DOI:
10.1039/D2RA01077A
(Paper)
RSC Adv., 2022,
12, 14377-14383
Investigations on the thermoelectric and thermodynamic properties of Y2CT2 (T = O, F, OH)
Received
18th February 2022
, Accepted 8th April 2022
First published on 12th May 2022
Abstract
Using the first-principle calculations combined with the Boltzmann transport theory, we studied the thermoelectric properties of Y2CT2 (T = O, F, OH) MXenes. Specifically, the Seebeck coefficient, thermal and electrical conductivities under constant relaxation time approximation were calculated. Results show that for p-type carriers, Y2CO2 has the largest power factor of up to 0.0017 W m−1 K−2 when the carrier concentration is 4.067 × 1013 cm−2 at 900 K, at the same temperature, for n-type carriers, the concentration is 9.376 × 1013 cm−2, the power factor in Y2C(OH)2 is 0.0026 W m−1 K−2. In particular, the figure of merit in Y2CF2 is 1.38 at 900 K because of its low thermal conductivity, indicating that it can be considered a potential medium-temperature thermoelectric material. In addition, the thermodynamics properties within 32 GPa and 900 K, such as bulk modulus, heat capacity and thermal expansion, are also estimated using the quasi-harmonic Debye model. Our results may offer some valuable hints for the potential application of Y2CT2 (T = O, F, OH) in the thermoelectric field.
1 Introduction
Due to the increase in energy consumption and severe environmental problems, it is of great significance to explore sustainable and ecologically sound energy resources. Thermoelectric materials (TE) can realize the conversion directly between heat and electricity.1 This unique feature makes the expectation from thermoelectric materials deal with the global energy crisis.2 The performance of thermoelectric materials is evaluated by the dimensionless quality factor ZT, which is defined as, 3 where S is the Seebeck coefficient, σ is the electrical conductivity, κ is the sum of electronic thermal conductivity κe and the lattice thermal conductivity κl, and T is the temperature.4,5 The thermopower is evaluated by the power factor S2σ.6,7 Generally, both, high S2σ and low κ lead to secure high ZT value.8,9 In the past few decades, a series of high-efficiency thermoelectric materials have been discovered.10 Among them, the typical thermoelectric materials with ZT > 1 have Bi2Te3, Si1−xGex, and PbTe.11 Unfortunately, the Carnot efficiency of thermoelectric materials is only 10% in practical applications, which has a great difference from traditional refrigeration with a Carnot efficiency of 30%.12 Although scientists have developed many methods to improve the performance of thermoelectric materials, they, however, still face many challenges; ZT is difficult to increase due to the coupling between thermoelectric factors.
3 where S is the Seebeck coefficient, σ is the electrical conductivity, κ is the sum of electronic thermal conductivity κe and the lattice thermal conductivity κl, and T is the temperature.4,5 The thermopower is evaluated by the power factor S2σ.6,7 Generally, both, high S2σ and low κ lead to secure high ZT value.8,9 In the past few decades, a series of high-efficiency thermoelectric materials have been discovered.10 Among them, the typical thermoelectric materials with ZT > 1 have Bi2Te3, Si1−xGex, and PbTe.11 Unfortunately, the Carnot efficiency of thermoelectric materials is only 10% in practical applications, which has a great difference from traditional refrigeration with a Carnot efficiency of 30%.12 Although scientists have developed many methods to improve the performance of thermoelectric materials, they, however, still face many challenges; ZT is difficult to increase due to the coupling between thermoelectric factors.
Nanostructuring is one of the effective ways to improve ZT.13,14 In 1993, Hicks and Dresselhaus expected that the quantum confinement effect of electronic carriers in low-dimensional materials can significantly improve the power factor,14 and they found that there is a ZT value of up to 2.62 at 923 K15 in a two-dimensional SnSe single crystal, indicating it may be possible that they may be promising thermoelectric materials.16 MXenes is a new type of two-dimensional material, which has been extensively studied for its potential applications in the fields of energy storage and conversion, environment and catalysis, separation membranes, medicine, optics and electronics.17 MXenes are formed by etching the A element from the MAX phase in an aqueous hydrofluoric acid solution.18 MAX phases with the chemical formula Mn+1AXn (n = 1, 2 or 3) have a layered structure, where M is an early transition metal, A is a group of elements, and X is nitrogen or carbon.19 During the etching process, A atoms are usually replaced by O, F and/or OH groups, resulting in functionalized MXenes Mn+1XnT2.20 Research on the thermoelectric properties of MXene materials mainly focuses on MXenes with semiconductor properties, such as Sc2CT2 (T = O, F, OH)21 and M2CO2 (M = Ti, Zr, Hf),22 and surface functionalization can convert the metal abundance of MXenes into semiconductor properties,23 and research shows that surface-functionalized Y2C has semiconductor properties;24 thus, in this work, we mainly studied the electronic structure and thermoelectric properties of the surface-functionalized Y2C.
2 Calculation details
All calculations are performed based on the first principles. We firstly performed the structural optimization of Y2CT2 (T = O, F, OH) by using Vienna Ab initio Simulation Package (VASP).25,26 The exchange-correlation functionals were treated by the generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE).27 The energy cutoff 500 eV and the Monkhorst–Pack k-mesh 14 × 14 × 1 were adopted. The thresholds of 0.005 eV Å−1 for the force convergence and 10−6 eV for the energy convergence were used. After relaxing the geometry structure, we calculated the band structure for Y2CT2 (T = O, F, OH) from the WIEN2k code, a full-potential linearized augmented plane-wave method.28 The value of energy cutoff RMT × Kmax was taken to be 7, where RMT is the smallest atomic sphere radius in the unit cell of Y2CT2 (T = O, F, OH), and Kmax is the magnitude of the largest K vector. More-dense k-point mesh of 15 × 15 × 1 was adopted in order to ensure the convergence of the electronic transport properties.
We solved the semi-classical Boltzmann transport equation within the constant relaxation time approximation (τ = 10−14 s)29 for calculating the Seebeck coefficient, electrical conductivity, and thermal conductivity. While the relaxation time τ generally depends on the electronic wave vector, energy, and scattering mechanism, reliable results for the transport properties have been obtained within the constant relaxation time approximation for a variety of materials.30–32 A rigid band model was used to simulate doping by shifting the chemical potential (BoltzTraP code).33 For this purpose, the electronic band structure was calculated on a very fine 60 × 60 × 1 k-mesh.
3 Results and discussion
3.1 Electronic properties
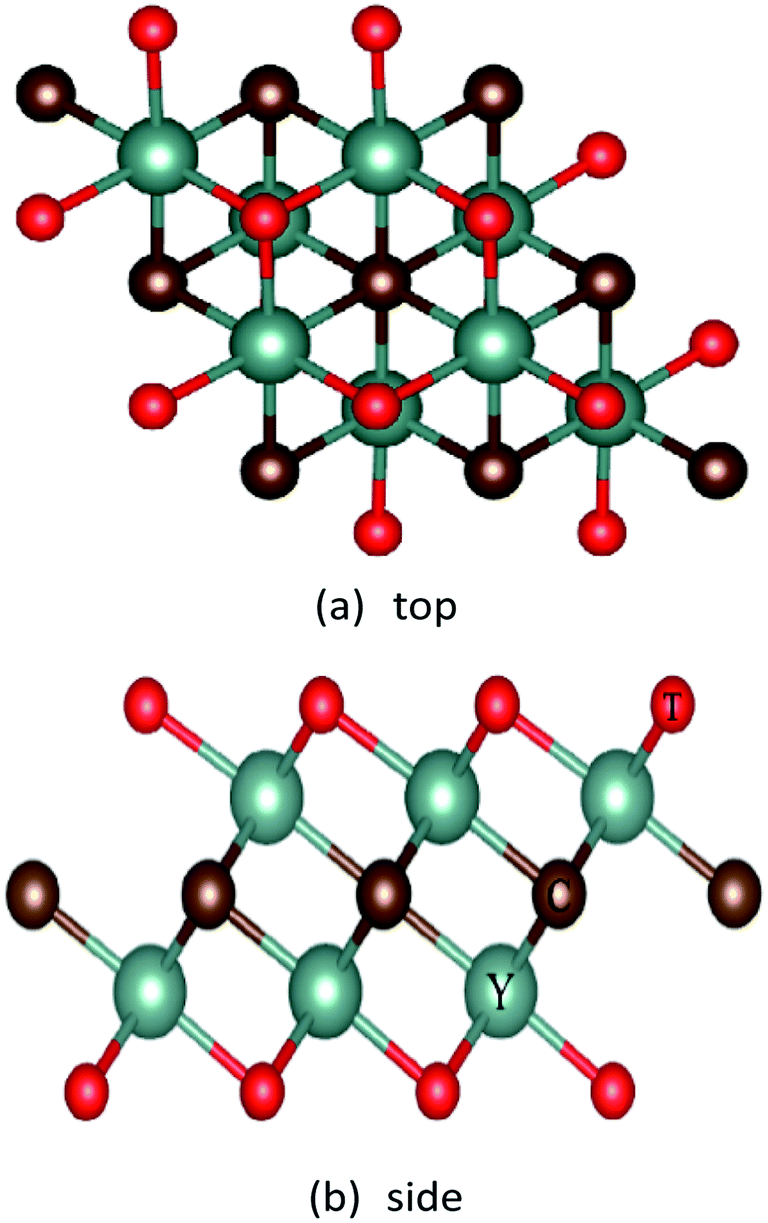
First, we built the structural model Y2CT2 (T = O, F, OH) as shown in Fig. 1, the three-dimensional periodic boundary conditions were combined with the 20 Å thick vacuum region along the c direction to produce a single layer structure, and optimize their structure to obtain the ground state. The lattice parameters at equilibrium are presented in Table 1. It was found that the O and OH functionalization leads to longer Y–C in Y2CO2 and Y–T in Y2C(OH)2 respectively, this difference is also reflected in the smaller bond angle of ∠CYC in Y2CO2 and ∠TYT in Y2C(OH)2, because the proximity of O and OH to C leads to the redistribution of charges, which weakens the bond strength of Y–C in Y2CO2 and Y–T in Y2C(OH)2.
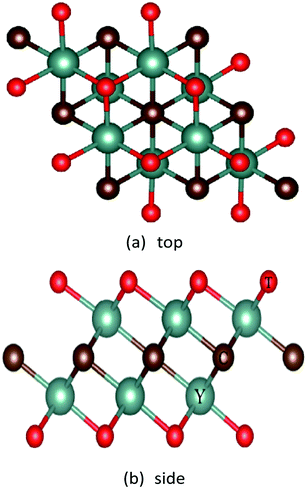 |
| | Fig. 1 Top and side views of the Y2CT2 (T = O, F and OH). | |
Table 1 Calculated lattice constants (Å), bond length (Å), bond angle and band gap (eV)
| |
a |
Y–C |
C–T |
Y–T |
∠TYT |
∠CYC |
Band gap |
| Y2CO2 |
3.54 |
2.70 |
3.25 |
2.18 |
108.81 |
81.76 |
3.63 |
| Y2CF2 |
3.58 |
2.48 |
3.28 |
2.18 |
108.80 |
92.38 |
1.17 |
| Y2C(OH)2 |
3.60 |
2.49 |
3.39 |
2.45 |
94.38 |
92.34 |
0.74 |
Since their structures are the same, the three functionalized Y2C studied here have similar energy band structures, as shown in Fig. 2. According to Fig. 2, an indirect band gap in Y2CO2 and Y2CF2was observed, the maximum valence band is at the Γ point, and the minimum conduction band was at the M point. In Y2C(OH)2, a direct band gap was found, and the maximum valence band and the minimum conduction band were both at Γ point. For O, F and OH functionalization, the band gap decreases sequentially. In addition, each compound exhibits asymmetry between the valence band and the conduction band, so the effective masses of p-type and n-type carriers are different. For the three compounds, Y-d, the C-p and T-s states form the valence band edge and the conduction band edge, among them, C-p contributes the most to the valence band edge, and Y-d contributes the most to the conduction band edge, as shown in Fig. 3.
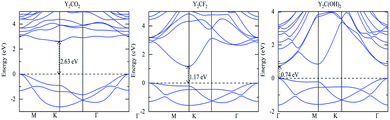 |
| | Fig. 2 Electronic band structures of Y2CT2 (T = O, F and OH). | |
 |
| | Fig. 3 Total and orbital densities of states of Y2CT2 (T = O, F and OH). | |
3.2 Thermoelectric properties
Flat bands near the Fermi level are beneficial to the thermoelectric properties of the material because they lead to higher S34, that is, the greater the effective mass of the carrier, the greater is the S. The conduction band near the Fermi level of Y2CO2 is flatter than the valence band, which can be seen in Fig. 2. Therefore, for Y2CO2, the effective mass of n-type carriers is greater than that of p-type carriers, while Y2CF2 and Y2C(OH)2 have opposite phenomena. As shown in Fig. 2 and Table 2. It can be observed from Fig. 4(a)–(f) that S for Y2CO2 n-type carriers is larger than S for p-type carriers, while Y2CF2 and Y2C(OH)2 are opposite. For all compounds, S increases with. Temperature and decreases with carrier concentration, which is consistent with Pisarenko's relationship35| |
 | (1) |
where m* and ρ are the effective mass and carrier concentration, respectively. However, for the Y2CF2 and Y2C(OH)2, although high temperature should be beneficial to S, due to bipolar conduction (a large number of minority carriers), the carrier concentration will decrease as the temperature rises, and the smaller the band gap, the greater this effect, this is consistent with the reported results in ref. 20.
Table 2 Effective mass m* (me) for p and n-types Y2CT2 (T = O, F and OH)
| |
Y2CO2 |
Y2CF2 |
Y2C(OH)2 |
| p-type |
1.19 |
2.53 |
2.17 |
| n-type |
1.79 |
0.57 |
1.21 |
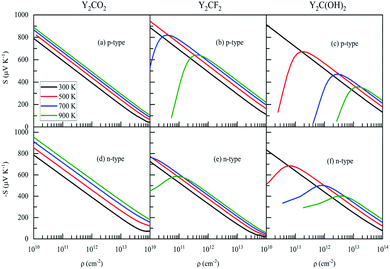 |
| | Fig. 4 Carrier concentration dependence of Seebeck coefficients (S) for (a)–(c) p-type and (d)–(f) n-type Y2CT2 (T = O, F and OH). | |
As expected, σ increases with ρ within a given temperature (300–900 K) and concentration (1010 to 1014 cm−2), as shown in Fig. 5(a)–(f). Obviously, for Y2CF2 and Y2C(OH)2, the conductivity of n-type carriers is greater than that of p-type carriers because n-type carriers are superior to p-type carriers due to the dispersive conduction band near the Fermi-level conductivity. For all compounds, the conductivity decreased with increasing temperature. The change of κe is the same as that of σ, (see Fig. 5(g)–(l)). The difference is that κe increases with temperature. It can be clearly seen that for the three substances, the electronic thermal conductivity increases exponentially, this trend indicates that they have better thermal conductivity and can be used for heat dissipation in electronic devices. According to Fig. 6(a)–(f), the S2σ of the three compounds show similar trends. The S2σ value increased with increasing temperature. For the S2σ of Y2CO2 and Y2CF2 p-type carriers, the value is always greater than the S2σ value of the n-type carrier in a given carrier concentration (1010 to 1014 cm−2) and temperature (300–900 K), which is determined by the effective carrier mass and the degree of dispersion of the band at the Fermi level. For the Y2CO2, Y2CF2 and Y2C(OH)2, the maximum values of S2σ appear in the vicinity of 2.0 × 1013, 8.0 × 1013, and 6.6 × 1013, respectively.
 |
| | Fig. 5 Carrier concentration dependence of electrical conductivities (σ) for (a)–(c) p-type and (d)–(f) n-type Y2CT2 (T = O, F and OH) and electronic thermal conductivities (κe) for (g)–(i) p-type and (j)–(l) n-type Y2CT2 (T = O, F and OH). | |
 |
| | Fig. 6 Carrier concentration dependence of thermoelectric power factors (S2σ) for (a)–(c) p-type and (d)–(f) n-type Y2CT2 (T = O, F and OH). | |
For the lattice thermal conduction, the most commonly used method is solving the Boltzmann transfer equation (BTE), which involves the calculation of phonon frequency, group velocity, and harmonic and non-harmonic interatomic force constants (IFCs).36,37 In order to consider the three phonon scattering processes, which need to be used to calculate the third-order anharmonic IFCs. Deinze et al.38 used density functional perturbation theory (DFPT) to obtain third-order IFCs to study the phonon linewidth. In recent years, this method has been successfully used to solve BTE and predict the lattice thermal conductivity of materials.39–41 This method requires the calculation of the electronic structure of multiple large supercells, and each supercell has a different set of atomic displacements, so it requires a lot of computing resources. A variety of simple and low-cost calculation methods have been developed to calculate the lattice thermal conductivity of materials. Among these, the Slack42 is one of the more effective methods. In this work, the lattice thermal conductivity was calculated using the method proposed by Slack using the Debye temperature and the Grüneisen parameter:
| |
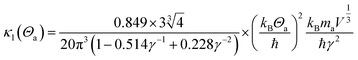 | (2) |
where
V is the volume of the unit cell,
ma is the average atomic mass and,
γ is the Grüneisen parameter.
Θa is the acoustic-mode Debye temperature,
42 which is different from the traditional Debye temperature
ΘD,
Θa is based on the assumption that the optical phonon mode in the crystal does not contribute to the lattice thermal conductivity, and only the acoustic mode is taken into account. It can be calculated from the traditional Debye temperature
Θa42| |
 | (3) |
where
n is the number of atoms in the primitive cell, the thermal conductivity at temperature is estimated by
42| |
 | (4) |
It can be seen from the above formula that the key to calculating κl lies in the Grüneisen parameter γ and the acoustic model Debye temperature Θa. In this work, we used the elastic constant to calculate the transverse and longitudinal sound velocities, and finally obtain the Grüneisen parameter γ and the acoustic model Debye temperature Θa. The elastic constant, bulk modulus, shear modulus, Young's modulus and Poisson's ratio are shown in Table 3, in which the elastic constant satisfies the Born stability criterion of Born-Huang's lattice dynamics theory of hexagonal two-dimensional structure,43 i.e., C11>|C12|, (C11 + C12)C33 > 2C132, and C66 > 0. The transverse sound velocity νt and longitudinal sound velocity νl can be calculated via the following formulas.44
| |
 | (5) |
| |
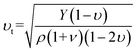 | (6) |
average sound velocity
νm can be calculated from
νt and
νl,

, replace
νt,
νl and
νm with expressions for
γ and
ΘD:
45,46| |
 | (7) |
| |
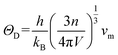 | (8) |
Table 3 Y2CT2 (T = O, F and OH) average atomic mass ma, cell volume V, the number of atoms n in the primitive cell, elastic constant Cij, Bulk modulus, Young's modulus, Shear modulus, Poisson's ratio, transverse sound velocity νt, longitudinal sound velocity νl, average sound velocity νm, Debye temperature ΘD, acoustic Debye temperature Θa, Grüneisen parameter γ and lattice thermal conductivity κl (at room temperature)
| |
Y2CO2 |
Y2CF2 |
Y2CO2 |
| n |
5 |
5 |
7 |
| ma (in amu) |
44.36 |
45.56 |
31.98 |
| V (Å3) |
288.41 |
268.19 |
318.39 |
| C11 (GPa) |
111.27 |
81.47 |
70.30 |
| C12 (GPa) |
50.48 |
18.71 |
15.07 |
| C13 (GPa) |
1.20 |
2.05 |
−2.41 |
| C33 (GPa) |
2.97 |
0.87 |
1.60 |
| C66 (GPa) |
30.11 |
30.91 |
26.45 |
| B (GPa) |
19.56 |
12.12 |
8.88 |
| Y (GPa) |
32.29 |
19.00 |
16.44 |
| G (GPa) |
13.18 |
7.67 |
6.90 |
| ν |
0.23 |
0.24 |
0.19 |
| νt (×103 m s−1) |
3.21 |
2.33 |
2.05 |
| νl (×103 m s−1) |
5.39 |
3.98 |
3.31 |
| νm (×103 m s−1) |
3.56 |
2.59 |
2.26 |
| ΘD (K) |
274.16 |
204.17 |
211.33 |
| Θa (K) |
160.33 |
119.40 |
110.47 |
| γ |
1.39 |
1.45 |
1.25 |
| κl (W m−1 K−1) |
7.16 |
2.70 |
2.20 |
νt, νl, νm, γ and ΘD are calculated as shown in Table 3. By substituting these parameters into formula (4), the lattice thermal conductivity at room temperature as shown in Table 3 and the κl at 300–900 K are shown in Fig. 7. From Fig. 7, for the three compounds, the lattice thermal conductivity decreases with increasing temperature, and the lattice thermal conductivity of Y2CO2 is the largest at the same temperature (300–900 K), followed by Y2CF2, and Y2C(OH)2 is the smallest, which means that for the phonon pair of Y2CO2 at the same temperature (300–900 K) the thermal conductivity has the largest contribution and Y2C(OH)2 has the smallest contribution.
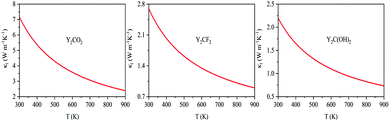 |
| | Fig. 7 Lattice thermal conductivity (κl) as a function of temperature for Y2CT2 (T = O, F and OH). | |
Fig. 8 shows the characteristics of the dependence of ZT on carrier concentration. For the n-type carriers, the maximum ZT value at 900 K increases in the order of Y2CO2, Y2CF2 and Y2C(OH)2. For the p-type carriers since PF is larger and κ is smaller, the ZT value of Y2CF2 is larger than Y2CO2 and Y2C(OH)2. For the Y2CO2, PF is larger than Y2CF2 and Y2C(OH)2, but κ is larger than Y2CF2 and Y2C(OH)2, so ZT is smaller than Y2CF2 and similar to Y2C(OH)2.
 |
| | Fig. 8 Carrier concentration dependence of figures of merit (ZT) for (a)–(c) p-type and (d)–(f) n-type Y2CT2 (T = O, F and OH). | |
3.3 Thermodynamic properties
In Fig. 9(a)–(c), we show the volume of Y2CT2 (T = O, F and OH) as a function of temperature and pressure. It is found, that under a given pressure, the volume increases with temperature, and this trend decreases with the increase of pressure. On the other hand, at a certain temperature, the volume decreases with the increase of pressure, and this change decreases with increasing pressure, we can understand that the pressure compresses solids and temperature expand solids. The change bulk modulus (B) under temperature and pressure. It can be seen from the Fig. 9(d)–(f) that for the Y2CO2, Y2CF2 and Y2C(OH)2, the change of bulk modulus with temperature and pressure is opposite to the change in volume. The bulk modulus of the three functional groups increases with pressure at a given temperature, which means that the pressure increases the hardness. For the Y2CO2, Y2CF2 and Y2C(OH)2, the bulk modulus decreases with temperature, which means that the temperature decreases the hardness.
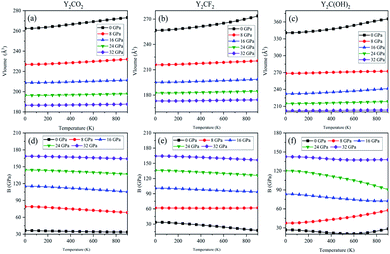 |
| | Fig. 9 Temperature dependence of (a)–(c) variations of volume (V) and (d)–(f) bulk modulus (B) for Y2CT2 (T = O, F and OH). | |
In addition, the heat capacity (CV) of Y2CT2 (T = O, F, OH) is calculated, which provides information about lattice vibration, phase transition, and measurement of molecular motion, and the changes in it with temperature and pressure are shown in Fig. 10(a)–(c). It can be seen from Fig. 10(a)–(c) that CV increases rapidly at lower temperature values from 0 K to 400 K. Above 400 K, it can be observed that CV slowly increases and becomes constant at 700 K, and finally close to the famous Dulong-Petit limit.47 Thermal expansion is the non-harmonic result of the interatomic force explained by the quasi-harmonic approximation, where it is assumed that the vibration frequency of the network has nothing to do with temperature, but depends on the volume. Fig. 10(d)–(f) shows the change in Y2CT2 (T = O, F, OH) thermal expansion α under constant pressure. For low temperatures up to 300 K, α at a given pressure (proportional to T3) increases sharply with the increase in temperature (most obvious at 0 GPa). Starting from 400 K, the increase becomes gentle. It is obvious that the lower the pressure, the greater the rate of increase. For the three functionalizations, in general, at a given temperature, the thermal expansion decreases with the increase in pressure.
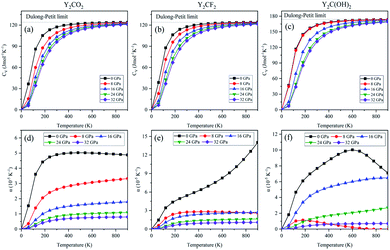 |
| | Fig. 10 Temperature dependence of (a)–(c) variations of heat capacity (Cv) and (d)–(f) thermal expansion (α) for Y2CT2 (T = O, F and OH). | |
4 Conclusions
We studied the thermoelectric properties of the two-dimensional functionalized Y2C by considering the transmission of electrons and phonons and studied their electronic structures using first-principles methods, and the Boltzmann transmission equation of electrons was solved. For the transport of phonons, we use the Slack model considering the high-temperature limit to study the contribution of phonons to the thermal conductivity to quantify the study of electrons and lattice pairs. The functional groups O, F and OH opened the band gaps of 2.63 eV, 1.17 eV and 0.74 eV at the Fermi level of Y2C, respectively. The effective masses of p-type and n-type carriers were calculated through the band structure, thus confirming the difference in the Seebeck coefficient. For the p-type carriers, when the carrier concentration was 4.067 × 1013 cm−2, the maximum power factor of Y2CO2 could reach 0.0017 W m−1 K−2 at 900 K, and at the same temperature, for the n-type carriers, the concentration was 9.376 × 1013 cm−2, the power factor in Y2C(OH)2 was 0.0026 W m−1 K−2. Through the study of electronic thermal conductivity and lattice thermal conductivity, it was found that the contribution of phonons to thermal conductivity occupies the main part. In particular, the quality factor of Y2CF2 was 1.38 at 900 K, because of its low thermal conductivity, indicating that it can be considered as a potential medium-temperature thermoelectric material. In addition, the thermodynamic properties in the range of 32 GPa and 900 K, such as bulk modulus, heat capacity, and thermal expansion, are also estimated using the quasi-harmonic Debye model. Our results may provide some valuable hints for the potential application of Y2CT2 (T = O, F, OH) in thermal electric fields. At present, the experimental research on the thermoelectric properties of MXenes materials is still in the research stage, this paper provides a good theoretical basis for experimental research.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is also supported by Natural Science Foundation of Gansu Province (No. 21JR7RA294).
References
- X. Shi and L. Chen, Nat. Mater., 2016, 15, 691–692 CrossRef CAS PubMed.
- X.-L. Shi, J. Zou and Z.-G. Chen, Chem. Rev., 2020, 120, 7399–7515 CrossRef CAS PubMed.
- F. J. DiSalvo, Science, 1999, 285, 703–706 CrossRef CAS PubMed.
- W. Liu, X. Yan, G. Chen and Z. Ren, Nano Energy, 2012, 1, 42–56 CrossRef CAS.
- A. Minnich, M. S. Dresselhaus, Z. Ren and G. Chen, Energy Environ. Sci., 2009, 2, 466–479 RSC.
- P. Qiu, X. Shi and L. Chen, Energy Storage Mater., 2016, 3, 85–97 CrossRef.
- A. M. Dehkordi, M. Zebarjadi, J. He and T. M. Tritt, Mater. Sci. Eng., R, 2015, 97, 1–22 CrossRef.
- R. Moshwan, L. Yang, J. Zou and Z.-G. Chen, Adv. Funct. Mater., 2017, 27, 1703278 CrossRef.
- X. Zhang and L.-D. Zhao, J. Materiomics, 2015, 1, 92–105 CrossRef.
- S. Twaha, J. Zhu, Y. Yan and B. Li, Renewable Sustainable Energy Rev., 2016, 65, 698–726 CrossRef CAS.
- T. M. Tritt, Science, 1999, 283, 804–805 CrossRef CAS.
- F. J. DiSalvo, Science, 1999, 285, 703–706 CrossRef CAS PubMed.
- L. Hicks and M. Dresselhaus, MRS Online Proc. Libr., 1993, 326, 413–418 CrossRef.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 12727 CrossRef CAS PubMed.
- L.-D. Zhao, S.-H. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Nature, 2014, 508, 373–377 CrossRef CAS PubMed.
- Y. Zhou and L.-D. Zhao, Adv. Mater., 2017, 29, 1702676 CrossRef PubMed.
- G. Yury and B. Anasori, Acs Nano, 2019, 13, 8491–8494 CrossRef PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- Z. Sun, Int. Mater. Rev., 2011, 56, 143–166 CrossRef CAS.
- A. Sinha, H. Zhao, Y. Huang, X. Lu, J. Chen and R. Jain, et al., TrAC, Trends Anal. Chem., 2018, 105, 424–435 CrossRef CAS.
- S. Kumar and U. Schwingenschlögl, Phys. Rev. B, 2016, 94, 035405 CrossRef.
- A. N. Gandi, H. N. Alshareef and U. Schwingenschlögl, Chem. Mater., 2016, 28, 1647–1652 CrossRef CAS.
- M. Khazaei, A. Ranjbar, M. Arai, T. Sasaki and S. Yunoki, J. Mater. Chem. C, 2017, 5, 2488–2503 RSC.
- L. Hong, R. F. Klie and S. Ögüt, Phys. Rev. B, 2016, 93, 115412 CrossRef.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- K. B. John, P. Perdew and M. Ernzerhof, Phys. Rev. Lett., 1997, 77, 1396 Search PubMed.
- P. Blaha, WIEN2k, An Augmented Plane Wave+ Local Orbitals Program for Calculating Crystal Properties, ed. K. Schwarz, Techn. Universität Wien, Austria, 2001 Search PubMed.
- M. Khazaei, M. Arai, T. Sasaki, M. Estili and Y. Sakka, Phys. Chem. Chem. Phys., 2014, 16, 7841–7849 RSC.
- P. B. Allen, W. E. Pickett and H. Krakauer, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 7482 CrossRef CAS PubMed.
- W. W. Schulz, P. B. Allen and N. Trivedi, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 10886 CrossRef CAS PubMed.
- L. Chaput, P. Pécheur, J. Tobola and H. Scherrer, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085126 CrossRef.
- G. K. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67–71 CrossRef CAS.
- G. Mahan and J. Sofo, Proc. Natl. Acad. Sci., 1996, 93, 7436–7439 CrossRef CAS PubMed.
- G. J. Snyder and E. S. Toberer, Materials for sustainable energy: a collection of peer-reviewed research and review articles from Nature Publishing Group, 2011, pp. 101–110 Search PubMed.
- D. A. Broido, M. Malorny, G. Birner, N. Mingo and D. Stewart, Appl. Phys. Lett., 2007, 91, 231922 CrossRef.
- W. Li, N. Mingo, L. Lindsay, D. A. Broido, D. A. Stewart and N. A. Katcho, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 195436 CrossRef.
- G. Deinzer, G. Birner and D. Strauch, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 144304 CrossRef.
- A. Ward, D. Broido, D. A. Stewart and G. Deinzer, Phys. Rev. B, 2009, 80, 125203 CrossRef.
- A. Ward and D. Broido, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085205 CrossRef.
- Q. Zhang, F. Cao and K. Lukas, et al., J. Am. Chem. Soc., 2012, 134, 17731–17738 CrossRef CAS PubMed.
- R. Bruls, H. Hintzen and R. Metselaar, J. Appl. Phys., 2005, 98, 126101 CrossRef.
- I. Waller, Acta Crystallogr., 1956, 9, 837–838 CrossRef.
- O. L. Anderson, J. Phys. Chem. Solids, 1963, 24, 909–917 CrossRef CAS.
- O. Anderson, J. Phys. Chem. Solids, 1959, 12, 41–52 CrossRef CAS.
- D. Sanditov, A. Mashanov and M. Darmaev, Tech. Phys., 2009, 54, 1398–1401 CrossRef CAS.
- A. Petit and P. Dulong, Ann. Chim. Phys., 1981, 10, 395 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Zi-Qi Sun and
Zi-Meng Zhang
*,
Zi-Qi Sun and
Zi-Meng Zhang
 3 where S is the Seebeck coefficient, σ is the electrical conductivity, κ is the sum of electronic thermal conductivity κe and the lattice thermal conductivity κl, and T is the temperature.4,5 The thermopower is evaluated by the power factor S2σ.6,7 Generally, both, high S2σ and low κ lead to secure high ZT value.8,9 In the past few decades, a series of high-efficiency thermoelectric materials have been discovered.10 Among them, the typical thermoelectric materials with ZT > 1 have Bi2Te3, Si1−xGex, and PbTe.11 Unfortunately, the Carnot efficiency of thermoelectric materials is only 10% in practical applications, which has a great difference from traditional refrigeration with a Carnot efficiency of 30%.12 Although scientists have developed many methods to improve the performance of thermoelectric materials, they, however, still face many challenges; ZT is difficult to increase due to the coupling between thermoelectric factors.
3 where S is the Seebeck coefficient, σ is the electrical conductivity, κ is the sum of electronic thermal conductivity κe and the lattice thermal conductivity κl, and T is the temperature.4,5 The thermopower is evaluated by the power factor S2σ.6,7 Generally, both, high S2σ and low κ lead to secure high ZT value.8,9 In the past few decades, a series of high-efficiency thermoelectric materials have been discovered.10 Among them, the typical thermoelectric materials with ZT > 1 have Bi2Te3, Si1−xGex, and PbTe.11 Unfortunately, the Carnot efficiency of thermoelectric materials is only 10% in practical applications, which has a great difference from traditional refrigeration with a Carnot efficiency of 30%.12 Although scientists have developed many methods to improve the performance of thermoelectric materials, they, however, still face many challenges; ZT is difficult to increase due to the coupling between thermoelectric factors.








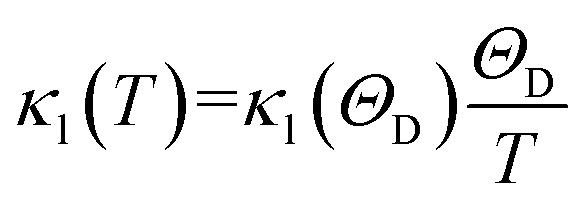


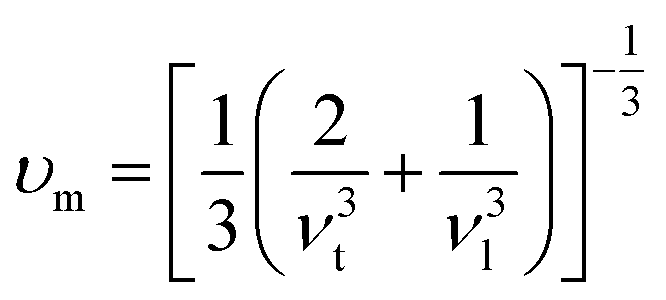 , replace νt, νl and νm with expressions for γ and ΘD:45,46
, replace νt, νl and νm with expressions for γ and ΘD:45,46




