
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical investigation of fused N-methyl-dithieno-pyrrole derivatives in the context of acceptor–donor–acceptor approach†
Tridip Chutia and
Dhruba Jyoti Kalita *
*
Department of Chemistry, Gauhati University, Guwahati-781014, India. E-mail: dhrubajyoti.kalita@gauhati.ac.in
First published on 12th May 2022
Abstract
In this work we have theoretically investigated the optoelectronic properties of a series of acceptor–donor–acceptor type molecules by employing density functional theory formalism. We have used 1,1-dicyano-methylene-3-indanone as the acceptor unit and a fused N-methyl-dithieno-pyrrole as the donor unit. We have calculated the values of dihedral angle, inter-ring bond length, bond length alteration parameters, HOMO–LUMO gap, ionization potential, electron affinity, partial density of states, reorganization energies for holes and electrons, charge transfer rate for holes and electrons of the seven types of compounds designed via molecular engineering. Calculated IP and EA values manifest that PBDB-C2 shows excellent charge transportation compared to others. Absorption spectra of the designed compounds have been studied using the time-dependent density functional theory method. From the calculation of reorganization energy it is confirmed that our designed molecules behave more likely as donor materials. Our calculated results also reveal that compounds with electron donating substituents at the acceptor units show higher value of λmax. Absorption spectra of donor/acceptor blends show similar trends with the isolated compounds. Observed lower exciton binding energy values for all the compounds indicate facile charge carrier separation at the donor/acceptor interface. Moreover, the negative values of Gibb's free energy change also indicate the ease of exciton dissociation of all the designed compounds. The photovoltaic characteristics of the studied compounds infer that all the designed compounds have the potential to become suitable candidate for the fabrication of organic semiconductors. However, PBDB-C2 and PBDB-C4 with the highest PCE of 18.25% can become the best candidate for application in photovoltaics.
1 Introduction
Organic material based optoelectronic devices have gain attention over the conventional Si-based devices due to their low weight, mechanical flexibility, low cost production etc. These materials are widely used in the field of photovoltaics such as solar cells, organic light emitting diodes (OLED), organic field effect transistors etc.1–3 It has already been reported that the use of organic π conjugated semiconducting materials as well as dye sensitizers in photovoltaics increases the power conversion efficiency (PCE) of solar cells up to 13%.4,5 Despite of their low power conversion efficiencies compared to inorganic materials based photovoltaics, these materials are still promising candidates for photovoltaic applications due to their light weight and easy synthesizability.2,4,6–9Fullerene based organic solar cells (OSCs) have attracted much attention due to their optimistic optoelectronic properties such as high electron affinity and mobility, remarkable power conversion efficiency (PCE) etc. In spite of all these remarkable optoelectronic properties it has many intrinsic disadvantages including weak absorption of solar radiation, poor chemical and electronic tunability, tedious purification, high production costs, difficult fabrication to modify their energy levels and absorption.1,2,6,7,10 To overcome these issues, non-fullerene (NF) small-molecule acceptors with wide and efficient absorption, facile synthesis, and more finely tunable energy levels have been developed rapidly in past few years. Numerous NF small-molecule acceptors have been reported on the basis of a variety of π-conjugated moieties viz. perylene diimide (PDI), naphthalene diimide (NDI), fluoro-ranthene-fused diimide, diketopyrrolopyrrole (DPP) etc. It is reported that some NF-based OSCs, exhibit better performance than fullerene-based control devices.11–14 It has been already reported that the use of non fullerene based semiconducting materials in photovoltaics have increased the PCE up to 18.5%.12,15,16 Among these, small-molecule acceptors with the acceptor–donor–acceptor (A–D–A) backbone architecture, similar to the widely used and rather successful small-molecule donors, have drawn interest due to their easily tunable energy levels and high device performances. Besides, conjugated small molecules have attracted much attention due to their well defined structures and hence less batch to batch variation, easy synthetic control, high mobility and open circuit voltage, high purity, versatile chemical structures and easy energy level control.9,11,17,18 A–D–A small molecules have drawn attention in solution-processed organic solar cells also due to the diversity of structures, easy control of energy levels.17,18 Moreover,the presence of conjugated π-electron system in organic compounds show interesting optical and electrical properties.
The hole and electron injection ability and transportation ability of the polymer plays a crucial role in determining the performance of an optoelectronic device. For efficient charge transportation, the fundamental band gap i.e., difference between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of polymers should lie within a suitable range so that electrons and holes can be injected to the polymer surface from the metal electrode.2,6,19,20 It is possible to improve the PCE of the OSCs by designing and synthesizing of small molecule acceptors. Presently, A–π–D–π–A type of small molecule in which an electron rich donor unit (D) is linked to two electron deficient acceptor (A) groups via two π-bridges is one of the most hopeful molecule design strategies.21
The structural and opto-electronic properties, carrier mobilities and solubility parameters etc. of A–π–D–π–A type molecules can be easily tuned by modifying the three parts viz. D, π and the A of the materials.21 Suitable modification of the end-capped groups can lead to the successful and efficient tuning of the absorption energy levels which in turn helps in the achievement of high performance of the OSCs.8,22
In this work we have chosen a series of A–D–A type molecules which has the potential to act as candidate for optoelectronic devices. We have used 1,1-dicyano-methylene-3-indanone as the acceptor unit and a fused N-methyl-dithieno-pyrrole as the donor unit.23 We have chosen N-methyl-dithieno-pyrrole as the donor unit due to its suitable photophysical properties viz. high planarity, low band gap, high absorption coefficient etc.24 The donor–acceptor representation of our investigated systems have been provided in Fig. 1. We have carried out substitution at the 7th and 8th positions of the acceptor unit with different groups viz. –F, –OCH3 etc. and their sketches have been provided in Fig. 2. In this regard, we have investigated the effect of electron donating and electron withdrawing substituents at different positions (7th and 8th) in the acceptor unit. It has been observed from the literature that substituents at 7th and 8th position of the acceptor unit leads to high planarity and high electron mobility.25 We have studied different properties viz. dihedral angle, inter-ring length (J), bond length alteration (BLA) parameter (Δr), band gap (ΔH–L values), absorption properties (λmax), reorganization energy etc. For better understanding of the electronic properties of our designed compounds we have taken a standard fused ring electron acceptor (FREAs, a76)26 unit and placed it in the length of 3.5 cm with all the compounds. The representative stacking form has been provided in Fig. 1 of ESI.†
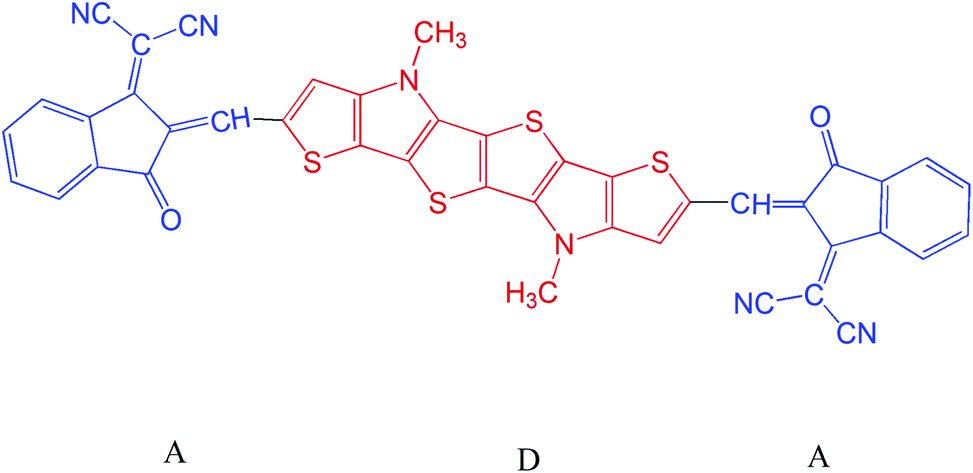 | ||
| Fig. 1 Representation of acceptor–donor–acceptor unit. | ||
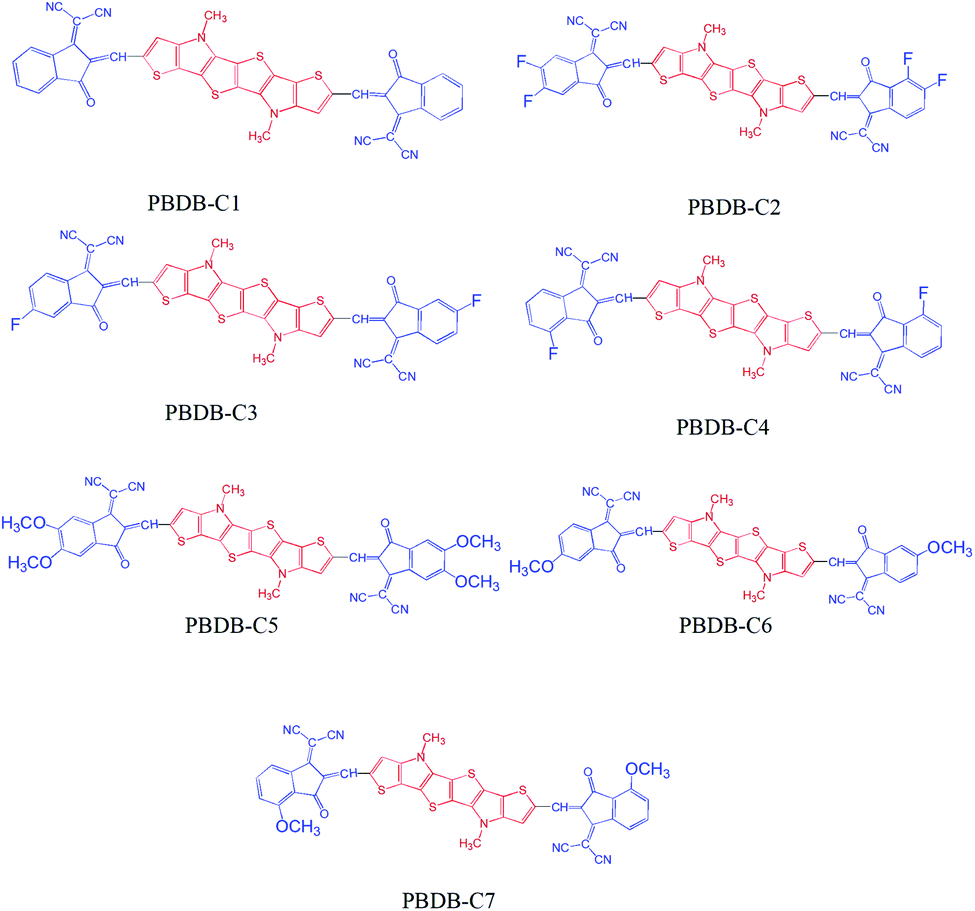 | ||
| Fig. 2 Sketches of the studied compounds. | ||
2 Computation methodology
All the calculations have been carried out using the Gaussian 09 (ref. 27) program package. Geometry optimization has been carried out both in the gas and solvent phase (THF) by applying polarizable continuum model (CPCM)28 employing the density functional theory (DFT) at 6-311G(d,p)/B3LYP-D3 level of theory.1,2,19,20,28–31To calculate the absorption properties of our studied compounds we have employed time-dependent density functional theory (TD-DFT) formalism19,20,28,32,33 using the long range corrected functional CAM-B3LYP1,20,34–36 at the same basis set level both in the gas and in THF solvent phase. The absorption spectra have been calculated for 30 singlet–singlet transitions of the optimized ground state. To get better understanding of the electronic transitions occurring in the molecules, the isodensity plots of frontier molecular orbitals have been generated using the Gauss View 5.0.9 software.
Geometry optimization at the neutral, cationic and anionic states, energy calculations of all the compounds have been carried out at B3LYP-D3/6-31G(d,p) level of theory.2,37 Various properties like IP, EA, reorganization energies (λ) for all the studied compounds have been calculated on the basis of above calculations. To gather information about the polarizability, dipole moment of the compounds have been computed in both the gas and solvent phase at the same level of theory.38–41
3 Validation of applied methodology and theoretical details
3.1 Validation of applied methodology
To attain better accuracy of calculated results it is necessary to choose appropriate methodology. We have performed a test calculation with compound s6 reported in the literature which has structural resemblance with our designed compounds.42 In this regard we have performed our ground state calculations by employing DFT formalism with six different functionals viz. B3LYP, B3LYP-D3, B3PW91, PBEPBE, HSEH1PBE, CAM-B3LYP, and 6-311G(d,p) basis set.2,42–45 The excited state calculations have been carried out with the time dependent-DFT (TD-DFT) method using the functionals CAM-B3LYP, ωB97XD and M06-2X-D3 and 6-311G(d,p) basis set.42,46–48 We have compared the calculated energies of HOMO and LUMO, ΔH–L and λmax values with the reported results. The results of our calculation have been presented in Table S2 in the ESI.† It has been observed from the calculation that HSEH1PBE and B3LYP-D3 functionals show good agreement with the reported results in the ground state. On the other hand, in the excited state calculations M06-2X-D3 and CAM-B3LYP functionals show good correlation with the reported results. Hence, to keep the computational cost under control we have carried out our calculations using B3LYP-D3/6-31G(d,p) and CAM-B3LYP/6-31G(d,p) functionals for ground and excited state respectively.3.2 Theoretical details
The vertical and adiabatic ionization potential (IP) and electron affinity calculations (EA) of the studied compounds have been carried out using eqn (1)–(4) respectively. The vertical IP and EA values of the compounds are determined from the calculations of the total energy on the neutral and ionic systems.1,2,49| IP(v) = E+(M°) − E°(M°) | (1) |
| IP(a) = E+(M+) − E°(M°) | (2) |
| EA(v) = E°(M°) − E−(M°) | (3) |
| EA(a) = E°(M°) − E−(M−) | (4) |
Reorganization energy (λ) is one of the key parameters which governs the charge transfer rate. It is similar to the activation energy barrier of a hole/electron transfer process in semiconductors. Usually the reorganization energy (λ) values have two contributions, the outer sphere and the inner sphere. The inner sphere part arises from the geometry relaxation process when a charge is accepted or released by the molecule.1,2 The polarization of the surrounding medium or relaxation of nucleus/electrons is responsible for the outer sphere reorganization energy. In this paper we have considered only the inner sphere contribution.
The reorganization energy for holes, λh and electrons, λe values are calculated using eqn (5) and (6) respectively.1,20,53–57
| λh = [E+(M°) − E+(M+)] + [E°(M+) − E°(M°)] | (5) |
| λe = [E°(M−) − E°(M°)] + [E−(M°) − E−(M−)] | (6) |
The value of charge transfer rate (kct) can be described by Marcus theory. The expression for kct is given by eqn (7) as follows:1,19,20,48,55,57–61
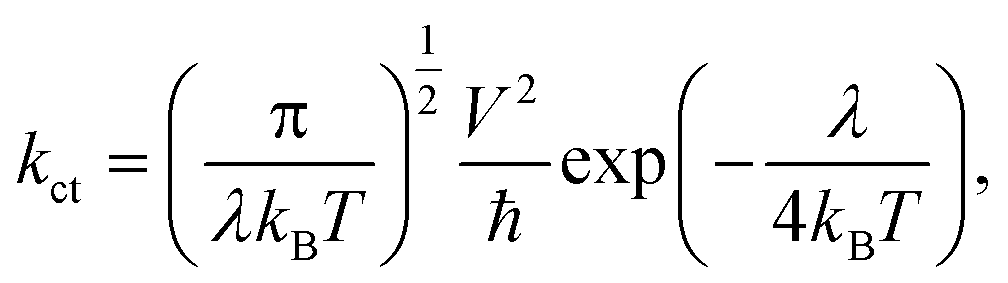 | (7) |
The electronic coupling matrix element (V) (often called transfer integral) are calculated by using eqn (8) (ref. 1, 19, 48 and 58–60)
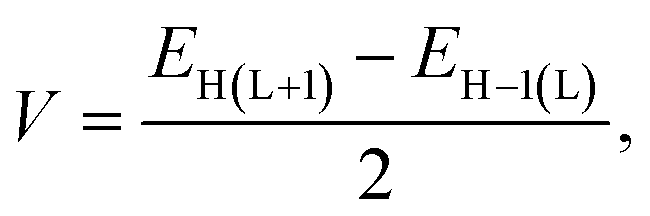 | (8) |
The theoretical power conversion efficiency (η) of a photovoltaic device can be generally expressed using eqn (9):62,63
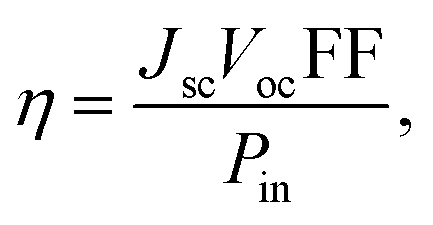 | (9) |
The Voc can be calculated using eqn (10):64,65
| eVoc = (|EDonorHOMO − EAcceptorLUMO|) − 0.3 eV, | (10) |
It is reported that the Jsc of a device depends on the intensity and spectral range of solar absorption. Jsc is a function of the external quantum efficiency (ηEQE) of the device and photon number S(λ) over the whole frequency region. It can be expressed as:67,68
| Jsc = q∫ηEQE(λ)S(λ)d(λ), | (11) |
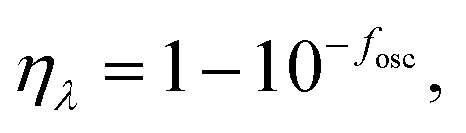 | (12) |
The FF can be calculated using eqn (13) (ref. 64 and 65)
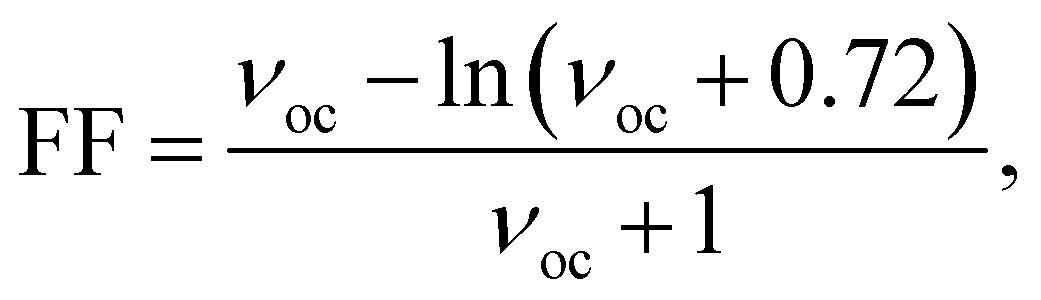 | (13) |
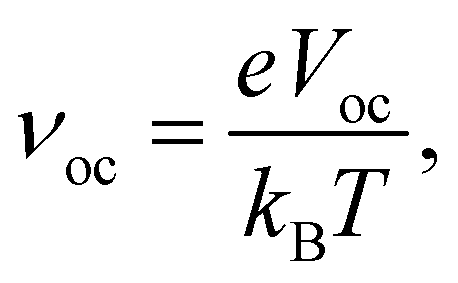 | (14) |
4 Results and discussion
Frequency calculation of all the studied compounds reveal their stability as no imaginary frequency is observed. The optimized structures have been represented in Fig. 1 of ESI.†4.1 Structural properties
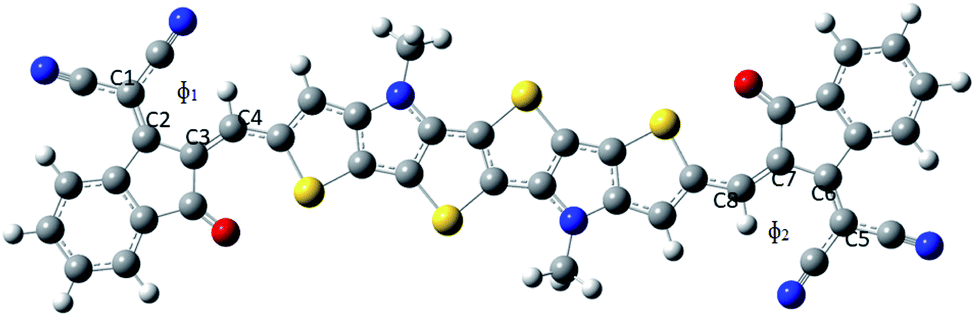 | ||
| Fig. 3 Representation of dihedral angle in a molecule where ϕ1 = C1C2C3C4 and ϕ2 = C5C6C7C8. | ||
The dihedral angles of the studied compounds (both gas as well as solvent phase) are reported in Table 1. It has been observed from Table 1 that the substitution of the electron withdrawing group –F at the acceptor part in both gas as well as solvent phase, leads to the lower value of dihedral angle (ϕ1 = 0.00362°, ϕ2 = 0.00047°). Owing to the small size of F it will undergo less steric hindrance with the thiophene group which in turn results into lower dihedral angle. Bond length alteration (BLA) parameter and inter-ring length (J) also affects the dihedral angle. BLA parameter and J values are also presented in Table 1. It is apparent from Table 1 that compound PBDB-C2 (gas phase) possesses the least value of both dihedral angles and BLA parameter. On the other hand, PBDB-C5 in gas phase shows the highest dihedral angle (ϕ1 = 2.03646°, ϕ2 = 0.59247°). Owing to the comparatively large size of –OMe group it will undergo repulsion with the thiophene group to the greater extent which results in the higher dihedral angle. We have also observed that the inter-ring length of PBDB-C5 is the highest among all the studied compounds. In solvent phase, PBDB-C6 exhibits minimum dihedral angle (ϕ1 = 0.58018°, ϕ2 = 0.54479°) and PBDB-C4 exhibits maximum dihedral angle (ϕ1 = 1.86340°, ϕ2 = 1.87129°). The same generalized reason can be accounted for these two compounds as well.
| Compounds | Phase | ϕ1 (°) | ϕ2 (°) | J1 (Å) | J2 (Å) | Javg (Å) | Δr (Å) |
|---|---|---|---|---|---|---|---|
| PBDB-C1 | Gas | 0.24272 | −0.23503 | 1.41683 | 1.41687 | 1.41685 | 0.0500 |
| Solvent | 1.64558 | −1.60283 | 1.41224 | 1.41224 | 1.41224 | 0.0465 | |
| PBDB-C2 | Gas | −0.00362 | 0.00047 | 1.41596 | 1.41596 | 1.41596 | 0.0480 |
| Solvent | 1.14349 | −1.15675 | 1.41114 | 1.41114 | 1.41114 | 0.0440 | |
| PBDB-C3 | Gas | −0.07407 | 0.07127 | 1.41626 | 1.41626 | 1.41626 | 0.0510 |
| Solvent | −1.51851 | 1.55026 | 1.41156 | 1.41155 | 1.41156 | 0.0455 | |
| PBDB-C4 | Gas | −0.08668 | 0.08548 | 1.41593 | 1.41593 | 1.41593 | 0.0540 |
| Solvent | 1.86340 | −1.87129 | 1.41119 | 1.41119 | 1.41119 | 0.0454 | |
| PBDB-C5 | Gas | −2.03646 | −0.59247 | 1.41894 | 1.41907 | 1.41901 | 0.0520 |
| Solvent | −0.75517 | −0.22485 | 1.41465 | 1.41486 | 1.41476 | 0.0475 | |
| PBDB-C6 | Gas | 0.13722 | −0.11558 | 1.41782 | 1.41844 | 1.41813 | 0.0540 |
| Solvent | −0.58018 | 0.54479 | 1.41436 | 1.41355 | 1.41396 | 0.0500 | |
| PBDB-C7 | Gas | 0.03845 | −0.02006 | 1.41805 | 1.41805 | 1.41805 | 0.0520 |
| Solvent | −1.20375 | 1.20209 | 1.41376 | 1.41377 | 1.41377 | 0.0465 |
Inter-ring bond length is linearly dependent on the dihedral angles. From Table 1 it is observed that there is significantly less variation in J values of the studied small molecules. They can be differentiated within the fractions of unit. From Table 1 it has been observed that, the average inter-ring length of PBDB-C4 has the least value. Besides, from Table 1, we can conclude that J values of all the compounds in gas phase are not significantly different. This can be attributed to their low value of dihedral angles. It is already known that with decrease in J values, the conjugation increases. With increase in conjugation, the energies of HOMO increases while that of LUMO decreases which results into lowering of band gap.20 PBDB-C4 has the least value of band gap as compared to others. In case of solvent phase, PBDB-C2 has the lowest value of J. The same reason can be accounted for this compound as well. Hence, we can conclude that the conjugation will be more for compounds PBDB-C2 and PBDB-C4 in both gas and solvent phases respectively.
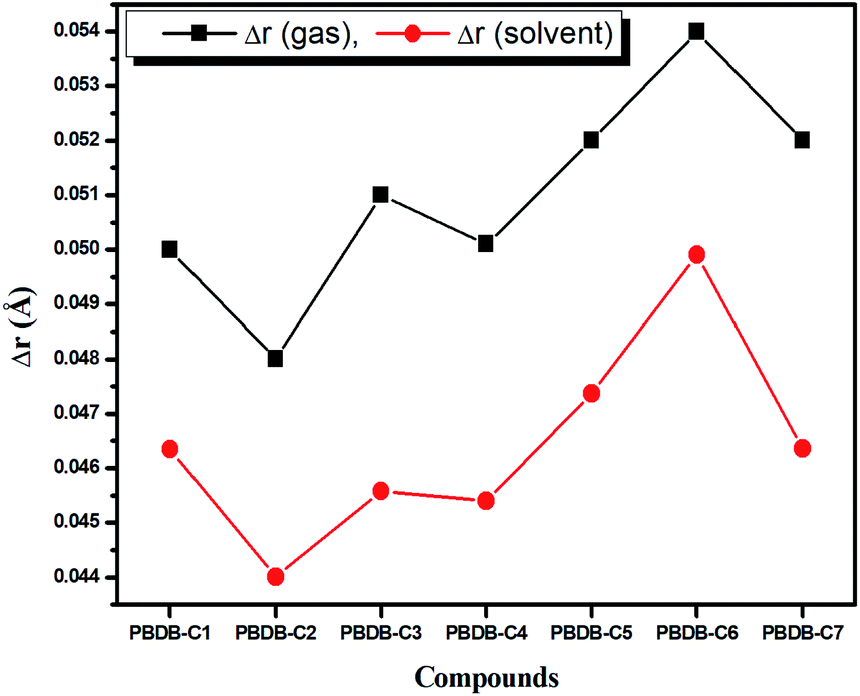 | ||
| Fig. 4 Plot of BLA parameters of the studied compounds in gas and solvent phase. | ||
BLA parameter (Δr) is defined as the difference between the average carbon–carbon single and double bond distances.71 Lesser the value of Δr, the greater will be the extent of conjugation.1,2 From Fig. 4 it is observed that PBDB-C2 possesses the lowest value of Δr in both the gas and solvent phase. This is due to the presence of electron withdrawing (–F) group at the acceptor part. However, PBDB-C6 possesses the highest value of Δr in both phases. Moreover, it is apparent from Fig. 4 that the Δr values of the compounds in the gas phase have higher value than that in the solvent phase. These values indicate the presence of high extent of conjugation in all the compounds in solvent phase than in the gas phase.
4.2 Frontier molecular orbitals and band gaps
Frontier molecular orbital (FMO) is found very much useful in gathering information about the optical and electronic properties of organic molecules. Theoretically, the band gap between the frontier orbitals, ΔH–L value is computed as the difference between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).28,31 Many properties including the spectroscopic properties of the compounds can be explained by scrutinizing the ΔH–L values. The energies of HOMO, LUMO and ΔH–L values of the studied compounds in both gas and solvent phase are reported in the Table 2. From Table 2 it is observed that the ΔH–L values are reduced in all the compounds possessing −I group. This happens due to the stabilization of the LUMO energy levels. Among the studied compounds, PBDB-C6 exhibits the highest ΔH–L value in both gas as well as solvent phase. This is because of the destabilization of the LUMO energy levels of the compounds due to the presence of –OMe group having strong +I effect. ΔH–L values of the compounds in the gas phase is higher than in the solvent phase. From Table 2 we can demonstrate that the HOMO orbitals in the non polar THF solvent is stabilized to a lesser extent than in the gas, which results into greater ΔH–L values in gas phase.| Compounds | Phase | HOMO (eV) | LUMO (eV) | ΔH–L (eV) |
|---|---|---|---|---|
| PBDB-C1 | Gas | −5.45 | −3.40 | 2.05 |
| Solvent | −5.32 | −3.34 | 1.98 | |
| PBDB-C2 | Gas | −5.60 | −3.57 | 2.03 |
| Solvent | −5.37 | −3.42 | 1.95 | |
| PBDB-C3 | Gas | −5.54 | −3.48 | 2.06 |
| Solvent | −5.35 | −3.37 | 1.98 | |
| PBDB-C4 | Gas | −5.49 | −3.47 | 2.02 |
| Solvent | −5.35 | −3.40 | 1.95 | |
| PBDB-C5 | Gas | −5.27 | −3.20 | 2.07 |
| Solvent | −5.23 | −3.25 | 1.98 | |
| PBDB-C6 | Gas | −5.35 | −3.25 | 2.1 |
| Solvent | −5.26 | −3.25 | 2.01 | |
| PBDB-C7 | Gas | −5.29 | −3.25 | 2.04 |
| Solvent | −5.25 | −3.29 | 1.96 |
The pictorial representation of frontier molecular orbitals of PBDB-C4 is provided in Fig. 5 and 6 and the rest of the compounds have been provided in Fig. 3 and 4 of the ESI.† From these figures it is clear that HOMOs of the compounds mostly concentrate in the donor unit via delocalization of π-electron cloud leaving the acceptor part empty. This supports the electron donating nature of the donor unit. On the contrary, the LUMOs of the compounds spread over the entire molecule leaving the donor part partially empty. It is also apparent from these figures that there is no considerable difference in the LUMO molecular orbitals of the compounds in the gas and the solvent phase.
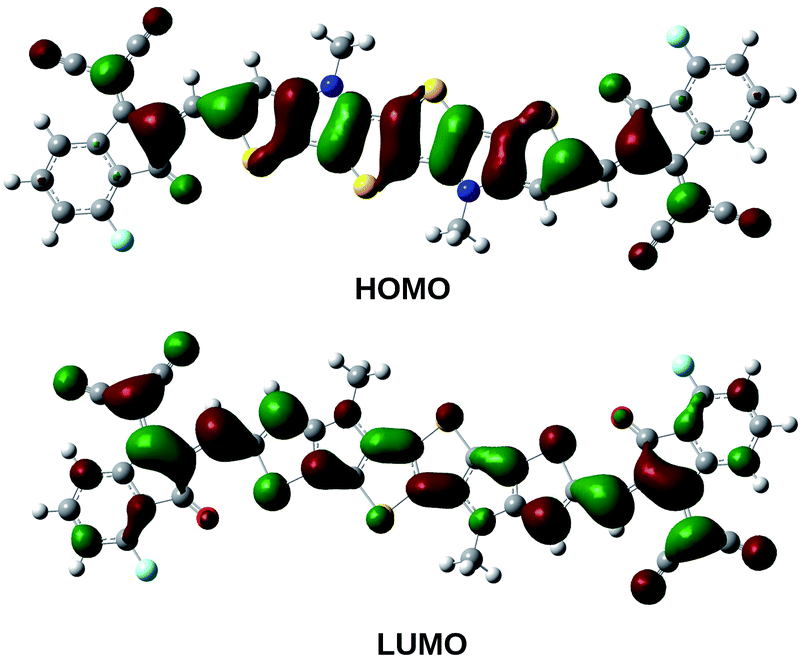 | ||
| Fig. 5 Frontier molecular orbital diagram (HOMO–LUMO) of PBDB-C4 in the gas phase. | ||
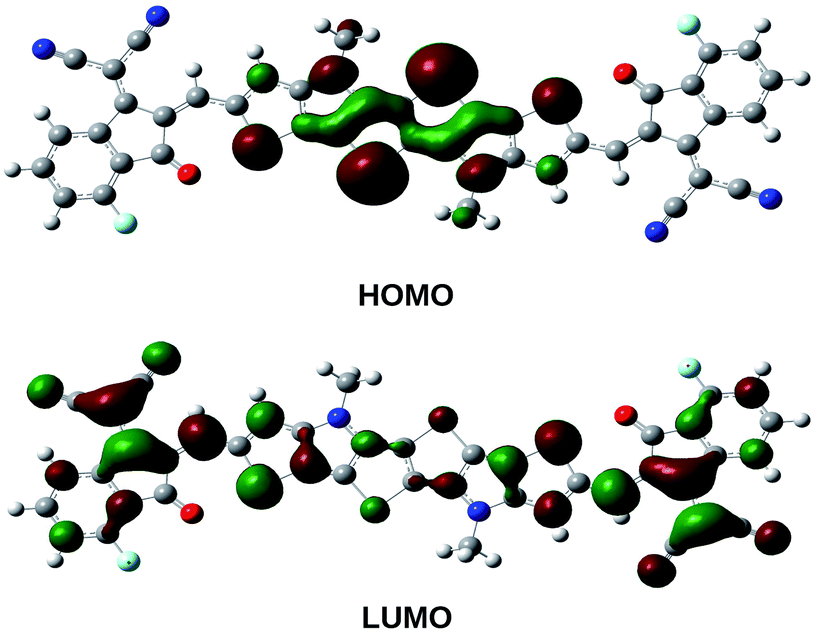 | ||
| Fig. 6 Frontier molecular orbital diagram (HOMO–LUMO) of PBDB-C4 in the solvent phase. | ||
4.3 Density of states
To investigate the correlation between the FMOs and electronic structures we have calculated the partial density of states (PDOS) of all the studied compounds and the respective plots of some of the compounds are presented in Fig. 7 and their spectral data have been provided in Table S3 and Fig. 5 in the ESI.† It has been observed from Table S3† and Fig. 7 that the donor unit offers maximum contribution to the HOMOs and the acceptor unit offers maximum contribution to the LUMOs. From Table S3† it is observed that the donor unit of PBDB-C6 has the highest contribution toward HOMOs (69%) and the acceptor unit of compounds PBDB-C4 and PBDB-C7 have the highest contribution toward LUMOs (64%) among all the investigated compounds.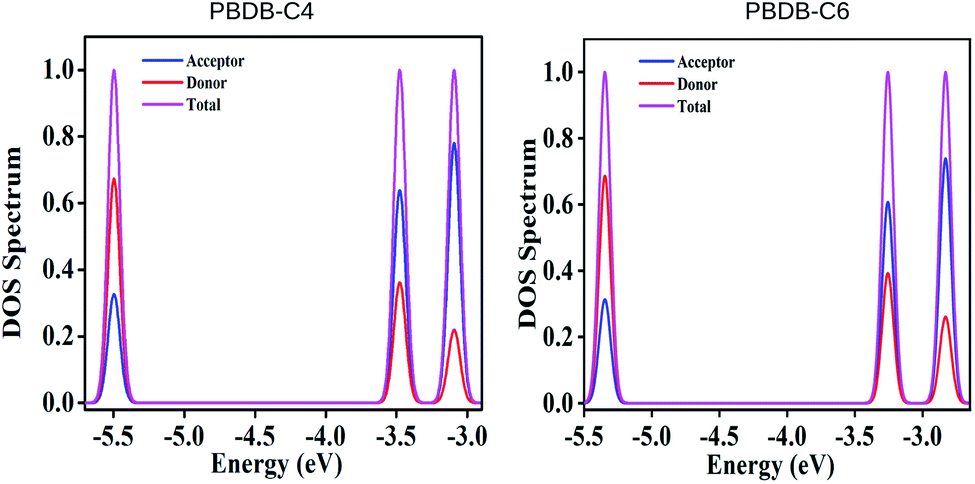 | ||
| Fig. 7 Plot of density of states of some of the studied compounds. | ||
In compounds PBDB-C2, PBDB-C3 and PBDB-C4, the acceptor unit is substituted with the electron withdrawing –F group whereas in compounds PBDB-C5, PBDB-C6 and PBDB-C7, the acceptor unit is substituted with the electron donating –OCH3 groups. Compounds PBDB-C4 and PBDB-C6 are substituted by –F and –OCH3 groups at the 7th position of the acceptor unit and offers maximum contribution toward LUMO energy levels. However, the donor unit shows almost similar contribution toward HOMO energy levels except PBDB-C6. Moreover, we have also observed that substituents attached at the 7th position of the acceptor unit exhibits lower ΔH–L values. Thus it can be concluded that attachment of electron withdrawing –F and and electron donating –OCH3 groups at the acceptor part increases the contribution toward LUMO energy levels and also enhances the contribution of donor unit toward HOMO energy levels. This observation reveals that the PDOS spectra gives a clear vision of the nature of HOMO and LUMO energy levels.
4.4 Ionization potential and electron affinity
Ionization potential (IP) and electron affinity (EA) of organic molecules are important parameters which give information about the charge-injection and charge transport properties of a molecule.49,72 We have calculated the vertical and adiabatic IP and EA values for the compounds in gas phase employing the functional B3LYP. The calculated values of IP and EA are presented in Table 3.| Compounds | IP (eV) | EA (eV) | ||
|---|---|---|---|---|
| Adiabatic | Vertical | Adiabatic | Vertical | |
| PBDB-C1 | 6.25 | 6.34 | 2.64 | 2.56 |
| PBDB-C2 | 6.39 | 6.48 | 2.82 | 2.73 |
| PBDB-C3 | 6.33 | 6.42 | 2.73 | 2.63 |
| PBDB-C4 | 6.29 | 6.38 | 2.71 | 2.63 |
| PBDB-C5 | 6.01 | 6.13 | 2.51 | 2.51 |
| PBDB-C6 | 6.13 | 6.22 | 2.52 | 2.52 |
| PBDB-C7 | 6.08 | 6.16 | 2.50 | 2.42 |
It is evident from Table 3 that the electron withdrawing substituents increase the IP and EA values, whereas the reverse trend is observed for electron donating substituents. PBDB-C5 exhibits the lowest IP values (both v and a) and PBDB-C7 exhibits the lowest EA (both v and a) values. Similarly PBDB-C2 exhibits the highest IP and EA (both v and a) values. The presence of the electron donating –OMe group in compounds PBDB-C5 and PBDB-C7 and the electron withdrawing –F group in PBDB-C2 mandate this variation in IP and EA values. The presence of –OMe and –F groups at the acceptor part increase and decrease the electron density of HOMOs of the respective compounds. These observations are supported by the frontier orbital diagram of our studied compounds represented in Fig. 8. This figure clearly indicates that the electron density in the acceptor part is minimum in PBDB-C2 and maximum in compounds PBDB-C5 and PBDB-C7 compared to the unsubstituted compound (PBDB-C1). Thus, from the above study it can be inferred that in PBDB-C2, the charge transportation is more favorable than the other compounds.
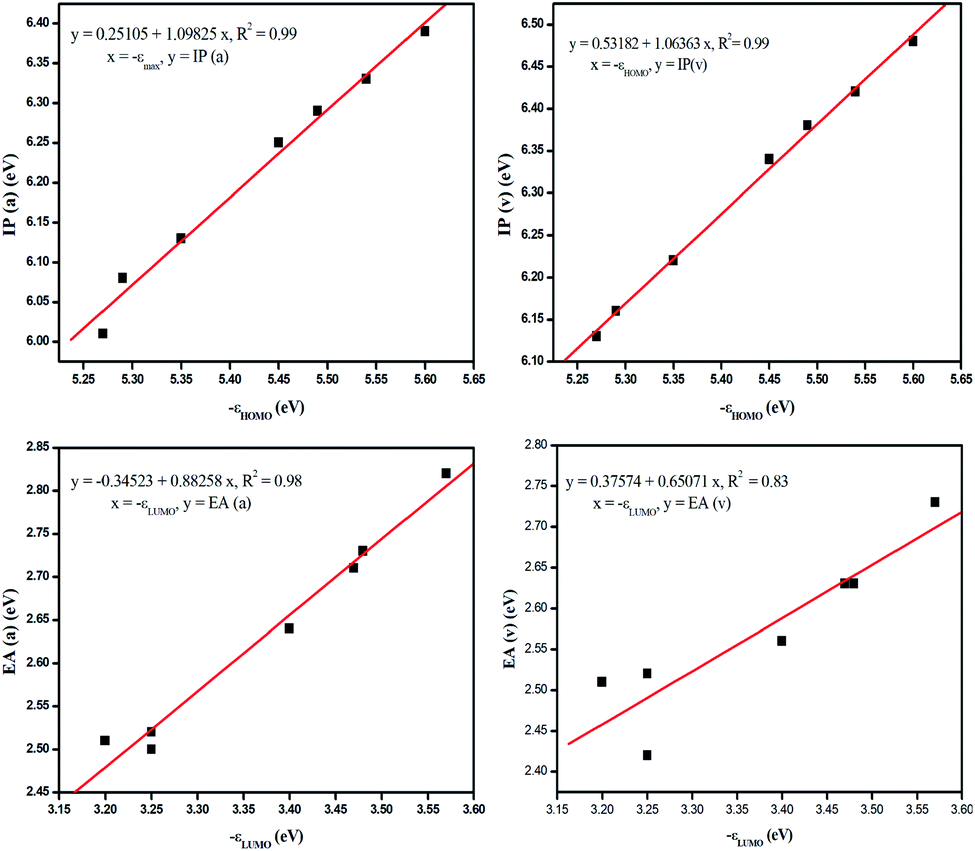 | ||
| Fig. 8 Correlation diagram between IP and HOMO energy and EA and LUMO energy in the gas phase calculated using B3LYP functional. | ||
For the calculation of IP(a, v) and EA(a, v) in solvent phase we have also employed the B3LYP functional and the results are presented in Table 4. From Tables 3 and 4 we have observed that the IP(a, v) and EA(a, v) values of the compounds follow the same trend for both the phases (gas and solvent phase). From Tables 3 and 4 it is observed that the vertical EA values are lower than the adiabatic counterparts. On the other hand, the vertical IP values are higher than their adiabatic counterparts for all the compounds. It is also observed from Tables 3 and 4 that the IP(v, a) values in the solvent phase are lower than that of the gas phase and EA(v, a) values in the solvent phase are higher than that of the gas phase. It is already mentioned that both the electronic and steric effects contribute to the EA values. As a consequence, compounds containing electron donating group possesses the least EA value due to extended delocalization of π-electron cloud created by the –OMe group. All the compounds containing the electron donating groups have smaller EA values and thus meets our expectations.
| Compounds | IP (eV) | EA (eV) | ||
|---|---|---|---|---|
| Adiabatic | Vertical | Adiabatic | vertical | |
| PBDB-C1 | 5.21 | 5.24 | 3.43 | 3.41 |
| PBDB-C2 | 5.26 | 5.29 | 3.51 | 3.48 |
| PBDB-C3 | 5.24 | 5.27 | 3.46 | 3.43 |
| PBDB-C4 | 5.24 | 5.26 | 3.48 | 3.46 |
| PBDB-C5 | 5.12 | 5.15 | 3.34 | 3.31 |
| PBDB-C6 | 5.15 | 5.18 | 3.35 | 3.31 |
| PBDB-C7 | 5.14 | 5.17 | 3.38 | 3.36 |
The correlation between the IP and the HOMO energy levels and EA and LUMO energy levels calculated using B3LYP functional in the gas as well as solvent phase can be clearly understood with the linear fitting plots which are depicted in Fig. 8. This figure clearly describes a good correlation between the mentioned parameters.
4.5 Effect of solvent polarity on the dipole moment (μ)
Dipole moment (μ) is defined as the product of magnitude of the charge and the distance between the centres of the positive and negative charges. It is a measure of the polarity of the organic dyes. A higher value of μ indicates the polar nature of the particular dye. The calculated ground state and excited state dipole moment (μg and μe) of the compounds in the gas as well as in the THF phases are reported in Table 5. From this table it is found that the ratio of the excited state to the ground state dipole moment is greater than one. The μg values range from 0.0017 D to 4.5182 D and μe values range from 0.0020 D to 4.5621 D. Similarly, in the THF solvent, μg values ranges from 0.0066 D to 5.9855 D and μe values ranges from 0.0067 D to 6.1457 D. These values clearly indicate that the compounds are more polar in the excited state and stabilized by the polar environment. In the excited state a transfer of electron densities from HOMO to LUMO takes place which in turn leads to the significant change in the dipole moment between ground and the excited state. However, in the solvent phase we observe an increase in μ values due to the polarity of the solvent. From Table 5 it is observed that all the compounds exhibits the higher value of μ in the solvent phase than in gas phase. From the μ values it can be concluded that PBDB-C5 has better push–pull ability both in the ground and excited state in both the phases.| Compounds | Gas | Solvent | ||
|---|---|---|---|---|
| Ground state (μg) | Excited state (μe) | Ground state (μg) | Excited state (μe) | |
| PBDB-C1 | 0.0017 | 0.0020 | 0.0066 | 0.0067 |
| PBDB-C2 | 0.0007 | 0.0009 | 0.0019 | 0.0020 |
| PBDB-C3 | 0.0022 | 0.0023 | 0.0055 | 0.0056 |
| PBDB-C4 | 0.0005 | 0.0007 | 0.0012 | 0.0013 |
| PBDB-C5 | 4.5182 | 4.5621 | 5.9855 | 6.1457 |
| PBDB-C6 | 2.3961 | 3.1026 | 3.0221 | 3.1776 |
| PBDB-C7 | 0.0004 | 0.0008 | 0.0014 | 0.0016 |
4.6 Reorganization energy and charge transfer rate
To attain better performance of the optoelectronic devices, the designed materials must establish a relationship between their structural and the charge transfer properties. Reorganization energy, λ is one of the key parameters which governs the charge transfer rate. It is similar to the activation energy barrier of a hole/electron transfer process in semiconductors. λ needs to be minimized for the compounds to offer efficient performance of the optoelectronic devices. Therefore, it is necessary to have a low value of λ for high charge transfer rate. If λh < λe the material behaves as a hole transporter and if λe < λh the material behaves as an electron transporter. The calculated values of λh and λe of the studied compounds in gas phase are reported in Table 6.| Compounds | λh (eV) | λe (eV) |
|
|
|
|
|---|---|---|---|---|---|---|
| PBDB-C1 | 0.1736 | 0.1784 | 0.39 | 0.20 | 11.27 | 28.00 |
| PBDB-C2 | 0.1818 | 0.1830 | 0.385 | 0.195 | 9.92 | 25.00 |
| PBDB-C3 | 0.1848 | 0.1987 | 0.385 | 0.21 | 9.55 | 23.90 |
| PBDB-C4 | 0.1755 | 0.1712 | 0.39 | 0.19 | 10.96 | 27.50 |
| PBDB-C5 | 0.2406 | 0.2345 | 0.405 | 0.195 | 5.39 | 13.45 |
| PBDB-C6 | 0.1847 | 0.2115 | 0.395 | 0.21 | 10.05 | 20.62 |
| PBDB-C7 | 0.1788 | 0.1807 | 0.400 | 0.185 | 11.12 | 23.26 |
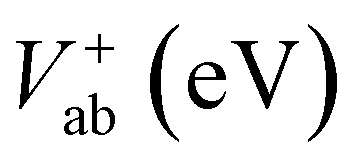
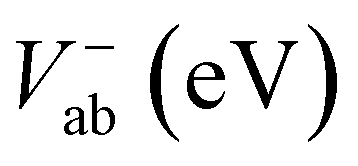
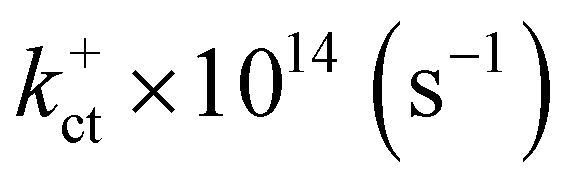
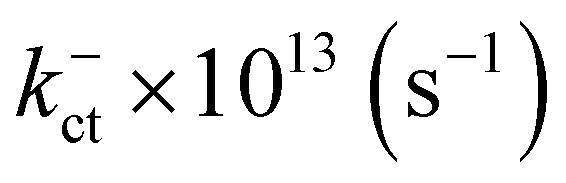
Electronic and steric effects of the substituents play the dominant role in the determination of λ. In both cationic and anionic geometries, compounds having –OMe functional group has the higher value of λ than that of compounds having –F group. This is due to bigger size of the –OMe group. As the size of the substituents increases, steric effects tend to increase and results into higher value of λ. All the compounds except PBDB-C5 and PBDB-C4 exhibit lower λh values than λe values. It is observed from Table 6 that compound PBDB-C5 exhibits the highest value of λ for both cationic and anionic geometries. The highest dihedral angle value of PBDB-C5 accounts for the observed maximum λ value. Moreover, the presence of electron donating group (–OMe) in PBDB-C5 increases the electron density in the acceptor part and hence charge transportation between the donor and acceptor is not facile. Similarly, in both the geometries PBDB-C4 has the lowest reorganization energy (both λh and λe). This is due to the presence of the strong electron withdrawing –F group in the acceptor part that pulls the electron density towards itself. The lowering of λ values leads to more charge separation. We have also calculated the charge transfer rate (kct) of the compounds using eqn (7) and respective values are reported in Table 6. From Table 6 it is observed that PBDB-C5 exhibits the lowest value of kct due to its higher value of λ among all the studied compounds. However, PBDB-C1 exhibits the highest value of kct among all. The same reason accounts for this observation as well. It has been observed from Table 6 that the Vab values of the compounds in the cationic geometries are higher than that of the anionic geometries. Moreover, kct values of the compounds in cationic geometries are also higher. Similarly, all the compounds show lower λ values in the cationic state. Therefore, we can conclude that our studied compounds are primarily hole transporting in nature.
4.7 Excited state calculation
The λmax values along with their corresponding oscillator strength (fosc) and excitation energies (Eg) for both the phase are represented in Tables 7 and 8. From these tables we have observed that the λmax values of all the compounds in the solvent phase are higher than that of the gas phase. From Tables 7 and 8 we have observed a blue shift for the compounds substituted with electron donating groups at the acceptor part. On the other hand for the compounds substituted with electron withdrawing group at the acceptor part we have observed red shift. Compound PBDB-C4 exhibits the highest value of λmax in both gas and solvent phase. This may be attributed to the observed smallest ΔH–L value, high hole mobility, lowest inter-ring bond length and greater delocalization of π-electron cloud for this compound. It has already been observed that with increase in conjugation, the energies of HOMO increases while that of LUMO decreases which results into narrow band gap and hence maximum absorption. However, PBDB-C6 exhibits the lowest λmax value in both the phases. This is due to the observed highest ΔH–L value for this compound. However, in the solvent phase all the compounds get red shifted around 41–46 nm. Moreover, BLA parameter is also crucial to explain the variation of λmax values. BLA parameter is inversely related to absorption wavelength and we have obtained the highest value of BLA parameter for PBDB-C6.
| Compounds | λmax (nm) | Eg (eV) | fosc | Configuration | Orbital contribution (%) |
|---|---|---|---|---|---|
| PBDB-C1 | 555 | 2.23 | 3.19 | H → L | 86.00 |
| 373 | 3.32 | 0.09 | H → L+2 | 68.00 | |
| PBDB-C2 | 559 | 2.21 | 3.21 | H → L | 86.00 |
| 382 | 3.24 | 0.10 | H → L+2 | 64.00 | |
| PBDB-C3 | 555 | 2.23 | 3.21 | H → L | 86.00 |
| 377 | 3.29 | 0.07 | H → L+2 | 66.00 | |
| PBDB-C4 | 561 | 2.21 | 3.17 | H → L | 86.00 |
| 379 | 3.26 | 0.14 | H → L+2 | 69.00 | |
| PBDB-C5 | 552 | 2.25 | 3.34 | H → L | 86.00 |
| 385 | 3.22 | 0.08 | H → L+2 | 56.00 | |
| PBDB-C6 | 548 | 2.26 | 3.27 | H → L | 86.00 |
| 376 | 3.29 | 0.03 | H−1 → L | 44.00 | |
| PBDB-C7 | 557 | 2.23 | 3.14 | H → L | 85.00 |
| 378 | 3.28 | 0.21 | H → L+2 | 57.00 |
| Compounds | λmax (nm) | Eg (eV) | fosc | Configuration | Orbital contribution (%) |
|---|---|---|---|---|---|
| PBDB-C1 | 599 | 2.0684 | 3.4477 | H → L | 85.00 |
| 301 | 4.1103 | 0.4252 | H−2 → L+1 | 18.00 | |
| PBDB-C2 | 605 | 2.0480 | 3.4575 | H → L | 85.00 |
| 308 | 4.0216 | 0.4967 | H−5 → L+1 | 17.00 | |
| PBDB-C3 | 599 | 2.0693 | 3.4721 | H → L | 85.00 |
| 305 | 4.0585 | 0.5889 | H−2 → L+1 | 23.00 | |
| PBDB-C4 | 606 | 2.0458 | 3.4360 | H → L | 85.00 |
| 301 | 4.1075 | 0.6751 | H−5 → L+1 | 25.00 | |
| PBDB-C5 | 593 | 2.0880 | 3.5645 | H → L | 85.00 |
| 351 | 3.5235 | 0.4673 | H−3 → L | 24.00 | |
| PBDB-C6 | 590 | 2.1001 | 3.5251 | H → L | 85.00 |
| 342 | 3.6151 | 0.7905 | H−4 → L | 32.00 | |
| PBDB-C7 | 599 | 2.0667 | 3.3969 | H → L | 85.00 |
| 289 | 4.2767 | 0.8339 | H−4 → L+2 | 19.00 |
It has been observed from this calculation that all the compounds show broad absorption spectra. In this regard compound PBDB-C4 shows the maximum broadness in absorption spectra with the highest λmax value in both the phases. Conversely, compound PBDB-C6 shows most intense peak in both the phases (Fig. 9).
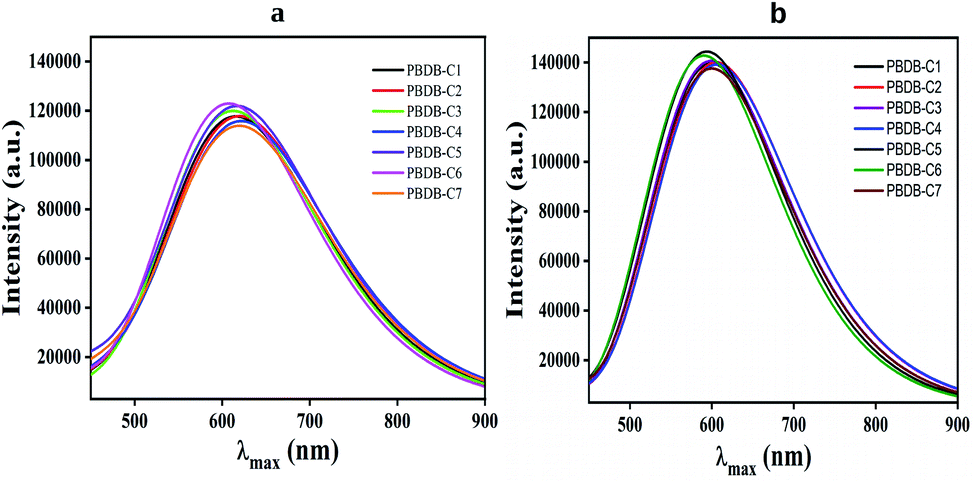 | ||
| Fig. 9 Plot of absorption spectra of all the compounds (a) gas phase (b) solvent phase. | ||
4.8 Properties of donor/acceptor (D/A) active layers
| Parameters (eV) | PBDB-C1 | PBDB-C2 | PBDB-C3 | PBDB-C4 | PBDB-C5 | PBDB-C6 | PBDB-C7 |
|---|---|---|---|---|---|---|---|
| ΔH–L | 2.06 | 1.92 | 2.00 | 1.95 | 2.03 | 2.09 | 2.03 |
| E1 | 1.18 | 1.60 | 1.66 | 1.64 | 1.70 | 1.76 | 1.71 |
| Eb | 0.88 | 0.32 | 0.33 | 0.31 | 0.33 | 0.34 | 0.31 |
| ΔG | −4.27 | −4.28 | −4.30 | −4.20 | −4.06 | −4.21 | −4.08 |
It is already reported that the suitable value of Eb for organic semiconducting materials is in the range 0.2 to 1 eV.73 However, the lower value of Eb signifies facile dissociation of excitons into free charge particles.74 From Table 9 it has been observed that the Eb values lie in the reported range for all compounds. Moreover, it is observed that all the compounds exhibit lower value of Eb except PBDB-C1. This implies a more convenient exciton dissociation in all compounds (except PBDB-C1).
The ground state electrons of the donor material get excited upon irradiation of light of suitable wavelength and transfer of these excited electrons to acceptor takes place when Gibb's free energy change (ΔG) becomes negative. The dissociation of electron–hole pairs into free charges takes place at the D/A interface and it spreads in the acceptor where, the electrons and holes should be collected by the respective electrodes. The value of ΔG can be calculated from the Rehm–Weller equation as follows:
| ΔG = IP(D) − EA(A) − E1 − ΔEb | (15) |
| Compounds | λmax (nm) | Eg (eV) | fosc | Configuration | Orbital contribution (%) |
|---|---|---|---|---|---|
| FREA/PBDB-C1 | 554 | 2.24 | 2.95 | H → L | 86.00 |
| 451 | 2.75 | 2.35 | H → L+1 | 68.00 | |
| FREA/PBDB-C2 | 597 | 2.07 | 1.37 | H → L | 83.00 |
| 470 | 2.63 | 2.38 | H → L+2 | 41.00 | |
| FREA/PBDB-C3 | 582 | 2.13 | 1.56 | H → L | 82.00 |
| 459 | 2.70 | 2.13 | H−1 → L+1 | 28.00 | |
| FREA/PBDB-C4 | 590 | 2.10 | 1.56 | H → L | 82.00 |
| 488 | 2.54 | 1.76 | H → L+1 | 70.00 | |
| FREA/PBDB-C5 | 573 | 2.16 | 1.64 | H → L | 81.00 |
| 470 | 2.64 | 2.50 | H → L+1 | 42.00 | |
| FREA/PBDB-C6 | 561 | 2.21 | 1.93 | H → L | 82.00 |
| 454 | 2.73 | 2.39 | H−1 → L+1 | 49.00 | |
| FREA/PBDB-C7 | 575 | 2.16 | 1.78 | H → L | 81.00 |
| 455 | 2.72 | 2.57 | H−1 → L+1 | 52.00 |
| Compounds | λmax (nm) | Eg (eV) | fosc | Configuration | Orbital contribution (%) |
|---|---|---|---|---|---|
| FREA/PBDB-C1 | 596 | 2.08 | 3.32 | H → L | 85.00 |
| 464 | 2.67 | 1.30 | H−1 → L+1 | 36.00 | |
| FREA/PBDB-C2 | 634 | 1.96 | 1.95 | H → L | 82.00 |
| 510 | 2.43 | 0.40 | H → L+2 | 59.00 | |
| FREA/PBDB-C3 | 620 | 2.00 | 2.23 | H → L | 82.00 |
| 504 | 2.46 | 0.97 | H → L+1 | 55.00 | |
| FREA/PBDB-C4 | 627 | 1.98 | 2.22 | H → L | 82.00 |
| 505 | 2.45 | 1.83 | H → L+1 | 62.00 | |
| FREA/PBDB-C5 | 608 | 2.04 | 2.394 | H → L | 82.00 |
| 490 | 2.53 | 2.30 | H → L+1 | 39.00 | |
| FREA/PBDB-C6 | 594 | 2.09 | 2.66 | H → L | 83.00 |
| 471 | 2.63 | 1.51 | H−1 → L+1 | 34.00 | |
| FREA/PBDB-C7 | 609 | 2.04 | 2.44 | H → L | 82.00 |
| 491 | 2.52 | 0.28 | H → L+1 | 39.00 |
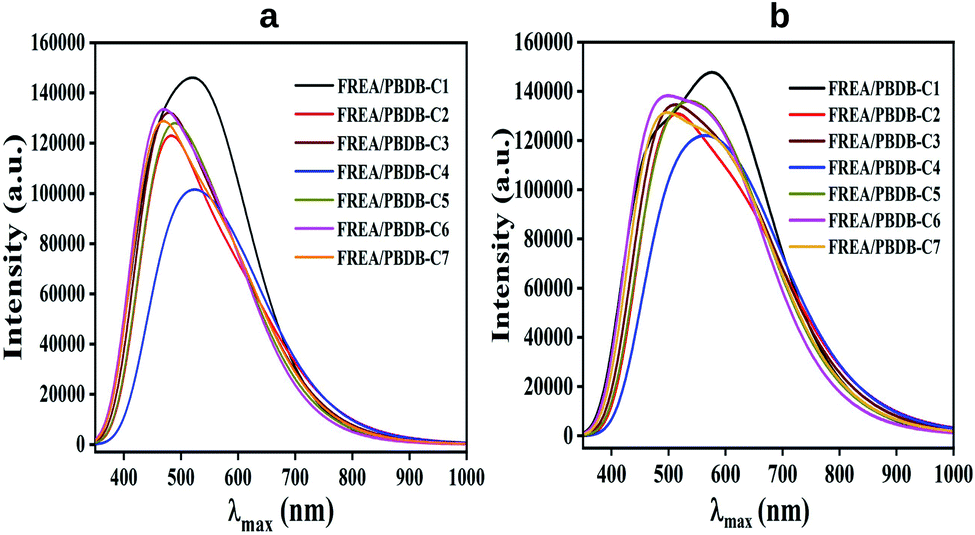 | ||
| Fig. 10 Plot of absorption spectra of D/A blends in gas and solvent phase. | ||
4.9 Photovoltaic performance
To evaluate the photovoltaic performance of our studied compounds we have calculated the short-circuit current density (Jsc), open-circuit voltage (Voc) and theoretical power conversion efficiency (PCE) (η) of the studied compounds. The obtained results have been reported in Table 12.| Compounds | Jsc (mA cm−2) | eVoc (eV) | νoc | FF | PCE (%) |
|---|---|---|---|---|---|
| PBDB-C1 | 10.07 | 1.9 | 73.07 | 0.928 | 17.75 |
| PBDB-C2 | 10.47 | 1.88 | 72.30 | 0.927 | 18.25 |
| PBDB-C3 | 10.06 | 1.9 | 73.07 | 0.928 | 17.75 |
| PBDB-C4 | 10.47 | 1.88 | 72.30 | 0.927 | 18.25 |
| PBDB-C5 | 9.80 | 1.92 | 73.85 | 0.929 | 17.48 |
| PBDB-C6 | 9.58 | 1.93 | 74.23 | 0.929 | 17.18 |
| PBDB-C7 | 10.07 | 1.9 | 73.07 | 0.928 | 17.76 |
It has been observed from Table 12 that the Jsc values show an increasing trend with the decreasing band gaps (ΔH–L). From this table it is observed that compounds PBDB-C1, PBDB-C3, PBDB-C5, PBDB-C6 and PBDB-C7 exhibits slightly lower value of Jsc due to higher band gap values. Moreover, among all the studied compounds, Jsc values are observed to be the highest (10.47 mA cm−2) for compounds PBDB-C2 and PBDB-C4 and lowest (9.58 mA cm−2) for PBDB-C6. It has also been observed from Table 12 that PBDB-C2 and PBDB-C4 exhibits the highest theoretical PCE (18.25%) among all the studied compounds. However, PBDB-C6 shows the lowest value of theoretical PCE (17.18%) due to the lowest Jsc value. These observations indicate that PBDB-C2 and PBDB-C4 can be chosen as potential candidates for application in photovoltaics. Besides, we can further emphasize that all the designed small molecules have the potential to act as ideal donor molecules for the fabrication of photovoltaic devices.
5 Conclusion
In this paper, we have tried to design some small potential donor molecules to fabricate efficient OSC. Besides, we have tried to tune their optoelectronic properties with the help of molecular engineering. The above analysis emphasises that the geometrical framework mainly depends on the steric hindrance played by the bulky substituents. Under this consideration, compound PBDB-C2 has the lowest dihedral angle and PBDB-C5 has the highest dihedral angle value in the gas phase. In the solvent phase PBDB-C6 possesses the lowest value of dihedral angle whereas, PBDB-C4 has the highest dihedral angle. From the calculation of dihedral angles of our studied compounds we find that PBDB-C2 in the gas phase and PBDB-C6 in the solvent phase will possess maximum planarity. In addition to this we have observed that parameter inter-ring bond length plays the vital role in tuning optoelectronic properties. From the inter-ring bond length study we can conclude that compound PBDB-C4 (gas phase) and PBDB-C2 (solvent phase) will be the best as they exhibit comparatively short inter-ring bond length.During study of electronic properties, we have investigated ΔH–L value, reorganization energy (λ), charge transfer rate (kct) and the electronic coupling matrix element (V). From these observed parameters we can conclude that our studied compounds primarily act as hole transporting materials. It is observed from the calculation of ΔH–L values that PBDB-C4 exhibits the lowest and PBDB-C6 exhibits the highest value of ΔH–L among all the studied compounds in both the phases. From this analysis we can conclude that compounds substituted with electron withdrawing group (–F) possess lower values of ΔH–L.
The absorption properties study reveals that PBDB-C4 exhibits the highest value of λmax in both gas and solvent phase. We have also studied the effect of solvent on the absorption properties of the molecules which illustrates that λmax values get red shifted in the solvent phase. Moreover, substitution by the electron withdrawing groups at the acceptor part leads to the red shift.
Absorption properties of the D/A blends manifest that all the compounds exhibit lower Eb values which in turn indicates their facile charge carrier separation at the D/A interface. However, the negative ΔG values also indicate the extent of ease of exciton dissociation of all the designed compounds. Moreover, the photovoltaic characteristics of the studied compounds reveal that PBDB-C2 and PBDB-C4 with the highest theoretical PCE of 18.21% can be chosen as potential candidate for application in OSCs.
In short, we can conclude that the attachment of electron withdrawing groups at the acceptor part of our studied molecules favors better tuning of optoelectronic properties. In a nutshell, we can conclude that our designed compounds may act as potential donor materials to fulfill the purpose of OSCs at the D/A interface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Department of Science and Technology (SB/FT/CS-077/2013), India for the financial support. The authors would like to acknowledge the University Grants Commission for UGC-BSR Research start-up-grant (NO.F.30.-122/2015(BSR)), Gauhati University for providing the research facilities and financial support.Notes and references
- S. Ahmed and D. J. Kalita, J. Chem. Phys., 2018, 149, 234906 CrossRef PubMed.
- H. Sahu and A. N. Panda, Phys. Chem. Chem. Phys., 2014, 16, 8563–8574 RSC.
- J. Lee, S. M. Lee, S. Chen, T. Kumari, S.-H. Kang, Y. Cho and C. Yang, Adv. Mater., 2019, 31, 1804762 CrossRef PubMed.
- Y. Yang, J. Wang, H. Xu, X. Zhan and X. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 18984–18992 CrossRef CAS PubMed.
- S. A. Siddique, M. Arshad, S. Naveed, M. Y. Mehboob, M. Adnan, R. Hussain, B. Ali, M. B. A. Siddique and X. Liu, RSC Adv., 2021, 11, 27570–27582 RSC.
- H. Sahu and A. N. Panda, Macromolecules, 2013, 46, 844–855 CrossRef CAS.
- A. E. Becquerel, C. R. Acad. Sci., 1839, 9, 561 Search PubMed.
- S. A. Siddique, S. Naveed, M. U. Alvi, M. Y. Mehboob, B. Ali, A. Rauf, M. B. A. Siddique, R. Hussain, M. Arshad and X. Liu, Comput. Theor. Chem., 2021, 1205, 39246–39261 Search PubMed.
- S. A. Siddique, M. B. A. Siddique, R. Hussain, X. Liu, M. Y. Mehboob, Z. Irshad and M. Adnan, Comput. Theor. Chem., 2020, 1191, 113045 CrossRef CAS.
- S. Ahmed, R. Dutta and D. J. Kalita, Chem. Phys. Lett., 2019, 730, 14–25 CrossRef CAS.
- B. Kan, H. Feng, X. Wan, F. Liu, X. Ke, Y. Wang, Y. Wang, H. Zhang, C. Li, J. Hou and Y. Chen, J. Am. Chem. Soc., 2017, 139, 4929–4934 CrossRef CAS PubMed.
- R. Wang, D. Zhang, S. Xie, J. Wang, Z. Zheng, D. Wei, X. Sun, H. Zhou and Y. Zhang, Nano Energy, 2018, 51, 736–744 CrossRef CAS.
- J. Yang, W.-L. Ding, Q.-S. Li and Z.-S. Li, J. Phys. Chem. Lett., 2022, 13, 916–922 CrossRef CAS PubMed.
- M. I. Khan, J. Iqbal, S. J. Akram, Y. A. El-Badry, M. Yaseen and R. A. Khera, J. Mol. Graphics Modell., 2022, 113, 108162 CrossRef CAS PubMed.
- X. Song, K. Zhang, R. Guo, K. Sun, Z. Zhou, S. Huang, L. Huber, M. Reus, J. Zhou and M. Schwartzkopf, et al., Adv. Mater., 2022, 2200907 CrossRef PubMed.
- L. Zhao, H. Ji, S. Li, X. Miao, Q. Shi, C. Zhu, W. Wang, S. Zhao, D. Huang and X. Dong, Org. Electron., 2022, 106495 CrossRef CAS.
- Y. Chen, X. Wan and G. Long, Acc. Chem. Res., 2011, 46, 2645–2655 CrossRef PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC.
- R. Dutta and D. J. Kalita, Eur. Phys. J. D, 2017, 71, 87–99 CrossRef.
- R. Dutta and D. J. Kalita, Comput. Theor. Chem., 2018, 1132, 42–49 CrossRef CAS.
- P. Wen, Z. Gao, R. Zhang, A. Li, F. Zhang, J. Li, J. Xie, Y. Wu, M. Wu and K. Guo, J. Mater. Chem. C, 2017, 5, 6136–6143 RSC.
- X. Yin, Q. An, J. Yu, Z. Xu, P. D. Deng, Y. Geng, B. Zhou, F. Zhang and W. Tang, Dyes Pigm., 2017, 140, 512–519 CrossRef CAS.
- Y.-L. Wang, L. Quan-Song and Z.-S. Li, Comput. Mater. Sci., 2019, 156, 252–259 CrossRef CAS.
- S. C. Rasmussen and S. J. Evenson, Prog. Polym. Sci., 2013, 38, 1773–1804 CrossRef CAS.
- J. Huang, H. Tang, C. Yan and G. Li, Cell Rep. Phys. Sci., 2021, 2, 100292 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 1–19 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ã. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision E.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- T. Sutradhar and A. Misra, J. Phys. Chem. A, 2018, 122, 4111–4120 CrossRef CAS PubMed.
- J. G. Roldan, A. Garzon, M. Moral, G. Garcia, T. Ruiz, M. P. F. Liencres, A. Navarro and M. F. Gomez, J. Chem. Phys., 2014, 140, 044908–044913 CrossRef PubMed.
- A. Garzon, J. Granadino, G. Garcia, M. Moral and M. F. Gomez, J. Chem. Phys., 2013, 138, 154902–154908 CrossRef.
- P. I. Koskin, A. E. Mostovich, E. Benassi and S. M. Kazantsev, J. Phys. Chem. C, 2017, 121, 23359–23369 CrossRef.
- M. Moral, G. Garcia, J. G. Roldan and M. F. Gomez, J. Chem. Phys., 2016, 144, 154902–154912 CrossRef PubMed.
- M. L. Hammock, A. N. Sokolov, R. M. Stoltenberg, B. D. Naab and Z. Bao, ACS Nano, 2012, 6, 3100–3108 CrossRef CAS.
- L. L. Estrella, P. M. Balanay and H. D. Kim, J. Phys. Chem. A, 2016, 120, 5917–5927 CrossRef CAS.
- T. I. Lima, S. A. d. Prado, B. L. J. Martins, P. H. d. O. Neto, A. M. Ceschin, W. F. d. Cunha and D. A. d. S. Filho, J. Phys. Chem. A, 2016, 120, 4944–4950 CrossRef PubMed.
- R. Dutta, B. Dey and D. J. Kalita, Chem. Phys. Lett., 2018, 707, 101–107 CrossRef CAS.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- J. M. Khoshkholgh, R. M. Abolhassani and F. Marsusi, Spectrochim. Acta, Part A, 2017, 181, 24–29 CrossRef.
- B. Carsten, M. J. Szarko, J. H. Son, W. Wang, L. Lu, F. He, S. B. S. Rolczynski, J. S. Lou, X. L. Chen and L. Yu, J. Am. Chem. Soc., 2011, 133, 20468–20475 CrossRef CAS PubMed.
- H. J. Son, B. Carsten, I. H. Jung and L. Yu, Energy Environ. Sci., 2012, 5, 7221–7676 RSC.
- J. M. Khoshkholgh, R. M. Abolhassani and F. Marsusi, Spectrochim. Acta, 2017, 181, 24–29 CrossRef PubMed.
- Y. L. Wang, Q. S. Li and Z. S. Li, Comput. Mater. Sci., 2019, 156, 252–259 CrossRef CAS.
- A. G. Baboul, L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 1999, 110, 7650–7657 CrossRef CAS.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- S. Ahmed and D. J. Kalita, J. Chem. Phys., 2018, 149, 234906 CrossRef.
- C. Walter, V. Kraemer and B. Engels, Int. J. Quantum Chem., 2017, 117, e25337 CrossRef.
- L. Goerigk, H. Kruse and S. Grimme, ChemPhysChem, 2011, 12, 3421–3433 CrossRef CAS PubMed.
- W.-Q. Dend and W. A. Goddard, J. Phys. Chem. B, 2004, 108, 8614–8621 CrossRef.
- C.-G. Zhan, J. A. Nichols and D. A. Dixon, J. Phys. Chem. A, 2003, 107, 4184–4195 CrossRef CAS.
- Y. Wen and Y. Liu, Adv. Mater., 2010, 22, 1331–1345 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2006, 127, 2339–2350 CrossRef PubMed.
- S. Chai, S.-H. Wen, J.-D. Huang and K.-L. Han, J. Comput. Chem., 2011, 32, 3218–3225 CrossRef CAS PubMed.
- P. Cias, C. Slugovc and G. Gescheidt, J. Phys. Chem. A, 2011, 115, 14519–14525 CrossRef CAS PubMed.
- M. Moral, A. Garzon, G. Garcia, J. M. Granadino-Roldan and M. Fernandez-Gomez, J. Phys. Chem. C, 2015, 119, 4588–4599 CrossRef CAS.
- L. L. Fu, H. Geng, G. Wang, Y. A. Duan, Y. Geng, Q. Peng, R. Zhu, T. Xiao, W. Wang and Y. Liao, Theor. Chem. Acc., 2018, 137, 1–11 Search PubMed.
- Y. Faree, M. Raissi, A. Fihey, Y. Pellegrin and E. Blart, Dyes Pigm., 2018, 148, 154–166 CrossRef.
- M. Moral, A. Garzon, J. Canales-Vazquez and J. C. Sancho-Garcia, J. Phys. Chem. C, 2016, 120, 24583–24596 CrossRef CAS.
- T. Qin, W. Zajaczkowski, W. Pisula, M. Baumgarten, M. Chen, M. Gao, W. Wilson, C. D. Easton, K. Mullen and S. E. Watkins, J. Am. Chem. Soc., 2014, 136, 6049–6055 CrossRef CAS.
- R. A. Marcus, J. Phys. Chem., 1963, 67, 679–701 Search PubMed.
- S. Zhang, X. Yang, Y. Numata and L. Han, Energy Environ. Sci., 2013, 6, 1443–1464 RSC.
- S. Ahmed, S. R. Bora, T. Chutia and D. J. Kalita, Phys. Chem. Chem. Phys., 2021, 23, 13190–13203 RSC.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- A. Irfan and A. Mahmood, J. Cluster Sci., 2018, 29, 359–365 CrossRef CAS.
- R. Zaier and S. Ayachi, Mater. Today Commun., 2021, 27, 102370 CrossRef CAS.
- C.-R. Zhang, X.-Y. Li, Y.-L. Shen, Y.-Z. Wu, Z.-J. Liu and H.-S. Chen, J. Phys. Chem. A, 2017, 121, 2655–2664 CrossRef CAS PubMed.
- R. Dutta, S. Ahmed and D. J. Kalita, Mater. Today Commun., 2020, 22, 100731 CrossRef CAS.
- R. Dutta, S. Kalita and D. J. Kalita, Comput. Theor. Chem., 2018, 1142, 39–44 CrossRef CAS.
- S. R. Bora and D. J. Kalita, RSC Adv., 2021, 11, 39246–39261 RSC.
- N. M. Baroja, J. Garin, J. Orduna, R. Andreu, M. Blesa, B. Villacampa, R. Alicante and S. Franco, J. Org. Chem., 2012, 77, 4634–4644 CrossRef PubMed.
- M. Weidelener, D. C. Wessendorf, J. Hanisch, E. Ahlswede, G. Gotz, M. Linden, G. Schulz, M. E. Osteritz, A. Mishra and P. Bauerle, Chem. Commun., 2013, 49, 10865–10867 RSC.
- Y.-L. Wang, Q.-S. Li and Z.-S. Li, Phys. Chem. Chem. Phys., 2017, 19, 23444 RSC.
- Y.-L. Wang, Q.-S. Li and Z.-S. Li, Comput. Mater. Sci., 2019, 156, 252–259 CrossRef CAS.
- H. Cha, S. Wheeler, S. Holliday, S. D. Dimitrov, A. Wadsworth, H. H. Lee, D. Baran, I. McCulloch and J. R. Durran, Adv. Funct. Mater., 2018, 28, 1704389 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: XYZ coordinates of the studied compounds, optimized structures of the compounds, energies of HOMO and LUMO, ΔH–L and λmax values of the reference compound, plots of density of states and spectral data of the PDOS spectra, frontier molecular orbital diagrams and optimized structures of the donor–acceptor blends. See https://doi.org/10.1039/d2ra01820a |
| This journal is © The Royal Society of Chemistry 2022 |