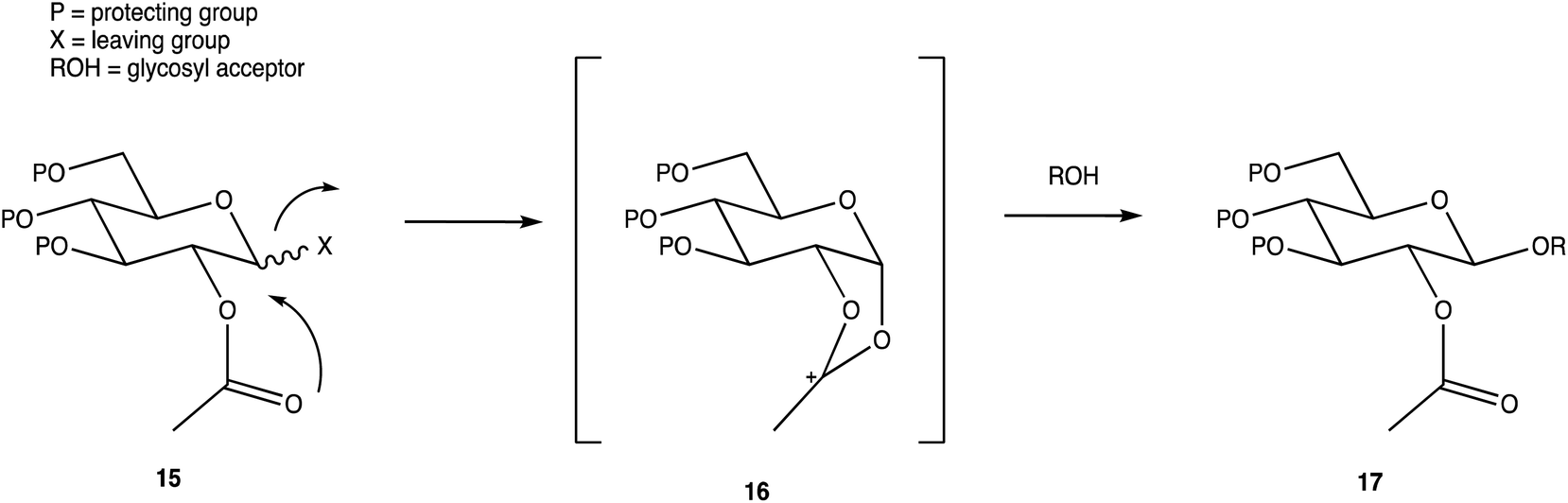 | ||
| Scheme 3 Stereoselective glycosylation with influence from a C2 acyl neighbouring group. | ||
Fig. 2 displays several glycosides (18–30) synthesised using unique SPSs, produced for an array of purposes. Human milk oligosaccharides (HMOs) are a popular area of research and derivatives were constructed by Moore et al., specifically on β-amino HMOs (18–20), to assess their ability to inhibit bacterial growth.156 Mandal and Chheda were able to provide a method of synthetically obtaining 21, which is difficult to isolate from natural sources, in sufficient quantities for immunochemical studies.157 The human body's interaction with glycosides is well documented, and the synthesis of such, and their derivatives, is frequently carried out, whether to study an OSs interaction with proteins,158 to determine enzyme kinetics (22, 23),159 or as Cui et al. showed, to chemically map an antibody binding site through chemical modification of the terminal disaccharide epitope (24–29).160 OS-based therapies are already being used as treatments; the cardiac glycoside digitoxin (30), comprising of a steroidal core with a linked trisaccharide which for over 2 centuries has been used to treat heart failure,161–163 and the synthesis of its analogues has been explored for anticancer properties, and heparin, an anticoagulant whose mimetics can play important roles in inflammation, cancer treatments and management of sepsis.164–166
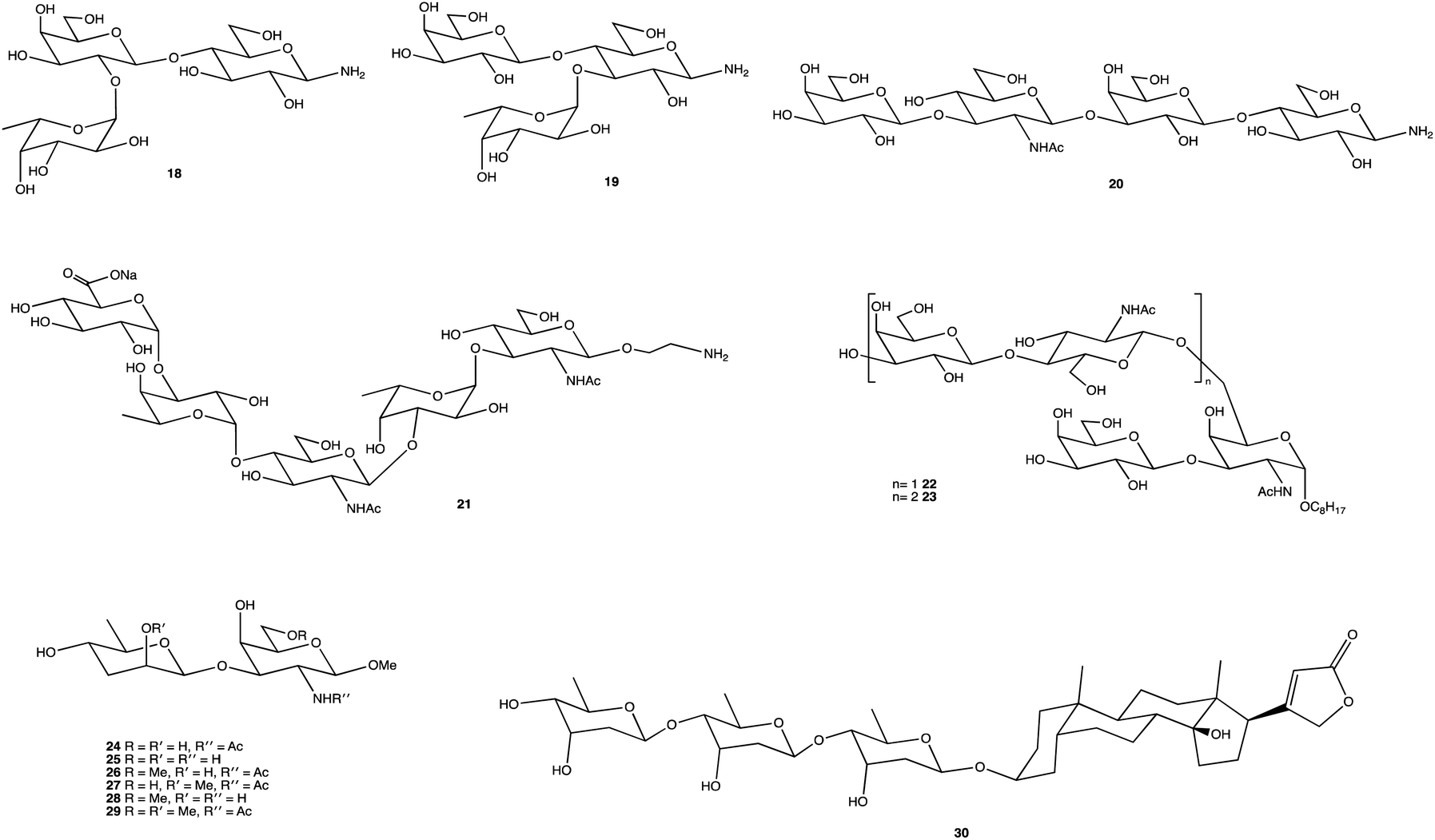 | ||
| Fig. 2 Structures of chemically synthesised glycosides 18–30. | ||
The synthesis of clean, isomerically pure OSs is vital to ensuring the reliability of research, with an overwhelming number of factors to consider including reagents, exclusive SPSs, and purification methods. With over a hundred years of chemical glycosylations documented, the harsher conditions used historically are unfavourable to some protection strategies such as those involving acid-labile groups, but with peaking interest in glycosides over the previous year's, thankfully better and greener methods are being developed167,168 and will continue to be produced until the field of glycoscience has been sufficiently explored.
An alternative option for OS assembly is through the use of glycosyltransferases (GTs); an enzymatic family abundant in nature that can provide regio- and stereo-specific glycosylations for their chosen activities.169,170 GTs are classified into families according to sequence homology and structure, which are highly conserved within the family along with its mechanism, but can hold enzymatic activities for different glycosyl donor and acceptors.131,171 A wealth of information has been acquired on GTs over the last few decades, but much is still unknown,172 with the most recent advances published within the Carbohydrate-Active enZYmes database (https://www.cazy.org) including information on families, structures, and activities of GTs – a set of 114 families currently (February 2022). Use of these enzymes is arguably favourable to the chemosynthetic route due to the reactions being easier, cleaner, and greener given the milder conditions used.173 The cloning and expression of enzymes from a variety of sources is well established, but problems with the stability, expression level, and solubility of GTs in aqueous solvents exist due to these enzymes being predominantly membrane-bound which limits their functionality and use in this field. It is possible to alleviate the issue of solubility or produce soluble subunits containing the enzymatically active site through codon optimisation,174,175 the use of molecular chaperones, or to obtain the holoenzyme through use of specific or expensive detergents to disrupt the bacterial membrane thereby releasing the enzyme, hopefully in a bioactive state.176 Sugiyama et al. were successful in producing a glycosynthase, a mutant glycosidase capable of transglycosylation using inexpensive donors (Scheme 4).177,178 Through codon optimisation, inversion of a fucosidase to a fucosynthase allowed for fucosylation of lacto-N-tetraose 31 producing branched OS 32 containing a terminal disaccharide (fuc-(α)-1,2gal) present in blood H-antigens. They showed that the synthase could also introduce the H-antigen onto a complete glycoprotein, demonstrating the significance of a highly regioselective protein in glycobiology. From the protected glycoside 35, Böcker et al. were able to produce the tetrasaccharide 38 through sequential glycosylations; using a β-1,4-galactosyltransferase (GalT), a GT from the family 7 (GT7), and 33 for the addition of galactose, and a β-1,3-N-acetyl-glucosaminyltransferase (GlcNAcT) (GT8) and 34 for the addition of GlcNAc (Scheme 5).179 These were the limiting steps biocatalytically and chemical synthesis was necessary for deprotection of 38 to 39, and subsequent reaction of the amino group to give tetrasaccharide 40, highlighting the symbiosis of these two fields to yield desired compounds; 40 was subsequently coupled to BSA and its binding affinity for the human galactin-3, a galactose specific lectin, was performed. In contrast, Unverzagt highlights possible complications with chemical synthesis whilst constructing dodecamer 43, as shown in Scheme 6, in which the final stage is performed using GTs. With the objective to integrate 43 into glycopeptides, getting from heptasaccharide-asparagine conjugate 42 chemically would involve complicated SPSs for both the OS and the peptide. The use of specific GTs and reagents, GT7 with 33 followed by GT80 with 41, made it easier and cleaner and again emphasises the benefits of combined chemoenzymatic synthesis
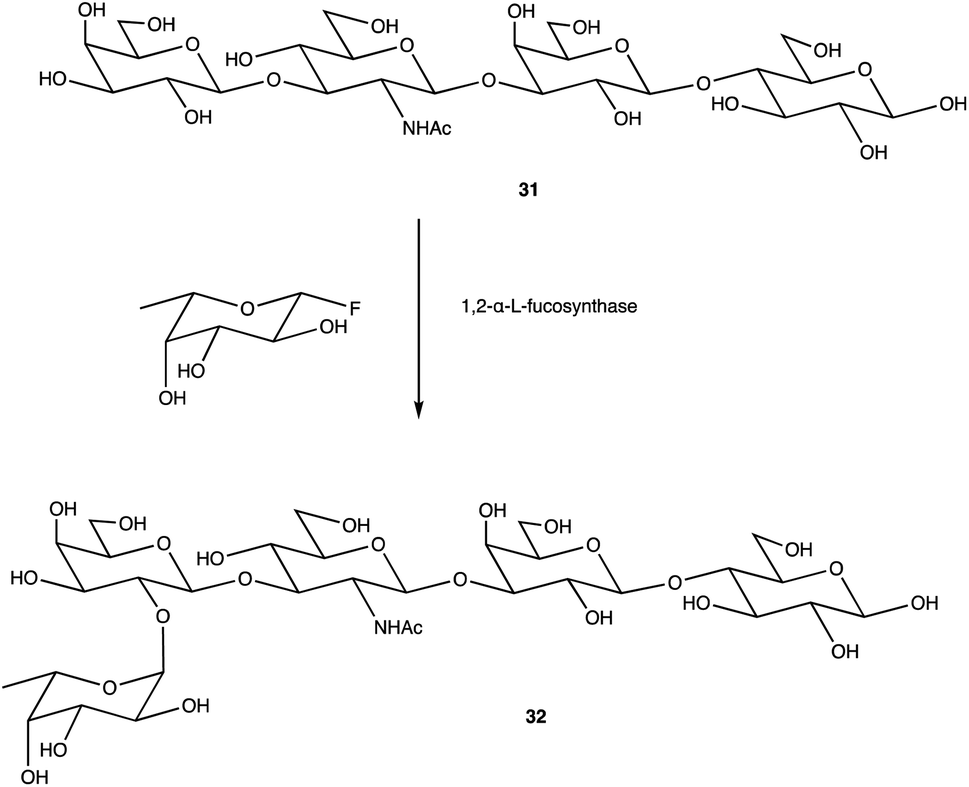 | ||
| Scheme 4 Fucosylation of lacto-N-tetraose using a 1,2-α-L-fucosynthase to yield desired terminal disaccharide. | ||
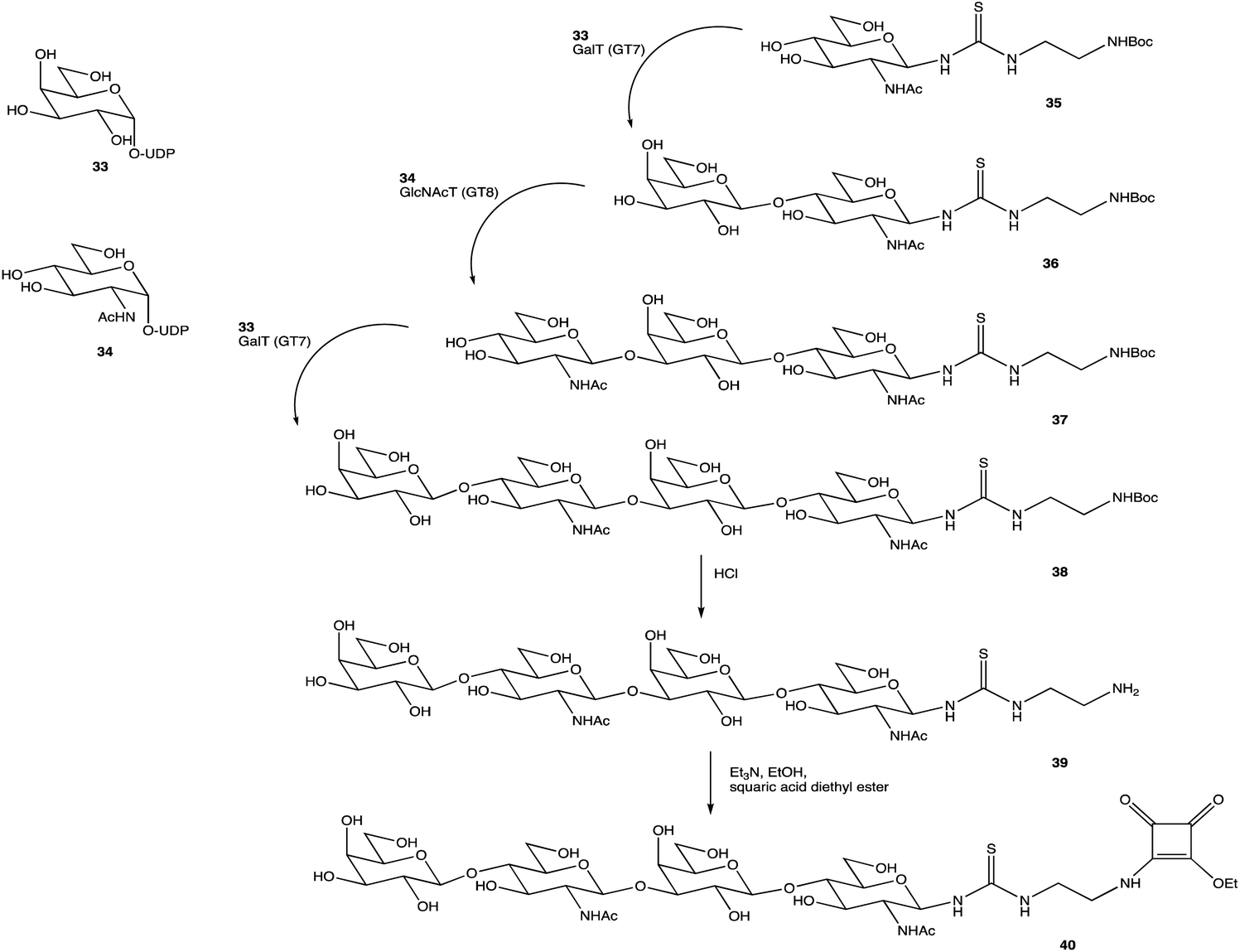 | ||
| Scheme 5 Assembly of OS 38 by consecutive glycosylation with GTs, before chemical synthesis to obtain 40. | ||
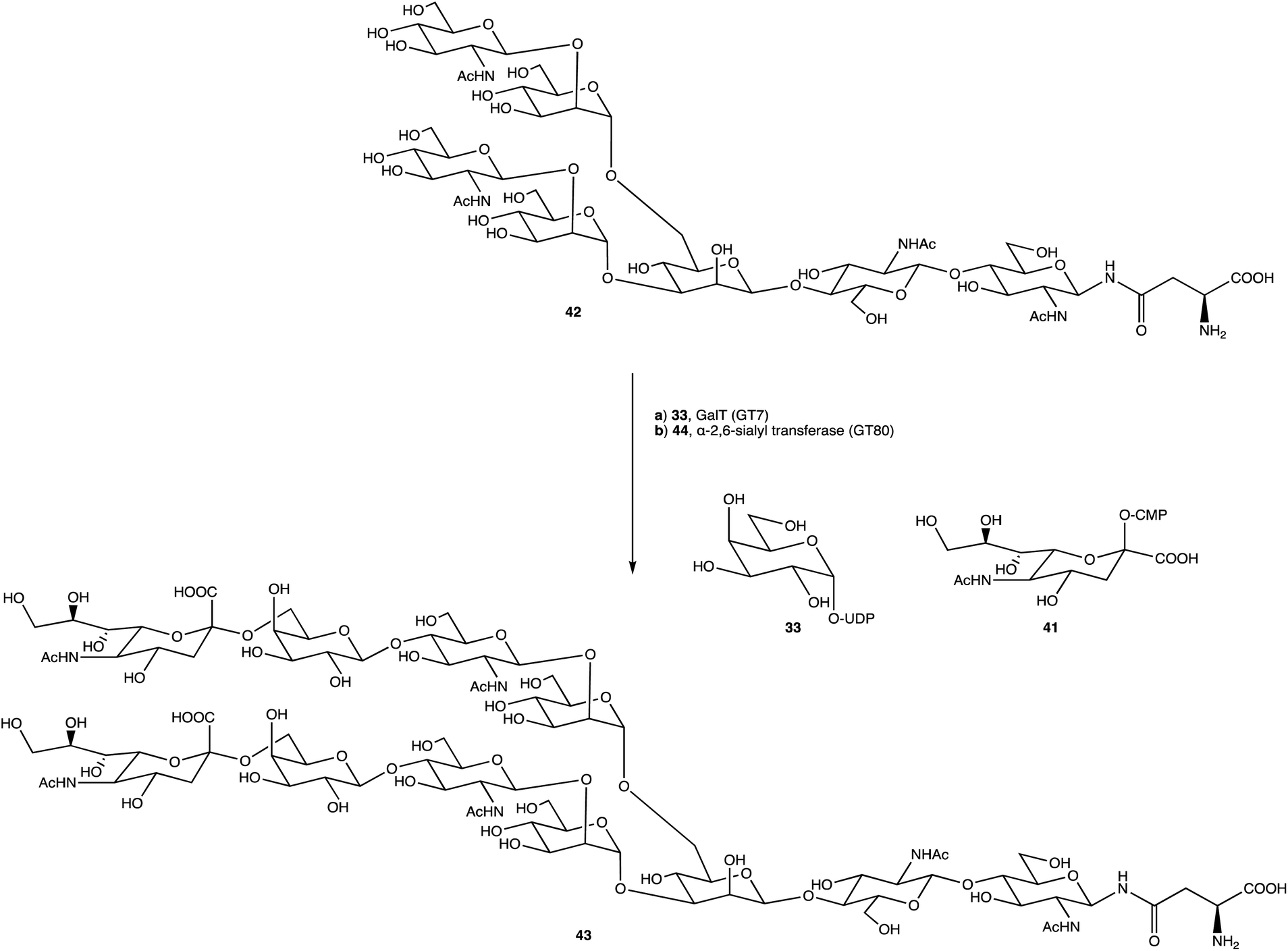 | ||
| Scheme 6 Dodecamer 43 formed through glycosylations with GT7 and GT80. | ||
There is a known historical problem of expressing GTs due the hydrophobic nature of them and isolating them from the membrane may require further purification increasing costs, affecting yields, and can ultimately fail to produce a bioactive enzyme or a yield that is economically viable for processes other than basic research. Work needs to be done in this area to develop new routes to help express and solubilise these proteins with assistance from artificial intelligence algorithms and other predictive expression tools, or else provide an alternative or better method for delivering functioning membrane-bound proteins. This would give access to a range of GTs for biocatalytic purposes, opening the field and make it possible to explore this OS space further without limitations. An ability to obtain more GTs could be one key aspect to opening this research, allowing us to create clean and uniquely glycosylated systems for a range of uses, potentially even at the keystroke level of simplicity inherent to modern ASO synthesis and research.
One field that can aid in the exploration of complex OS assembly is that of automated oligosaccharide synthesis (AOSS). Automated synthesis is an established field for both peptides and nucleotides and has enhanced research in these areas by making precise oligomers widely available and highly affordable,180 yet despite two decades of oligopeptide and oligonucleotide automated synthesis, success in the OS field, although desirable, has been limited. Two popular methods of chemical OS assembly include the use of resins or polymers as a solid supports attached to the glycosyl donor or acceptor, as first described by Schuerch and Frechet to synthesise a trisaccharide.181 Using an automated solid-phase approach, Seeberger et al. succeeded in constructing a 50-mer of repeating units, a polymannoside,182 and other approaches have been extensively reviewed by Seeberger and Haase,144 Castagner and Seeberger,180 and Bennett.183 The second method is one pot synthesis which can be successful, as demonstrated by Xiao et al. in 2020.184 For automated one pot synthesis, programmes assist in a streamlined assembly of an OS using designed ‘building block’ monosaccharides without purification of intermediates.165,185 The increasing requirement for unique anomerically pure products will likely see a growing interest in the more specific automated enzymatic synthesis of OSs which is still limited by the restricted access to enzymes along with stability issues;186 it would be prudent to assume that if more stable GTs were expressed, there could be huge advancement in an enzyme driven AOSS. In depth reviews discussing the chemical and enzymatic AOSS space are well summarised by Panza et al.187 and Wen et al.188 respectively.
Commercial AOSS is also beginning to become available with The Glyconeer® (https://glycouniverse.com/glyconeer), a system which uses fully or partially protected monosaccharides along with solid phase resins to build OSs on request. Its effectiveness has been demonstrated by Seeberger et al.189 The whole system is designed to operate under argon gas to reduce potential hydrolytic side reactions, reaction temperatures can be controlled, and with the smart use of inbuilt UV detectors, products can be monitored. Limitations with this instrument do exist; there are few linkers and resins, and currently only six distinct monosaccharides available to use, although these do cover most conventional glycan building blocks. This instrument is a huge step in the right direction, however one capable of harmonious use of chemical and biocatalytic reagents could catalyse any complex OS synthesis and help develop the field of glycomics.
Conclusion
Historically, the naked delivery of oligonucleotides hasn't proven successful either experimentally or commercially, but hepatic delivery of NATs has been solved with the success of the GalNAc-based DDS. With the precedent GalNAc has set for NAT delivery, precise targeting with alternative complex OS-based DDSs elsewhere, allowing the distribution of therapeutics to other specified target tissues and tissue-specific cell types is warranted. The recent global adoption of LNPs and mRNA-based therapeutics emphasises the need for better targeting outside the vaccine and hepatic space, and GalNAc has opened the door to demonstrating the kinds of benefits that can be achieved. The huge potential promised for NATs is now being realised, and we speculate that if the paradigm set out by GalNAc is achievable for alternative complex OSs beyond GalNAc, the ability to provide bespoke OS-based DDSs will allow delivery of tailored NATs to targets both in vitro and in vivo, allowing sugars to play a significant role in this sweet revolution.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors are grateful for the financial support of High Force Research in this research.References
- S. Hyde, K. Southern, U. Gileadi, E. Fitzjohn, K. Mofford, B. Waddell, H. Gooi, C. Goddard, K. Hannavy, S. Smyth, J. Egan, F. Sorgi, L. Huang, A. Cuthbert, M. Evans, W. Colledge, C. Higgins, A. Webb and D. Gill, Gene Ther., 2000, 7, 1156–1165 CrossRef CAS PubMed.
- P. C. Nopoulos, Dialogues Clin. Neurosci., 2016, 18, 91–98 CrossRef PubMed.
- J. K. Watts, R. H. Brown and A. Khvorova, Neurotherapeutics, 2019, 16, 245–247 CrossRef PubMed.
- S. A. Moschos, A. E. Williams and M. A. Lindsay, Biochem. Soc. Trans., 2007, 35, 807–810 CrossRef CAS PubMed.
- K. Takakura, A. Kawamura, Y. Torisu, S. Koido, N. Yahagi and M. Saruta, Int. J. Mol. Sci., 2019, 20, 3331 CrossRef CAS PubMed.
- J. B. Opalinska and A. M. Gewirtz, Nat. Rev. Drug Discovery, 2002, 1, 503–514 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- C. M. O'Driscoll, A. Bernkop-Schnürch, J. D. Friedl, V. Préat and V. Jannin, Eur. J. Pharm. Sci., 2019, 133, 190–204 CrossRef PubMed.
- K. G. Rajeev, J. K. Nair, M. Jayaraman, K. Charisse, N. Taneja, J. O'Shea, J. L. S. Willoughby, K. Yucius, T. Nguyen, S. Shulga-Morskaya, S. Milstein, A. Liebow, W. Querbes, A. Borodovsky, K. Fitzgerald, M. A. Maier and M. Manoharan, ChemBioChem, 2015, 16, 903–908 CrossRef CAS PubMed.
- A. J. Debacker, J. Voutila, M. Catley, D. Blakey and N. Habib, Mol. Ther., 2020, 28, 1759–1771 CrossRef CAS PubMed.
- J. Chery, Postdoc J. a J. Postdr. Res. Postdr. Aff., 2016, 4, 35–50 Search PubMed.
- E. J. Sontheimer and R. W. Carthew, Cell, 2005, 122, 9–12 CrossRef CAS PubMed.
- C. Frøkjær-Jensen, N. Jain, L. Hansen, M. W. Davis, Y. Li, D. Zhao, K. Rebora, J. R. M. Millet, X. Liu, S. K. Kim, D. Dupuy, E. M. Jorgensen and A. Z. Fire, Cell, 2016, 166, 343–357 CrossRef PubMed.
- S. Veldhoen, S. Laufer and T. Restle, Int. J. Mol. Sci., 2008, 9, 1276–1320 CrossRef CAS PubMed.
- G. M. Lenk, P. Jafar-Nejad, S. F. Hill, L. D. Huffman, C. E. Smolen, J. L. Wagnon, H. Petit, W. Yu, J. Ziobro, K. Bhatia, J. Parent, R. J. Giger, F. Rigo and M. H. Meisler, Ann. Neurol., 2020, 87, 339–346 CrossRef CAS PubMed.
- Y. Weng, Q. Huang, C. Li, Y. Yang, X. Wang, J. Yu, Y. Huang and X.-J. Liang, Mol. Ther. Nucleic Acids, 2020, 19, 581–601 CrossRef CAS PubMed.
- K. Murakami and M. Miyagishi, Biomed. Rep., 2014, 2, 509–512 CrossRef CAS PubMed.
- P. C. Zamecnik and M. L. Stephenson, Proc. Natl. Acad. Sci. USA, 1978, 75, 280–284 CrossRef CAS PubMed.
- M. L. Stephenson and P. C. Zamecnik, Proc. Natl. Acad. Sci. USA, 1978, 75, 285–288 CrossRef CAS PubMed.
- S. D. Putney, S. J. Benkovic and P. R. Schimmel, Proc. Natl. Acad. Sci. USA, 1981, 78, 7350–7354 CrossRef CAS PubMed.
- S. Freier, Nucleic Acids Res., 1997, 25, 4429–4443 CrossRef CAS PubMed.
- W. C. Song, K.-R. Kim, M. Park, K. E. Lee and D.-R. Ahn, Biomater. Sci., 2017, 5, 412–416 RSC.
- J. H. Chan, S. Lim and W. F. Wong, Clin. Exp. Pharmacol. Physiol., 2006, 33, 533–540 CrossRef CAS PubMed.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. USA, 1987, 84, 7413–7417 CrossRef CAS PubMed.
- A. Khaled, S. Guo, F. Li and P. Guo, Nano Lett., 2005, 5, 1797–1808 CrossRef CAS PubMed.
- J. B. Connolly, Gene Ther., 2002, 9, 1730–1734 CrossRef CAS PubMed.
- S. A. Moschos, L. Usher and M. A. Lindsay, Pharmacol. Ther., 2017, 169, 83–103 CrossRef CAS PubMed.
- S. F. Dowdy, Nat. Biotechnol., 2017, 35, 222–229 CrossRef CAS PubMed.
- R. Kole, A. R. Krainer and S. Altman, Nat. Rev. Drug Discovery, 2012, 11, 125–140 CrossRef CAS PubMed.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- M. M. Zhang, R. Bahal, T. P. Rasmussen, J. E. Manautou and X. Zhong, Biochem. Pharmacol., 2021, 189, 114432 CrossRef CAS PubMed.
- H. Xiong, R. N. Veedu and S. D. Diermeier, Int. J. Mol. Sci., 2021, 22, 3295 CrossRef CAS PubMed.
- D. Hattab, A. M. Gazzali and A. Bakhtiar, Pharmaceutics, 2021, 13, 1009 CrossRef CAS PubMed.
- K. Dhuri, C. Bechtold, E. Quijano, H. Pham, A. Gupta, A. Vikram and R. Bahal, J. Clin. Med., 2020, 9, 2004 CrossRef CAS PubMed.
- J. Kim, C. Hu, C. Moufawad El Achkar, L. E. Black, J. Douville, A. Larson, M. K. Pendergast, S. F. Goldkind, E. A. Lee, A. Kuniholm, A. Soucy, J. Vaze, N. R. Belur, K. Fredriksen, I. Stojkovska, A. Tsytsykova, M. Armant, R. L. DiDonato, J. Choi, L. Cornelissen, L. M. Pereira, E. F. Augustine, C. A. Genetti, K. Dies, B. Barton, L. Williams, B. D. Goodlett, B. L. Riley, A. Pasternak, E. R. Berry, K. A. Pflock, S. Chu, C. Reed, K. Tyndall, P. B. Agrawal, A. H. Beggs, P. E. Grant, D. K. Urion, R. O. Snyder, S. E. Waisbren, A. Poduri, P. J. Park, A. Patterson, A. Biffi, J. R. Mazzulli, O. Bodamer, C. B. Berde and T. W. Yu, N. Engl. J. Med., 2019, 381, 1644–1652 CrossRef CAS PubMed.
- E. Marrosu, P. Ala, F. Muntoni and H. Zhou, Mol. Ther. Nucleic Acids, 2017, 8, 416–427 CrossRef CAS PubMed.
- V. Mathew and A. K. Wang, Drug Des. Devel. Ther., 2019, 13, 1515–1525 CrossRef CAS PubMed.
- C. D. Novina and P. A. Sharp, Nature, 2004, 430, 161–164 CrossRef CAS PubMed.
- A. A. Barba, S. Bochicchio, A. Dalmoro and G. Lamberti, Pharmaceutics, 2019, 11, 360 CrossRef CAS PubMed.
- Y. Dong, D. J. Siegwart and D. G. Anderson, Adv. Drug Deliv. Rev., 2019, 144, 133–147 CrossRef CAS PubMed.
- A. D. Keefe, S. Pai and A. Ellington, Nat. Rev. Drug Discovery, 2010, 9, 537–550 CrossRef CAS PubMed.
- K. Sridharan and N. J. Gogtay, Br. J. Clin. Pharmacol., 2016, 659–672 CrossRef PubMed.
- H. Frangoul, D. Altshuler, M. D. Cappellini, Y.-S. Chen, J. Domm, B. K. Eustace, J. Foell, J. de la Fuente, S. Grupp, R. Handgretinger, T. W. Ho, A. Kattamis, A. Kernytsky, J. Lekstrom-Himes, A. M. Li, F. Locatelli, M. Y. Mapara, M. de Montalembert, D. Rondelli, A. Sharma, S. Sheth, S. Soni, M. H. Steinberg, D. Wall, A. Yen and S. Corbacioglu, N. Engl. J. Med., 2021, 384, 252–260 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- D. D. Rao, J. S. Vorhies, N. Senzer and J. Nemunaitis, Adv. Drug Deliv. Rev., 2009, 61, 746–759 CrossRef CAS PubMed.
- A. D. Springer and S. F. Dowdy, Nucleic Acid Therapeut., 2018, 28, 109–118 CrossRef CAS PubMed.
- L. Xu and T. Anchordoquy, J. Pharm. Sci., 2011, 100, 38–52 CrossRef CAS PubMed.
- K. B. Kaufmann, H. Büning, A. Galy, A. Schambach and M. Grez, EMBO Mol. Med., 2013, 5, 1642–1661 CrossRef CAS PubMed.
- M. Thomas, J. J. Lu, J. Chen and A. M. Klibanov, Adv. Drug Deliv. Rev., 2007, 59, 124–133 CrossRef CAS PubMed.
- Y. Cao, Y. F. Tan, Y. S. Wong, M. W. J. Liew and S. Venkatraman, Mar. Drugs, 2019, 17, 381 CrossRef CAS PubMed.
- V. Study Group, Am. J. Ophthalmol., 2002, 133, 484–498 CrossRef.
- E. S. Gragoudas, A. P. Adamis, E. T. Cunningham, M. Feinsod, D. R. Guyer and VEGF Inhibition Study, Ocular Neovascularization Clinical Trial Group, N. Engl. J. Med., 2004, 351, 2805–2816 CrossRef CAS PubMed.
- J. R. Mendell, L. R. Rodino-Klapac, Z. Sahenk, K. Roush, L. Bird, L. P. Lowes, L. Alfano, A. M. Gomez, S. Lewis, J. Kota, V. Malik, K. Shontz, C. M. Walker, K. M. Flanigan, M. Corridore, J. R. Kean, H. D. Allen, C. Shilling, K. R. Melia, P. Sazani, J. B. Saoud, E. M. Kaye and Eteplirsen Study Group, Ann. Neurol., 2013, 74, 637–647 CrossRef CAS PubMed.
- M. Haché, K. J. Swoboda, N. Sethna, A. Farrow-Gillespie, A. Khandji, S. Xia and K. M. Bishop, J. Child Neurol., 2016, 31, 899–906 CrossRef PubMed.
- M. D. Benson, M. Waddington-Cruz, J. L. Berk, M. Polydefkis, P. J. Dyck, A. K. Wang, V. Planté-Bordeneuve, F. A. Barroso, G. Merlini, L. Obici, M. Scheinberg, T. H. Brannagan, W. J. Litchy, C. Whelan, B. M. Drachman, D. Adams, S. B. Heitner, I. Conceição, H. H. Schmidt, G. Vita, J. M. Campistol, J. Gamez, P. D. Gorevic, E. Gane, A. M. Shah, S. D. Solomon, B. P. Monia, S. G. Hughes, T. J. Kwoh, B. W. McEvoy, S. W. Jung, B. F. Baker, E. J. Ackermann, M. A. Gertz and T. Coelho, N. Engl. J. Med., 2018, 379, 22–31 CrossRef CAS PubMed.
- D. Adams, A. Gonzalez-Duarte, W. D. O'Riordan, C.-C. Yang, M. Ueda, A. V. Kristen, I. Tournev, H. H. Schmidt, T. Coelho, J. L. Berk, K.-P. Lin, G. Vita, S. Attarian, V. Planté-Bordeneuve, M. M. Mezei, J. M. Campistol, J. Buades, T. H. Brannagan, B. J. Kim, J. Oh, Y. Parman, Y. Sekijima, P. N. Hawkins, S. D. Solomon, M. Polydefkis, P. J. Dyck, P. J. Gandhi, S. Goyal, J. Chen, A. L. Strahs, S. V. Nochur, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. A. Gollob and O. B. Suhr, N. Engl. J. Med., 2018, 379, 11–21 CrossRef CAS PubMed.
- M. Balwani, E. Sardh, P. Ventura, P. A. Peiró, D. C. Rees, U. Stölzel, D. M. Bissell, H. L. Bonkovsky, J. Windyga, K. E. Anderson, C. Parker, S. M. Silver, S. B. Keel, J.-D. Wang, P. E. Stein, P. Harper, D. Vassiliou, B. Wang, J. Phillips, A. Ivanova, J. G. Langendonk, R. Kauppinen, E. Minder, Y. Horie, C. Penz, J. Chen, S. Liu, J. J. Ko, M. T. Sweetser, P. Garg, A. Vaishnaw, J. B. Kim, A. R. Simon, L. Gouya and ENVISION Investigators, N. Engl. J. Med., 2020, 382, 2289–2301 CrossRef CAS PubMed.
- S. F. Garrelfs, Y. Frishberg, S. A. Hulton, M. J. Koren, W. D. O'Riordan, P. Cochat, G. Deschênes, H. Shasha-Lavsky, J. M. Saland, W. G. Van’t Hoff, D. G. Fuster, D. Magen, S. H. Moochhala, G. Schalk, E. Simkova, J. W. Groothoff, D. J. Sas, K. A. Meliambro, J. Lu, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. M. Gansner, T. L. McGregor, J. C. Lieske and ILLUMINATE-A Collaborators, N. Engl. J. Med., 2021, 384, 1216–1226 CrossRef CAS PubMed.
- M. Shirley, Drugs, 2021, 81, 875–879 CrossRef CAS PubMed.
- J. K. W. Lam, M. Y. T. Chow, Y. Zhang and S. W. S. Leung, Mol. Ther. Nucleic Acids, 2015, 4, e252 CrossRef CAS PubMed.
- C. Lorenzer, M. Dirin, A.-M. Winkler, V. Baumann and J. Winkler, J. Contr. Release, 2015, 203, 1–15 CrossRef CAS PubMed.
- T. R. Damase, R. Sukhovershin, C. Boada, F. Taraballi, R. I. Pettigrew and J. P. Cooke, Front. Bioeng. Biotechnol., 2021, 9, 1–24 Search PubMed.
- I. J. Goldstein, R. C. Hughes, M. Monsigny, T. Osawa and N. Sharon, Nature, 1980, 285, 66 CrossRef.
- M. H. Ravindranath, H. H. Higa, E. L. Cooper and J. C. Paulson, J. Biol. Chem., 1985, 260, 8850–8856 CrossRef CAS PubMed.
- R. J. Stockert, A. G. Morell and I. H. Scheinberg, Science, 1974, 186, 365–366 CrossRef CAS PubMed.
- D. Roggenbuck, M. G. Mytilinaiou, S. V. Lapin, D. Reinhold and K. Conrad, Autoimmun. Highlights, 2012, 3, 119–125 CrossRef CAS PubMed.
- R. J. Stockert and A. G. Morell, Hepatology, 2007, 3, 750–757 CrossRef PubMed.
- J. L. S. Willoughby, A. Chan, A. Sehgal, J. S. Butler, J. K. Nair, T. Racie, S. Shulga-Morskaya, T. Nguyen, K. Qian, K. Yucius, K. Charisse, T. J. C. van Berkel, M. Manoharan, K. G. Rajeev, M. A. Maier, V. Jadhav and T. S. Zimmermann, Mol. Ther., 2018, 26, 105–114 CrossRef CAS PubMed.
- J. U. Baenziger and Y. Maynard, J. Biol. Chem., 1980, 255, 4607–4613 CrossRef CAS PubMed.
- R. T. Lee, P. Lin and Y. C. Lee, Biochemistry, 1984, 23, 4255–4261 CrossRef CAS PubMed.
- Y. C. Lee and R. T. Lee, Acc. Chem. Res., 1995, 28, 321–327 CrossRef CAS.
- A. L. Schwartz, D. Rup and H. F. Lodish, J. Biol. Chem., 1980, 255, 9033–9036 CrossRef CAS PubMed.
- M. Spiess, Biochemistry, 1990, 29, 10009–10018 CrossRef CAS PubMed.
- T. P. Prakash, M. J. Graham, J. Yu, R. Carty, A. Low, A. Chappell, K. Schmidt, C. Zhao, M. Aghajan, H. F. Murray, S. Riney, S. L. Booten, S. F. Murray, H. Gaus, J. Crosby, W. F. Lima, S. Guo, B. P. Monia, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2014, 42, 8796–8807 CrossRef CAS PubMed.
- D. T. Connolly, R. R. Townsend, K. Kawaguchi, M. K. Hobish, W. R. Bell and Y. C. Lee, Biochem. J., 1983, 214, 421–431 CrossRef CAS PubMed.
- H. Löhr, U. Treichel, T. Poralla, M. Manns, K. M. Zum Büschenfelde and B. Fleischer, Hepatology, 1990, 12, 1314–1320 CrossRef PubMed.
- G. Y. Wu and C. H. Wu, J. Biol. Chem., 1987, 262, 4429–4432 CrossRef CAS PubMed.
- C. Plank, K. Zatloukal, M. Cotten, K. Mechtler and E. Wagner, Bioconjug. Chem., 1992, 3, 533–539 CrossRef CAS PubMed.
- L. A. J. M. Sliedregt, P. C. N. Rensen, E. T. Rump, P. J. van Santbrink, M. K. Bijsterbosch, A. R. Valentijn, G. A. van der Marel, J. H. van Boom, T. J. van Berkel and E. A. L. Biessen, J. Med. Chem., 1999, 42, 609–618 CrossRef CAS PubMed.
- A. R. P. M. Valentijn, G. A. van der Marel, L. A. J. M. Sliedregt, T. J. C. van Berkel, E. A. L. Biessen and J. H. van Boom, Tetrahedron, 1997, 53, 759–770 CrossRef CAS.
- P. C. N. Rensen, L. A. J. M. Sliedregt, M. Ferns, E. Kieviet, S. M. W. van Rossenberg, S. H. van Leeuwen, T. J. C. van Berkel and E. A. L. Biessen, J. Biol. Chem., 2001, 276, 37577–37584 CrossRef CAS PubMed.
- Y. C. Lee, R. R. Townsend, M. R. Hardy, J. Lönngren, J. Arnarp, M. Haraldsson and H. Lönn, J. Biol. Chem., 1983, 258, 199–202 CrossRef CAS PubMed.
- M. A. Maier, C. G. Yannopoulos, N. Mohamed, A. Roland, H. Fritz, V. Mohan, G. Just and M. Manoharan, Bioconjug. Chem., 2003, 14, 18–29 CrossRef CAS PubMed.
- J. J. Hangeland, J. T. Levis, Y. C. Lee and P. O. P. Ts'o, Bioconjug. Chem., 1995, 6, 695–701 CrossRef CAS PubMed.
- P. C. N. Rensen, S. H. van Leeuwen, L. A. J. M. Sliedregt, T. J. C. van Berkel and E. A. L. Biessen, J. Med. Chem., 2004, 47, 5798–5808 CrossRef CAS PubMed.
- E. A. L. Biessen, D. M. Beuting, H. C. P. F. Roelen, G. A. van de Marel, J. H. Van Boom and T. J. C. Van Berkel, J. Med. Chem., 1995, 38, 1538–1546 CrossRef CAS PubMed.
- O. Khorev, D. Stokmaier, O. Schwardt, B. Cutting and B. Ernst, Bioorg. Med. Chem., 2008, 16, 5216–5231 CrossRef CAS PubMed.
- K. Schmidt, T. P. Prakash, A. J. Donner, G. A. Kinberger, H. J. Gaus, A. Low, M. E. Østergaard, M. Bell, E. E. Swayze and P. P. Seth, Nucleic Acids Res., 2017, 45, 2294–2306 CrossRef CAS PubMed.
- G. A. Kinberger, T. P. Prakash, J. Yu, G. Vasquez, A. Low, A. Chappell, K. Schmidt, H. M. Murray, H. Gaus, E. E. Swayze and P. P. Seth, Bioorg. Med. Chem. Lett., 2016, 26, 3690–3693 CrossRef CAS PubMed.
- C. R. Brown, S. Gupta, J. Qin, T. Racie, G. He, S. Lentini, R. Malone, M. Yu, S. Matsuda, S. Shulga-Morskaya, A. V Nair, C. S. Theile, K. Schmidt, A. Shahraz, V. Goel, R. G. Parmar, I. Zlatev, M. K. Schlegel, J. K. Nair, M. Jayaraman, M. Manoharan, D. Brown, M. A. Maier and V. Jadhav, Nucleic Acids Res., 2020, 48, 11827–11844 CrossRef CAS PubMed.
- Y. Huang, Mol. Ther. Nucleic Acids, 2017, 6, 116–132 CrossRef CAS PubMed.
- J. K. Nair, J. L. S. Willoughby, A. Chan, K. Charisse, M. R. Alam, Q. Wang, M. Hoekstra, P. Kandasamy, A. Kelvin, S. Milstein, N. Taneja, J. O'Shea, S. Shaikh, L. Zhang, R. J. van der Sluis, M. E. Jung, A. Akinc, R. Hutabarat, S. Kuchimanchi, K. Fitzgerald, T. Zimmermann, T. J. C. van Berkel, M. A. Maier, K. G. Rajeev and M. Manoharan, J. Am. Chem. Soc., 2014, 136, 16958–16961 CrossRef CAS PubMed.
- R. Z. Yu, M. J. Graham, N. Post, S. Riney, T. Zanardi, S. Hall, J. Burkey, C. S. Shemesh, T. P. Prakash, P. P. Seth, E. E. Swayze, R. S. Geary, Y. Wang and S. Henry, Mol. Ther. Nucleic Acids, 2016, 5, e317 CrossRef CAS PubMed.
- L. J. Scott, Drugs, 2020, 80, 335–339 CrossRef PubMed.
- E. Sardh, P. Harper, M. Balwani, P. Stein, D. Rees, D. M. Bissell, R. Desnick, C. Parker, J. Phillips, H. L. Bonkovsky, D. Vassiliou, C. Penz, A. Chan-Daniels, Q. He, W. Querbes, K. Fitzgerald, J. B. Kim, P. Garg, A. Vaishnaw, A. R. Simon and K. E. Anderson, N. Engl. J. Med., 2019, 380, 549–558 CrossRef PubMed.
- S. A. Rosenberg, P. Aebersold, K. Cornetta, A. Kasid, R. A. Morgan, R. Moen, E. M. Karson, M. T. Lotze, J. C. Yang, S. L. Topalian, M. J. Merino, K. Culver, A. D. Miller, R. M. Blaese and W. F. Anderson, N. Engl. J. Med., 1990, 323, 570–578 CrossRef CAS PubMed.
- S. Razi Soofiyani, B. Baradaran, F. Lotfipour, T. Kazemi and L. Mohammadnejad, Adv. Pharm. Bull., 2013, 3, 249–255 Search PubMed.
- C. L. Halbert, J. M. Allen and A. D. Miller, J. Virol., 2001, 75, 6615–6624 CrossRef CAS PubMed.
- S. Daya and K. I. Berns, Clin. Microbiol. Rev., 2008, 21, 583–593 CrossRef CAS PubMed.
- D. Wang, P. W. L. Tai and G. Gao, Nat. Rev. Drug Discovery, 2019, 18, 358–378 CrossRef CAS PubMed.
- M. A. F. V Gonçalves, Virol. J., 2005, 2, 43 CrossRef PubMed.
- I. Khan, K. Saeed and I. Khan, Arab. J. Chem., 2019, 12, 908–931 CrossRef CAS.
- S. Azarmi, W. H. Roa and R. Löbenberg, Adv. Drug Deliv. Rev., 2008, 60, 863–875 CrossRef CAS PubMed.
- C. Schleh, B. Rothen-Rutishauser and W. G. Kreyling, Eur. J. Pharm. Biopharm., 2011, 77, 350–352 CrossRef CAS PubMed.
- F. Salahpour Anarjan, Nano-Struct. Nano-Objects, 2019, 19, 100370 CrossRef CAS.
- S. A. Torres-Pérez, C. E. Torres-Pérez, M. Pedraza-Escalona, S. M. Pérez-Tapia and E. Ramón-Gallegos, Front. Oncol., 2020, 10, 1–10 CrossRef PubMed.
- V. Iranpur Mobarakeh, M. H. Modarressi, P. Rahimi, A. Bolhassani, E. Arefian, F. Atyabi and R. Vahabpour, Int. J. Biol. Macromol., 2019, 129, 305–315 CrossRef CAS PubMed.
- R. Koynova, Lipid Insights, 2008, 2, LPI.S864 CrossRef.
- Y. Zhao and L. Huang, Physiol. Behav., 2014, 176, 13–36 Search PubMed.
- Y. Uemura, T. Naoi, Y. Kanai and K. Kobayashi, Pharm. Dev. Technol., 2019, 24, 263–268 CrossRef CAS PubMed.
- J. A. Katakowski, G. Mukherjee, S. E. Wilner, K. E. Maier, M. T. Harrison, T. P. DiLorenzo, M. Levy and D. Palliser, Mol. Ther., 2016, 24, 146–155 CrossRef CAS PubMed.
- T. P. Prakash, W. F. Lima, H. M. Murray, S. Elbashir, W. Cantley, D. Foster, M. Jayaraman, A. E. Chappell, M. Manoharan, E. E. Swayze and S. T. Crooke, ACS Chem. Biol., 2013, 8, 1402–1406 CrossRef CAS PubMed.
- A. Naderi Sohi, J. Kiani, E. Arefian, A. Khosrojerdi, Z. Fekrirad, S. Ghaemi, M. K. Zim, A. Jalili, N. Bostanshirin and M. Soleimani, Vaccines, 2021, 9, 1007 CrossRef CAS PubMed.
- D. Kim, Y. Wu, G. Shim and Y.-K. Oh, Pharmaceutics, 2021, 13, 490 CrossRef CAS PubMed.
- T. T. Win-Shwe and H. Fujimaki, Int. J. Mol. Sci., 2011, 12, 6267–6280 CrossRef CAS PubMed.
- G. Oberdörster, E. Oberdörster and J. Oberdörster, Environ. Health Perspect., 2005, 113, 823–839 CrossRef PubMed.
- S. I. Sinis, K. I. Gourgoulianis, C. Hatzoglou and S. G. Zarogiannis, Environ. Toxicol. Pharmacol., 2019, 67, 29–34 CrossRef CAS PubMed.
- J. Lojk, J. Repas, P. Veranič, V. B. Bregar and M. Pavlin, Toxicology, 2020, 432, 152364 CrossRef CAS PubMed.
- L. Gonzalez-Moragas, A. Roig and A. Laromaine, Adv. Colloid Interface Sci., 2015, 219, 10–26 CrossRef CAS PubMed.
- Q. Wu, W. Wang, Y. Li, Y. Li, B. Ye, M. Tang and D. Wang, J. Hazard. Mater., 2012, 243, 161–168 CrossRef CAS PubMed.
- J. Moon, J. Il Kwak, S. W. Kim and Y. J. An, Environ. Pollut., 2017, 220, 46–52 CrossRef CAS PubMed.
- A. Wamucho, J. M. Unrine, T. J. Kieran, T. C. Glenn, C. L. Schultz, M. Farman, C. Svendsen, D. J. Spurgeon and O. V. Tsyusko, Environ. Pollut., 2019, 254, 113078 CrossRef CAS PubMed.
- Y. L. Hu and J. Q. Gao, Int. J. Pharm., 2010, 394, 115–121 CrossRef CAS PubMed.
- M. Kumar and S. A. Moschos, Mol. Ther., 2017, 25, 2604–2606 CrossRef CAS PubMed.
- S. H. Kim, S. Jeon, D. K. Lee, S. Lee, J. Jeong, J. S. Kim and W. S. Cho, Nanotoxicology, 2020, 14, 468–478 CrossRef CAS PubMed.
- H. Parhiz, J. S. Brenner, P. Patel, T. E. Papp, H. Shahnawaz, Q. Li, R. Shi, M. Zamora, A. Yadegari, O. A. Marcos-Contreras, A. Natesan, N. Pardi, V. V. Shuvaev, R. Kiseleva, J. Myerson, T. Uhler, R. S. Riley, X. Han, M. J. Mitchell, K. Lam, J. Heyes, D. Weissman and V. Muzykantov, J. Contr. Release, 2022, 344, 50–61 CrossRef CAS PubMed.
- H. Bové, E. Bongaerts, E. Slenders, E. M. Bijnens, N. D. Saenen, W. Gyselaers, P. Van Eyken, M. Plusquin, M. B. J. Roeffaers, M. Ameloot and T. S. Nawrot, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed.
- M. J. R. Ruigrok, H. W. Frijlink and W. L. J. Hinrichs, J. Contr. Release, 2016, 235, 14–23 CrossRef CAS PubMed.
- R. Goswami, G. Subramanian, L. Silayeva, I. Newkirk, D. Doctor, K. Chawla, S. Chattopadhyay, D. Chandra, N. Chilukuri and V. Betapudi, Front. Oncol., 2019, 9, 1–25 CrossRef PubMed.
- P. M. Coutinho, E. Deleury, G. J. Davies and B. Henrissat, J. Mol. Biol., 2003, 328, 307–317 CrossRef CAS PubMed.
- T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed.
- M. Weishaupt, S. Eller and P. H. Seeberger, Methods Enzymol., 2010, 478, 463–484 CAS.
- M. Benkoulouche, R. Fauré, M. Remaud-Siméon, C. Moulis and I. André, Interface Focus, 2019, 9, 20180069 CrossRef PubMed.
- W. Koenigs and E. Knorr, Ber. Dtsch. Chem. Ges., 1901, 34, 957–981 CrossRef.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- C. García-Oliva, A. H. Cabanillas, A. Perona, P. Hoyos, Á. Rumbero and M. J. Hernáiz, Chem.–Eur. J., 2020, 26, 1588–1596 CrossRef PubMed.
- C. I. C. Crucho, P. Correia-Da-Silva, K. T. Petrova and M. T. Barros, Carbohydr. Res., 2015, 402, 124–132 CrossRef CAS PubMed.
- T. Buskas, S. Ingale and G.-J. Boons, Glycobiology, 2006, 16, 113R–136R CrossRef CAS PubMed.
- H. Geyer and R. Geyer, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 1853–1869 CrossRef CAS PubMed.
- S. J. Moons, R. A. Mensink, J. P. J. Bruekers, M. L. A. Vercammen, L. M. Jansen and T. J. Boltje, J. Org. Chem., 2019, 84, 4486–4500 CrossRef CAS PubMed.
- J. Guo and X.-S. Ye, Molecules, 2010, 15, 7235–7265 CrossRef CAS PubMed.
- D. Crich, Acc. Chem. Res., 2010, 43, 1144–1153 CrossRef CAS PubMed.
- P. H. Seeberger and W. C. Haase, Chem. Rev., 2000, 100, 4349–4393 CrossRef CAS PubMed.
- G. Wei, G. Gu and Y. Du, J. Carbohydr. Chem., 2003, 22, 385–393 CrossRef CAS.
- D. M. Whitfield and S. P. Douglas, Glycoconj. J., 1996, 13, 5–17 CrossRef CAS PubMed.
- S. Xing and J. L. Gleason, Org. Biomol. Chem., 2015, 13, 1515–1520 RSC.
- A. V Demchenko and G. Boons, Tetrahedron Lett., 1998, 39, 3065–3068 CrossRef.
- W. J. Sanders, D. D. Manning, K. M. Koeller and L. L. Kiessling, Tetrahedron, 1997, 53, 16391–16422 CrossRef CAS.
- M. M. Nielsen and C. M. Pedersen, Chem. Rev., 2018, 118, 8285–8358 CrossRef CAS PubMed.
- P. R. Andreana and D. Crich, ACS Cent. Sci., 2021, 7, 1454–1462 CrossRef CAS PubMed.
- L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed.
- R. Johnsson, M. Ohlin and U. Ellervik, J. Org. Chem., 2010, 75, 8003–8011 CrossRef CAS PubMed.
- M. Sakagami and H. Hamana, Tetrahedron Lett., 2000, 41, 5547–5551 CrossRef CAS.
- T. Nukada, A. Berces, M. Z. Zgierski and D. M. Whitfield, J. Am. Chem. Soc., 1998, 120, 13291–13295 CrossRef CAS.
- R. E. Moore, K. M. Craft, L. L. Xu, S. A. Chambers, J. M. Nguyen, K. C. Marion, J. A. Gaddy and S. D. Townsend, J. Org. Chem., 2020, 85, 16128–16135 CrossRef CAS PubMed.
- P. K. Mandal and P. R. Chheda, Tetrahedron Lett., 2015, 56, 900–902 CrossRef CAS.
- V. V. Severov, I. M. Belyanchikov, G. V. Pazynina and N. V. Bovin, Russ. J. Bioorg. Chem., 2007, 33, 122–138 CrossRef CAS PubMed.
- A. K. Misra, M. Fukuda and O. Hindsgaul, Bioorg. Med. Chem. Lett., 2001, 11, 2667–2669 CrossRef CAS PubMed.
- L. Cui, C.-C. Ling, J. Sadowska and D. R. Bundle, Carbohydr. Res., 2014, 383, 1–13 CrossRef CAS PubMed.
- P. J. Hauptman and R. A. Kelly, Circulation, 1999, 99, 1265–1270 CrossRef CAS PubMed.
- T. M. Beale and M. S. Taylor, Org. Lett., 2013, 15, 1358–1361 CrossRef CAS PubMed.
- M. Jensen, S. Schmidt, N. U. Fedosova, J. Mollenhauer and H. H. Jensen, Bioorg. Med. Chem., 2011, 19, 2407–2417 CrossRef CAS PubMed.
- J.-C. Lee, X.-A. Lu, S. S. Kulkarni, Y.-S. Wen and S.-C. Hung, J. Am. Chem. Soc., 2004, 126, 476–477 CrossRef CAS PubMed.
- S. Dey, H.-J. Lo and C.-H. Wong, J. Am. Chem. Soc., 2019, 141, 10309–10314 CrossRef CAS PubMed.
- Q. Ma, U. Cornelli, I. Hanin, W. P. Jeske, R. J. Linhardt, J. M. Walenga, J. Fareed and J. M. Lee, Curr. Pharm. Des., 2007, 13, 1607–1616 CrossRef CAS PubMed.
- H. Hinou, N. Saito, T. Maeda, M. Matsuda, Y. Kamiya and S.-I. Nishimura, J. Carbohydr. Chem., 2011, 30, 575–586 CrossRef CAS.
- M. Tojino, Y. Hirose and M. Mizuno, Tetrahedron Lett., 2013, 54, 7124–7126 CrossRef CAS.
- G. M. Watt, P. A. Lowden and S. L. Flitsch, Curr. Opin. Struct. Biol., 1997, 7, 652–660 CrossRef CAS PubMed.
- T. M. Gloster, Curr. Opin. Struct. Biol., 2014, 28, 131–141 CrossRef CAS PubMed.
- K. M. Ruane, G. J. Davies and C. Martinez-Fleites, Proteins: Struct., Funct., Bioinf., 2008, 73, 784–787 CrossRef CAS PubMed.
- L. Chao and S. Jongkees, Angew. Chem., Int. Ed., 2019, 58, 12750–12760 CrossRef CAS PubMed.
- J. Zhang, C. Chen, M. R. Gadi, C. Gibbons, Y. Guo, X. Cao, G. Edmunds, S. Wang, D. Liu, J. Yu, L. Wen and P. G. Wang, Angew. Chem., Int. Ed., 2018, 57, 16638–16642 CrossRef CAS PubMed.
- J. Lauber, R. Handrick, S. Leptihn, P. Dürre and S. Gaisser, Microb. Cell Factories, 2015, 14, 3 CrossRef PubMed.
- T. Ito, R. Sadamoto, K. Naruchi, H. Togame, H. Takemoto, H. Kondo and S.-I. Nishimura, Biochemistry, 2010, 49, 2604–2614 CrossRef CAS PubMed.
- N. B. Schwartz and L. Rodén, J. Biol. Chem., 1975, 250, 5200–5207 CrossRef CAS PubMed.
- Y. Sugiyama, A. Gotoh, T. Katoh, Y. Honda, E. Yoshida, S. Kurihara, H. Ashida, H. Kumagai, K. Yamamoto, M. Kitaoka and T. Katayama, Glycobiology, 2016, 26, 1235–1247 CAS.
- L. F. Mackenzie, Q. Wang, R. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 1998, 120, 5583–5584 CrossRef CAS.
- S. Böcker, D. Laaf and L. Elling, Biomolecules, 2015, 5, 1671–1696 CrossRef PubMed.
- B. Castagner and P. H. Seeberger, Top. Curr. Chem., 2007, 278, 289–309 CrossRef CAS.
- C. Schuerch and J. M. Frechet, J. Am. Chem. Soc., 1971, 93, 492–496 CrossRef CAS.
- K. Naresh, F. Schumacher, H. S. Hahm and P. H. Seeberger, Chem. Commun., 2017, 53, 9085–9088 RSC.
- C. S. Bennett, Org. Biomol. Chem., 2014, 12, 1686–1698 RSC.
- X. Xiao, J. Zeng, J. Fang, J. Sun, T. Li, Z. Song, L. Cai and Q. Wan, J. Am. Chem. Soc., 2020, 142, 5498–5503 CrossRef CAS PubMed.
- X. S. Ye and C. H. Wong, J. Org. Chem., 2000, 65, 2410–2431 CrossRef CAS PubMed.
- C.-H. Hsu, S.-C. Hung, C.-Y. Wu and C.-H. Wong, Angew. Chem., Int. Ed., 2011, 50, 11872–11923 CrossRef CAS PubMed.
- M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS PubMed.
- L. Wen, G. Edmunds, C. Gibbons, J. Zhang, M. R. Gadi, H. Zhu, J. Fang, X. Liu, Y. Kong and P. G. Wang, Chem. Rev., 2018, 118, 8151–8187 CrossRef CAS PubMed.
- H. S. Hahm, M. K. Schlegel, M. Hurevich, S. Eller, F. Schuhmacher, J. Hofmann, K. Pagel and P. H. Seeberger, Proc. Natl. Acad. Sci. USA, 2017, 114, E3385–E3389 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |