DOI:
10.1039/D2RA01956F
(Paper)
RSC Adv., 2022,
12, 14368-14376
Rational design of M–N4–Gr/V2C heterostructures as highly active ORR catalysts: a density functional theory study†
Received
26th March 2022
, Accepted 2nd May 2022
First published on 12th May 2022
Abstract
Inspired by the composites of N-doped graphene and transition metal-based materials as well as MXene-based materials, heterostructures (M–N4–Gr/V2C) of eight different transition metals (M = Ti, Cr, Mn, Fe, Co, Ni, Cu, and Zn) doped with nitrogen-coordinated graphene and V2C as potential catalysts for the oxygen reduction reaction (ORR) using density functional theory (DFT) were designed and are described herein. The calculations showed that the heterostructure catalysts (except for Zn–N4–Gr/V2C) were thermodynamically stable. Ni–N4–Gr/V2C and Co–N4–Gr/V2C showed higher activities towards the ORR, with overpotentials as low as 0.32 and 0.45 V, respectively. Excellent catalytic performance results were observed from the change in electronic structure caused by the strong interaction between V2C and the graphene layers as well as the synergistic effect between the MN4 groups and the graphene layers. This study further provides insights into the practical application of ORR catalysts for MXene systems through the modulation of the electronic structure of two-dimensional materials.
1 Introduction
In recent years, the development and utilization of clean energy have received global attention due to the serious environmental pollution caused by fossil fuels. Proton exchange membrane fuel cells (PEMFCs), as clean and efficient energy conversion devices, have attracted increasing attention due to their high energy transformation efficiency, high power density, and low emissions.1,2 The cathode oxygen reduction reaction (ORR) is key to determining the fuel cell performance and energy transfer efficiency of PEMFCs, and the slow dynamics process is a major limiting factor in their efficiency.3,4 Currently, the most widely used cathode catalysts in PEMFCs are Pt/C catalysts,5 but their high cost, low stability, and low activity hinder their large-scale commercialization. Therefore, to reduce the use of Pt metals, noble metal-free catalysts should be screened and designed for the ORR.
Two-dimensional (2D) transition metal carbides and nitrides, known as MXenes, constitute a large family of 2D materials.6 To date, MXenes have attracted significant attention for their potential applications in energy storage,7 sensors,8–10 and catalysis.11–13 Specifically, MXenes with fast charge transfer properties, adjustable chemical-specific surface area, and high electrical conductivity offer broad prospects for electrochemical energy applications. They are also considered as promising carriers and prominent conductivity enhancers to obtain synergistic effects for fast charge transfer kinetics.14,15 Zhu et al., have synthesized superlattice-like heterostructure of Fe–N–C with MXene by experiments with a positive onset potential of 0.92 V in the electrocatalytic oxygen reduction reaction (ORR) and a durability of 20 h in alkaline electrolytes.16 Zhang et al., obtained Ni1/Ni2- and Fe1/Ni2-modified MXene-based double atom catalysts (DACs) as excellent bifunctional ORR/OER catalysts by introducing non-precious metals such as Fe/Co/Ni on the MXene surface through first-principles calculations.17 Zhou et al., by first-principles calculations for 3d, 4d and 5d transition metal single atoms immobilized on the surface of 2D titanium carbides (Ti2CT and Ti3C2T) as active sites, derived good ORR activity for single-atom Cu-doped Ti2CO2 catalysts with an overpotential of 0.25 V.18 V2C exhibited good performance as a carrier material for catalysts in the ORR.
Nitrogen atom-doped graphene19–21 is a potential alternative to Pt-based catalysts due to its high activity and low cost. To design highly stable catalysts, they must be stabilized using novel supports. 2D MXenes with abundant nonmetallic groups include a graphene layer stabilized by the strong interaction forces of nitrogen coordination, which not only modulates the catalytic properties of the selected transition metals but also affects the intrinsic activity of the 2D MXenes.22–25 Constructing heterogeneous structures is a powerful strategy for designing high-performance materials, where phenomena such as charge transfer can be observed.26,27 Geng et al. synthesized a large-area Mo2C/MXene on graphene templates,28 and this heterogeneous structure was highly active for electrocatalysis via the hydrogen evolution reaction (HER) with a lower starting voltage than that of Mo2C-only electrodes. Zhou et al. constructed G/Ti2C, G/V2C, G/Nb2C, and G/Mo2C heterostructures by theoretical simulations, among which G/V2C and G/Mo2C readily underwent electrocatalysis with an ORR overpotential of 0.36 V.29
Using their synergistic effects, the composites of N-doped graphene and transition metal-based materials are one of the main electrocatalysts for HER, OER, and ORR.30–32 Due to the excellent conductivity and charge transfer kinetics of V2C MXene,9,33,34 We are inspired to investigate the possibility of using it as a potential carrier material for N-doped graphene to develop efficient ORR catalysts. Considering the nature of transition metals and the effective charge transfer kinetics of MXene, the heterostructure of N-doped graphene and transition metal-based material composites with V2C (M–N4–Gr/V2C) were investigated as a superior ORR catalyst. The electrocatalytic properties of eight different transition metals (M = Ti, Cr, Mn, Fe, Co, Ni, Cu, and Zn) doped with nitrogen ligands were investigated for graphene with V2C heterostructures. Their electronic properties, free energies, and volcano curves were obtained using DFT calculations. The calculations showed that the MN4 group was the active center of the reaction intermediate and Ni–N4–Gr/V2C had a promising ORR catalytic effect. The ORR effect of the eight catalysts was investigated by analyzing the interactions between the MN4 groups and the V2C substrates through the charge transfer mechanism. The density of states (DOS) indicated that increased ORR activity arose from the moderate hybridization between the d-orbital of the Ni atom and the p-orbital of O. The volcano curves indicate that Ni–N4–Gr/V2C and Co–N4–Gr/V2C are located at the peak of the volcano, resulting in the highest activity with moderate interaction between the reaction intermediate and the catalyst. This study provides a novel approach for the design of ORR catalysts through the modulation of the electronic structure of 2D MXene materials and inspiration for the practical application of high-performance electrocatalysts in MXene systems.
2 Calculation method
2.1 Method
All calculations were performed using the Vienna Ab initio Simulation Package (VASP),35 wherein the Perdew–Burke–Ernzerhof (PBE) generalization of the generalized gradient approximation (GGA) was used to describe the exchange relation effect.36 A plane-wave basis set with a cutoff energy of 550 eV and a projector-augmented wave (PAW) potential were used.37 To simulate the heterogeneous structure of graphene and MXene, we used 2 × 2 × 1 graphene supercell and 1 × 3 × 1 V2C supercell while swapping the a and b coordinates of graphene. Specifically, the lattice mismatch ratio is 1.85% (Table 1). The initial layer spacing of the heterogeneous structure was set to 3 Å. A vacuum layer of 20 Å was applied in the vertical direction so that the lattice strain of graphene has a negligible effect on the binding properties and catalytic activity.15 The model structure is fully optimized in the constrained supercell using a total energy of 10−4 eV and a force of 0.02 eV Å−1. The weak van der Waals (vdW) interactions between the graphene layers and MXene can be described using Grimme's DFT-D3 dispersion correction scheme.38 The Gamma scheme was used to sample the Brillouin zone, with a k-point grid of 2 × 3 × 1 for geometric optimization and 4 × 6 × 1 for electronic structure calculations. The partial charge density and charge transfer were evaluated by Bader charge analysis.39 The DOS was calculated to analyze the atomic bonding, interatomic interactions, and electrical conductivity in the reaction following adsorption. The minimum energy pathway was obtained using the climbing image nudged elastic band method40 and MD simulations with a time step of 2 fs using density functional tight-binding.41
Table 1 Lattice mismatch ratio (δ) and interlayer distance (d) of the graphene/MXene heterostructures
| System |
a (Å) |
b (Å) |
Heterostructure (Å) |
δ (%) |
d (Å) |
| G |
2.46 |
4.26 |
4.92 × 8.52 (2 × 2) |
1.85% |
2.10 |
| V2C |
5.01 |
2.89 |
5.01 × 8.67 (1 × 3) |
2.2 Calculations
The adsorption energies (Eads) were calculated for all intermediates during the ORR reactions (*O, *O2, *OH, and *OOH):| | |
Eads = Ead/S − Ead − ES
| (1) |
where Ead/S, Ead, and ES represent the energies of the MXene substrate, free adsorbate, and adsorbate on the pure MXene substrate, respectively. A negative Eads indicates that the adsorbed molecules are energetically favored to adsorb on the catalyst surface.
The free energy diagram of the reaction was calculated according to the method developed by Nørskov et al. The reaction free energy is the difference between the initial and final states, and the change in the Gibbs free energy at each step of the ORR is given by:
| | |
ΔG = ΔEDFT + ΔZPE − TΔS + ΔGU + ΔGpH
| (2) |
where Δ
EDFT, ΔZPE, and Δ
S are the changes in the reaction energy, zero-point energy, and entropy from the initial state to the final state, respectively, as calculated by DFT.
T is the standard room temperature (298.15 K). The vibrational frequencies were calculated by density general function theory, and the ZPE correction values and entropies were obtained. The vibrational frequencies and entropies of the gas-phase molecules can be found in the NIST database.
42 Δ
GU is the free energy contribution associated with the applied electrode potential
U, while Δ
GpH is the correction for the H
+ free energy by concentration. Δ
GpH = ln
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
10 × pH ×
kBT, where
kB is the Boltzmann constant, and pH = 0 under acidic conditions. The standard hydrogen electrode (SHE) method was used to calculate the ORR overpotential (
η*ORR), and we considered the 4e
− associate pathway of the ORR under acidic conditions, which is the main mechanism of transition metal-doped graphene with V
2C/MXene heterostructured catalysts.
| | |
* + O2(g) + H+ + e− → *OOH
| (3) |
| | |
*OOH + H+ + e− → *O + H2O(l)
| (4) |
| | |
*OH + H+ + e− → H2O(l) + *
| (6) |
where * represents the adsorption sites on the catalyst surface and *OOH, *O, and *OH are oxygen intermediates. The Gibbs free energy was calculated for each ORR step (
Table 2), and the ORR overpotential is given by:
| | |
ηORR = 1.23 − ΔGmin/e
| (7) |
where Δ
Gmin is the minimum Gibbs free energy for the four reaction steps given by
eqn (3)–(6) and 1.23 V is the equilibrium potential of water at pH 0 and 298.15 K.
Table 2 Gibbs free energy of oxygen-containing intermediates (O, OH, and OOH) ΔG*O, ΔG*OH, and ΔG*OOH; η*ORR represents the overpotential in the ORR process
| System |
ΔG*O |
ΔG*OH |
ΔG*OOH |
η*ORR |
| Ti–N4–Gr/V2C |
−1.67 |
−1.49 |
2.01 |
— |
| Cr–N4–Gr/V2C |
−0.01 |
−0.22 |
2.94 |
1.45 |
| Mn–N4–Gr/V2C |
1.10 |
0.19 |
3.31 |
0.98 |
| Fe–N4–Gr/V2C |
1.65 |
0.54 |
3.54 |
0.69 |
| Co–N4–Gr/V2C |
2.26 |
0.78 |
3.80 |
0.45 |
| Ni–N4–Gr/V2C |
2.75 |
0.91 |
3.96 |
0.32 |
| Cu–N4–Gr/V2C |
2.73 |
0.73 |
3.84 |
0.50 |
The expression was used to calculate the formation energy of the M–N4–Gr/V2C defect Ef:
| | |
Ef = EM–N4–C/V2C + 6μC − (Eg/V2C + 4μN + μM)
| (8) |
where
EM–N4–Gr/V2C and
Eg/V2C are the respective energies with M–N
4–Gr/V
2C and g/V
2C defects within the optimized geometry of graphene; the numbers 4 and 6 denote the number of doped N and C atoms removed from the pristine graphene sheet to form M–N
4–Gr defects;
μC is the chemical potential of carbon as the total energy per carbon atom of defect-free graphene;
39,43 μN is the chemical potential of N or half of the total energy of a N
2 molecule; and
μM is the chemical potential of an isolated transition metal atom.
2.3 Models
In this study, the heterostructure formed by transition metal-doped graphene with V2C was used to improve the activity of the cathode catalyst. Four N atoms were chosen to modify graphene, and the transition metal atoms were placed in the center of the N atoms, resulting in an M–N4–Gr/V2C heterostructure. A total of eight structures were studied: Ti–N4–Gr/V2C, Cr–N4–Gr/V2C, Mn–N4–Gr/V2C, Fe–N4–Gr/V2C, Co–N4–Gr/V2C, Ni–N4–Gr/V2C, Cu–N4–Gr/V2C, and Zn–N4–Gr/V2C (Fig. 1a).
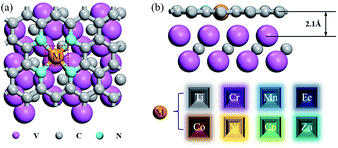 |
| | Fig. 1 (a) Top view and (b) side view of M-doped N-coordinated graphene supported by V2C/MXene, and their interlayer distances; the pink, cyan, and gray balls denote V, N, and C atoms, respectively. | |
3 Results and discussion
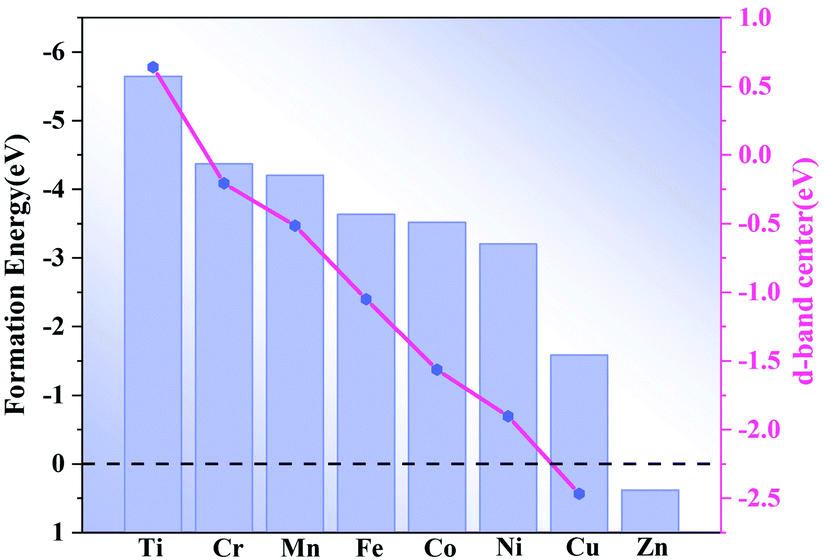
Composites of N-doped graphene and transition metal-based materials that form heterostructures with MXene are known to act as bifunctional catalysts for ORR and OER.14 To further motivate the catalytic activity of single atom-doped N-liganded graphene as an active site, we formed heterostructures of M-doped N-liganded graphene with V2C. The synthesis of such heterostructures is feasible, as recent experiments generated MoC2 films directly on the graphene surface via chemical deposition as well as on the graphene nanoribbons.24 The distance between our designed graphene and V2C heterostructure is 2.1 Å (Fig. 1b) and resulted in an energy of −10.13 eV, indicating strong coupling between the interfaces. After the transition metal was modified, the formation energy was calculated (shown in Fig. 2), where a negative value indicated the greater stability of the structure, and only the Zn-doped N-liganded graphene heterostructure was found to be unstable. The other transition metals follow the sequence according to stability intensity: Ti–N4–Gr/V2C > Co–N4–Gr/V2C > Fe–N4–Gr/V2C > Ni–N4–Gr/V2C > Cr–N4–Gr/V2C > Mn–N4–Gr/V2C > Cu–N4–Gr/V2C, with the results ranging from −5.64 to −1.59 eV.
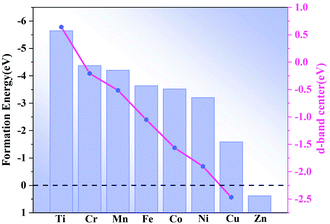 |
| | Fig. 2 Formation energy and d-band center of different transition metal heterostructures M–N4–Gr/V2C (M = Ti, Cr, Mn, Fe, Co, Ni, Cu, and Zn). | |
The transition metal d-band center is a useful parameter for predicting the catalytic performance of a catalyst. The calculated d-band center (dε) is shown in Fig. 2, and the results indicate that the dε of Ti–N4–Gr/V2C, Cr–N4–Gr/V2C, and Mn–N4–Gr/V2C are close to the Fermi level (Ef). The absorption would, therefore, be too strong for the intermediate and may deactivate the catalyst. In addition, the dε of Cu–N4–Gr/V2C and Zn–N4–Gr/V2C are far from the Ef, resulting in an adsorption capacity too weak for the ORR. Unlike Ti-, Cr-, Mn-, and Cu-based systems, Fe-, Ni-, and Co-based systems have moderate dε values and are expected to show good catalytic activity for the ORR.
3.1 ORR thermodynamic process of the M–N4–Gr/V2C heterojunction catalyst
Due to the weak adsorption of O2 and *OOH on pristine graphene and transition metal atom-doped graphene, such as M–N–Gr, the ORR always via the associative mechanism rather than the dissociative mechanism. ORR needs to overcome the high energy barrier on the surface.44–46 Therefore, in our calculations, we calculated the catalytic activity of M–N4–Gr/V2C using the binding pathway of ORR (eqn (3)–(6)). To understand the catalytic effect of the M–N4–Gr/V2C catalyst, we calculated the thermodynamic processes to obtain the Gibbs free energy of the 4e− path at U = 0 and 1.23 V as well as the theoretical voltage to evaluate the catalytic performance of the M–N4–Gr/V2C catalyst. It should be noted that the overpotential (η*ORR) for the assessment of catalytic activity is determined by the maximum free energy difference (ΔG) in the 4e− step, indicating that a lower free energy may not always lead to better catalytic performance. The ORR process for Ti–N4–Gr/V2C, Cr–N4–Gr/V2C, and Mn–N4–Gr/V2C had high overpotentials, resulting in poor catalytic activity, while the η*ORR values of Fe–N4–Gr/V2C, Co–N4–Gr/V2C, Ni–N4–Gr/V2C, and Cu–N4–Gr/V2C were relatively low at 0.69, 0.45, 0.32, and 0.50 V, respectively. As shown in Fig. 3, at an electrode potential of zero (U = 0 V), the free energies of the basic reduction steps for the Mn–N4–Gr/V2C, Fe–N4–Gr/V2C, Co–N4–Gr/V2C, Ni–N4–Gr/V2C, and Cu–N4–Gr/V2C catalysts and their theoretical potentials decreased, indicating that the ORR process was self-initiated. Experimental and theoretical calculations have revealed that changes in the adsorption of oxygen-containing intermediates are key to rate limiting in catalytic reactions.47 The hydrogenation of OH to water increased significantly in the Ti–N4–Gr/V2C and Cr–N4–Gr/V2C catalysts, which is not favorable for the ORR process. The hydrogenation *OH to H2O is the rate-determining step for Ti–N4–Gr/V2C, Cr–N4–Gr/V2C, Mn–N4–Gr/V2C, Fe–N4–Gr/V2C, Co–N4–Gr/V2C, Ni–N4–Gr/V2C, and Cu–N4–Gr/V2C. Among them, Ni–N4–Gr/V2C and Co–N4–Gr/V2C exhibited high ORR performance with theoretical overpotentials of 0.32 and 0.45 V, respectively; Co–N4–Gr/V2C showed a catalytic activity comparable to that of the Pt/C catalyst.
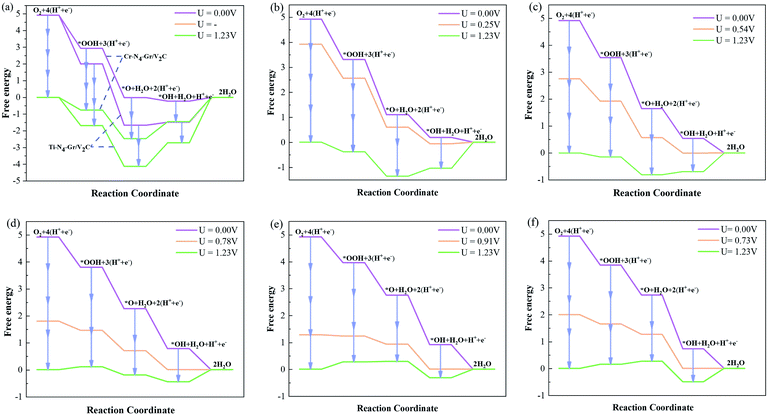 |
| | Fig. 3 Free energy diagrams of ORR intermediates on (a) Ti–N4–Gr/V2C, Cr–N4–Gr/V2C, (b) Mn–N4–Gr/V2C, (c) Fe–N4–Gr/V2C, (d) Co–N4–Gr/V2C, (e) Ni–N4–Gr/V2C, and (f) Cu–N4–Gr/V2C surfaces at zero electrode potential (purple lines), working potential (orange lines), and equilibrium potential (green lines). | |
The free energy of adsorption of oxygen-containing intermediates of ORR is shown in Table 2. The results showed that the free energies of adsorption of O on Ti–N4–Gr/V2C (−1.67 eV) and Cr–N4–Gr/V2C (−0.01 eV) were significantly lower than on the other reactive groups (1.65–2.75 eV). As mentioned above, the strong binding of O prevents the subsequent reaction process. This is illustrated in the energy diagram in Fig. 3a, where the rate-determining steps of the ORR on M–N4–Gr/V2C are the generation of *OH and *OOH from *O.
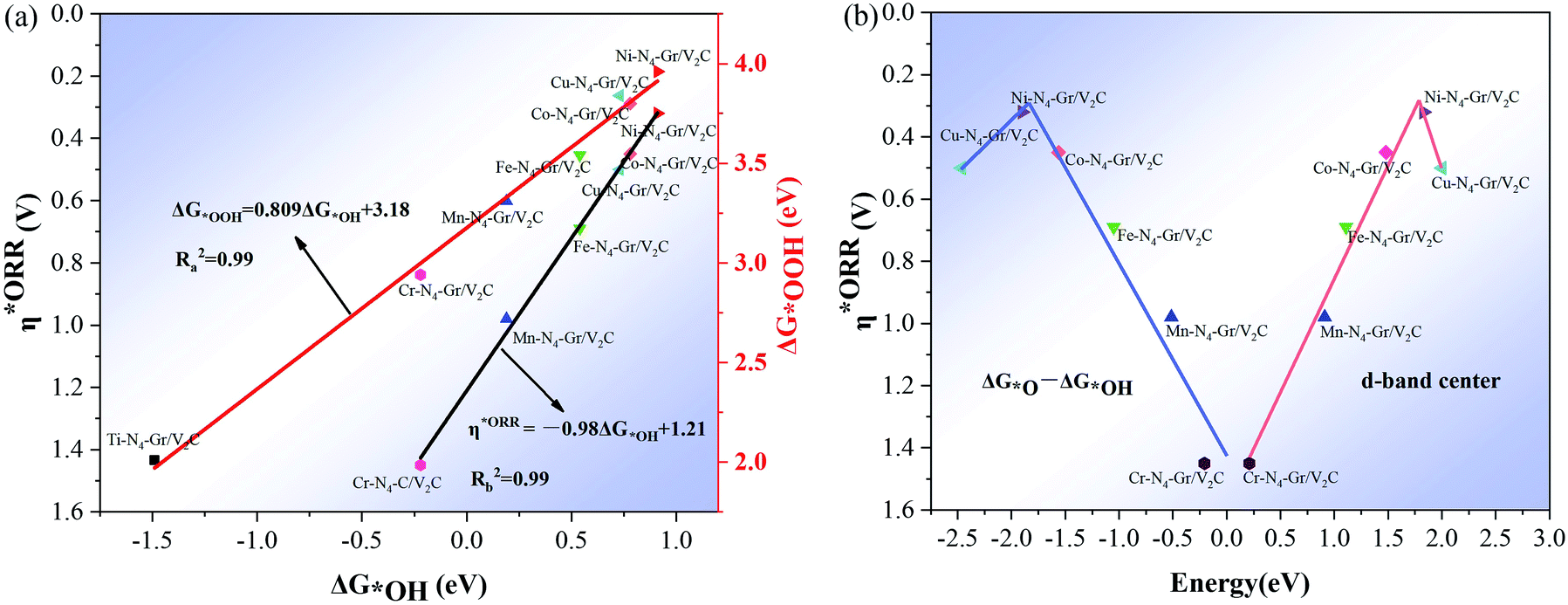
The linear relationship of the Gibbs free energy between different oxygen-containing intermediates was obtained during the calculation of the 4e− reaction, further revealing the performance of ORR on these heterogeneous structured catalysts. As shown in Fig. 4a, a strong linear relationship exists between the oxygenated intermediate *OOH (G*OOH) and *OH (ΔG*OH) Gibbs free energies, center as descriptors, where the Ni–N4–Gr/V2C catalyst is located at the top of the volcano plot and demonstrates the highest activity. A comparison with the corresponding data on the Pt(111) surface revealed that Ni–N4–Gr/V2C has a high catalytic performance for ORR. The d-band center revealed improved catalytic activity between −1.5 and −2.5, with Ni–N4–Gr/V2C showing the highest catalytic activity.
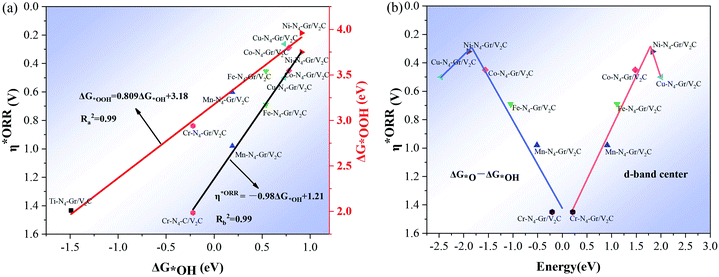 |
| | Fig. 4 (a) Scaling relationships of OOH (ΔG*OOH) in proportion to the adsorption free energy of OH (ΔG*OH) and the overpotential η*ORR in proportion to OH (ΔG*OH); (b) volcano plot of η*ORR versus the change in Gibbs free energy (ΔG*O − ΔG*OH) and η*ORR versus d-band center curves. | |
3.2 Effect of V2C as the substrate material on M–N4–Gr
To study the effect of V2C as a substrate carrier material on M–N4–Gr, we calculated the projected DOS (Fig. 5). In the absence of V2C as the substrate material, Ti–N4–Gr and Cr–N4–Gr catalysts exhibit a small amount of electron crossing at the Fermi energy level and possess significant electron leaps at the Fermi energy level, indicating their very weak conductivity.
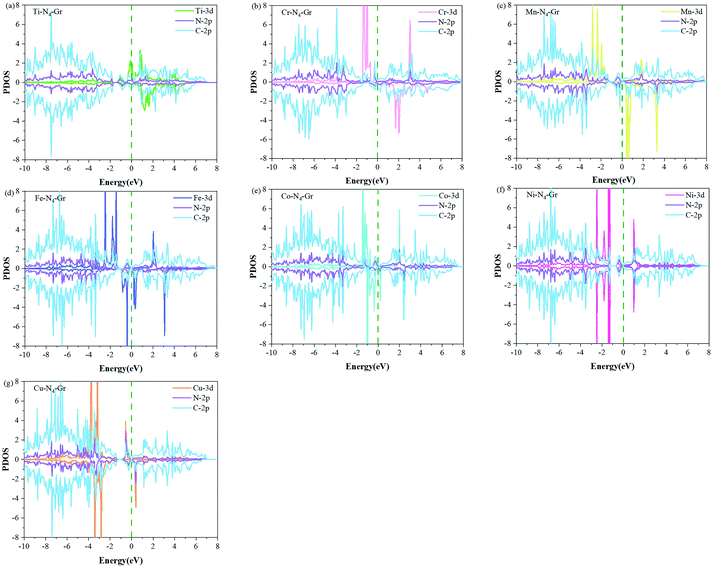 |
| | Fig. 5 PDOS plots of M-3d, N-2p, and C-2p for (a–g) M–N4–C (M = Ti, Cr, Mn, Fe, Co, Ni, and Cu). The dotted line at zero represents the Fermi energy level. | |
Notably, Ni–N4–Gr exhibits semiconducting properties through its paramagnetism, a perfectly symmetric electron population in the spin-up and spin-down orbitals, and lack of an electron leap at the Fermi energy level. However, in the presence of V2C substrate as the material (Fig. S1†), the M–N4–Gr/V2C catalyst undergoes a large number of electron transitions at both Fermi energy levels, while Ni–N4–Gr changes from paramagnetic to the metallic nature of Ni–N4–Gr/V2C with strong electron transfer ability. This further indicates that V2C as the carrier material can improve the conductivity of the catalyst and thus the catalytic activity. Moreover, the Bader charges of the transition metals with and without the V2C base material were calculated. In the absence of V2C, M experiences losses in charge transfer to N in M–N4–Gr, while this charge loss gradually decreases with the change of M. The charge transfer range is 1.66–0.86e. In the presence of V2C as the substrate material, the charge transfer of M in M–N4–Gr/V2C decreased significantly to 1.39–0.61e (Table S1†), indicating that the adsorption is favored when oxygen-containing intermediates are adsorbed, further facilitating the catalytic reactions.
3.3 Effects of changes in electronic structure on ORR activity
The adsorption of reactants on the catalyst surface is a prerequisite for the ORR. We investigated the adsorption properties of oxygen-containing intermediates on different transition metal surfaces. Table 3 shows the adsorption energy of O2 (Eads-O2), the distance between adsorbed oxygen atom (dO–O), the distance between transition metal and oxygen atoms (dM–O), and the number of charges obtained by O2 from M–N4–Gr/V2C (Q − O2). The adsorption energy and O–O bond length of O2 were in the ranges of −0.46 to −4.25 eV and 1.28 to 0.46 Å, respectively, indicating its ability to adsorb stably on the surface. In the reaction process, the binding of an effective catalytic site to an intermediate cannot be too strong or it would be not conducive to further reduction reactions. After the stabilization of O2 adsorption, the O–O bond lengths were measured and were found to all be elongated compared to the bond length of O2 itself (1.21 Å). In addition, O2 and *OOH got different charges from the transition metal M, and the corresponding charge transfer ranges were 0.39–0.97e and 0.49–0.58e, respectively, and it can be found that O2 and *OOH at Ni–N4–Gr/V2C from M get the least charge, indicating that they are relatively weakly adsorbed on the surface and can be desorbed in the reaction, which is favorable for the ORR reaction. O2 possessed different charges, mainly due to charge transfer from the MN4 group to O2, indicating that the M–N4 group was the active center.
Table 3 Adsorption energy of oxygen (Eads-O2), intermolecular distance of adsorbed oxygen (dO–O), distance between transition metal and O atoms (dM–O), number of charge transfers obtained by O2 (Q − O2) and OOH (Q − OOH (e)) from the M–N4–Gr/V2C substrate (Q − O2)
| Properties |
Eads-O2 (eV) |
dO–O (Å) |
dM–O (Å) |
Q − O2 (e) |
Q − OOH (e) |
| Ti–N4–Gr/V2C |
−4.25 |
1.46 |
1.75 |
0.97 |
0.56 |
| Cr–N4–Gr/V2C |
−2.16 |
1.42 |
1.68 |
0.85 |
0.57 |
| Mn–N4–Gr/V2C |
−1.17 |
1.42 |
1.83 |
0.86 |
0.83 |
| Fe–N4–Gr/V2C |
−0.90 |
1.30 |
1.93 |
0.51 |
0.52 |
| Co–N4–Gr/V2C |
−0.53 |
1.29 |
2.08 |
0.56 |
0.51 |
| Ni–N4–Gr/V2C |
−0.46 |
1.28 |
2.14 |
0.39 |
0.49 |
| Cu–N4–Gr/V2C |
−0.71 |
1.29 |
1.95 |
0.43 |
0.53 |
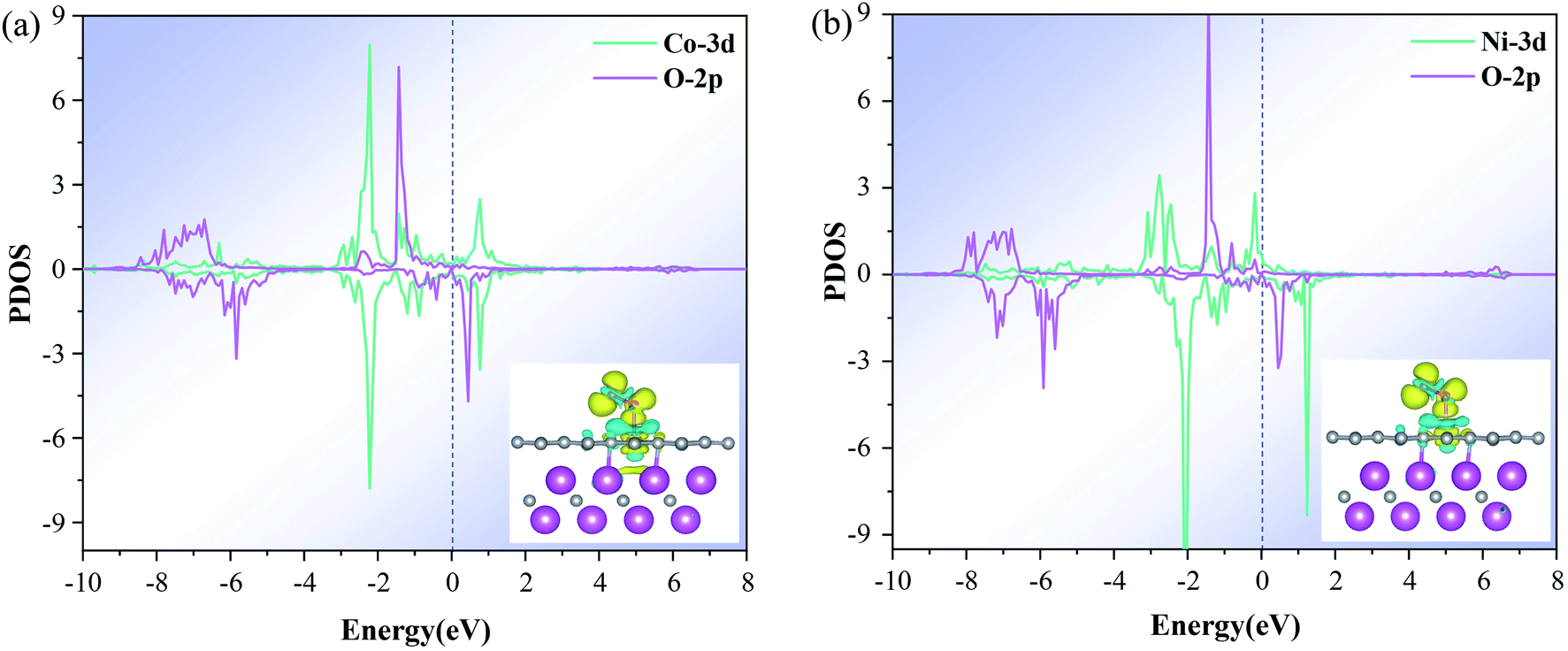
The electronic properties of O2 adsorbed on the M–N4–Gr/V2C catalysts were calculated to reveal the high reactivity and ORR activity of the constructed heterostructure catalysts. First, the partial density of states (PDOS) of all catalysts were found to exhibit strong DOS at the Fermi energy level (Ef), and the M-d orbitals were strongly coupled to the O-p orbitals, indicating the high M-3d electron concentration within the catalyst (Fig. S2†). These high 3d states provided enough electrons for the intermediates, which led to extremely strong interactions and the difficult release of the intermediates. In contrast, the Co–N4–Gr/V2C and Ni–N4–Gr/V2C catalysts showed marked improvement with moderate binding compared to the other catalysts. The Co-3d and Ni-3d states not only had significant peak overlap with the O-2p orbitals at the Fermi energy level (Fig. 6) but also have moderate interactions at the catalyst and intermediate. Second, in the analysis of the distribution of exchange charges, the charge on the substrate gradually decreases, transferring to the transition metal and further transferring to the O2 molecule. As shown in Table 3, the O2 molecule gains a significant amount of charge, making it easier for O2 to adsorb on the catalyst, with a charge transfer range of 0.39–0.97e. Meanwhile, the O–O bond length is significantly stretched compared to the initial length of 1.21 Å, indicating the favored reduction of O2. Finally, for O2 adsorbed in the Co–N4–Gr/V2C and Ni–N4–Gr/V2C distributions of exchange charges (Fig. 6a and b insets), the blue and yellow regions represent the depletion and accumulation of electrons, respectively. The differences in charge density indicate that the positive charges are mainly concentrated in the middle of the two O atoms perpendicular to the O–O bond, while the negative charges are mainly concentrated on the two O atoms, leading to repulsive interactions between them.48 In general, the negative charge accumulated on the O atom helps to trigger the dissociation of O2. O2 molecules receive their charge from the M–N4 group, which is considered as the reactive group, and its electron donating capacity is a key factor affecting the catalytic activity of the system.
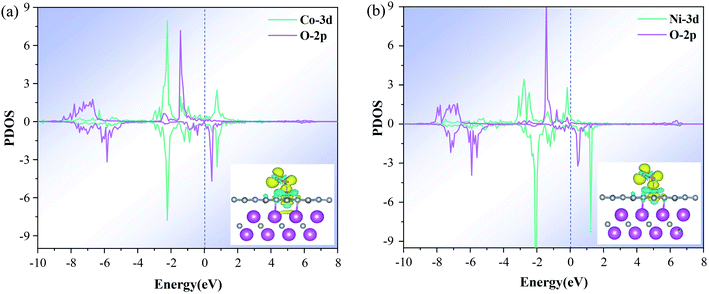 |
| | Fig. 6 PDOS of O2 adsorbed on (a) Co–N4–Gr/V2C and (b) Ni–N4–Gr/V2C catalysts with M 3d and O 2p. The inset is the difference charge density between O2 and Co–N4–Gr/V2C or Ni–N4–Gr/V2C with an isovalue of 0.054e Å−3. The blue and yellow bubbles represent positive and negative charges, respectively. | |
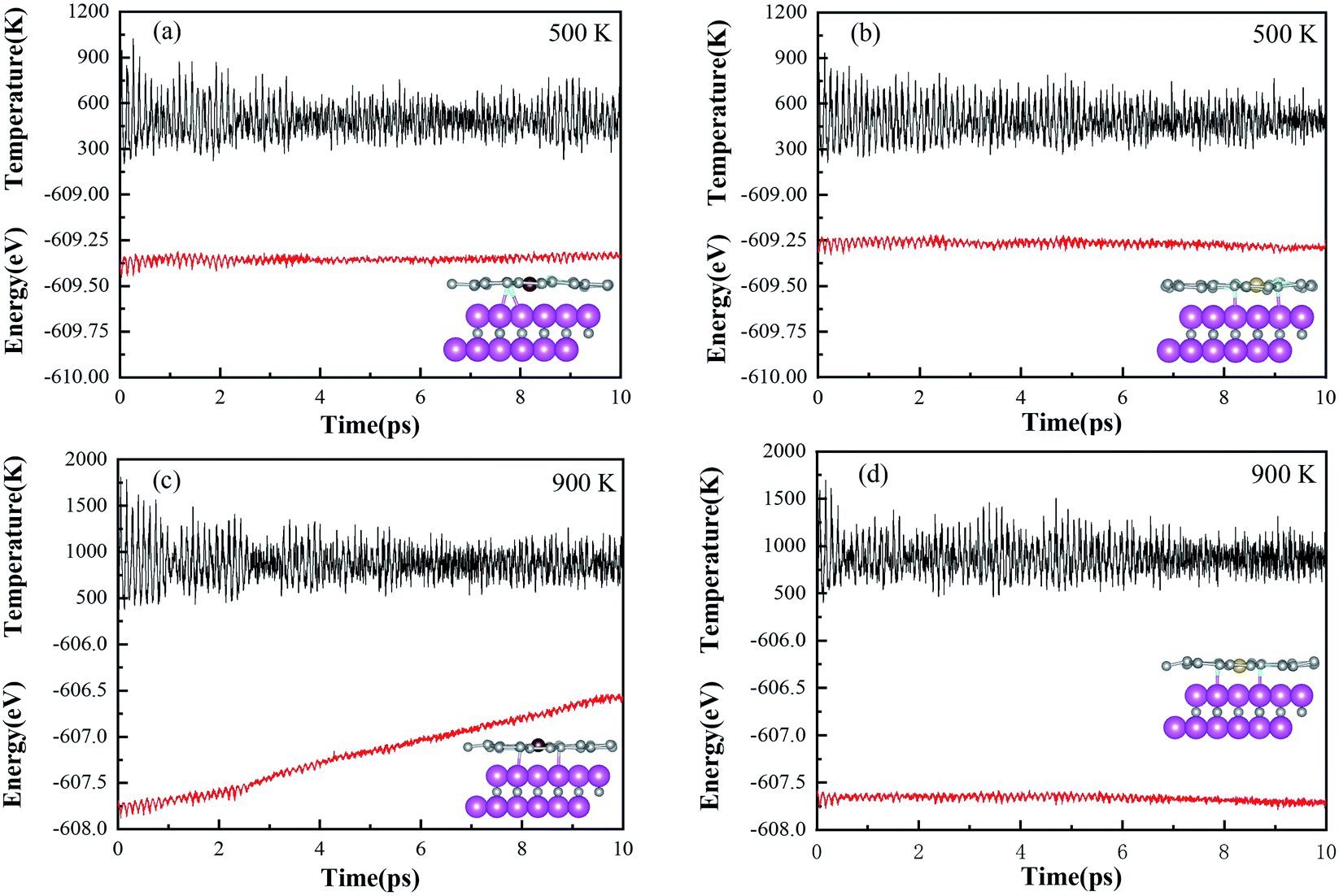
The designed Ni–N4–Gr/V2C and Co–N4–Gr/V2C catalysts were subjected to dynamics testing (Fig. 7). The results showed that the energy of the Ni–N4–Gr/V2C catalyst oscillated back and forth over a range and no bonds were broken or formed between the atoms during the simulation, indicating that the Ni–N4–Gr/V2C catalyst was kinetically stable. In contrast, the Co–N4–Gr/V2C catalyst showed significant energy fluctuations at 900 K, which may destabilize the catalyst.
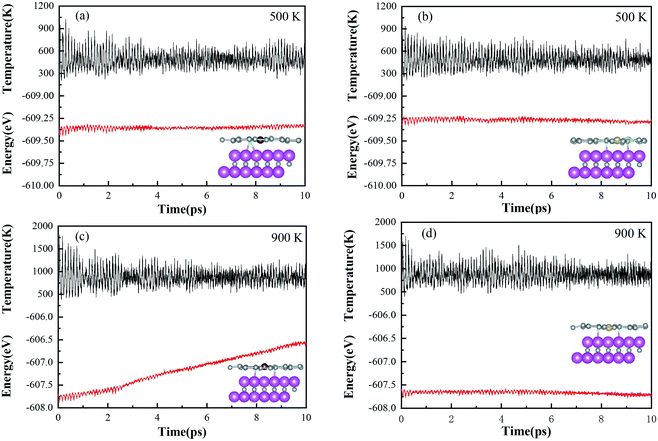 |
| | Fig. 7 Change in total energy of the Co–N4–Gr/V2C catalyst after 10 ps of MD simulation at (a) 500 K and (c) 900 K. Change in total energy of the Ni–N4–Gr/V2C catalyst after 10 ps of MD simulation at (b) 500 K and (d) 900 K. | |
4 Conclusion
We simulated the heterostructures of eight different transition metals (M = Ti, Cr, Mn, Fe, Co, Ni, Cu, and Zn) doped with nitrogen-coordinated graphene with V2C as potential ORR catalysts using DFT calculations and investigated the heterostructure stability, active volcano map, electrical conductivity, adsorption energy of the intermediates, and electronic structure of the transition metals. It was found that the heterostructure catalysts were thermodynamically stable, except for Zn–N4–Gr/V2C. Moreover, it was thermodynamically determined that Ni–N4–Gr/V2C is a potential ORR catalyst with an overpotential of 0.32 V, and the d-band center as a descriptor has a good volcano curve relationship with the overpotential. In the presence of the V2C substrate, there is a strong electron jump at the Fermi energy level, improving the conductivity of the catalyst. The Ni–N4–Gr/V2C catalysts also demonstrated excellent thermodynamic stability at 500 and 900 K. The results are significant for the rational design of transition metal-doped N-liganded graphene/MXene heterostructure catalysts with high ORR performance.
Author contributions
Yunjian Chen: conceptualization, data curation, formal analysis, investigation, methodology, software, visualization. Qi Jiang: investigation. Xue Bai: formal analysis, project administration, supervision. Pengyue Shan: supervision, visualization. Tong Liu: investigation, project administration, writing – review & editing. Yazhou Wang: resources, writing – original draft. Hong Cui: funding acquisition, resources, supervision, writing – review & editing. Rong Feng: supervision, validation. Qin Kang: supervision, validation. Zhiyong Liang: supervision, validation. Hongkuan Yuan: resources, software. All authors have given their approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the National Natural Science Foundation of China (No. 61701288, 51706128), Basic Research Plan of Natural Science in Shaanxi Province (No. 2021JM-485), Key Scientific Research Project of Shaanxi Provincial Education Department (No. 20JS019) and Postgraduate Innovation Project of Shaanxi University of Technology (No. SLGYCX2125).
References
- A. Nouri-Khorasani, A. Bonakdarpour, B. Fang and D. P. Wilkinson, ACS Appl. Mater. Interfaces, 2022, 14, 9084–9096 CrossRef PubMed.
- L. Lu, B. Wang, D. Wu, S. Zou and B. Fang, Nanoscale, 2021, 13, 3709–3722 RSC.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- J. Snyder, T. Fujita, M. W. Chen and J. Erlebacher, Nat. Mater., 2010, 9, 904–907 CrossRef CAS PubMed.
- B. Fang, L. Daniel, A. Bonakdarpour, R. Govindarajan, J. Sharman and D. P. Wilkinson, Small, 2021, 17, 2102288 CrossRef CAS PubMed.
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- S. J. Kim, H.-J. Koh, C. E. Ren, O. Kwon, K. Maleski, S.-Y. Cho, B. Anasori, C.-K. Kim, Y.-K. Choi, J. Kim, Y. Gogotsi and H.-T. Jung, ACS Nano, 2018, 12, 986–993 CrossRef CAS PubMed.
- E. Lee, A. VahidMohammadi, Y. S. Yoon, M. Beidaghi and D.-J. Kim, ACS Sens., 2019, 4, 1603–1611 CrossRef CAS PubMed.
- Y. Ma, N. Liu, L. Li, X. Hu, Z. Zou, J. Wang, S. Luo and Y. Gao, Nat. Commun., 2017, 8, 1207 CrossRef PubMed.
- Z. W. Seh, K. D. Fredrickson, B. Anasori, J. Kibsgaard, A. L. Strickler, M. R. Lukatskaya, Y. Gogotsi, T. F. Jaramillo and A. Vojvodic, ACS Energy Lett., 2016, 1, 589–594 CrossRef CAS.
- Z. Li and Y. Wu, Small, 2019, 15, 1804736 CrossRef PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- X. Wu, Z. Wang, M. Yu, L. Xiu and J. Qiu, Adv. Mater., 2017, 29, 1607017 CrossRef PubMed.
- Z. Zhang, H. Li, G. Zou, C. Fernandez, B. Liu, Q. Zhang, J. Hu and Q. Peng, ACS Sustainable Chem. Eng., 2016, 4, 6763–6771 CrossRef CAS.
- L. Jiang, J. Duan, J. Zhu, S. Chen and M. Antonietti, ACS Nano, 2020, 14, 2436–2444 CrossRef CAS PubMed.
- B. Wei, Z. Fu, D. Legut, T. C. Germann, S. Du, H. Zhang, J. S. Francisco and R. Zhang, Adv. Mater., 2021, 33, 2102595 CrossRef CAS PubMed.
- Q. Peng, J. Zhou, J. Chen, T. Zhang and Z. Sun, J. Mater. Chem. A, 2019, 7, 26062–26070 RSC.
- Y. Xie, C. Zhang, X. He, J.-W. Su, T. Parker, T. White, M. Griep and J. Lin, Appl. Surf. Sci., 2019, 464, 344–350 CrossRef CAS.
- Y. Xie, C. Zhang, J.-W. Su, H. Deng, C. Zhang and J. Lin, ChemSusChem, 2019, 12, 473–479 CrossRef CAS PubMed.
- A. Kumar, S. Ibraheem, T. Anh Nguyen, R. K. Gupta, T. Maiyalagan and G. Yasin, Coord. Chem. Rev., 2021, 446, 214122 CrossRef CAS.
- X. Zhang, J. Lei, D. Wu, X. Zhao, Y. Jing and Z. Zhou, J. Mater. Chem. A, 2016, 4, 4871–4876 RSC.
- P. Li, J. Zhu, A. D. Handoko, R. Zhang, H. Wang, D. Legut, X. Wen, Z. Fu, Z. W. Seh and Q. Zhang, J. Mater. Chem. A, 2018, 6, 4271–4278 RSC.
- J. Qu, J. Xiao, H. Chen, X. Liu, T. Wang and Q. Zhang, Chin. J. Catal., 2021, 42, 288–296 CrossRef CAS.
- J. Zhou, G. Liu, Q. Jiang, W. Zhao, Z. Ao and T. An, Chin. J. Catal., 2020, 41, 1633–1644 CrossRef CAS.
- S. Shen, Z. Wang, Z. Lin, K. Song, Q. Zhang, F. Meng, L. Gu and W. Zhong, Adv. Mater., 2022, 34, 2110631 CrossRef CAS PubMed.
- Z. Wang, B. Xiao, Z. Lin, Y. Xu, Y. Lin, F. Meng, Q. Zhang, L. Gu, B. Fang and S. Guo, Angew. Chem., 2021, 133, 23576–23581 CrossRef.
- D. Geng, X. Zhao, Z. Chen, W. Sun, W. Fu, J. Chen, W. Liu, W. Zhou and K. P. Loh, Adv. Mater., 2017, 29, 1700072 CrossRef PubMed.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10, 10876–10883 RSC.
- D. U. Lee, P. Xu, Z. P. Cano, A. G. Kashkooli, M. G. Park and Z. Chen, J. Mater. Chem. A, 2016, 4, 7107–7134 RSC.
- Y. Nie, L. Li and Z. Wei, Chem. Soc. Rev., 2015, 44, 2168–2201 RSC.
- J. Deng, P. Ren, D. Deng, L. Yu, F. Yang and X. Bao, Energy Environ. Sci., 2014, 7, 1919–1923 RSC.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- H. He, Q. Xia, B. Wang, L. Wang, Q. Hu and A. Zhou, Chin. Chem. Lett., 2020, 31, 984–987 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- Y. Yang, K. Li, Y. Meng, Y. Wang and Z. Wu, New J. Chem., 2018, 42, 6873–6879 RSC.
- X. Xiong, Y. Li, Y. Jia, Y. Meng, K. Sun, L. Zheng, G. Zhang, Y. Li and X. Sun, Nanoscale, 2019, 11, 15900–15906 RSC.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, Y. Li, L. Xiao, J. Zhang and M. An, Phys. Chem. Chem. Phys., 2020, 22, 28297–28303 RSC.
- Z. Wang, S. Shen, Z. Lin, W. Tao, Q. Zhang, F. Meng, L. Gu and W. Zhong, Adv. Funct. Mater., 2022, 2112832 CrossRef CAS.
- A. Staykov, D. Derekar and K. Yamamura, Int. J. Quantum Chem., 2016, 116, 1486–1492 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Hong Cui
ab,
Hong Cui