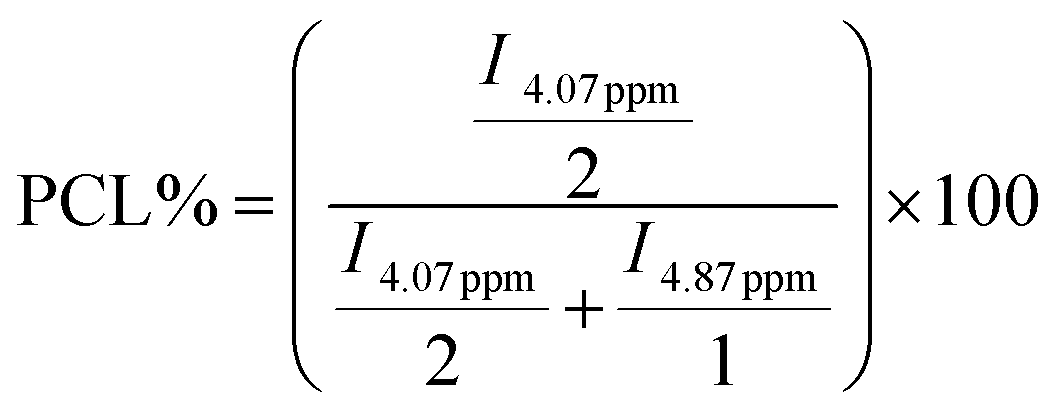DOI:
10.1039/D2RA01861F
(Paper)
RSC Adv., 2022,
12, 18154-18163
Synthesis of copolyesters based on substituted and non-substituted lactones towards the control of their crystallinity and their potential effect on hydrolytic degradation in the design of soft medical devices†
Received
22nd March 2022
, Accepted 3rd June 2022
First published on 21st June 2022
Abstract
A series of copolymers based on ε-caprolactone (ε-CL) in combination with lactone monomers substituted with alkyl groups (4 and 6 carbon atoms), specifically δ-decalactone (δ-DL), ε-decalactone (ε-DL) and δ-dodecalactone (δ-DD), as well as a copolymer using two substituted lactone monomers with alkyl groups (ε-DL and δ-DD) were synthesized in different molar ratios. The objective of the synthesis of these copolymers was to evaluate the effects of branching in the polymer backbone on the crystallinity and the thermal properties of the synthesized materials. All copolymers were obtained via ring-opening polymerization with high conversion values for both comonomers using neodymium isopropoxide (Nd(i-Pr)3) as the initiator, and their compositions were determined by 1H NMR and 13C NMR. The molar masses (Mn and Mw) and distributions were obtained by GPC measurements. Such measurements showed that a majority of the copolymers exhibited dispersities (Ɖ) in the range of 1.2–1.6 and Mn in the range of 15–40 kDa. First- and second-order transitions such as melting, crystallization and glass transition, as well as the crystallization degree (melting enthalpy), were determined by DSC analysis. Copolymers based on ε-CL developed interesting behaviors, wherein the copolymers with higher percentages of this monomer exhibited semicrystalline behavior, while the copolymers with a higher percentage of the comonomers ε-DL, δ-DL or δ-DD showed amorphous behavior. In contrast, the copolymers synthesized using both monomers from the alkyl group-substituted lactone developed fully amorphous features, regardless of their composition. These changes in the crystalline features of the synthesized copolymers suggest that the content of short branchings on the copolymer backbone will significantly modify their rates of hydrolytic degradation and their potential use in the development of different soft medical devices.
1. Introduction
In the last two decades, aliphatic polyesters have been widely studied because they exhibit notable biodegradable and biocompatible features.1–5 Therefore, they tend to be used in biomedical applications such as tissue engineering, fracture supports in bones (screws, staples, nails), and surgical sutures.6–9 In addition, polyesters based on branched lactone monomers like the monomers (δ-DL, ε-DL and δ-DD) proposed in this work have applications in drug delivery systems, soft electrically conducting composites and thermoplastic elastomers, among others.10–12 This kind of material can be prepared by two principal methods: (a) the condensation polymerization of bifunctional (or multi-functional) monomers, with the main difficulty being producing low molar mass and broad-dispersity polyesters, and (b) the ring-opening polymerization (ROP) of cyclic lactones, which affords polyesters with higher molar mass values, narrower dispersities and stereochemical control.13–16 Moreover, the ROP methodology can be applied across a broad range of cyclic monomers, such as lactones with or without alkyl side groups, lactide, glycolide and their derivatives.3,17–21
Polycaprolactone (PCL) possesses interesting performance in biomedical applications due to its good balance of mechanical properties, high strain values, processability, biodegradability and biocompatibility. Nevertheless, PCL requires long periods of time to achieve complete degradation (2–3 years) when it has a high crystallinity degree.22–24 In this regard, Tsira-ard et al. reported the synthesis of copolymers based on ε-CL with other cyclic monomers, namely (L-LA) and glycolide (GL) (Mn = 100 kDa, Ɖ = 2.17), wherein the thermal and mechanical properties observed in PCL and the hydrolytic degradation performance observed in poly-L-lactide (PLLA) and polyglycolide (PGA) were combined as a result of the random sequence of monomers with the composition of L-LA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ε-CL:GL = 74:15:1.25 In another related work, Loriot et al.26 reported the synthesis, characterization and hydrolytic degradation of PCL plus δ-valerolactone random copolymers in bulk at 140 °C, employing tin(II) octoate as the catalyst and studying the molecular weight (from 6200 to 22500 g mol−1) as the main variable. In this work, a correlation between the molecular weight and the hydrolysis rate was demonstrated by HPLC-MS and the reported copolymers could be promising for application as controlled release films. In addition, control of the crystallization degree is another interesting strategy for setting up the degradation rate in PCL; control can be achieved by incorporating comonomers with short side chain alkyl groups into the PCL backbone, which could have an impact on the amorphous/crystalline features of the resulting copolymers.
:ε-CL:GL = 74:15:1.25 In another related work, Loriot et al.26 reported the synthesis, characterization and hydrolytic degradation of PCL plus δ-valerolactone random copolymers in bulk at 140 °C, employing tin(II) octoate as the catalyst and studying the molecular weight (from 6200 to 22500 g mol−1) as the main variable. In this work, a correlation between the molecular weight and the hydrolysis rate was demonstrated by HPLC-MS and the reported copolymers could be promising for application as controlled release films. In addition, control of the crystallization degree is another interesting strategy for setting up the degradation rate in PCL; control can be achieved by incorporating comonomers with short side chain alkyl groups into the PCL backbone, which could have an impact on the amorphous/crystalline features of the resulting copolymers.
A few works related to the development of copolymers involving ε-CL with alkyl side chain cyclic esters have been published, as well as corresponding studies on the influence of their chemical structure on their morphological parameters and thermal and mechanical properties. For instance, Albertson et al.27 synthesized the copolymers poly(ε-caprolactone-co-ε-decalactone-co-ε-caprolactone) and poly(L-lactide-co-ε-decalactone-co-L-lactide) with different molar ratios of the comonomers to demonstrate that it is possible to determine the degradation path through the heterogeneity control of the amorphous phase, and this feature was the result of secondary interactions between their blocks. The control of the amorphous phase was completely dependent on the copolymer composition, with the most amorphous copolymers being those rich in ε-decalactone. Orozco-Castellanos et al.28 reported the synthesis of biodegradable esters, specifically poly(ε-caprolactone) and poly(ε-caprolactone-co-γ-butyrolactone) (PCB) with Mn ∼ 4–5.5 kDa and Ɖ ∼ 1.2–1.4. They studied the hydrolytic degradation features and the hydrocortisone loading/delivery in these polymers, wherein the best degradation performance was observed in the copolymer PCB, which showed a higher degradation rate of 1.72% day−1 in comparison to the 1.43% day−1 observed in the PCL homopolymer. A similar study was reported by Li et al.29 using a combination of ε-CL and β-butyrolactone (β-BL) as comonomers in different molar ratios of εCL:β-BL from 100:0 (Mn = 29.5 kDa, Ɖ = 1.8) to 60:40 (Mn = 15 kDa, Ɖ = 1.4), wherein the enzymatic degradation rates were evaluated in Pseudomonas lipase and the best weight loss performance was observed in the copolymer with the highest percentage of β-BL due to its methyl side group favoring the decoupling of the polyester main chains, causing more hole sites where water permeation could occur. Zhao et al.30 developed block copolymers by the sequential addition polymerization of ω-pentadecalactone (PDL) as the first block, and L-lactide (LLA), ε-CL or δ-valerolactone (VL) as the second blocks, employing an organic catalytic tandem system of cyclic trimeric phosphazene base (CTPB) and benzyl alcohol (BnOH), reaching molar mass values of Mn = 7.1–44.1 kDa and a dispersity of Ɖ = 1.74–2.21. It is worth mentioning that less basic conditions of CTPB/BnOH allowed the achievement of the block conformation in the copolymers of PDL-CL and PDL-VL with their catalytic activity measured using the turnover frequency (TOF) of up to 600 h−1. Another work that involved ROP for the synthesis of copolymers based on PDL and lactones with different ring sizes (γ-butyrolactone, δ-valerolactone and ε-caprolactone) was reported by Walther and Naumann,31 wherein N-heterocyclic olefins and a Lewis acid (MgCl2 or LiCl) were used as a catalyst system to obtain copolymers with high conversion (85–97%), molar masses up 40 kDa and dispersities of 1.5–1.8; ROP reactions were also achieved in a short time (∼15 min). It should be noted that the reduction of transesterification is a consequence of the convenient modulation of polymerization favored by the presence of a Lewis acid cocatalyst. Xu et al.32 reported the preparation of block copolymer based on ε-CL and a “functional ε-CL” named γ-(carbamic acid benzyl ester)-ε-caprolactone in order to alter the degradation and mechanical properties of the copolymers. In this work, it was demonstrated that the content and the distribution of pendant side chains in the resulting copolymers can adjust the nucleation of macromolecular chains to modulate the lamellar thickness and crystallinity in PCL/block copolymer films. It was concluded that such variations of macromolecular structures result in distinct changes in the mechanical properties during degradation.
Likewise, Schneiderman et al.33 synthesized telechelic statistical copolymers based on ε-CL and ε-decalactone (ε-DL) in different molar ratios (ε-CL = 0.66–1.0), wherein the reactivity ratios (rCL = 5.9; rDL = 0.03) firstly favored the polymerization of ε-CL followed by that of ε-DL. Thereafter, these copolymers underwent chain extension by the addition of lactide to the reacting system to obtain a triblock arrangement of poly(lactide)-b-poly(ε-caprolactone-co-ε-decalactone)-b-poly(lactide). Herein, an increase in ε-DL content favored reductions in the crystallization degree (33–42%) and the melting temperature (49–54 °C) as well as changes in the mechanical properties such as the Young's modulus (E = 0.45–31 MPa), stress (σ = 0.6–2.9 MPa) and strain (ε = 218–2100%).
Following these works, we have realized the synthesis of polyesters with different monomer compositions and changes in their structural features. We look forward to contributing to the development of a series of copolymers that can exhibit diversity in their chemical, thermal, and mechanical properties supported by variations in the molar ratios of monomers such as ε-caprolactone, δ-decalactone, ε-decalactone and δ-dodecalactone to obtain polyesters via ROP using neodymium isopropoxide as an organometallic catalyst. The alkyl-substituted comonomers may enhance perturbations in lateral interactions between carboxylic functional groups on the polyester backbone and thus reach a balance in the melting and crystallization temperatures, the glass transition and the crystallization degree. Therefore, these screenings in different polyester formulations can help us to understand their structure–property performance for application in biodegradable medical devices with acceptable degradation rates, which are a function of the crystallization degree studied in this work.
2. Methodology
2.1 Reagents
Monomers such as δ-decalactone (Ventos), ε-caprolactone, ε-decalactone and δ-dodecalactone (Sigma-Aldrich) were purified using a distillation system that includes Na0 as a drying agent, a vacuum pressure of 15 mm Hg and 160 °C in order to achieve the complete removal of humidity and oxygen. Neodymium isopropoxide (Nd(i-Pr)3) catalyst (Sigma-Aldrich) was used as received and was handled under glovebox conditions. Toluene and methanol solvents were provided by J. T. Baker. Toluene was used as a solvent in the polymer isolation procedure, and it was washed with H2SO4, dried in CaCl2, stirred under reflux conditions with LiAlH4 and distilled using a sodium/benzophenone complex, while methanol was used as received.
2.2 Characterization and equipment
The chemical structures and compositions of the copolymers were determined by proton and carbon nuclear magnetic resonance (1H NMR, 13C NMR) at room temperature using a Bruker Avance III 400 MHz spectrometer and the solvent employed was deuterated chloroform (CDCl3). Additionally, characteristic functional groups in copolymers were confirmed through Fourier transform infrared spectroscopy (FTIR) (attenuated total reflectance (ATR) mode), recorded from 4000 to 400 cm−1 (25 scans) using an FTIR spectrometer from Thermo-Scientific (Nicolet iS5). Gel permeation chromatography was used in the determination of the molar mass and dispersity of the copolymers, employing an Agilent PL-GPC50 instrument calibrated with polystyrene standards, THF (HPLC grade) as the eluent, a diffraction index detector, a 5 μm column type C and a pressure of 2.34 MPa.
The thermal stability in the copolymers was tested using a thermogravimetric analyzer from TA Instruments (Q500), wherein the heating rate was 10 °C min−1 from 30 to 600 °C under a N2 atmosphere. The thermal transitions (Tg, Tc and Tm) and crystallization percentage were obtained using a differential scanning calorimeter from TA Instruments (DSC-2500) at heating and cooling rates of 10 °C min−1 from −70 to 100 °C under a N2 atmosphere.
2.3 Polyester copolymer synthesis
Polyesters were synthesized via organocatalyzed ROP employing combinations of cyclic esters (Scheme 1) under bulk reaction conditions using 30 mL glass vials with magnetic stirrers and sealed with rubber caps. These vessels were previously exposed to 3 argon/vacuum cycles at 85 °C to achieve the total removal of oxygen and humidity, taking into consideration that the initiator (Nd(i-Pr)3) is liable to deactivation in the presence of these reagents. Thereafter, monomers (ε-CL, δ-DL, ε-DL, δ-DD) were loaded in the corresponding molar ratio for each copolymer in series I–IV, as shown in Table 1; the loading of raw materials was done using syringes with stainless steel gauges and the reaction mixture was stirred until it reached the target temperature (4 °C). Afterwards, 1 mL Nd(i-Pr)3 (3.55 mM) in toluene was added dropwise, and the reaction was kept under stirring at 4 °C for 168 hours. It is worth mentioning that the molar ratio between the monomer and initiator (I) (M:I ratio) was fixed at 150:1 to obtain copolymers with a targeted number average molar mass (Mn) of 17–30 kDa. Then, the crude product was dissolved in 50 mL toluene and precipitated in 500 mL cold methanol (repeated twice), filtered, and dried under vacuum at 200 mm Hg and 50 °C.
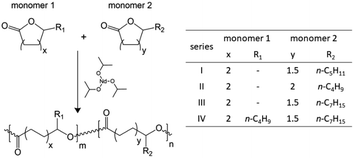 |
| | Scheme 1 Synthetic route of the polyester series (I–IV) by the ROP methodology. | |
Table 1 Nomenclature and molar ratios in the synthesis of the polyester copolymer series (I–IV)
| Series I |
ε-CL |
δ-DL |
Series II |
ε-CL |
ε-DL |
| Molar percentage. |
| I-1 |
100a |
0 |
II-1 |
100 |
0 |
| I-2 |
80 |
20 |
II-2 |
80 |
20 |
| I-3 |
60 |
40 |
II-3 |
60 |
40 |
| I-4 |
50 |
50 |
II-4 |
50 |
50 |
| I-5 |
40 |
60 |
II-5 |
40 |
60 |
| I-6 |
20 |
80 |
II-6 |
20 |
80 |
| I-7 |
0 |
100 |
II-7 |
0 |
100 |
| Series III |
ε-CL |
δ-DD |
Series IV |
ε-DL |
δ-DD |
| III-1 |
100 |
0 |
IV-1 |
100 |
0 |
| III-2 |
80 |
20 |
IV-2 |
80 |
20 |
| III-3 |
60 |
40 |
IV-3 |
60 |
40 |
| III-4 |
50 |
50 |
IV-4 |
50 |
50 |
| III-5 |
40 |
60 |
IV-5 |
40 |
60 |
| III-6 |
20 |
80 |
IV-6 |
20 |
80 |
| III-7 |
0 |
100 |
IV-7 |
0 |
100 |
3. Results and discussion
3.1 Polyester copolymer synthesis
The polyester series I–IV were synthesized via ROP using Nd(i-Pr)3 as a catalyst and were all analyzed to confirm their chemical structure and to corroborate the absence of monomer residues by 1H NMR (ESI, Fig. S1†), and the determination of conversion in the copolymers and their monomer fractions were conducted by 1H and 13C NMR. For instance, Fig. 1 shows five spectra corresponding to the ε-CL and ε-DL monomers as well as the two homopolymers (II-1, II-7) and the copolymer 50:50 (II-4), taking the molar percentage as a reference. On analyzing the spectrum of II-4, the characteristic signal of CH2 protons in the position α to the O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group, which is located at 4.07 ppm in the copolymer (signal a′), shifted to the low-frequency region in comparison to the signal a in the cyclic monomer ε-CL (4.15 ppm). In contrast, the equivalent CH proton in the position α to the O–CO group in II-7 is positioned at 4.87 ppm (signal c′) in contrast to the methine proton in the precursor (4.12 ppm, signal c). These changes in chemical shifts in the protons are due to differences in their chemical environments, mainly affecting the side alkyl group attached to the methine proton in ε-DL.34 It should be mentioned that in the polyester spectra (II-1, II-4 and II-7) shown in Fig. 1, the characteristic signals of the monomers ε-CL and ε-DL are not observed; therefore, the series of copolymers in this work satisfy the purity requirements for subsequent analysis.
O group, which is located at 4.07 ppm in the copolymer (signal a′), shifted to the low-frequency region in comparison to the signal a in the cyclic monomer ε-CL (4.15 ppm). In contrast, the equivalent CH proton in the position α to the O–CO group in II-7 is positioned at 4.87 ppm (signal c′) in contrast to the methine proton in the precursor (4.12 ppm, signal c). These changes in chemical shifts in the protons are due to differences in their chemical environments, mainly affecting the side alkyl group attached to the methine proton in ε-DL.34 It should be mentioned that in the polyester spectra (II-1, II-4 and II-7) shown in Fig. 1, the characteristic signals of the monomers ε-CL and ε-DL are not observed; therefore, the series of copolymers in this work satisfy the purity requirements for subsequent analysis.
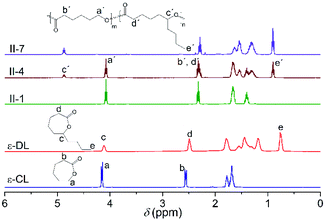 |
| | Fig. 1 1H NMR spectra of the monomers and three polyesters corresponding to series II. | |
Additional characterization concerning the chemical structure was done through FTIR, as shown in Fig. 2, wherein the FTIR spectra of the same polyesters discussed in Fig. 1 are presented. Herein, the polyesters possess two characteristic absorption bands related to CO and C–O bonds in the carboxylate group. It should be noted that homopolymer II-1 exhibits a CO signal at 1722 cm−1 located in the lower wavelength position with respect to the ε-DL derivatives (II-4 and II-7), which present a CO absorption band at 1730 cm−1. This wavenumber shift is known as the bathochromic effect, which is attributed to the presence of alkyl side chains on the II-7 backbone segment in these copolymers.35 In the case of the C–O absorption band, this is located at 1240 cm−1 with a lower intensity in the branched polyesters (II-4 and II-7). Furthermore, the absorption band that corresponds to C–C(O)–O is observed at 1185 cm−1 (II-1) and 1165 cm−1 (II-4 and II-7), and these changes in the wavenumber and shape of the peaks are supported by changes in the chemical structure, specifically the presence of alkyl side groups from the ε-DL comonomer. Additionally, an increase in the intensity of the absorption band at ∼2930 cm−1 in II-4 and II-7 was caused by the higher percentage of alkyl lateral groups present in the branched comonomer ε-DL.
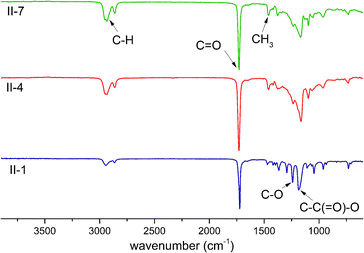 |
| | Fig. 2 FTIR spectra in the ATR mode of three polyesters corresponding to series II. | |
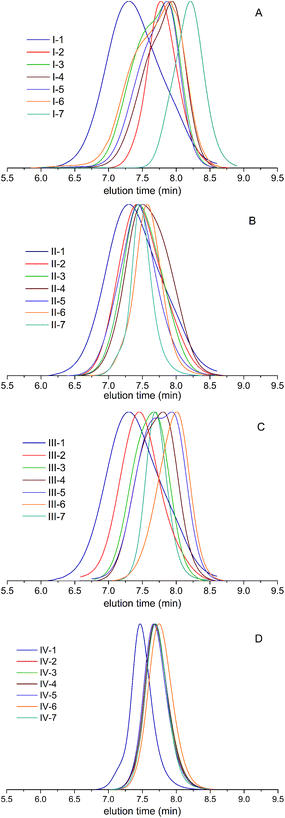
1H and 13C NMR analyses have been used to determine the real comonomer molar percentage in each copolymer (Tables 2–5). This determination comes from the 1H NMR integration values (I) of chemical shifts at 4.07 ppm, related to the O–CH2 protons in PCL, and at 4.87 ppm, associated with the O–CH protons in the δ-DL, ε-DL, and δ-DD copolymer derivatives; the corresponding calculations were done using eqn (1) and (2). In the case of series IV, the similarity of branched monomers caused the O–CH protons in the 1H NMR spectra to be observed as a single broad signal, making the integration assignment impossible for them. Therefore, the 13C NMR integration values of the chemical shifts of CO at 173.28 ppm, related to the vicinal carbonyl group CH proton in ε-DL, and 173.09 ppm, associated with CO in the δ-DD derivatives, were used to obtain the comonomer percentage according to eqn (3) and (4).
| |
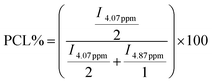 | (1) |
| |
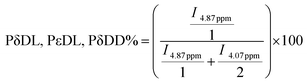 | (2) |
| |
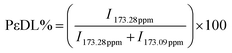 | (3) |
| |
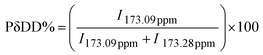 | (4) |
Table 2 Comonomer molar percentages, Mn, Mw and Ɖ in series I
| Series I |
ε-CL (%) |
δ-DL (%) |
Mn (kDa) |
Mw (kDa) |
Ɖ |
| I-1 |
100 |
0 |
37.70 |
84.16 |
2.23 |
| I-2 |
98.49 |
1.51 |
20.05 |
26.31 |
1.31 |
| I-3 |
97.62 |
2.38 |
24.49 |
38.98 |
1.59 |
| I-4 |
97.39 |
2.61 |
19.62 |
31.60 |
1.61 |
| I-5 |
89.10 |
10.90 |
23.55 |
37.89 |
1.61 |
| I-6 |
47.78 |
52.22 |
22.30 |
45.48 |
2.04 |
| I-7 |
0 |
100 |
9.01 |
11.36 |
1.26 |
Table 3 Comonomer molar percentages, Mn, Mw and Ɖ in series II
| Series II |
ε-CL (%) |
ε-DL (%) |
Mn (kDa) |
Mw (kDa) |
Ɖ |
| II-1 |
100 |
0 |
37.70 |
84.16 |
2.23 |
| II-2 |
79.57 |
20.43 |
36.19 |
58.63 |
1.62 |
| II-3 |
63.90 |
36.10 |
36.73 |
54.47 |
1.48 |
| II-4 |
62.12 |
37.88 |
29.34 |
46.96 |
1.60 |
| II-5 |
43.34 |
56.63 |
42.34 |
63.98 |
1.51 |
| II-6 |
30.80 |
69.20 |
34.37 |
43.41 |
1.26 |
| II-7 |
0 |
100 |
43.31 |
51.46 |
1.19 |
Table 4 Comonomer molar percentages, Mn, Mw and Ɖ in series III
| Series III |
ε-CL (%) |
δ-DD (%) |
Mn (kDa) |
Mw (kDa) |
Ɖ |
| III-1 |
100 |
0 |
37.70 |
84.16 |
2.23 |
| III-2 |
92.24 |
7.76 |
37.42 |
59.92 |
1.60 |
| III-3 |
90.95 |
9.05 |
31.37 |
43.18 |
1.38 |
| III-4 |
89.78 |
10.22 |
24.86 |
35.55 |
1.43 |
| III-5 |
89.97 |
10.03 |
21.32 |
33.43 |
1.57 |
| III-6 |
68.90 |
31.10 |
15.56 |
20.35 |
1.31 |
| III-7 |
0 |
100 |
26.87 |
31.12 |
1.16 |
Table 5 Comonomer molar percentages, Mn, Mw and Ɖ in series IV
| Series IV |
ε-DL (%) |
δ-DD (%) |
Mn (kDa) |
Mw (kDa) |
Ɖ |
| IV-1 |
100 |
0 |
43.31 |
51.46 |
1.19 |
| IV-2 |
83.25 |
16.75 |
26.47 |
31.15 |
1.18 |
| IV-3 |
54.75 |
45.25 |
26.63 |
31.50 |
1.18 |
| IV-4 |
44.44 |
55.56 |
26.21 |
31.20 |
1.19 |
| IV-5 |
33.33 |
66.67 |
27.34 |
32.35 |
1.18 |
| IV-6 |
10.71 |
89.29 |
23.42 |
27.81 |
1.19 |
| IV-7 |
0 |
100 |
26.87 |
31.12 |
1.16 |
Fig. 3A–D show the calculated percentages of the comonomers in series I–IV, where it can be observed that the contribution of ε-CL in the first three series is higher than that of the other alkyl-substituted comonomers due to ε-CL being more reactive than the alkyl-substituted comonomers, and so the PCL blocks have higher percentages than the various established formulations.33 Regarding series IV, the constitutive branched monomers seem to possess similar reactivity, where the contribution of each comonomer is close to the theoretical feed in each copolymer.
 |
| | Fig. 3 Percentages of comonomers in the copolymers of series I–IV. | |
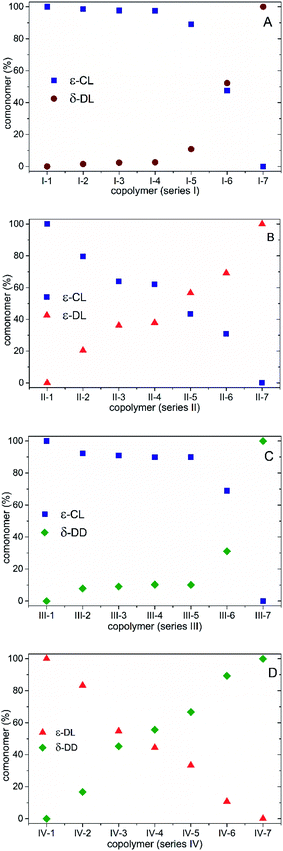
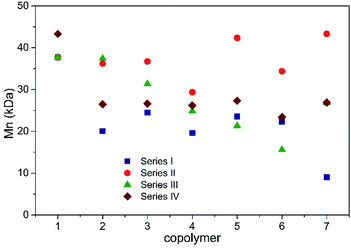
The molar mass values and their corresponding dispersities were recorded through gel permeation chromatography (GPC) analysis for the polyesters synthesized in the four series, and their Mn values ranged from 9 to 43 kDa, as shown in Fig. 4, and their Ɖ values ranged from 1.16 to 2.23 (Tables 2–5). It is important to mention that these Mn and Ɖ values may be overestimated as a result of using polystyrene standards to determine them.36 With regards to the chromatograms for series I–IV shown in Fig. 5, the shorter elution time observed for the PCL homopolymer denotes the highest Mn values for I-1, II-1 and III-1 as compared to the copolymers with a lower presence of the ε-CL comonomer and the branched homopolymers (I-7, II-7 and III-7). Likewise, the PCL homopolymer presented the broadest molar mass dispersion (Ɖ = 2.23), and this behavior could be related to the highest reactivity of ε-CL as compared to the corresponding alkyl side chain lactones (δ-DL, ε-DL and δ-DD). Furthermore, a previous report focused on the synthesis of copolymers based on ε-CL and ε-DL, wherein the determination of the reactivity ratio of these two monomers provides information for determining the existence of the block arrangement and the propagation rate was favored in the non-alkyl-substituted lactone (ε-CL).33 In the case of the copolymers based on two alkyl side chain lactones, ε-DL and δ-DD (Fig. 5D), a narrowed and monomodal molar mass distribution was observed, with propagation favored in IV-1, which corresponds to the ε-DL homopolymer. Additionally, series IV has a low conversion performance (ESI, Fig. S2†), wherein the side chain groups may limit the propagation rates and influence the low molar mass values. Likewise, the reactivity ratios in series I (ε-CL–δ-DL), II (ε-CL–ε-DL), III (ε-CL–δ-DD) and IV (ε-DL–δ-DD) were calculated, taking into consideration the proposed reaction conditions. It is worth mentioning that a detailed screening of the chemical arrangement in these copolymers will contribute to the understanding of subsequent hydrolytic degradation tests.
 |
| | Fig. 4 Average number molar mass (Mn) in the polyesters from series I–IV. | |
 |
| | Fig. 5 GPC analysis in the copolymer series I–IV. | |
3.2 Thermal and crystallization degree analysis
The copolymer series I–IV were analyzed by TGA in order to determine their thermal stability. Copolymers I-4, II-4, III-4 and IV-4 were taken as representative samples because they possess similar theoretical molar ratios of around 50:50, and the main idea of these analyses was to gain a general overview of the thermal degradation performance of the polyesters, taking into consideration a subsequent study of the thermal transitions through DSC characterization. The TGA analyses were conducted, taking a weight loss of 5% as a reference, reaching values from 165 to 266 °C, as shown in Fig. 6.
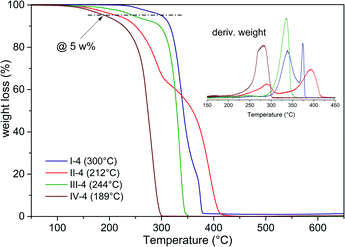 |
| | Fig. 6 Weight loss in the copolymers via TGA. | |
Once we confirmed that there was no thermal degradation in any copolymers at ∼100 °C, the range of temperatures for DSC characterization was established by heating and cooling cycles from −70 to 100 °C. Variations in Tc, Tm and Tg values were observed (Tables 6–9), as well as in the magnitude and shape of the endotherms and exotherms. It should be mentioned that the values of Tc and Tm were taken at the peak maximum for each transition. Therefore, the thermal performance in the copolymers was dependent on changes in the molar ratios and the chemical nature of the monomers. For instance, in the thermograms of series I (ε-CL and δ-DL) it was observed that the major contribution of ε-CL (I-1, I-2) leads to endotherms at higher temperature values with monomodal transition curves, as shown in Fig. 7A and A′. In the case of I-3 to I-6, endotherms with two or more peaks were developed, which means that several polymeric populations coexist in a wide temperature range, but in the cooling process, this behavior was observed only in I-5 and I-6 (higher content of δ-DL). Likewise, I-7 only developed a glass transition, meaning that this comonomer merely contributes to amorphous domains. In series II (ε-CL and ε-DL) (Fig. 7B and B′), a decreasing tendency in Tm and ΔHf was observed, while the percentage of ε-DL was augmented, as shown in Fig. 7B, and this kind of performance was previously reported by Fernandez et al. during the synthesis of copolymers based on ω-pentadecalactone and ε-DL.37 Copolymers II-2 to II-6 exhibited two transitions in the heating process (Tg and Tm); besides, there were broad endotherms and exotherms with two peaks that may be related to the coexistence of different crystalline/amorphous arrangements, suggesting the presence of a wide range of polymeric chain sizes consistent with the broad molar mass dispersity (Ɖ) determined by GPC analysis. In series III, behavior like that shown in series I was observed, wherein an increase in δ-DD tends to decrease the enthalpy of fusion (ΔHf) as well the Tm and Tc values (Table 8). Regarding the copolymers synthesized from ε-DL and δ-DD, those in series IV only developed glass transitions in all cases without evidence of first-order transitions such as melting (Fig. 7D) or crystallization (Fig. 7D′), as shown by Stamm et al. who performed the synthesis of polyesters based on ε-DL.38 Therefore, all copolymers (IV-1 to IV-7) exhibited an amorphous arrangement with Tg in a narrow temperature range.
Table 6 Thermal properties and crystallization degrees in series I
| Series I |
Tm (°C) |
Tc (°C) |
Tg (°C) |
ΔHf (J g−1) |
Xca (%) |
| Crystallization degree. |
| I-1 |
57.2 |
34.8 |
— |
80.5 |
58.2 |
| I-2 |
54.9 |
34.2 |
— |
65.8 |
49.5 |
| I-3 |
50.0 |
32.4 |
— |
68.3 |
50.8 |
| I-4 |
48.9 |
31.4 |
— |
58.5 |
44.8 |
| I-5 |
50.5 |
31.0 |
— |
60.3 |
48.7 |
| I-6 |
47.1 |
10.2 |
−64.6 |
22.8 |
16.6 |
| I-7 |
— |
— |
−57.4 |
— |
— |
Table 7 Thermal properties and crystallization degrees in series II
| Series II |
Tm (°C) |
Tc (°C) |
Tg (°C) |
ΔHf (J g−1) |
Xc (%) |
| II-1 |
57.2 |
34.8 |
— |
80.5 |
58.2 |
| II-2 |
53.2 |
19.1 |
−61.5 |
50.7 |
36.7 |
| II-3 |
50.3 |
14.9 |
−59.7 |
42.5 |
33.8 |
| II-4 |
47.9 |
6.3 |
−57.8 |
36.8 |
27.2 |
| II-5 |
41.2 |
— |
−58.3 |
16.3 |
12.9 |
| II-6 |
28.8 |
— |
−60.5 |
3.6 |
2.8 |
| II-7 |
— |
— |
−54.6 |
— |
— |
Table 8 Thermal properties and crystallization degrees in series III
| Series III |
Tm (°C) |
Tc (°C) |
Tg (°C) |
ΔHf (J g−1) |
Xc (%) |
| III-1 |
57.2 |
34.8 |
— |
80.5 |
58.2 |
| III-2 |
55.9 |
25.6 |
— |
72.7 |
52.8 |
| III-3 |
53.7 |
25.9 |
— |
67.6 |
53.5 |
| III-4 |
53.8 |
22.2 |
— |
65.7 |
52.9 |
| III-5 |
50.9 |
28.6 |
— |
65.1 |
53.0 |
| III-6 |
49.2 |
16.3 |
−67.3 |
44.9 |
32.5 |
| III-7 |
— |
— |
−61.3 |
— |
— |
Table 9 Thermal properties and crystallization degrees in series IV
| Series IV |
Tm (°C) |
Tc (°C) |
Tg (°C) |
ΔHf (J g−1) |
Xc (%) |
| IV-1 |
— |
— |
−54.6 |
— |
— |
| IV-2 |
— |
— |
−56.5 |
— |
— |
| IV-3 |
— |
— |
−58.8 |
— |
— |
| IV-4 |
— |
— |
−59.2 |
— |
— |
| IV-5 |
— |
— |
−59.9 |
— |
— |
| IV-6 |
— |
— |
−61.6 |
— |
— |
| IV-7 |
— |
— |
−61.3 |
— |
— |
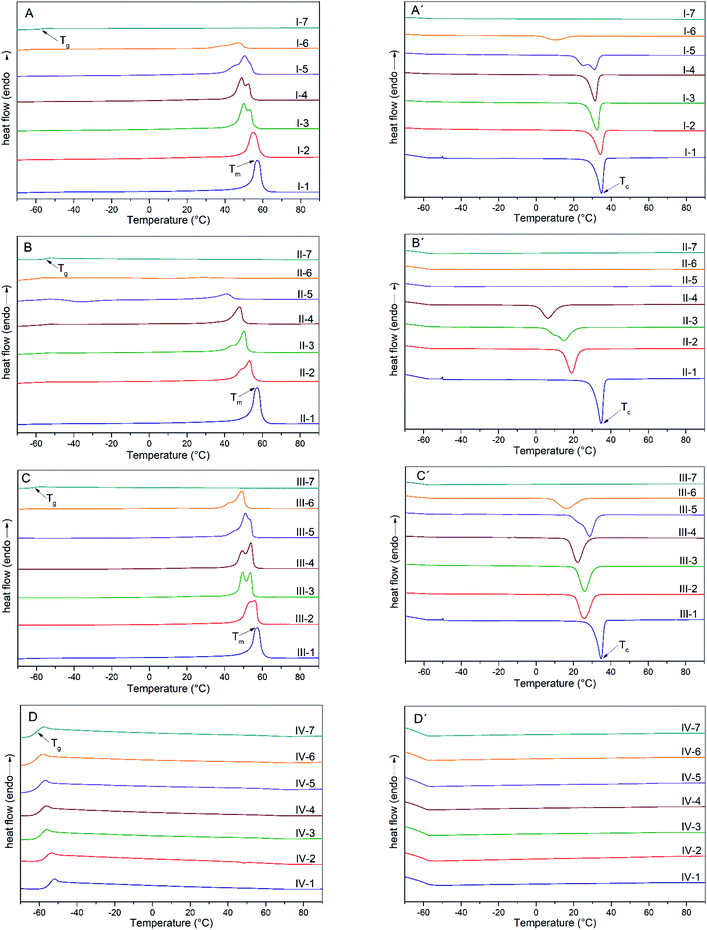 |
| | Fig. 7 Heating and cooling thermograms in the copolymer series I–IV. | |
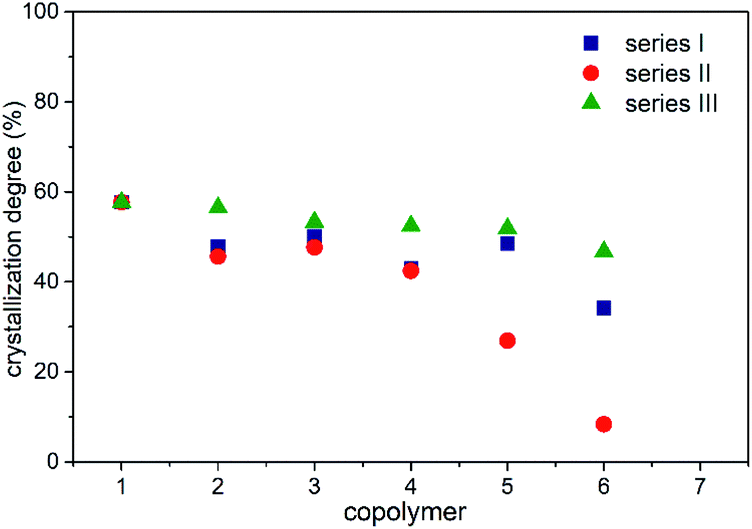
The crystallization degrees (Xc) in series I–III were calculated using eqn (5), where ΔHf corresponds to the enthalpy of fusion recorded via DSC in each copolymer (Tables 6–8), ΔHfref comes from PCL at a 100% crystalline arrangement (139.5 J g−1),33,39 and fPCL is related to the molar fraction of PCL, which was determined using the integral values collected from 1H NMR analysis, as displayed in Tables 2–4. In this work, the addition of branched comonomers (δ-DL, ε-DL and δ-DD) was suggested as an alternative solution for decoupling interactions between carboxylic groups in the PCL backbone. Herein, it is confirmed that the crystallization degree decreases as the percentage of alkyl-substituted comonomers increases, as shown in Fig. 8. It should be noted that series IV is not shown in Fig. 8 because the corresponding copolymers did not develop a semicrystalline arrangement, and this is supported by the absence of an endothermal transition in the DSC analysis (Fig. 7D).
| |
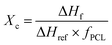 | (5) |
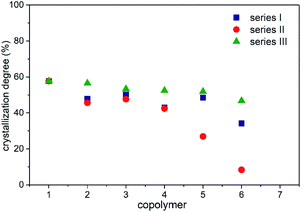 |
| | Fig. 8 Crystallization degree in series I–III. | |
4. Conclusions
In this work, four series of copolymers based on substituted and non-substituted lactones were synthesized via the ROP methodology using neodymium isopropoxide as a catalyst, with high conversion values, molar masses ranging from 9 to 43 kDa, and acceptable distributions. It is worth mentioning that changes in the composition of the comonomers impacted the thermal properties of the copolymers, denoted as broad endotherms with two different polyester populations, and increased the percentage of branched comonomers as well. The effect of branched monomers on the degree of crystallization is especially worth mentioning, where a decrease in Xc was observed when side alkyl groups were present in abundance. In series I–III, a loss of crystalline domains was observed as a consequence of the lessening of the side interactions between the carboxylic groups. Therefore, the contribution in this work was focused on the development of polyesters with different chemical natures and morphological features. It should be noted that these promising polyesters will be used in hydrolytic degradation tests in order to validate the impact of the crystallization degree on their degradation rates. Likewise, these copolymers may be applicable in the development of medical devices, which require materials with specific degradation periods. Additionally, these polyesters were exposed to hydrolytic degradation conditions and showed acceptable performance under these conditions. The biodegradable polymers may be improved by changing the reaction parameters or using new cyclic ester comonomers that exhibit degradation features.
Author contributions
G. R. D.: investigation, review, and editing; A. C. G. Z.: investigation, review, and editing; F. J. E. M.: validation, review and editing, methodology; M. S. H.: methodology, validation; M. A. J. T.: investigation, validation, writing–original draft; R. D. L.: conceptualization, resources, review and editing, supervision; H. R. L. G.: conceptualization, funding acquisition, project administration, review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge the technical support of Alejandro Díaz, Ricardo Mendoza, Guadalupe Mendez, Judith Cabello and Maricela García. M. A. J. T. acknowledges the financial support from Consejo Nacional de Ciencia y Tecnología (Conacyt) through the Postdoctoral grants program and Conacyt-Project: 76219 and CEMAI. The authors acknowledge Conacyt for the financial support granted at Ciencia Básica project A1-S-34241 through the program Ciencia Básica Projects SEP – Conacyt 2018–2019. G. R. D. acknowledges the financial support received from Consejo Nacional de Ciencia y Tecnología (Conacyt) through the post-grade scholarship program. R. D. L. acknowledges the support from CIQA through project 6609/6621.
References
- C. Ellingford, P. K. Samantaray, S. Farris, T. McNally, B. Tan, Z. Sun, W. Huang, Y. Ji and C. Wan, J. Appl. Polym. Sci., 2022, 139, 51617 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- P. K. Samantaray, A. Little, D. M. Haddleton, T. McNally, B. Tan, Z. Sun, W. Huang, Y. Ji and C. Wan, Green Chem., 2020, 22, 4055–4081 RSC.
- P. K. Samantaray, A. Little, A. M. Wemyss, E. Iacovidou and C. Wan, ACS Sustainable Chem. Eng., 2021, 9, 9151–9164 CrossRef CAS.
- P. K. Samantaray, C. Ellingford, S. Farris, D. O'Sullivan, B. Tan, Z. Sun, T. McNally and C. Wan, ACS Sustainable Chem. Eng., 2022, 10, 1267–1276 CrossRef CAS.
- C. Zhang and S. Agarwal, Biodegrade. Polyesters, ed. S. Fakirov, Wiley, Weinheim, 1st edn, 2015, ch. 1, pp. 1–24 Search PubMed.
- I. Manavitehrani, A. Fathi, H. Badr, S. Daly, A. Negahi Shirazi and F. Dehghani, Polym, 2016, 8, 1–32 CAS.
- R. P. Brannigan and A. P. Dove, Biomater. Sci., 2017, 5, 9–21 RSC.
- M. Gigli, M. Fabbri, N. Lotti, R. Gamberini, B. Rimini and A. Munari, Eur. Polym. J., 2016, 75, 431–460 CrossRef CAS.
- K. K. Bansal, J. Gupta, A. Rosling and J. M. Rosenholm, Saudi Pharm. J., 2018, 26, 358–368 CrossRef PubMed.
- S. Lee, K. Lee, J. Jang, J. S. Choung, W. J. Choi, G.-J. Kim, Y.-W. Kim and J. Shin, Polymer, 2017, 112, 306–317 CrossRef CAS.
- K. Krukiewicz, J. Britton, D. Więcławska, M. Skorupa, J. Fernandez, J.-R. Sarasua and M. J. P. Biggs, Sci. Rep., 2021, 11, 1295 CrossRef CAS PubMed.
- S. Agarwal, Biodegrade. Polyesters, ed. S. Fakirov, Wiley, Weinheim, 1st edn, 2015, ch. 2, pp. 25–45 Search PubMed.
- Q. Song, C. Pascouau, J. Zhao, G. Zhang, F. Peruch and S. Carlotti, Prog. Polym. Sci., 2020, 110, 101309 CrossRef CAS.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS PubMed.
- C. Thomas, F. Peruch and B. Bibal, RSC Adv., 2012, 2, 12851–12856 RSC.
- C. K. Williams, Chem. Soc. Rev., 2007, 36, 1573–1580 RSC.
- S. Metkar, V. Sathe, I. Rahman, B. Idage and S. Idage, Chem. Eng. Commun., 2019, 206, 1159–1167 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- A. Little, A. M. Wemyss, D. M. Haddleton, B. Tan, Z. Sun, Y. Ji and C. Wan, Polym, 2021, 13, 2458 CAS.
- F.-J. Lai, S.-C. Yu, Y.-C. Chang, T.-Y. Wu, K.-H. Wu, Y.-L. Chang, S. Ding, H.-Y. Chen and C.-H. Lai, J. Polym. Sci., 2020, 58, 1400–1409 CrossRef CAS.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- A. Heimowska, M. Morawska and A. Bocho-Janiszewska, Pol. J. Chem. Technol., 2017, 19, 120–126 CrossRef CAS.
- M. Bartnikowski, T. R. Dargaville, S. Ivanovski and D. W. Hutmacher, Prog. Polym. Sci., 2019, 96, 1–20 CrossRef CAS.
- M. Srisa-ard, R. Molloy, N. Molloy, J. Siripitayananon and M. Sriyai, Polym. Int., 2001, 50, 891–896 CrossRef CAS.
- M. Loriot, I. Linossier, K. Vallée-Réhel and F. Faÿ, J. Appl. Polym. Sci., 2016, 133, 43007 CrossRef.
- V. Arias, P. Olsén, K. Odelius, A. Höglund and A.-C. Albertsson, Polym. Chem., 2015, 6, 3271–3282 RSC.
- L. M. Orozco-Castellanos, A. Marcos-Fernández and A. Martínez-Richa, Polym. Adv. Technol., 2011, 22, 430–436 CrossRef CAS.
- S. Li, M. Pignol, F. Gasc and M. Vert, Macromolecules, 2004, 37, 9798–9803 CrossRef CAS.
- N. Zhao, C. Ren, Y. Shen, S. Liu and Z. Li, Macromolecules, 2019, 52, 1083–1091 CrossRef.
- P. Walther and S. Naumann, Macromolecules, 2017, 50, 8406–8416 CrossRef CAS.
- M. Xu, C. Guo, H. Dou, Y. Zuo, Y. Sun, J. Zhang and W. Li, Polym. Chem., 2019, 10, 3786–3796 RSC.
- D. K. Schneiderman, E. M. Hill, M. T. Martello and M. A. Hillmyer, Polym. Chem., 2015, 6, 3641–3651 RSC.
- D. Kakde, V. Taresco, K. K. Bansal, E. P. Magennis, S. M. Howdle, G. Mantovani, D. J. Irvine and C. Alexander, J. Mater. Chem. B, 2016, 4, 7119–7129 RSC.
- E. Pretsch, P. Bühlmann and M. Badertscher, Structure Determination of Organic Compounds, ed. E. Pretsch, P. Bühlmann and M. Badertscher, Springer, Heidelberg, 4th edn, 2009, ch. 7, pp. 269–335 Search PubMed.
- H. R. Kricheldorf and S. Eggerstedt, Macromol. Chem. Phys., 1998, 199, 283–290 CAS.
- J. Fernández, A. Etxeberria, A. L. Varga and J.-R. Sarasua, Polymer, 2015, 81, 12–22 CrossRef.
- A. Stamm, A. Biundo, B. Schmidt, J. Brücher, S. Lundmark, P. Olsén, L. Fogelström, E. Malmström, U. T. Bornscheuer and P.-O. Syrén, ChemBioChem, 2019, 20, 1664–1671 CrossRef CAS PubMed.
- W. Lee, J. W. Chung and S.-Y. Kwak, Eur. Polym. J., 2022, 162, 110882 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Marco A. De Jesús-Téllez*,
Ramon Díaz de León* and
Hector Ricardo López-González*
,
Marco A. De Jesús-Téllez*,
Ramon Díaz de León* and
Hector Ricardo López-González*