
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel one-pot synthesis of a library of 2-aryloxy-1,4-naphthoquinone derivatives. Determination of antifungal and antibacterial activity†
Katherine Chaves-Carballo a,
Guy V. Lamoureux*a,
Alice L. Perez*a,
Alexandre Bella Cruzb and
Valdir Cechinel Filhob
a,
Guy V. Lamoureux*a,
Alice L. Perez*a,
Alexandre Bella Cruzb and
Valdir Cechinel Filhob
aCentro de Investigaciones en Productos Naturales and Escuela de Química, Universidad de Costa Rica, San Pedro 2060, San José, Costa Rica. E-mail: alice.perez@ucr.ac.cr
bNúcleo de Investigações Químico-Farmacêuticas (NIQFAR), CCS, Universidade do Vale do Itajaí (UNIVALI), Itajaí, SC, Brazil
First published on 23rd June 2022
Abstract
The development of new antibiotics and inexpensive antifungals is an important field of research. Based on the privileged pharmacophore of lawsone, a series of phenolic ether derivatives of 1,4-naphthoquinone were synthesized easily in one step in reasonable yields. All the new compounds were characterized and tested as potential antifungal and antibacterial agents against Candida albicans, Escherichia coli and Staphylococcus aureus. Compound 55 has significant antibacterial action (as good as or better than the controls) against E. coli and S. aureus. Against C. albicans, compounds 38, 46, 47 and 60 were the best candidates as antifungals. Using a qualitative structure–activity analysis, a correlation between molar mass and antimicrobial activity was identified, regardless of the substituent group on the phenolic moiety, except for 55 and 63, where electronic effects seem more important. An in silico evaluation of the absorption, distribution, metabolism and excretion (ADME) for 37, 50, 55 and 63 was made, indicating that the classic Lipinski's rule of five applies in all cases.
1. Introduction
The treatment of bacterial infections by commercial antibiotics in the past two decades has been threatened by the increasing incidence of multi-drug resistant pathogens. The same can be said of fungal infections by antifungal agents. The misuse and overuse of antibiotics have contributed significantly to this health issue, compromising the public health systems in many countries.1–3Among the most pernicious resistant microbes are Candida albicans, Staphylococcus aureus and Escherichia coli, which are three of the main microorganisms responsible for nosocomial infections by formation of biofilms on medical devices.4,5 E. coli and S. aureus are part of the ESKAPE group, an acronym for difficult to treat bacteria that stands for Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter species.6–9
As part of the search for new effective antibiotics and antimicrobials, natural products have played a significant role as structure–activity templates. One family of bioactive natural products, naphthoquinones, are a group of chromatic pigments found inside the vacuoles and as secondary metabolites in some families of plants (Verbenaceae, Bignoniaceae, Lythraceae), bacteria (Streptomyces) and fungi (Fusarium). These compounds provide protection against microorganisms, acting as pro-oxidants by the production of reactive oxygen species (ROS) through radical chain reactions, and as potential electrophiles when they react with nucleophilic centers in biological molecules to form covalent bonds.10–12 Examples of common naphthoquinones (Fig. 1) include lapachol 1, extracted as a yellow solid from the tree bark of the Tabebuia species,13,14 and lawsone 2, obtained from the leaves of Lawsonia inermis (henna) and some trees of the Lythraceae family.15,16
 | ||
| Fig. 1 Lapachol 1 and lawsone 2, naturally occurring naphthoquinones. | ||
Both lapachol and lawsone exhibit antimalarial, anti-cancer, antitrypanosomal, antifungal and antibacterial properties that have been used as natural remedies in indigenous communities,16,17 and both structures can be modified for the development of new drugs that will potentially present selective and efficient mechanisms against malaria, cancer, bacteria and fungi.8,18–23 Fieser and co-workers, in work that extended over a quarter century, determined the physical and chemical properties of lapachol and lawsone and some synthetic derivatives, in order to evaluate their activity against Plasmodium species.24–31 Based on Fieser's seminal work, a series of metal complexes18,32,33 and C2 and/or C3 naphthoquinone substituted derivatives, such as alkyl,34,35 nitrogen,36–43 sulfur,39,41,44–48 and oxygen-containing21,22,42,49–51 molecules have been proposed as potential antitumoral, antibacterial, antiparasitical, antiviral and antifungal agents.
Among the oxygen-containing compounds are aryloxynaphthoquinones, which include a phenolic or naphtholic moiety. Phenols are important secondary metabolites in plants, because they act as pigments, antioxidants and antimicrobial agents against molds, fungi and bacteria through the modification of the permeability of the cell membrane, modification of the cell wall rigidity or induced changes in intracellular functions.52–54 Based on the suspected activity of phenolic derivatives, Bolognesi and co-workers have reported the synthesis of a small library of quinone-phenol hybrid compounds, starting with 2-bromo-1,4-naphthoquinone and 2-bromo-1,4-anthraquinone, using dimethylformamide (DMF) as solvent at room temperature and K2CO3 as base. The synthesized compounds were tested to determinate their antiparasitic action against Trypanosoma cruzi, T. brucei rhodesiense and Leishmania donovani. Some of these 1,4-naphthoquinone phenolic ethers showed interesting activity.49 Also, the antitumoral action of these compounds was tested against human dermal fibroblasts, IGROV-1 (ovarian) and HT-29 (colon) adenocarcinoma cells; significant cytotoxic activity was displayed by certain quinone–phenol hybrids. The active compounds had an inhibitory effect on glycolysis and mitochondrial respiration.22
Vázquez and co-workers have also synthesized some 2-aryloxynaphthoquinones under basic conditions (K2CO3 as base and DMF at room temperature), as well as 7-aryloxyquinolinquinones and 6-aryloxyfuranaphthoquinones, to evaluate their antitrypanosoidal action against the epimastigote form of T. cruzi, using nifurtimox as reference compound. Most of the prepared compounds were more active than the reference drug, and those that were more potent, were also more selective in comparison with J-774 cells.42 In a second study by the same authors, a new set of aryloxyquinones, synthesized under the same reaction conditions, was tested against the epimastigote form of T. cruzi in the presence of nifurtimox as reference drug. They discovered that the majority of the synthesized products showed higher potency than the control compound, but only two of them showed high selectivity towards nonneoplastic monkey kidney cells (Vero).50
Detailed in this article is our contribution to naphthoquinone research; a library of thirty phenoxy-1,4-naphthoquinone derivatives was prepared via a novel one-pot synthesis from 2-bromo-1,4-naphthoquinone (2-BrNQ) combined with the corresponding phenols in the presence of a base (CsOH, Cs2CO3 or KF/Al2O3) and toluene as solvent, which proceeded through an nucleophilic substitution reaction. The main objective of the study was to evaluate the antimicrobial action of this group of compounds against C. albicans, E. coli and S. aureus, in order to determine a posible correlation between these results and basic structural properties of aryloxy-1,4-naphthoquinones: molar mass and the substitution pattern at the phenolic moiety. Furthermore, an in silico evaluation of the absorption, distribution, metabolism and excretion (ADME) of the most active compounds was applied, based on the Lipinski's rule of five to establish the druglikeness of the prepared compounds.
2. Results and discussion
2.1 Chemistry
Thirty derivatives of 2-phenoxy-1,4-naphthoquinones (Fig. 2) were synthesized via a one-pot nucleophilic substitution reaction between 2-bromo-1,4-naphthoquinone (2-BrNQ) 3 and the corresponding phenolate, which was previously deprotonated by bases such as Cs2CO3 64, CsOH 65 or KF/Al2O3 66 (Table 1). The synthesis was performed using toluene as solvent, and under a positive nitrogen atmosphere or in the presence of a cellulose thimble with CaH2 to reduce the incidence of moisture, allowing the reaction to go to completion. In general, the compounds of interest were obtained with low-to-excellent yields (Table 1).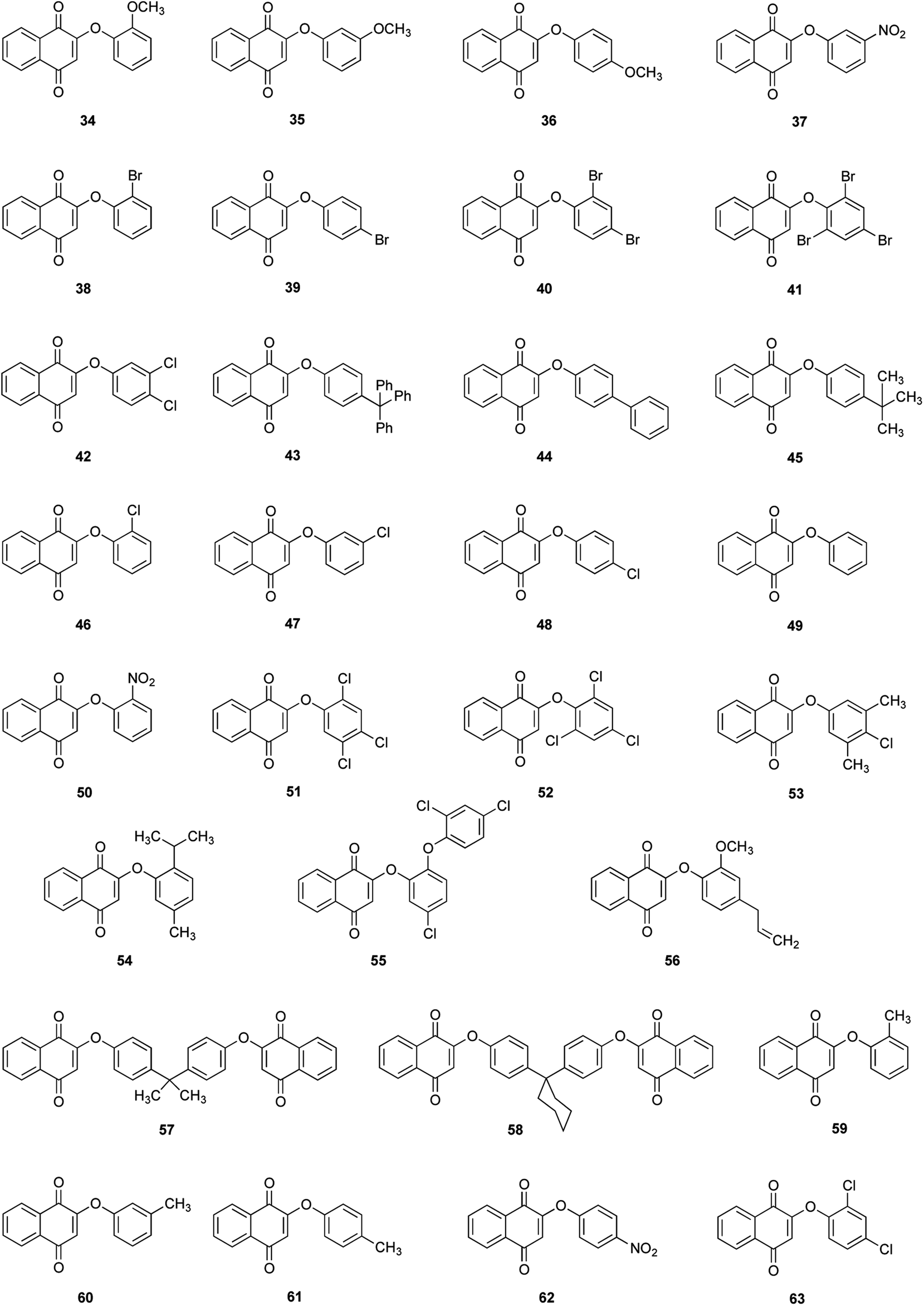 | ||
| Fig. 2 Synthesized phenolic ether derivatives of 1,4-naphthoquinones. | ||
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Base | Time (h) | Yield (%) | Purity (%) | Compound | Base | Time (h) | Yield (%) | Purity (%) |
| a N.D. = not determined. | |||||||||
| 34 | 64 | 2 | 68 | 99.9 | 49 | 64 | 4 | 77 | 98.6 |
| 35 | 64 | 2 | 52 | 95.9 | 50 | 64 | 4 | 30 | N.D. |
| 36 | 64 | 2 | 96 | 99.3 | 51 | 64 | 4 | 26 | 98.7 |
| 37 | 64 | 2 | 58 | 99.0 | 52 | 64 | 4 | 44 | 99.3 |
| 38 | 64 | 2 | 87 | 99.7 | 53 | 64 | 4 | 53 | 98.0 |
| 39 | 64 | 3 | 75 | 99.7 | 54 | 64 | 4 | 58 | 96.2 |
| 40 | 64 | 3 | 62 | 98.9 | 55 | 64 | 4 | 40 | 99.1 |
| 41 | 64 | 3 | 65 | 97.3 | 56 | 64 | 4 | 11 | 97.1 |
| 42 | 64 | 3 | 50 | 98.7 | 57 | 64 | 5 | 56 | 95.1 |
| 43 | 64 | 3 | 82 | 96.0 | 58 | 64 | 5 | 92 | 99.1 |
| 44 | 64 | 3 | 16 | 99.4 | 59 | 65 | 3 | 27 | 96.6 |
| 45 | 64 | 3 | 56 | 98.8 | 60 | 65 | 3 | 69 | 98.7 |
| 46 | 64 | 3 | 58 | 97.9 | 61 | 65 | 3 | 59 | 97.9 |
| 47 | 64 | 3 | 48 | 97.9 | 62 | 66 | 4 | 24 | 97.1 |
| 48 | 64 | 3 | 76 | 99.6 | 63 | 66 | 3 | 56 | 98.3 |
In all cases, a single compound was obtained: the product of the nucleophilic substitution at the ipso position, which was confirmed by X-ray crystal diffraction analysis (to be published) and by the presence of a signal around 6.0 ppm (singlet) in the 1H NMR spectrum of the crude reaction mixture, assigned to the hydrogen atom bonded to C3. This regioselectivity is a consequence of the transmission of the electronic effects of the carbonyl groups of the naphthoquinone moiety through the C2–C3 double bond, so that the 2-bromo-1,4-naphthoquinone can be regarded as a vinylogous acyl bromide.55 The C2 position has a better nucleofuge (–Br), which makes it a more reactive position towards a nucleophilic substitution reaction.
In terms of the kinetic reactivity and the thermodynamic stability of the phenolates, these factors are a result of the nature (electron donating or electron withdrawing) and the location of the substituents in the phenolic moiety.56,57 The nucleophilicity of the phenols is strongly influenced by their acidity, shape and polarizability, so the reaction conditions were chosen to enhance those properties and to maximize the recuperation of the product from the crude reaction mixture. For instance, the selection of the base depended on the pKa of the phenols, which is affected by the presence of electron-withdrawing (EW) or electron-donating (ED) groups (either by inductive or mesomeric effects); the former groups provide a more thermodynamically stable and less kinetically reactive phenolate than the latter. In this research, phenols containing one or more strongly electron-withdrawing (–NO2), weakly electron-withdrawing (–Cl, –Br), weakly electron-donating (alkyl and aromatic groups) and strongly electron-donating (–OCH3) groups were used, as well as sterically small and bulky substituents, so the resultant interval of reaction yields and reaction times depended on the nucleophilicity of each phenolate.58,59
The selection of toluene as the reaction solvent was based on the selective solubility of the aryloxy-1,4-naphthoquinone, 2-BrNQ and the phenol, but not the inorganic salts and the base residue. These solubility differences facilitated the work-up process, in terms of time and steps needed, given that the separation of the compound of interest from the reaction crude only involved a hot filtration step. Initially, aprotic solvents such as DMF and dimethylsulfoxide (DMSO) were considered, as previously reported in the literature,22,42,49,50 due to the greater solubility of the bases in comparison with non-polar solvents. However, the resulting crude reaction mixtures in DMF consisted of viscous syrups, which were not easy to concentrate or separate by filtration. The isolation of the product from these mixtures involved a series of unit operations (washing, extraction, filtration) that significantly lowered the yield.
The deprotonation of the phenol was the first step of the reaction. To promote this process, the phenol and the appropriate base were stirred under reflux in the reactor and left for 30 min under a dry nitrogen atmosphere or in the presence of a cellulose thimble with CaH2 (to remove trace water given the high atmospheric humidity levels under which the reactions were performed). As to the selection of the base, in the cases where intermediate or strongly electron-donating groups were present, CsOH or Cs2CO3 were chosen as bases. The preference of Cs2CO3 over CsOH rests on two criteria: (1) the former's lower hygroscopic character; even a trace of water produced subproducts due to attack on C2 in the naphthoquinone moiety to form lawsone, and (2) because carbonate is a weaker base with negligible nucleophilic character, compared to hydroxide ion.60–62 On the other hand, Al2O3/KF was reserved to the most acidic phenols. Although the pKa of HF is low (pKa = 3.14), it is suspected that KOH is produced in the early stages of deposition of KF over alumina, hence augmenting the basicity of the resulting material without the concomitant production of free hydroxide ion.63 K3PO4 and K2CO3 were also tested as bases, but due to the poor yields obtained and their low solubility in toluene, their use was discarded. In terms of solubility, cesium salts were preferred over other alkaline bases, given that the polarizability of the cesium ion increases their solubility in non-polar solvents.64
2.2 Biological activity of the naphthoquinone phenolic ethers
The results of the measured biological activity of the synthesized compounds (in MIC, μmol L−1) are shown in Table 2. From all the synthesized phenolic ethers, the triclosan derivative 55 was the most effective against S. aureus and E. coli (the MICs were comparable to the control compound, gentamicin sulphate, in a 1.7 and 0.97-fold, respectively), but not for C. albicans, so 55 shows significant antibacterial activity, but little antifungal activity against C. albicans. These results agree with the known bactericidal action of triclosan, attributed to the inhibition of the enoyl-acyl reductase protein transporting enzyme, which blocks lipid synthesis.65,66| Compound | Molar mass (g mol−1) | MIC (μmol L−1) | ||
|---|---|---|---|---|
| S. aureus | E. coli | C. albicans | ||
| 34 | 280.275 | 3568 | 892 | 3568 |
| 35 | 280.275 | 223 | 3568 | 446 |
| 36 | 280.275 | 1784 | 3568 | 3568 |
| 37 | 295.246 | 21.2 | 3387 | 3387 |
| 38 | 329.145 | 760 | 3038 | 190 |
| 39 | 329.145 | 3038 | 3038 | 3038 |
| 40 | 408.041 | 2450 | 2450 | 2450 |
| 41 | 486.937 | 2054 | 2054 | 2054 |
| 42 | 319.139 | 3133 | 3133 | 3133 |
| 43 | 492.563 | 2030 | 2030 | 2030 |
| 44 | 326.345 | 3064 | 3064 | 3064 |
| 45 | 306.355 | 3264 | 3264 | 3264 |
| 46 | 284.694 | 220 | 3512 | 220 |
| 47 | 284.694 | 878 | 3512 | 220 |
| 48 | 284.694 | 439 | 3512 | 439 |
| 49 | 250.249 | 250 | 250 | 4000 |
| 50 | 295.246 | 13 | 1693 | 85 |
| 51 | 353.584 | 2828 | 2828 | 2828 |
| 52 | 353.584 | 2828 | 2828 | 2828 |
| 53 | 312.747 | 3197 | 3197 | 3197 |
| 54 | 306.355 | 3262 | 3262 | 3262 |
| 55 | 445.679 | 0.34 | 0.56 | 2244 |
| 56 | 320.339 | 1561 | 3122 | 3122 |
| 57 | 540.562 | 925 | 1850 | 1850 |
| 58 | 580.626 | 431 | 1722 | 1722 |
| 60 | 264.276 | 473 | 3784 | 237 |
| 61 | 264.276 | 237 | 3784 | 473 |
| 62 | 295.246 | 212 | 3387 | 3387 |
| 63 | 319.139 | 49 | 3133 | 3133 |
| Gentamicin sulphate | 1488.8 | 0.52 | 0.52 | — |
| Ketoconazole | 541.43 | — | — | 0.56 |
In general, except for the triclosan derivative (55), the synthesized compounds exhibit a mild-to-low action towards the selected microorganisms, although the antibacterial effect is more significant than the antifungal, given the high MIC values against C. albicans. S. aureus, a Gram-positive bacteria, proved to be slightly more sensitive to the synthetized compounds than E. coli (a Gram-negative bacteria), accordingly to what is reported in the literature for some naphthoquinone derivatives.41,67 As to the phenolic moiety, the results agree with the reported sensitivity, which is slightly greater towards S. aureus. In terms of the reported phenolic antibacterial action, the sensitivity of these microorganisms towards the synthesized compounds agrees with what is reported for some phenols and polyphenols.52,68–70
A plot of the MIC versus the molar mass of each compound shows an interesting trend, in which the increase of lipophilic character, due to the greater non-polar section, seems to be the main factor (Fig. 3). Some exceptions are observed (mostly for S. aureus), which can be attributed to a greater impact of electronic effects, caused by the position of the substituent in the phenolic moiety and its electron-donor or electron-withdrawing nature.
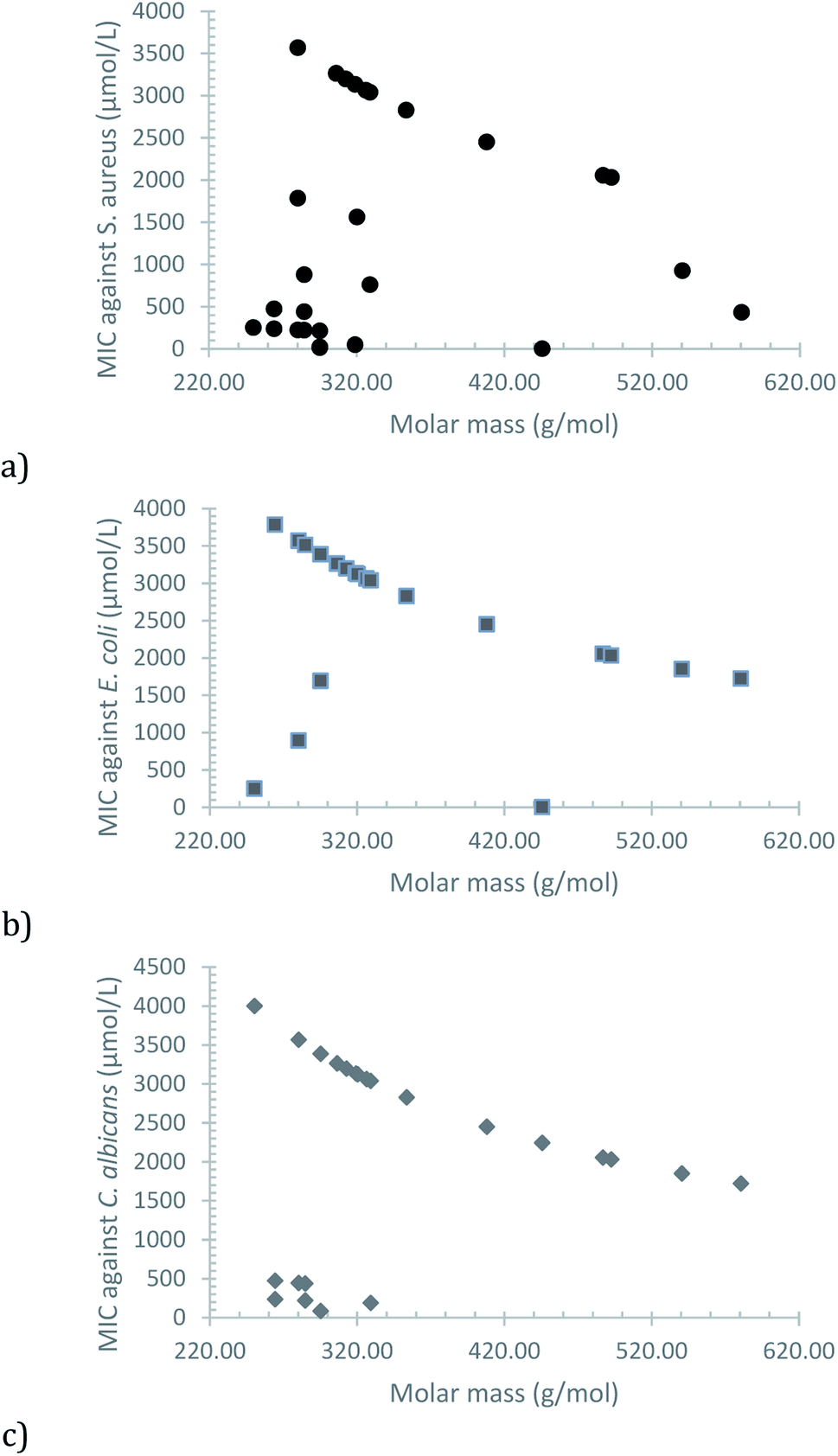 | ||
| Fig. 3 Observed correlation between MIC and molar mass of the synthesized compounds for (a) S. aureus, (b) E. coli and (c) C. albicans. | ||
To assess the effect of the substituent group in the phenolic moiety, in comparison to the unsubstituted compound (49), a ratio between the MIC of 49 and the corresponding MIC for each compound, was calculated (Table 3). In general, the presence of substituents seems to affect in a greater extent the action against C. albicans, given that all the substituted 2-aryloxynaphthoquinones display a greater MIC ratio, while for E. coli, only 55 significantly increases this ratio. For S. aureus, electron-withdrawing groups like –Cl and –NO2 demonstrate a greater ratio than electron-donating groups like –OCH3 or –CH3.
| Compound | Molar mass (g mol−1) | MIC49/MICcompound | ||
|---|---|---|---|---|
| S. aureus | E. coli | C. albicans | ||
| 34 | 280.275 | 0.07 | 0.28 | 1.12 |
| 35 | 280.275 | 1.12 | 0.07 | 8.97 |
| 36 | 280.275 | 0.14 | 0.07 | 1.12 |
| 37 | 295.246 | 11.79 | 0.07 | 1.18 |
| 38 | 329.145 | 0.33 | 0.08 | 21.05 |
| 39 | 329.145 | 0.08 | 0.08 | 1.32 |
| 40 | 408.041 | 0.10 | 0.10 | 1.63 |
| 41 | 486.937 | 0.12 | 0.12 | 1.95 |
| 42 | 319.139 | 0.08 | 0.08 | 1.28 |
| 43 | 492.563 | 0.12 | 0.12 | 1.97 |
| 44 | 326.345 | 0.08 | 0.08 | 1.31 |
| 45 | 306.355 | 0.08 | 0.08 | 1.23 |
| 46 | 284.694 | 1.14 | 0.07 | 18.18 |
| 47 | 284.694 | 0.28 | 0.07 | 18.18 |
| 48 | 284.694 | 0.57 | 0.07 | 9.11 |
| 49 | 250.249 | 1.00 | 1.00 | 1.00 |
| 50 | 295.246 | 19.23 | 0.15 | 47.06 |
| 51 | 353.584 | 0.09 | 0.09 | 1.41 |
| 52 | 353.584 | 0.09 | 0.09 | 1.41 |
| 53 | 312.747 | 0.08 | 0.08 | 1.25 |
| 54 | 306.355 | 0.08 | 0.08 | 1.23 |
| 55 | 445.679 | 735.29 | 446.43 | 1.78 |
| 56 | 320.339 | 0.16 | 0.08 | 1.28 |
| 57 | 540.562 | 0.27 | 0.14 | 2.16 |
| 58 | 580.626 | 0.58 | 0.15 | 2.32 |
| 60 | 264.276 | 1.05 | 0.07 | 8.46 |
| 61 | 264.276 | 0.53 | 0.07 | 16.88 |
| 62 | 295.246 | 1.18 | 0.07 | 1.18 |
| 63 | 319.139 | 5.10 | 0.08 | 1.28 |
In terms of the substituent's position in the phenolic moiety (o, m, or p), the three derivatives of –NO2 (37, 50, 62), –Cl (46, 47, 48) and –OCH3 (34, 35, 36) were considered (Table 4). For S. aureus, the presence of substituents in ortho and para positions exhibits a greater incidence in the activity as the electron-withdrawing effect increases, while for the meta position, the presence of weak EW groups like –Cl diminishes the activity, in comparison to –OCH3 and –NO2. As to the influence of the ortho and para positions on the activity, the former shows a greater effect than the latter. It is interesting to point out that the presence of two –Cl groups in ortho and para positions increases the activity in comparison to the monosubstituted compounds, as 63 is more effective towards S. aureus than 46 and 48.
| Substituent group | MIC (μmol L−1) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| S. aureus | E. coli | C. albicans | |||||||
| o | m | p | o | m | p | o | m | p | |
| –NO2 | 13 | 21.2 | 212 | 1693 | 3387 | 3387 | 85 | 3387 | 3387 |
| –Cl | 220 | 878 | 439 | 3512 | 3512 | 3512 | 220 | 220 | 439 |
| –OCH3 | 3568 | 223 | 1784 | 892 | 3568 | 3568 | 3568 | 446 | 3568 |
In the case of E. coli, the meta and para positions do not exert an important effect on the activity regardless of the substituent's nature, while for ortho, the presence of an ED group (–OCH3) lowers the MIC, in comparison to a strong EW as –NO2, or a weak EW group as –Cl. As to C. albicans, the presence of –NO2 exerts a more significant effect on the activity when it is located at the ortho position, while for –Cl, ortho and meta positions display the same effect, that is slightly greater than the para position. In the case of –OCH3, the location at the meta position produces an 8-fold increase in the activity when compared to the ortho or para positions.
3D Cartesian plots were made using Plotly Chart Studio online (https://chart-studio.plotly.com/feed/) to visualized 3D structural parameters. One of the correlations made is shown in Fig. 4. The value of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P were calculated using SwissADME web tool.71 As seen with the 2D correlations, there is no general tendency that can be drawn besides the fact that compound 55 seems singled out from the rest.
P were calculated using SwissADME web tool.71 As seen with the 2D correlations, there is no general tendency that can be drawn besides the fact that compound 55 seems singled out from the rest.
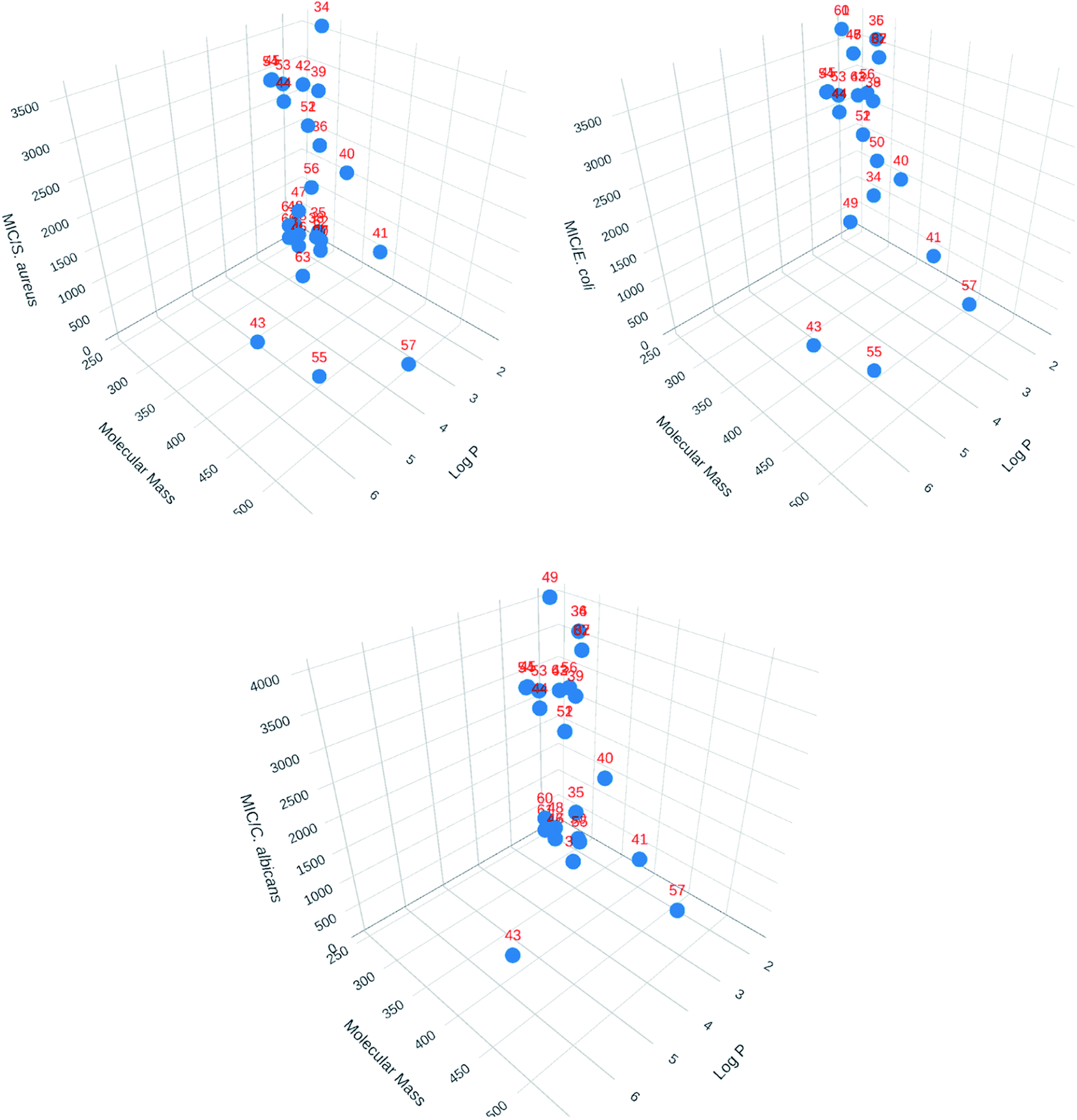 | ||
| Fig. 4 3D Cartesian correlations for the library of compounds (Plotly Chart Studio online, https://chart-studio.plotly.com/feed/). | ||
Evaluation in silico of the more active compounds (55, 37, 50 and 63) for their ADME (Absorption, Distribution, Metabolism and Excretion) properties using the SwissADME web tool71 was performed. The results obtained indicated the drug-likeness of all these phenol ethers, according to the classic Lipinski rule of five. The radar plots (Fig. 5), provided by SwissADME show how the degree of insaturation (INSATU) for a series of aromatic compounds seems to be the most important factor for the drug-likeness of this series (see the ESI† for more details). With further analysis, 55 shows intermediate solubility and lipophilic properties compared to the other three compounds. This behavior may explain the lower drug-likeness of 55 even though it is the most active of the whole group of molecules that have been synthesized.
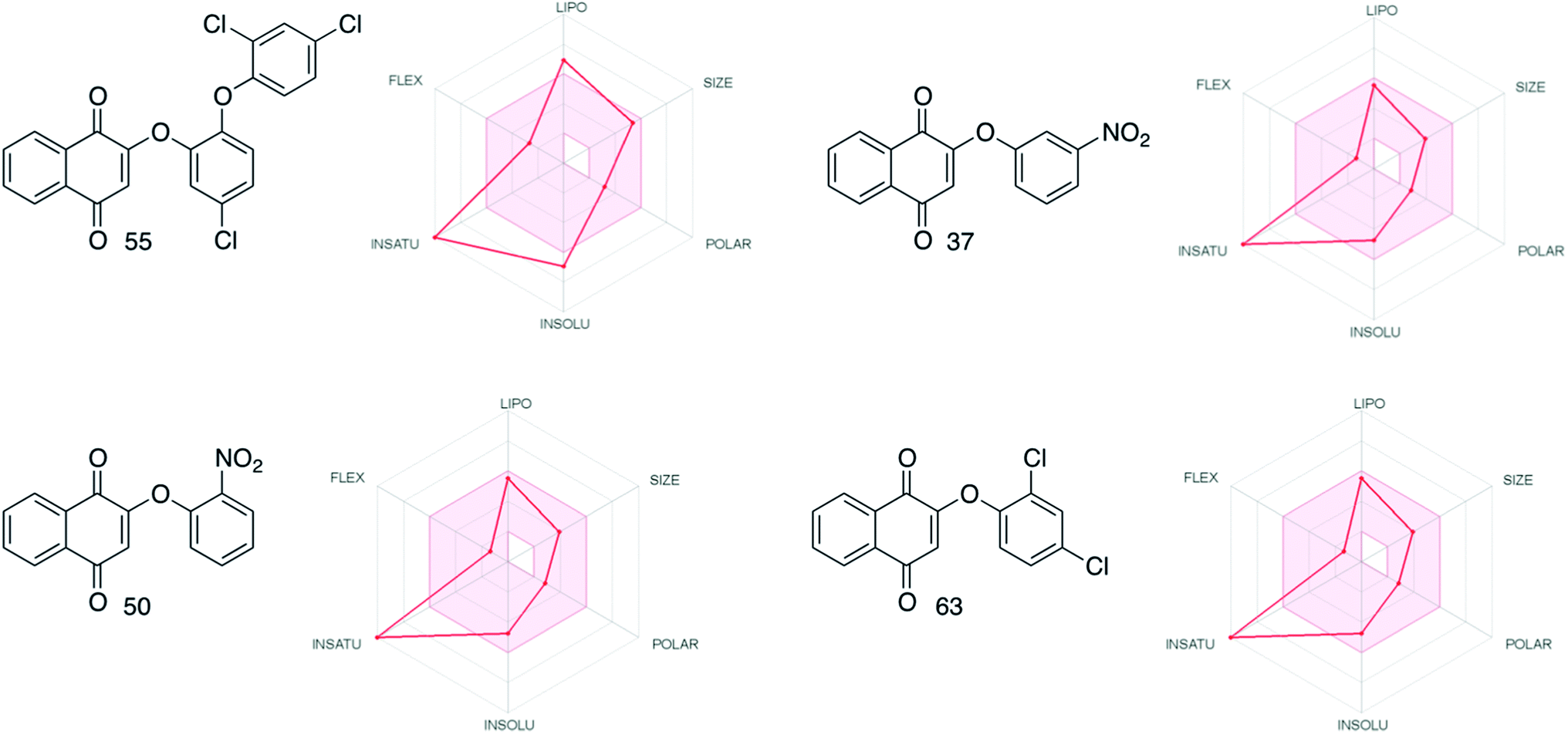 | ||
| Fig. 5 Radar plots comparison for the most active compounds (SwissADME web tool71). | ||
3. Conclusions
A series of 30 compounds with substituted aryloxy groups connected to the C2 position of 1,4-naphthoquinone were synthesized by an efficient procedure. All the compounds were characterized by spectroscopy and the purity was confirmed by HPLC. The activity of the compounds against selected microorganisms was slight to low, with the exception of the triclosan derivative 55, which has significant antibacterial action (as good as or better than the controls) against E. coli and S. aureus. This compound is being further investigated as a potent new antibacterial. In general, for all the compounds, the bactericidal activity is significantly greater against S. aureus than against E. coli. The fungicidal activity cannot be considered practical, although against C. albicans, compounds 38, 46, 47 and 60 were the best candidates. We feel that the antibiotic tendencies are worth pursuing with a wider range of compounds whereas the antifungal results show that this pharmacophore is not a viable target against fungi.In terms of quantitative structure–activity relationships (QSAR), there is no clear relationship between the activity and the structure of the compound in our library. We showed a general correlation between the activity and molar mass, which seems independent of type of substituent, but further QSAR need to be analyzed to clarify the correlation tendencies. It is interesting that moving away from ‘flatland’ and using more three-dimensional structures seems to benefit the activity; we are actively pursuing this lead.
The calculated ADME tendencies were ambiguous. Future research will take only the best candidates and run mouse and/or human liver microsomal preparations to determine experimental results. An expanded library of these bioactive compounds will be tested also against other biological strains or pathological microorganisms in the future, including an investigation of the mode of action of these types of compounds.
4. Experimental
4.1 Chemistry
All reagents (except eugenol, bisphenol A and bisphenol Z) were purchased from commercial suppliers [(Sigma-Aldrich-Merck)] and used without further purifications. A Radleys® tube carrousel reaction station (Radleys, UK), was used for the synthesis of the phenolic ethers. Thin-layer chromatography (TLC) was performed using silica gel Kieselgel 60 F254 (Merck, Darmstadt, Germany) precoated on aluminum sheets, with fluorescent indicator. Visualization of TLC plates was carried out by means of UV light or I2 staining. NMR spectra were recorded on a Bruker Ascend spectrometer (1H NMR at 600 MHz, 13C NMR at 150 MHz) at 25 °C, using tetramethylsilane as internal standard for 1H NMR spectra and CDCl3 as solvent for 1H NMR (7.26 ppm) and 13C NMR (77.2 ppm). All chemical shifts were reported in ppm, and the coupling constants (J), in Hz. IR spectra were recorded on a PerkinElmer 1000 FT and Varian 640-IR, on KBr pellets and Nujol® mulls. High resolution mass spectrometry spectra were measured on a quadrupole accelerated time-of-flight mass spectrometer (Synapt Acquity UPLC/TOF-MS, Waters). A PerkinElmer Series 200 liquid chromatographer with an UV/Vis detector was used, with a reverse phase C18 Discovery® (Supelco Analytical) chromatography column, dimensions of 25 cm × 4.6 mm and a particle size of 5 μm. The solvents employed (MeCN and MeOH) for the determination of the compounds' purity were HPLC grade (LiChrosolv, Merck). The melting point was determined using a Fisher-Johns melting point apparatus. Bisphenol A and bisphenol Z were synthesized according to the procedure described by Rahimi and Farhangzadeh (2001),72 and their characterization by 1H and 13C NMR agreed with the reported spectroscopic data. For the synthesis of 56, the corresponding mass of whole cloves (Syzygium aromaticum) was used to give approximately 1.25 mmol of eugenol, based on the reported average composition of essential oil.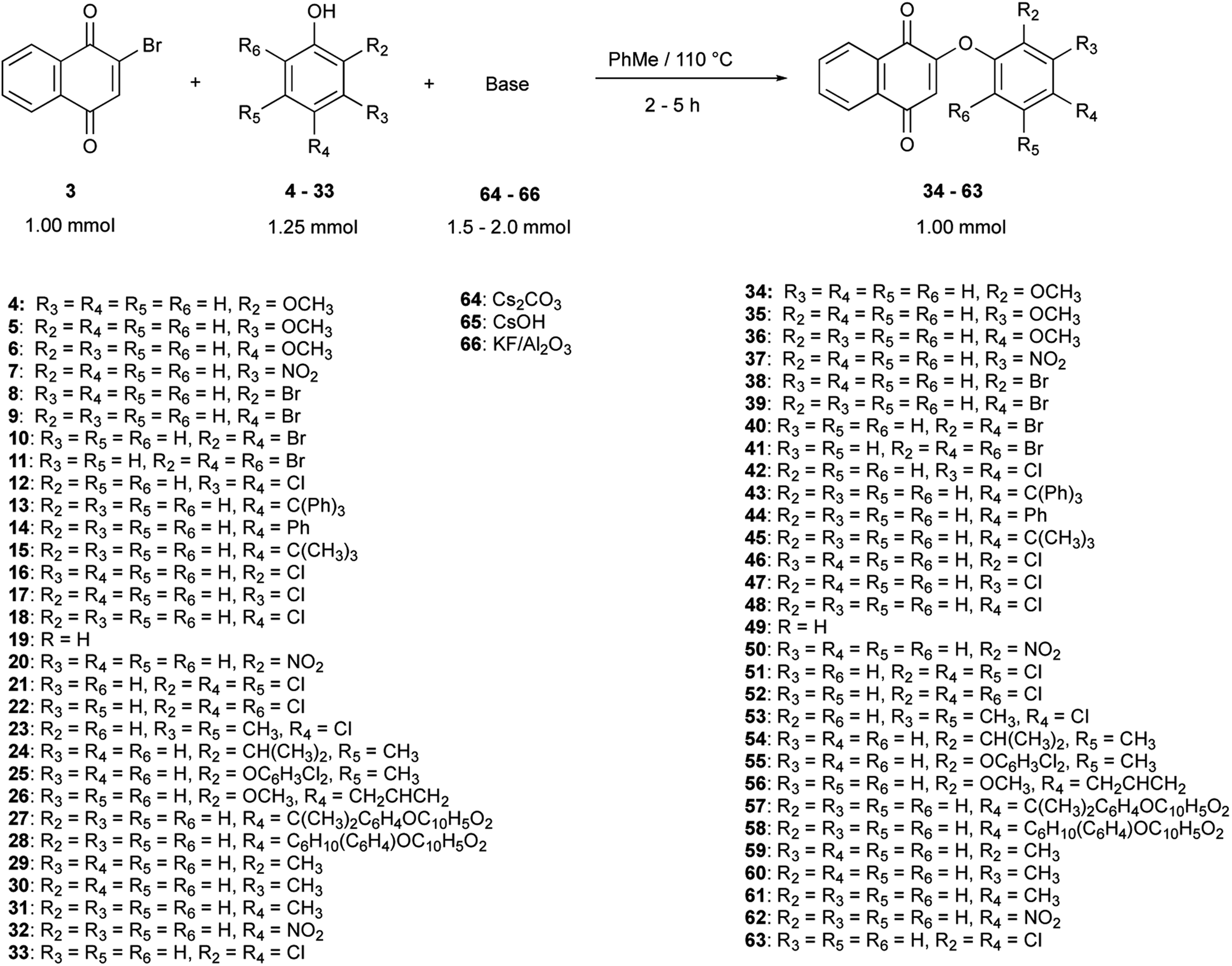 | ||
| Scheme 1 Synthesis of a series of 2-phenoxy-1,4-naphthoquinones (34–63). | ||
4.1.4.1 Hot extraction–filtration. After the reaction was deemed completed, 10 mL of hot methyl t-butyl ether (MTBE), isooctane, n-heptane, or dichloromethane (depending on the extraction) were added to the Radleys® reaction tube, and the mixture was then heated up to reflux. The hot solution (red to yellow in color) was filtered by gravity (Whatman #42 filter paper) and the filtrate is collected in a round bottom flask (RBF), while the inorganic salts (black to reddish solids) were retained in the tube or the filter paper. The extraction process was repeated until a clear filtrate was obtained (approx. 4 to 6 times). The filtrate was concentrated under vacuum with a rotary evaporator. The obtained solid product (red to yellow solid) was suspended in cold pentane to dissolve impurities of 2-BrNQ 3, and the suspension was vacuum filtered to obtain dry product.
4.1.4.2 Cold base wash. After the reaction was deemed completed, the crude reaction mixture was dissolved in MTBE or dichloromethane (15 mL) and the mixture was transferred to a Squibb separatory funnel. The solution was washed five times with 10 mL of cold NaOH 0.5 mol L−1 or a saturated cold solution of K2CO3 to deprotonate the unreacted phenol. Some phenolate solutions were red, blue, or purple. The combined organic phase was washed with three portions of distilled water to remove any excess of the basic solution, and the organic solution was dried over anhydrous sodium sulfate. The solvent was removed under vacuum with a rotary evaporator. The obtained solid product (red to yellow solid) was suspended in cold pentane to dissolve impurities of 2-BrNQ 3, and the suspension was vacuum filtered to obtain dry product.
4.1.4.3 Column chromatography. After the reaction was deemed completed, the crude reaction mixture was dissolved in dichloromethane and the solution was transferred to a silica gel (Kieselguhr, 230–400 mesh, Merck) chromatography column. The column was eluted with toluene, cyclohexane, MTBE, dichloromethane, or gradients of solvents, depending on the polarity of the compound and the amount of the phenol residue. The fractions were monitored by TLC and those that contained the product were combined and concentrated with a rotary evaporator. The obtained solid product (red to yellow solid) was suspended in cold pentane to dissolve impurities of 2-BrNQ 3, and the suspension was vacuum filtered to obtain dry product.
4.1.4.4 Recrystallization. The crude reaction mixture or the treated solid was dissolved in an appropriate hot solvent. The mixture was heated to boiling and then let to cool down to room temperature. Later, the mixture was cooled in a freezer at −20 °C and the suspension of crystals was then vacuum filtered. The obtained solid product (red to yellow solid) was suspended in cold pentane to dissolve impurities of 2-BrNQ 3, and the suspension was vacuum filtered to obtain dry product.
4.2 Determination of the purity of the synthesized compounds by HPLC
Solutions (500–1000 μmol L−1) of each prepared compound, the corresponding phenol and 2-BrNQ 3 were separately prepared in 10.00 mL volumetric flasks by the direct method, weighting the required mass of compound on the analytical balance to 4 decimals. In some cases, to complete the dissolution of all the solids, the flask with solvent was placed in an ultrasonic bath and sonicated until the solid dissolved completely. The solutions were filtered through a nylon syringe filters 4.6 cm × 0.20 μm (Agilent), and the filtrate was collected in a vial. 10 μL of the solution were injected in the liquid chromatograph. The elution was perform using an isocratic flow of 1 mL min−1 and total elution time of 25 minutes. The wavelengths used were 269 nm (36–45, 48, 49, 52, 53, 55–58, 62, 63), 274 nm (34, 51, 60) and 286 nm (35, 46, 47, 54, 59, 61). MeCN (100%) was used as eluent at 269 nm and 274 nm, and a mixture of 40% water/methanol at 286 nm. The purity was determined as the percentage ratio of the area of the compound's peak and the total area.4.3 Antimicrobial assays of the synthesized compounds against C. albicans, E. coli and S. aureus via the diffusion method73–75
The bacteria used were cultivated on Mueller-Hinton agar (MHA – Difco) at 35 °C for 24 h. Cell suspension in saline (0.86%) was adjusted to give a final concentration of 1.5 × 108 cell per mL, standardized with 0.5 on the McFarland scale (λ = 530 nm).76 The fungi were cultivated on Sabouraud dextrose agar (SDA-Difco). The yeast was prepared according to Pfaller et al. (1988),77 adjusting the suspension to give a final concentration of between 1.0 × 106 and 5.0 × 106 cell per mL, also standardized with 0.5 on the McFarland scale (λ = 530 nm).
Author contributions
The manuscript was written through contributions of all authors.Declarations of interest
The authors have no competing financial interests to declare.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the Vicerrectoría de Investigación of the University of Costa Rica (grant number 809-B2-027), Vicerrectoría de Investigación-CONICIT-CNPq (grant number 809-B0-500, CONICIT grant number IQ-0001-09), and the Núcleo de Investigações Químico-Farmacêuticas from UNIVALI for the biological activity essays. Also, we would like to recognize the support of RIBIOFAR/CYTED/CNPq during the realization of the present work.References
- World Health Organization, Antimicrobial resistance. Fact Sheets - Detail. 2020, cited 2020 Oct 3, available from: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance Search PubMed.
- CDC, Antibiotic resistance threats in the United States, 2019, Centers for Disease Control and Prevention, Atlanta, Georgia, 2019, available from: https://stacks.cdc.gov/view/cdc/82532 Search PubMed.
- CDC, Antibiotic/antimicrobial resistance, U.S. Department of Health and Human Services, Atlanta, GA, 2020, cited 2020 Apr 28, available from: https://www.cdc.gov/drugresistance/about.html Search PubMed.
- C. J. Nobile and A. D. Johnson, Candida albicans Biofilm and Human Disease, Annu. Rev. Microbiol., 2015, 59, 71–92 CrossRef PubMed
.
- Z. Khatoon, C. D. McTiernan, E. J. Suuronen, T. F. Mah and E. I. Alarcon, Bacterial biofilm formation on implantable devices and approaches to its treatment and prevention, Heliyon, 2018, 4(12), e01067, DOI:10.1016/j.heliyon.2018.e01067
- M. Lu, C. Yu, X. Cui, J. Shi, L. Yuan and S. Sun, Gentamicin synergises with azoles against drug-resistant Candida albicans, Int. J. Antimicrob. Agents, 2018, 51(1), 107–114, DOI:10.1016/j.ijantimicag.2017.09.012
- M. Lu, T. Li, J. Wan, X. Li, L. Yuan and S. Sun, Antifungal effects of phytocompounds on Candida species alone and in combination with fluconazole, Int. J. Antimicrob. Agents, 2017, 49(2), 125–136, DOI:10.1016/j.ijantimicag.2016.10.021
- J. S. Novais, C. S. Moreira, A. C. J. A. Silva, R. S. Loureiro, A. M. Sá Figueiredo and V. F. Ferreira, et al., Antibacterial naphthoquinone derivatives targeting resistant strain Gram-negative bacteria in biofilms, Microb. Pathog., 2018, 118, 105–114, DOI:10.1016/j.micpath.2018.03.024
- S. Jahani-Sherafat, M. Razaghi, V. D. Rosenthal, E. Tajeddin, S. Seyedjavadi and M. Rashidan, et al., Device-associated infection rates and bacterial resistance in six academic teaching hospitals of Iran: findings from the International Nocosomial Infection Control Consortium (INICC), J. Infect. Public Health, 2015, 8(6), 553–561, DOI:10.1016/j.jiph.2015.04.028
- L. I. López-López, D. S. Nery-Flores, Y. S. Silva-Belmares and A. Sáenz-Galindo, Naphthoquinones: biological properties and synthesis of lawsone and derivatives — a structured review, Vitae, 2014, 21(3), 248–258 Search PubMed
- Y. Kumagai, Y. Shinkai, T. Miura and A. K. Cho, The chemical biology of naphthoquinones and its environmental implications, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 221–247 CrossRef CAS PubMed
- D. O. Futuro, P. G. Ferreira, C. D. Nicoletti, L. P. Borba-Santos, F. C. Da Silva and S. Rozental, et al., The antifungal activity of naphthoquinones: an integrative review, An Acad. Bras Ciências, 2018, 90(1), 1187–1214 CrossRef CAS PubMed
- F. Epifano, S. Genovese, S. Fiorito, V. Mathieu and R. Kiss, Lapachol and its congeners as anticancer agents: a review, Phytochem. Rev., 2014, 13(1), 37–49 CrossRef
- J. R. Gómez-Castellanos, J. M. Prieto and M. Heinrich, Red Lapacho (Tabebuia impetiginosa)--a global ethnopharmacological commodity? J, Ethnopharmacol, 2009, 121(1), 1–13 CrossRef PubMed
- K. Ghédira and P. Goetz, Le henné Lawsonia inermis L. (Lythraceae), Phytothérapie, 2017, 15, 85–90 CrossRef
- G. Chaudhary, S. Goyal, P. Poonia and L. Linn, Lawsonia inermis Linnaeus: a phytopharmacological review, Int. J. Pharm. Sci. Drug Res., 2010, 2(2), 91–98 Search PubMed
- R. C. Da Silveira e Sá and M. Oliveira-Guerra, Reproductive toxicity of lapachol in adult male Wistar rats submitted to short-term treatment, Phytother Res., 2007, 21(7), 658–662 CrossRef PubMed
- M. I. F. Barbosa, R. S. Corrêa, K. M. De Oliveira, C. Rodrigues, J. Ellena and O. R. Nascimento, et al., Antiparasitic activities of novel ruthenium/lapachol complexes, J. Inorg. Biochem., 2014, 136, 33–39, DOI:10.1016/j.jinorgbio.2014.03.009
- N. Bao, J. Ou, W. Shi, N. Li, L. Chen and J. Sun, Highly Efficient Synthesis and Structure–Activity Relationships of a Small Library of Substituted 1,4-Naphthoquinones, Eur. J. Org. Chem., 2018, 2018(19), 2254–2258 CrossRef CAS
- R. Pradhan, From body art to anticancer activities: perspectives on medicinal properties of Henna, Curr. Drug Targets, 2012, 13(14), 1777–1798 CrossRef CAS PubMed
- S. El Hage, M. Ane, J. L. Stigliani, M. Marjorie, H. Vial and G. Baziard-Mouysset, et al., Synthesis and antimalarial activity of new atovaquone derivatives, Eur. J. Med. Chem., 2009, 44(11), 4778–4782 CrossRef CAS PubMed
- F. Prati, C. Bergamini, M. T. Molina, F. Falchi, A. Cavalli and M. L. Bolognesi, 2-Phenoxy-1,4-naphthoquinones: from a multitarget antitrypanosomal to a potential antitumor profile, J. Med. Chem., 2015, 58(16), 6422–6434, DOI:10.1021/acs.jmedchem.5b00748
- X. B. Shen, Y. Wang, X. Z. Han, L. Q. Sheng, F. F. Wu and X. Liu, Design, synthesis and anticancer activity of naphthoquinone derivatives, J. Enzym. Inhib. Med. Chem., 2020, 35(1), 773–785, DOI:10.1080/14756366.2020.1740693
- L. F. Fieser, The alkylation of hydroxynaphthoquinone I. Ortho-ethers, J. Am. Chem. Soc., 1926, 48(11), 2922–2937 CrossRef CAS
- L. F. Fieser, The alkylation of hydroxynaphthoquinone II. Carbon alkylation, J. Am. Chem. Soc., 1926, 48(12), 3201 CrossRef CAS
- L. F. Fieser and M. A. Peters, The potentials and the decomposition reactions of ortho quinones in acid solution, J. Am. Chem. Soc., 1931, 53(2), 793–805 CrossRef CAS
- L. F. Fieser, E. Berliner, F. J. Bondhus, F. C. Chang, W. G. Dauben and M. G. Ettlinger, et al., Naphthoquinone antimalarials. I. General survey, J. Am. Chem. Soc., 1948, 70(10), 3151–3155 CrossRef CAS PubMed
- L. F. Fieser, The tautomerism of hydroxyquinones, J. Am. Chem. Soc., 1928, 50(2), 439–465 CrossRef CAS
- L. F. Fieser and G. Ettlinger, Naphthoquinone Antimalarials. XV. Distribution between Organic Solvents and Aqueous Buffers, J. Am. Chem. Soc., 1948, 70(10), 3228–3232 CrossRef CAS
- L. F. Fieser and M. Fieser, Naphthoquinone antimalarials. XII. The Hooker oxidation reaction, J. Am. Chem. Soc., 1948, 70(10), 3215–3222 CrossRef CAS PubMed
- L. F. Fieser and E. Berliner, Naphthoquinone antimalarials. IV-XI Synthesis, J. Am. Chem. Soc., 1947, 70(10), 3174–3215 CrossRef PubMed
- P. M. García-Barrantes, G. V. Lamoureux, A. L. Pérez, R. N. García-Sánchez, A. R. Martínez and A. San Feliciano, Synthesis and biological evaluation of novel ferrocene-naphthoquinones as antiplasmodial agents, Eur. J. Med. Chem., 2013, 70, 548–557 CrossRef PubMed
- S. Salunke-Gawali, E. Pereira, U. A. Dar and S. Bhand, Metal complexes of hydroxynaphthoquinones: lawsone, bis-lawsone, lapachol, plumbagin and juglone, J. Mol. Struct., 2017, 1148, 435–458, DOI:10.1016/j.molstruc.2017.06.130
- M. Janeczko, O. M. Demchuk, D. Strzelecka, K. Kubiński and M. Masłyk, New family of antimicrobial agents derived from 1,4-naphthoquinone, Eur. J. Med. Chem., 2016, 124, 1019 CrossRef CAS PubMed
- L. W. Bieber, P. J. R. Nero and R. M. Generino, Regioselective alkylation of substituted quinones by trialkylboranes, Tetrahedron Lett., 1999, 40, 4473–4476 CrossRef CAS
- V. K. Tandon, D. B. Yadav, R. V. Singh, A. K. Chaturvedi and P. K. Shukla, Synthesis and biological evaluation of novel
(L)-alpha-amino acid methyl ester, heteroalkyl, and aryl substituted 1,4-naphthoquinone derivatives as antifungal and antibacterial agents, Bioorg. Med. Chem. Lett., 2005, 15(23), 5324–5328 CrossRef CAS
- C. G. T. Oliveira, F. F. Miranda, V. F. Ferreira, C. C. Freitas, R. F. Rabello and J. M. Carballido, et al., Synthesis and Antimicrobial Evaluation of 3-Hydrazino-Naphthoquinones as Analogs of Lapachol, J. Braz. Chem. Soc., 2001, 12(3), 339–345 CrossRef CAS
- I. Crespo, A. Cousido-siah, A. Podjarny, H. Pratsinis, D. Kletsas and F. X. Ruiz, et al., Design, synthesis, structure-activity relationships and X-ray structural studies of novel 1-oxo-pyrimido[4,5-c]quinoline-2-acetic acid derivatives as selective and potent inhibitors of human aldose reductase, Eur. J. Med. Chem., 2018, 152, 160–174 CrossRef CAS PubMed
- R. Almeida, W. Valença, L. Rosa, C. de Simone, S. de Castro and J. Barbosa, et al., Synthesis of quinone imine and sulphur-containing compounds with antitumor and trypanocidal activities: redox and biological implications, RSC Med. Chem., 2020, 18–20 Search PubMed
- M. A. Souza, S. Johann, L. A. R. dos Santos Lima, F. F. Campos, I. C. Mendes and H. Beraldo, et al., The antimicrobial activity of lapachol and its thiosemicarbazone and semicarbazone derivatives, Mem. Inst. Oswaldo Cruz, 2013, 108(3), 342–351 CrossRef CAS
- P. Ravichandiran, S. Sheet, D. Premnath, A. R. Kim and D. J. Yoo, 1,4-Naphthoquinone Analogues: Potent Antibacterial Agents and Mode of Action Evaluation, Molecules, 2019, 24(7), 1437 CrossRef CAS PubMed
- K. Vázquez, C. Espinosa-Bustos, J. Soto-Delgado, R. A. Tapia, J. Varela and E. Birriel, et al., New aryloxy-quinone derivatives as potential anti-chagasic agents: synthesis, trypanosomicidal activity, electrochemical properties, pharmacophore elucidation and 3D-QSAR analysis, RSC Adv., 2015, 5(80), 65153–65166, 10.1039/C5RA10122K
- U. Sharma, D. Katoch, S. Sood, N. Kumar, B. Singh and A. Thakur, et al., Synthesis, antibacterial and antifungal activity of 2-amino-1,4-naphthoquinones using silica-supported perchloric acid (HClO4-SiO2) as a mild, recyclable and highly efficient heterogeneous catalyst, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2013, 52(11), 1431–1440 Search PubMed
- T. Sreelatha, S. Kandhasamy, R. Dinesh, S. Shruthy, S. Shweta and D. Mukesh, et al., Synthesis and SAR study of novel anticancer and antimicrobial naphthoquinone amide derivatives, Bioorg. Med. Chem. Lett., 2014, 24(15), 3647–3651, DOI:10.1016/j.bmcl.2014.04.080
- C. K. Ryu, J. Y. Shim, M. J. Chae, I. H. Choi and S. H. Jeong, Synthesis and antifungal activity of 2/3-arylthio- and 2,3-bis(arylthio)-5-hydroxy-/5-methoxy-1,4-naphthoquinones, Eur. J. Med. Chem., 2005, 40(5), 438–444 CrossRef CAS PubMed
- L. I. López-López, E. Leyva and R. F. de la Cruz-García, Las naftoquinonas: Más que pigmentos naturales, Rev. Mex. Cienc. Farm., 2011, 42(1), 6–17 Search PubMed
- V. K. Tandon, H. K. Maurya, N. N. Mishra and P. K. Shukla, Design, synthesis and biological evaluation of novel nitrogen and sulfur containing hetero-1,4-naphthoquinones as potent antifungal and antibacterial agents, Eur. J. Med. Chem., 2009, 44(8), 3130–3137, DOI:10.1016/j.ejmech.2009.03.006
- V. K. Tandon, R. V. Singh and D. B. Yadav, Synthesis and evaluation of novel 1,4-naphthoquinone derivatives as antiviral, antifungal and anticancer agents, Bioorg. Med. Chem. Lett., 2004, 14(11), 2901–2904 CrossRef CAS PubMed
- M. L. Bolognesi, F. Lizzi, R. Perozzo, R. Brun and A. Cavalli, Synthesis of a small library of 2-phenoxy-1,4-naphthoquinone and 2-phenoxy-1,4-anthraquinone derivatives bearing anti-trypanosomal and anti-leishmanial activity, Bioorg. Med. Chem. Lett., 2008 Apr 1, 18(7), 2272–2276 Search PubMed
- C. Espinosa-Bustos, K. Vázquez, J. Varela, H. Cerecetto, M. Paulino and R. Segura, et al., New aryloxy-quinone derivatives with promising activity on Trypanosoma cruzi, Arch. Pharm., 2020, 353(1), 1–11 CrossRef PubMed
- J. M. Sánchez-Calvo, G. R. Barbero, G. Guerrero-Vásquez, A. G. Durán, M. Macías and M. A. Rodríguez-Iglesias, et al., Synthesis, antibacterial and antifungal activities of naphthoquinone derivatives: a structure–activity relationship study, Med. Chem. Res., 2016, 25(6), 1274–1285 CrossRef
- L. Bouarab-Chibane, V. Forquet, P. Lantéri, Y. Clément, L. Léonard-Akkari and N. Oulahal, et al., Antibacterial properties of polyphenols: characterization and QSAR (Quantitative structure-activity relationship) models, Front. Microbiol., 2019, 10, 829 CrossRef PubMed
- N. Guo, J. Liu, X. Wu, X. Bi and L. Yu, Antifungal activity of thymol against clinical isolates of fluconazole-sensitive and -resistant Candida albicans, J. Med. Microbiol., 2009, 58(Pt 8), 1074–1079 CrossRef CAS PubMed
- T. P. T. Cushnie and A. J. Lamb, Recent advances in understanding the antibacterial properties of flavonoids, Int. J. Antimicrob. Agents, 2011, 38(2), 99–107 CrossRef CAS PubMed
- R. C. Fuson, The Principle of Vinylogy, Chem. Rev., 1935, 16(1), 1–27, DOI:10.1021/cr60053a001
- M. T. Nguyen, E. S. Kryachko and L. G. Vanquickenborne, General and theoretical aspects of phenols, in The Chemistry of Phenols, Part 1, ed. Z. Rappaport, John Wiley & Sons Ltd., West Sussex, 2003, pp. 1–198 Search PubMed
- S. Pratihar and S. Roy, Nucleophilicity and site selectivity of commonly used arenes and heteroarenes, J. Org. Chem., 2010, 75(15), 4957–4963 CrossRef CAS PubMed
- A. R. Katritzky and R. D. Topsom, The sigma and pi inductive effects, J. Chem. Educ., 1971, 48(7), 427–430 CrossRef CAS
- M. B. Smith and J. March, March's Advanced Organic Chemistry, 6th edn, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007, p. 2374 Search PubMed
- U. Sankar, C. Raju and R. Uma, Cesium carbonate mediated exclusive dialkylation of active methylene compounds, Curr. Chem. Lett., 2012, 1, 123–132 CrossRef CAS
- N. A. Hawryluk and B. B. Snider, Alcohol inversion using cesium carboxylates and DMAP in toluene, J. Org. Chem., 2000 Dec 1, 65(24), 8379–8380 Search PubMed
- T. Flessner and S. Doye, Cesium carbonate: a powerful inorganic base in organic synthesis, J. Prakt. Chem., 1999, 341(2), 186–190 CrossRef CAS
- E. Gershonov, I. Columbus and Y. Zafrani, Facile hydrolysis-based chemical destruction of the warfare agents VX, GB, and HD by alumina-supported fluoride reagents, J. Org. Chem., 2009, 74(1), 329–338 CrossRef CAS PubMed
- A. Steinmetz, The broad scope of Cesium salts in Organic Chemistry, Acros Catalysts Cesium, 2015, pp. 4–5, https://studylib.net/doc/8821748/catalysts-cesium Search PubMed.
- G. D. Yadav and B. G. Motirale, Novelties of Solid−Liquid Phase Transfer Catalyzed Synthesis of Triclosan from Potassium 2,4-Dichlorophenolate and 2,5-Dichlorophenol, Ind. Eng. Chem. Res., 2008, 47(23), 9055–9060 CrossRef CAS
- C. W. Levy, A. Roujeinikova, S. Sedelnikova, P. J. Baker and J. B. Rafferty, Molecular basis of triclosan activity, Nature, 1999, 398(6726), 383–384 CrossRef CAS PubMed
- P. Ravichandiran, M. Masłyk, S. Sheet, M. Janeczko, D. Premnath and A. R. Kim, et al., Synthesis and Antimicrobial Evaluation of 1,4-Naphthoquinone Derivatives as Potential Antibacterial Agents, ChemistryOpen, 2019, 8(5), 589–600 CrossRef CAS PubMed
- H. J. Dorman and S. G. Deans, Antimicrobial agents from plants: antibacterial activity of plant volatile oils, J. Appl. Microbiol., 2000, 88(2), 308–316 CrossRef CAS PubMed
- C. Pavesi, L. A. Banks and T. Hudaib, Antifungal and antibacterial activities of eugenol and non-polar extract of Syzygium aromaticum L, J. Pharm. Sci. Res., 2018, 10(2), 337–339 CAS
- T. Taguri, T. Tanaka and I. Kouno, Antimicrobial
activity of 10 different plant polyphenols against bacteria causing food-borne disease, Biol. Pharm. Bull., 2004, 27(12), 1965–1969 CrossRef CAS PubMed
- A. Daina, O. Michielin and V. Zoete, SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 1–13, DOI:10.1038/srep42717
- A. Rahimi and S. Farhangzadeh, Kinetics Study of Bisphenol A Synthesis by Condensation Reaction, Iran. Polym. J., 2001, 10(1), 29–32 CAS
- T. Tristão, F. Campos-Buzzi, R. Corrêa, R. Cruz, V. Cechinel Filho and A. Bella Cruz, Antimicrobial and cytotoxicity potential of acetamido, amino and nitrochalcones, Arzneim. Forsch., 2012, 62, 590–594 CrossRef PubMed
- D. S. Dos Santos, J. V. Oberger, R. Niero, T. Wagner, F. Delle Monache and A. B. Cruz, et al., Seasonal phytochemical study and antimicrobial potential of Vetiveria zizanioides roots, Acta Pharm., 2014, 64(4), 495–501 CrossRef PubMed
- L. G. Faqueti, I. V. Farias, E. C. Sabedot, F. Delle Monache, A. San Feliciano and I. T. A. Schuquel, et al., Macrocarpal-like Compounds from Eugenia umbelliflora Fruits and Their Antibacterial Activity, J. Agric. Food Chem., 2015, 63(37), 8151–8155 CrossRef CAS PubMed
- Clinical and Laboratory Standards Institute, Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, Approved Standard M07-A8, CLSI, Wayne, PA, 2009 Search PubMed
- M. A. Pfaller, L. Burmeister, M. S. Bartlett and M. G. Rinaldi, Multicenter evaluation of four methods of yeast inoculum preparation, J. Clin. Microbiol., 1988, 26(8), 1437–1441 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01814d |
| This journal is © The Royal Society of Chemistry 2022 |