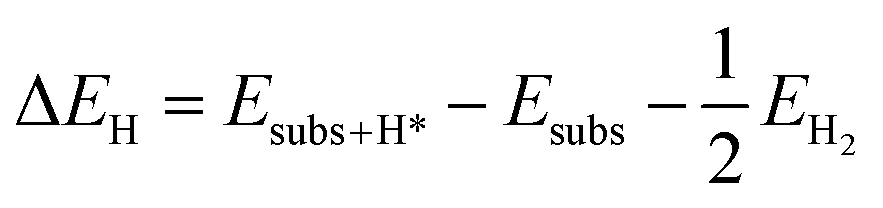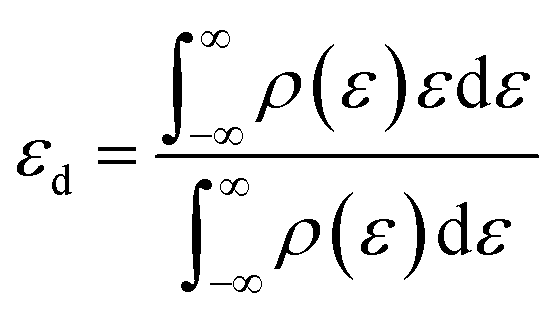DOI:
10.1039/D2RA01865A
(Paper)
RSC Adv., 2022,
12, 17257-17263
The effect of heteroatom doping on the active metal site of CoS2 for hydrogen evolution reaction
Received
23rd March 2022
, Accepted 1st June 2022
First published on 10th June 2022
Abstract
The exploration of cost-effective hydrogen evolution reaction (HER) electrocatalysts through water splitting is important for developing clean energy technology and devices. The application of CoS2 in HER has been drawing more and more attention due to its low cost and relatively satisfactory HER catalytic performance. And CoS2 was found to exhibit excellent HER catalytic performance after appropriate doping according to other experimental investigations. However, the theoretical simulation and the intrinsic catalytic mechanism of CoS2 remains insufficiently investigated. Therefore, in this study, density functional theory is used to investigate the HER catalytic activity of CoS2 doped with a heteroatom. The results show that Pt-, N- and O-doped CoS2 demonstrates smaller Gibbs free energies close to that of Pt, compared with the original CoS2 and CoS2 doped with other atoms. Furthermore, HER catalytic performance of CoS2 can be improved by tuning d-band centers of H adsorption sites. This study provides an effective method to achieve modified CoS2 for high-performance HER and to investigate other transition metal sulfides as HER electrode.
1 Introduction
The high combustion heat value, clean combustion products, and abundant resources make hydrogen (H2) a distinguished energy storage material. Producing hydrogen from the electrochemical decomposition of water (H2O), the commonly believed future energy carrier,1,2 through hydrogen evolution reaction (HER) has been proved to be a clean and renewable method. However, the hydrogen production catalyst precious metal platinum (Pt), the most efficient electrocatalyst for HER owing to its high stability, low overpotential (about 0.08 V), as well low Tafel slope,3 has not been applied on large scales due to its high cost and scarcity. Hence, it is quite necessary to find a new high-performance HER catalyst.
In recent decades, transition metal sulfides (TMSs), such as MoS2,4–10 VS2,11–15 CoS2,2,16–20 NiS2 (ref. 2, 7, 15, 21 and 22) have been attracting attention from researchers and industry practitioners. Though the effects of the aforesaid compounds as hydrogen production catalysts had been considered in some previous studies, the more recent ones show that some of them such as HER electrode material can exhibit outstanding electrocatalytic performance that is close to that of precious metals (such as Pt). To further optimize the performance of the TMSs, some other studies suggest the incorporating of a third heteroatom in a binary compound, which can improve the electrocatalytic activity of the catalyst.23
As a representative of pyrite-type transition metal sulfides, CoS2 is a low-cost catalytic material with excellent HER performance. Unfortunately, when doping CoS2 with a third heteroatom, no good descriptor can be found to describe the relationship between the structure and activity of the doped CoS2, and the inherent mechanism remains insufficiently explored.
It is noteworthy that Hoffmann et al. found that the center of the d-band formed by the TM turned out to be a good descriptor to predict the adsorption and reactivity of transition metal catalysts.24 Furthermore, an analogous model of d-band based on surface resonance states has also been developed in the transition metal carbides (TMCs),25 metal nitrides (MNs),26 and layered TMSs.27 This can significantly contribute to the analysis of structure–activity relationships in TMCs/TMSs, which can further help improve the design and high-throughput screening of catalysts. Many studies have also found that doping can change the energy band structure near the Fermi energy, so as to change the properties of materials.10,28,29 Therefore, we take interest in investigating the possibility to doped CoS2 for high-performance HER by tuning the d-band centers.
In this work, we study the effect of a heteroatom doping on HER electrochemical properties for CoS2 electrode material based on density functional theory and explore the structure–activity relationships. Herein, the models of the pristine CoS2 and doped CoS2 were built, the stability, the catalytic activity, and the d-band centers of the active metal sites of CoS2 doped with a heteroatom (Mn, Fe, Ni, Cu, Mo, Pt, Ru, C, N, O, P) are studied based on density functional theory (DFT). Our first-principles calculations exhibit that the structures of all doped CoS2 except C-doped CoS2 are stable, the catalytic behavior of CoS2 can be effectively enhanced after Pt, N, and O doping, and the HER catalysis of CoS2 can be changed by tailoring the d-band centers of active metal sites. Our studies are of great benefit to uncovering the intrinsic modulation mechanism of CoS2 for HER catalysis.
2 Computational methods
All density functional theory (DFT) simulations were employed using the Generalized Gradient Approximation-Perdew Burke Ernzerhof (GGA-PBE) was used for the exchange-correlation term in the Spanish Initiative for Electronic Simulations with Thousands of Atoms (SIESTA).30,31 The geometric optimization was employed using a conjugate gradient method until the maximum force was less than 0.02 eV Å−1. An energy cut-off for all simulations was set to be 150 Ry.
2.1 Bulk CoS2
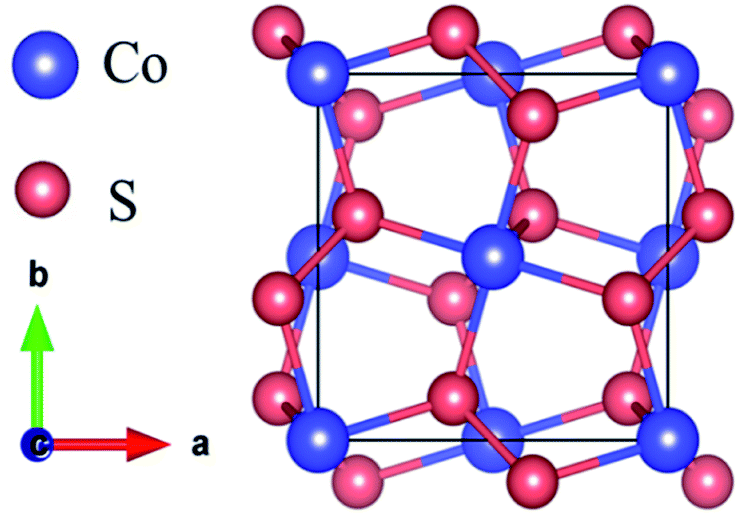
The k-point mesh is set as Γ-centered 7 × 7 × 7 to relax the CoS2 unit cell with a space group of Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . The calculated CoS2 unit cell parameter is 5.544 Å, which is highly consistent with the experimental value (5.538 Å).23,32 The optimized bulk structure is shown in Fig. 1, where the big and small balls represent Co and S atoms, respectively.
. The calculated CoS2 unit cell parameter is 5.544 Å, which is highly consistent with the experimental value (5.538 Å).23,32 The optimized bulk structure is shown in Fig. 1, where the big and small balls represent Co and S atoms, respectively.
 |
| | Fig. 1 The optimized structure of the bulk CoS2. | |
2.2 CoS2(001) surface
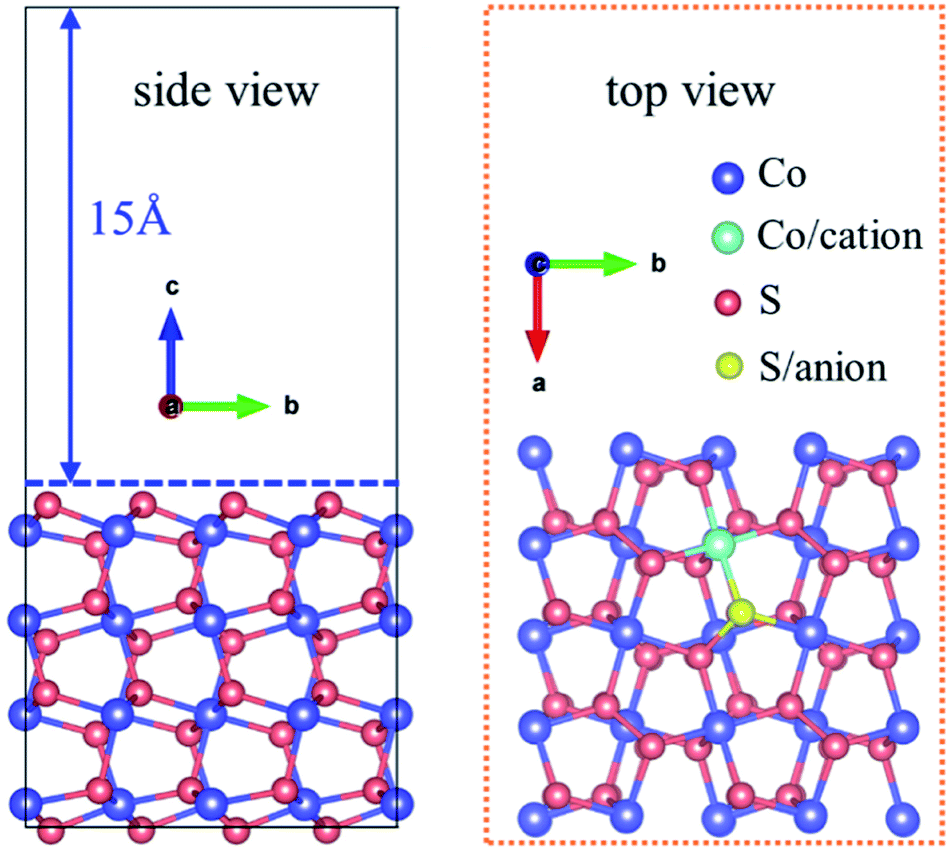
The (001) surface of CoS2 is selected as the optimization model owing to its stability,32 which is built as a 2 × 2 repeating surface unit cells consisting of four S–Co–S atomic layers and 8 CoS2 units. Cation-doped CoS2 is acquired by replacing one Co atom with one metal atom, and anion-doped CoS2 is acquired by replacing one S atom with one nonmetal atom. A vacuum layer with a thickness of 15 Å is introduced to reduce interaction between periodic images.33 The CoS2(001) crystalline structure is represented in Fig. 2.
 |
| | Fig. 2 The crystallographic structure of CoS2(001) surface. | |
2.3 Binding energy
The binding energies (Eb) were first calculated to measure the stability of CoS2 after doping, Eb is the difference between the cohesive energy (Ecoh) of cubic CoS2 and the embedding energy of the dopants (Eemb) on the CoS2 surface.34| | |
Eemb = Edopant+subs − Esubs − Eiso
| (2) |
where Ebulk is the total energy of an atom in the bulk, Eiso is the total energy of an isolated atom, Edopants+subs is the total energy of doped CoS2, and Esubs is the total energy of CoS2 with one vacancy.
2.4 Gibbs free energy of H adsorption
Nørskov et al.3 showed that with the absolute Gibbs free energy of hydrogen adsorption (ΔGH*) approximating zero, the binding between HER intermediates and the electrode surface was neither too strong nor too weak, demonstrating a high catalytic performance of HER. Hence, the (ΔGH*) as a significant factor of measuring the HER catalytic activity is quite essential to is calculated using reliable DFT simulations.35 In our work, the HER catalytic properties in acidic media were investigated. While considering the HER reaction process in acid, the Volmer reaction3 was selected under our calculations, because most reactions occur in protic solution or involve proton as the reactant:36 H+(sol) + e− → H*, where H* refers to H adsorbed on the active site, and many studies analyze the electrochemical catalytic properties of HER based on the Volmer reaction.3 The Gibbs free energy of hydrogen adsorption ((ΔGH*)) as a significant factor of measuring the HER catalytic activity is calculated based on the Volmer reaction using the DFT method. Its definition is as follows35| | |
ΔGH* = ΔEH + ΔEZPE − TΔS
| (4) |
where ΔEZPE is the difference in zero-point energy between the adsorption state of H and gas phase, while ΔS is the difference in entropy between the adsorption state of H and gas phase.3 ΔEH is the hydrogen adsorption energy. Given that the vibrational entropy of H* in the adsorbed state is small, the entropy of adsorption of −1/2H2 is simplified as ΔSH ≈ −1/2SH20, where SH20 is the entropy of H2 in the gas phase at standard conditions. Therefore, the overall corrections of ΔGH* are3
The hydrogen adsorption energy (ΔEH) on the electrode surfaces is defined as
| |
 | (6) |
where
Esubs+H* is the total energy of doped CoS
2(001) with H* adsorption,
Esubs is the total energy of doped CoS
2(001) without H* adsorption, and
EH2 is the total energy for one hydrogen molecule in the gas phase.
2.5 d-band centers
To further explore the possibility of enhancing electrochemical performance of CoS2 via doping, we calculated the d-band center of the metal at active site.
The d-band center εd is defined as
| |
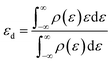 | (7) |
where
ρ are the densities of states and
ε are the energies.
3 Results and discussion
Some materials demonstrate good computational performance but are difficult to synthesize due to their aggregation- or clustering-related instability, which limits their application in experiments or in practice.37 So, it is important to first investigate the stability of CoS2 in order to explore the possibility of its synthesis. We use the binding energy (Eb) descriptor to measure the stability of doped CoS2, if, Eb < 0, the atoms shows more tendency of being incorporated into the CoS2(001) surface rather than clustering or aggregation, and then we regard that the material satisfies the stability criterion.34 The calculated binding energies are listed in Table 1. From Table 1 we can see, Eb of all cation and anion doped CoS2 except for C-doped CoS2 are less than zero, which demonstrates that it is energetically possible for one cation dopant to replace one Co atom in the CoS2(001) surface and for one anion dopant to replace one S atom in the CoS2(001) surface. However, the binding energy is 1.96 eV when one S atom in the CoS2(001) surface is substituted by one C atom, which is much higher than zero, indicating that when one C is incorporated into the CoS2(001), it tends to take the form of cluster or aggregation, which causes an unstable C-doped CoS2(001) structure. Therefore, C will not be considered as a heteroatom to form a HER catalyst.
Table 1 Binding energies of doped CoS2 (Eb, eV)
| Cation |
Model |
Mn |
Fe |
Ni |
Cu |
Mo |
Pt |
Ru |
| Eb/eV |
−2.89 |
−2.02 |
−1.67 |
−0.96 |
−1.78 |
−1.41 |
−1.97 |
| Anion |
Model |
C |
N |
O |
P |
| Eb/eV |
1.96 |
−1.67 |
−2.27 |
−1.52 |
After identifying the stability for a cation/anion heteroatom incorporated into the CoS2(001) surface, the state of the adsorbed H was discussed according to H adsorption energy. The negative value means that H is easy to bind to the CoS2(001).38–40 The value of adsorption energy for 11 doped-CoS2 are from −1.43 eV to −2.46 eV, which indicates that it is feasible to H bind to the surface of CoS2(001). Moreover, the catalytic performance for HER of the CoS2(001) with and without cation/anion heteroatom doping was investigated. To determine whether CoS2 is an outstanding HER catalyst, the Gibbs free energies (ΔGH*) for hydrogen adsorption at active metal sites were calculated based on the DFT method. Fig. 3(a) is the relaxed crystallographic structure of H adsorption at the Co site for the pure CoS2(001), in which the green ball refers to H atom. The Co–H bond length is 1.50 Å. The calculated |ΔGH*| of the pure CoS2(001) is 0.25 eV, as shown in Fig. 3(b), which is larger than that of the precious metal Pt (0.08 eV).3 In order to further probe the effect of heteroatom atoms on electrochemical HER catalytic performance for CoS2(001) as electrode material, the ΔGH* of cation- (Mn, Fe, Ni, Cu, Mo, Pt and Ru) and anion- (N, O and P) doped CoS2 are calculated using the DFT method. Fig. 4(a) and (c) show the calculated ΔGH* of H adsorption at the active metal sites for cation- and anion-doped CoS2(001), respectively. In Fig. 4(a) and (c), ΔGH* of Ni-doped CoS2(001) is −0.12 eV, that of Pt-doped CoS2(001) is 0.03 eV, that of N-doped CoS2(001) is 0.07 eV, and that of O-doped CoS2(001) is −0.01 eV. Compared with that (0.25 eV) of the pure CoS2 electrode, the |ΔGH*| of Ni-, Pt-, N-, and O-doped CoS2(001) electrode are much smaller when H is adsorbed at metal sites. It is noteworthy that |ΔGH*| of Pt-, N-, and O-doped CoS2(001) is smaller than that of Pt (0.08 eV), revealing their potential excellent electrochemical HER catalytic performance. And |ΔGH*| of O-doped CoS2(001) is closest to zero, which suggests that O-doped CoS2 show the best HER catalytic activity among these doped CoS2 material. Fig. 4(b) and (d) show the optimized structures of the Pt-doped CoS2(001) with H adsorption at Pt site, Pt–H bond length is 1.66 Å, and that of the O-doped CoS2(001) with H adsorption at Co site, Co–H bond length is 1.51 Å, respectively. To further explore why the electrochemical HER performance of CoS2 at active metal sites via doping can be enhanced, the d-band centers of the active metal sites were calculated using the DFT method. We plotted in Fig. 5 the density of states for active metal sites used to H adsorption and marked the d-band center at active metal site with a dashed line. The d-band center of Co active site for the pristine CoS2 is −2.06 eV and it is closest to the Fermi level. The d-band center of Cu active site for Cu-doped CoS2 is −6.01 eV and it is farthest to the Fermi level. Meanwhile, ΔGH* for Cu-doped CoS2 is 0.71 eV. To further probe the relationship between the d-band center of active metal site used to H adsorption and the Gibbs free energy of H adsorption on the active metal sites, a more detailed analysis diagram is plotted in Fig. 6. We can see from Fig. 6 that ΔGH* increases as the d-band centers of metal active sites decrease, which clearly illustrates that the binding between H adsorption atom and metal atom at the active site is weakened as the d band center is far away from the Fermi level. Our results just described are in line with investigations in the ΔGH* and d-band centers based on other materials.26 This study reveals an intrinsic mechanism that d-band center of active metal site moves downwards after doping, leading to the weakening of hydrogen adsorption, which facilitates the desorption of H from the surface of CoS2 as HER electrode material.
 |
| | Fig. 3 (a) Represents the H adsorption structure of the pristine CoS2, where H adsorbs at Co site. The green ball refers to H atom. (b) is ΔGH* diagram of the pristine CoS2. | |
 |
| | Fig. 4 (a) and (b) are ΔGH* diagram and the structure of Pt-doped CoS2(001) with H adsorption at Pt site, respectively. (c) and (d) are ΔGH* diagram and the structure of O-doped CoS2(001) with H adsorption at Co site, respectively. The blue, dark red, dark green, red balls and light green ball are Co, S, Pt, O and H atoms, respectively. Pt–H bond length is 1.66 Å and Co–O bond length is 2.21 Å. | |
 |
| | Fig. 5 The density of states at active metal sites. The dashed line marked the d-band center. The Fermi energy is set to zero. | |
 |
| | Fig. 6 The linear relationship between ΔGH* and d-band centers of H adsorption sites. | |
Based on our DFT calculations, we can find that Ni-, Pt-, O-, and N-doping can intrinsically promote the electrochemical HER catalytic performance of CoS2 and a line in ΔGH* and d-band centers of H adsorption sites is achieved. The Pt-, O-, and N-doped CoS2 show overpotentials of 0.03 V, 0.01 V, and 0.07 V, respectively, which are very close to values when using Pt, which is believed to be the best catalyst for the HER because of its small overpotential (about 0.08 V).3 Therefore, the CoS2, after doping by Pt, O, and N becomes a promising catalyst.
4 Conclusions
In summary, we studied HER catalytic activities of cation/anion-doped CoS2 as HER catalyst based on the DFT method. Our results reveal that the Pt-, N- and O-doped CoS2 possesses an outstanding HER catalytic activity comparable to Pt. And a negative linear relationship was identified between ΔGH* and d-band centers of active metal sites. This exhibits that d-band center of active metal site moves downwards after cation/anion doping, leading to the weakening of hydrogen adsorption and facilitating the H desorption from the surface of CoS2. Therefore, the intrinsic HER catalytic performance of CoS2 can be improved by tailoring the d-band center of the active metal site. Based on this study, we can further explore the relationship between the structure and activity of CoS2, which may shed light on the exploration of the potential use of other TMSs as HER electrode materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Fund Project of Chengdu Technological University under Grant No. 2020RC004.
References
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- B. Qiu, Y. Zhang, X. Guo, Y. Ma, M. Du, J. Fan, Y. Zhu, Z. Zeng and Y. Chai, J. Mater. Chem. A, 2022, 10, 719–725 RSC.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, ChemInform, 2005, 152, L5 Search PubMed.
- D. Saha, V. Patel, P. R. Selvaganapathy and P. Kruse, Nanoscale Adv., 2022, 4, 125–137 RSC.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- R. Li, J. Liang, T. Li, L. Yue, Q. Liu, Y. Luo, M. S. Hamdy, Y. Sun and X. Sun, Chem. Commun., 2022, 15, 633–644 Search PubMed.
- J. Tian, X. Xing, Y. Sun, X. Zhang, Z. Li, M. Yang and G. Zhang, Chem. Commun., 2022, 58, 557–560 RSC.
- S. Mansingh, K. K. Das and K. Parida, Sustainable Energy Fuels, 2021, 5, 1952–1987 RSC.
- H. Wang, X. Xiao, S. Liu, C.-L. Chiang, X. Kuai, C.-K. Peng, Y.-C. Lin, X. Meng, J. Zhao, J. Choi, Y.-G. Lin, J.-M. Lee and L. Gao, J. Am. Chem. Soc., 2019, 141, 18578–18584 CrossRef CAS PubMed.
- J. Lee, J. Heo, H. Y. Lim, J. Seo, Y. Kim, J. Kim, U. Kim, Y. Choi, S. H. Kim, Y. J. Yoon, T. J. Shin, J. Kang, S. K. Kwak, J. Y. Kim and H. Park, ACS Nano, 2020, 14, 17114–17124 CrossRef CAS PubMed.
- Y. Qu, H. Pan, C. Tat Kwok and Z. Wang, Phys. Chem. Chem. Phys., 2015, 17, 24820–24825 RSC.
- Y. Lin, M. Yu, X. Li, W. Gao, L. Wang, X. Zhao, M. Zhou, X. Yao, M. He and X. Zhang, J. Mater. Chem. C, 2021, 9, 15877–15885 RSC.
- J. Xu, Y. Zhu, B. Yu, C. Fang and J. Zhang, Inorg. Chem. Front., 2019, 6, 3510–3517 RSC.
- Z. Wang, K. Yu, R. Huang and Z. Zhu, CrystEngComm, 2021, 23, 5097–5105 RSC.
- N. Ran, B. Sun, W. Qiu, E. Song, T. Chen and J. Liu, J. Phys. Chem. Lett., 2021, 12, 2102–2111 CrossRef CAS PubMed.
- G. Chen, H. Li, Y. Zhou, C. Cai, K. Liu, J. Hu, H. Li, J. Fu and M. Liu, Nanoscale, 2021, 13, 13604–13609 RSC.
- Y. Dong, H. Sun and G. Liu, Sustainable Energy Fuels, 2021, 5, 4115–4125 RSC.
- Z. Shi, X. Qi, Z. Zhang, Y. Song, J. Zhang, C. Guo, W. Xu, K. Liu and Z. Zhu, Nanoscale, 2021, 13, 6890–6901 RSC.
- Y. Jiang, S. Gao, J. Liu, G. Xu, Q. Jia, F. Chen and X. Song, Nanoscale, 2020, 12, 11573–11581 RSC.
- Y. Zang, B. Yang, A. Li, C. Liao, G. Chen, M. Liu, X. Liu, R. Ma and N. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 41573–41583 CrossRef CAS PubMed.
- C. n. Karakaya, N. Solati, U. Savacı, E. Keleş, S. Turan, S. Çelebi and S. Kaya, ACS Catal., 2020, 10, 15114–15122 CrossRef CAS.
- C. Karakaya, N. Solati, U. Savacı, E. Keleş, S. Turan, S. Çelebi and S. Kaya, ACS Catal., 2020, 10, 15114–15122 CrossRef CAS.
- J. Zhang, Y. Liu, B. Xia, C. Sun, Y. Liu, P. Liu and D. Gao, Electrochim. Acta, 2018, 259, 955–961 CrossRef CAS.
- G. Papoian, J. K. Nørskov and R. Hoffmann, J. Am. Chem. Soc., 2000, 122, 4129–4144 CrossRef CAS.
- A. Vojvodic, A. Hellman, C. Ruberto and B. I. Lundqvist, Phys. Rev. Lett., 2009, 103, 146103 CrossRef CAS PubMed.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS PubMed.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, J. Phys. Chem. Lett., 2014, 5, 3884–3889 CrossRef CAS PubMed.
- A. Maibam, S. Krishnamurty and M. Ahmad Dar, Mater. Adv., 2022, 3, 592–598 RSC.
- A. Maibam, S. K. Das, P. P. Samal and S. Krishnamurty, RSC Adv., 2021, 11, 13348–13358 RSC.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, C. Sun, P. Xi, S. Peng, D. Gao and D. Xue, ACS Energy Lett., 2018, 3, 779–786 CrossRef CAS.
- J. Shi, Z. Wang and Y. Q. Fu, J. Phys. D: Appl. Phys., 2016, 49, 505601 CrossRef.
- W. I. Choi, B. C. Wood, E. Schwegler and T. Ogitsu, Adv. Energy Mater., 2015, 5, 1501423 CrossRef.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- D. Kim, J. Shi and Y. Liu, J. Am. Chem. Soc., 2018, 140, 9127–9131 CrossRef CAS PubMed.
- Q. Sun, Q. Wang, P. Jena and Y. Kawazoe, J. Am. Chem. Soc., 2005, 127, 14582–14583 CrossRef CAS PubMed.
- Y. Sun, A. Huang and Z. Wang, RSC Adv., 2019, 9, 26321–26326 RSC.
- G. Feng, M. V. Ganduglia-Pirovano, C.-F. Huo and J. Sauer, J. Phys. Chem. C, 2018, 122, 18445–18455 CrossRef CAS.
- A. Rasool, I. Anis, M. Dixit, A. Maibam, A. Hassan, S. Krishnamurty and M. A. Dar, Catal. Sci. Technol., 2022, 12, 310–319 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Tao Chen*a and
Xiaoli Sun*bc
a,
Tao Chen*a and
Xiaoli Sun*bc