DOI:
10.1039/D2RA01797K
(Review Article)
RSC Adv., 2022,
12, 18245-18265
Retracted Article: Recent advances in visible-light graphitic carbon nitride (g-C3N4) photocatalysts for chemical transformations
Received
19th March 2022
, Accepted 4th June 2022
First published on 21st June 2022
Abstract
Graphitic carbon nitride (g-C3N4) has emerged as a new research hotspot, attracting broad interdisciplinary attention in the form of metal-free and visible-light-responsive photocatalysts in the field of solar energy conversion and environmental remediation. These photocatalysts have evolved as attractive candidates due to their non-toxicity, chemical stability, efficient light absorption capacity in the visible and near-infrared regions, and adaptability as a platform for the fabrication of hybrid materials. This review mainly describes the latest advances in g-C3N4 photocatalysts for chemical transformations. In addition, the typical applications of g-C3N4-based photocatalysts involving organic transformation reactions are discussed (synthesis of heterocycles, hydrosulfonylation, hydration, oxygenation, arylation, coupling reactions, etc.).
 Praveen P. Singh | Currently, Praveen P. Singh is an Assistant Professor in the Department of Chemistry at the United College of Engineering and Research, Prayagraj, India. He obtained his BSc and M.Sc. in Organic Chemistry from T. D. P. G. College (V. B. S Purvanchal University) Jaunpur and DPhil. from the Department of Chemistry, University of Allahabad, India. His current research interests include the development of synthetic receptors for the recognition of biological target structures and the application of visible light chemical photocatalysis towards organic synthesis. |
 Vishal Srivastava | Currently, Vishal Srivastava is an Assistant Professor, Department of Chemistry, C.M.P. College, (Constituent P.G. College of Central University of Allahabad) Prayagraj, India. He completed his undergraduate (BSc) and post-graduate (M.Sc.) studies in Organic Chemistry and Doctoral Degree (DPhil.) in the Department of Chemistry, University of Allahabad, India. His current research involves the design of novel biologically active photoredox-catalysed synthetic organic compounds. |
1 Introduction
Currently, photocatalytic technology is considered as one of the most important approaches to address the global energy and environmental issues.1 Inspired by photosynthesis in plants2 (Fig. 1), synthetic organic chemists are always trying to develop new organic reactions that can be carried out under green, ecofriendly, and sustainable conditions.
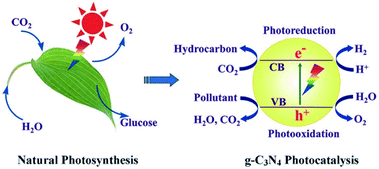 |
| | Fig. 1 Natural photosynthesis and the photocatalytic process using g-C3N4. Adapted with permission from ref. 2. Copyright 2013, Elsevier. | |
Polymeric g-C3N4 contains the earth-abundant carbon and nitrogen elements. Polymeric carbon nitride (PCN) is a highly active photocatalyst. The history of C3N4 polymers and their precursors can be traced back to 1834, where the embryonic form of melon, which is a linear polymer of interconnected tri-s-triazine through secondary nitrogen, was first developed by Berzelius and named by Liebig.3,4 The utilization of g-C3N4 in the field of heterogeneous catalysis started in 2006.5 Wang and co-workers, in 2009,6 reported the discovery of g-C3N4 as a metal-free conjugated semiconductor photocatalyst for H2 evolution, potentially shifting the research focus from inorganic to polymeric conjugated semiconductor photocatalysts.7
The photocatalytic activities of g-C3N4 are greatly influenced by its structure,8 including its electronic structure, crystal structure, nanostructure and/or heterostructure. In recent years, several synthetic techniques and effective alternative processes have been suggested for the optimization of the photoactivity of g-C3N4 with respect to nanostructure design, electronic structure modulation, crystal structure engineering and heterostructure construction. Both doping and copolymerization have been considered to be an efficient technique to incorporate external impurities in g-C3N4 for the modulation of its electronic structure and photocatalytic activity. In the context of nanostructure design, nanostructures with controllable morphologies not only facilitate mass transfer in catalysis but also accelerate the collection and separation of electron–hole pairs to drive relevant reactions.9 Different synthetic pathways have been developed for g-C3N4 materials with nanopores and nanoscale geometrical shapes including hard/soft templating strategies, supramolecular preorganization, solvothermal technology and exfoliation strategies. With respect to crystal-structure engineering, ionothermal synthesis reveals great opportunities for the synthesis of highly crystalline, more completely condensed g-C3N4 with enhanced photocatalytic activity.10a The synthesis of hetero-structured composites by coupling g-C3N4 with other semiconductors not only suppresses the recombination of photoinduced charges, but also endows the composites with enhanced properties or novel features as a result of their synergistic effects.10b Accordingly, the enlightened design of its structure at different levels can provide new insights into the creation of high-performance g-C3N4 materials11a–f for the efficient conversion of solar energy.
Polymeric g-C3N4 is an inexpensive and high-performance nanomaterial, exhibiting abundant scope for adaptable applications.11g Polymeric g-C3N4 nanocomposites have been extensively explored for photocatalytic degradation,11h–o environmental remediation,11p removal of heavy metals from contaminated water,11q high-dispersed photocatalyst for phenol mineralization and E. coli disinfection,11r photocatalytic hydrogen production and water detoxification,11s photocatalytic water splitting,11t photo-reduction11u of CO2 and efficient removal of tetracycline hydrochloride.11v
Polymeric g-C3N4 has been previously applied in the above-mentioned process but its application in the field of organic transformation has been limited. Visible light photocatalysis is a robust emerging field for organic transformation and highly appreciable. The combination of visible light photocatalysis with polymeric g-C3N4 has enabled organic chemists to carry out the functionalization of organic compounds. In continuation of our work on photocatalysed organic synthesis,12 this review mainly describes the latest advances in g-C3N4 photocatalysts for chemical transformations and may be particularly useful for those who are already involved in the area of heterocycles and interested in the use of visible light photocatalysis in combination with polymeric g-C3N4 for bioactive heterocyclic synthesis.
2 Graphitic carbon nitride photocatalyst
Several methods have been reported to synthesize g-C3N4, including chemical vapor deposition,13 plasma sputtering reaction deposition,14 solvothermal method,15 and solid-state reaction.16 However, these protocols are hindered by their strict conditions and the limited access to expensive instruments. Thus, due to its simplicity, high yield and cost effectiveness, thermal polycondensation has become the most popular method for the preparation of g-C3N4.17–19 Given that g-C3N4 only contains earth-abundant elements (carbon and nitrogen), it can be prepared with accessible raw materials at low cost. Several inexpensive N-rich precursors with a C–N core structure, such as melamine,20,21 cyanamide,22,23 dicyandiamide,24,25 urea,26,27 thiourea,28,29 and mixtures thereof,30 have been widely used to prepare g-C3N4 via the thermal polycondensation method (Fig. 2a).31 In the facile annealing process, the N-rich molecules are polymerized at the temperature of 450–660 °C to form g-C3N4. Yan et al. performed a thermogravimetric–differential scanning calorimetry (TGA-DSC) analysis to investigate the condensation processes of melamine.21 They observed that the sublimation and condensation of melamine occur at 297–390 °C, and the subsequent deamination and decomposition of the material happen at a higher temperature of 545–630 °C. The g-C3N4 synthesized from urea normally has a higher specific surface area than that synthesized from melamine,32 and g-C3N4 obtained from urea shows a larger bandgap than that prepared from thiourea.33 Zhang and co-workers studied the effect of annealing temperature on the properties of the obtained g-C3N4.28 They found that an increase of the temperature in the range of 450–600 °C could be favorable for the polycondensation of g-C3N4, and thus could improve the electron delocalization in its aromatic structure. However, a further increase in the temperature to over 650 °C could result in the decomposition of g-C3N4 into small particles with increased bandgaps. Therefore, the annealing temperature is a key parameter in the synthesis of g-C3N4 nanosheets.
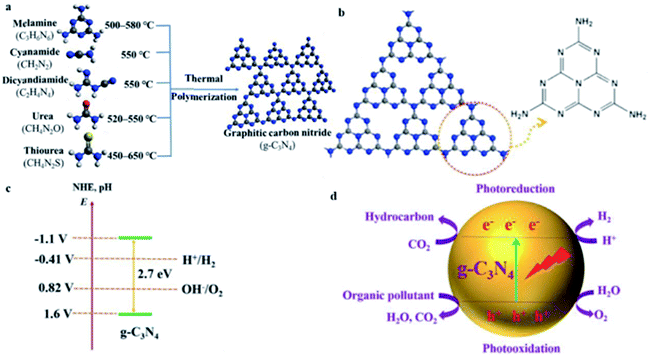 |
| | Fig. 2 (a) Schematic illustration of the process for the synthesis of g-C3N4. Adapted with permission from ref. 31. (b) Tri-s-triazine-based structure of g-C3N4. Adapted with permission from ref. 38. (c) Band structure of g-C3N4. Adapted with permission from ref. 45a. (d) Photocatalysis of g-C3N4. Adapted with permission from ref. 45b. | |
With a graphite-like layered structure, g-C3N4 exists in the form of 2D nanosheets based on s-triazine (Fig. 2b) or tri-s-triazine (Fig. 2b) tectonic units interconnected by tertiary amines.34 In this case, given that the nitride pore size and the electronic environment of the N atom are different, the energetic stability of the allotropes is different. Among the allotropes of C3N4, tri-s-triazine-based g-C3N4 is the most stable under ambient conditions.35–37 Benefitting from the aromatic C–N heterocycles and their high condensation, g-C3N4 with tri-s-triazine rings is thermally stable at a temperature of up to 600 °C in the air, as confirmed by thermogravimetric analysis.38 Furthermore, the strong interlayer van der Waals interactions render g-C3N4 chemically stable, which can resist many organic solvents, as well as acid and alkaline solutions.38,39 The 2D g-C3N4 nanosheets also provide a large specific surface area due to their graphite-like layered structure, with a theoretical value for monolayer g-C3N4 of up to 2500 m2 g−1,40 which is favorable for the adsorption of reactants in solution, and thus can facilitate surface reactions, including photocatalysis.
The presence of sp2-hybridized C and N results in the unique established π-conjugated electronic structures of g-C3N4. Wang et al. performed density functional theory (DFT) calculations,41 which revealed that the valence band (VB) maximum and the conduction band (CB) minimum consist predominantly of the N and C pz orbitals of g-C3N4, respectively, which has a direct bandgap of ∼2.7 eV. As depicted in Fig. 2c, the bandgap of g-C3N4 is around 2.7 eV, enabling the harvesting of visible light up to 460 nm. The conduction band (CB) position of g-C3N4 is approximately −1.1 eV and its valence band (VB) position is about +1.6 eV with reference to the normal hydrogen electrode, thereby meeting the requirements for photocatalytic water splitting to produce O2 and H2, respectively.41,42 Compared with TiO2, which has a bandgap of 3.0–3.2 eV and can only respond to UV light,43,44 g-C3N4 possesses more suitable band positions to enable water splitting and shows photocatalytic activity in the visible-light region. For the first time, in 2009,41 g-C3N4 was used as a metal-free photocatalyst for water splitting to generate H2 under visible-light irradiation. Since this pioneering work and because of its aforementioned intriguing properties, g-C3N4 has been attracting significant interest from the scientific community and has drawn worldwide attention in recent years. Furthermore, g-C3N4 exhibits photocatalytic activity for CO2 reduction,11u pollutant degradation,11p and photocatalytic degradation11h–o via photoredox processes (Fig. 2d).
The design of g-C3N4-based materials includes zero-dimensional quantum dots (QDs);46,47 one-dimensional (1D) nanowires,48,49 nanorods,50–52 nanotubes,53,54 and nanobelts;55,56 two-dimensional (2D) nanosheets57,58 and nanoplates;59 and 3D microspheres,60 micro-flowers,61 sandwich-like structures,62 and porous structures.63–65 For instance, combining theoretical calculation results with practical experimental findings, the structure and electronic and optical properties of modified g-C3N4 QDs were systematically and comprehensively explored by Bandyopadhyay et al.47 The preparation method is relatively simple for single-component 3D g-C3N4. For example, Chen et al. designed a bottom-up supramolecular self-assembly pathway to synthesize 3D porous g-C3N4 assembled from highly crystalline and ultrathin nanosheets, which achieved both stability and overall photocatalytic water splitting activity.66 The unique electronic structure of g-C3N4-based photocatalysts has prompted their applications in selective organic transformations,67 including oxidation, reduction, and coupling reactions. Given that these organic transformation reactions play a key role in industrial and fine chemical synthesis, it is very attractive for nontoxic g-C3N4-based photocatalysts driven by visible light to accomplish the corresponding reactions under mild conditions.
3 Functionalization of g-C3N4
3.1 Elemental doping of g-C3N4
Elemental doping involves the incorporation of heteroatoms or impurities (metal or nonmetal elements) in g-C3N4 to regulate its absorption, electronic, and physicochemical properties. There are two main types of elemental doping, i.e., nonmetal doping and metal doping. In the field of photocatalysis, bandgap engineering of g-C3N4 via the incorporation of anions and cations or the co-doping of both plays a predominant role in modulating its light absorption and redox band potentials for targeted photocatalytic applications. Hitherto, numerous studies on the doping of anions such as O,68–72 C,73,74 P,75–77 S,78–80 B,81–85 I,86,87 F88 and some combinations thereof89–91 in g-C3N4 have been extensively reported.
Besides metal elements, nonmetal elemental doping is also a promising way to govern the electronic structure of g-C3N4. Jiang and co-workers92 fabricated defective oxygen-doped porous C3N4 via the polymerization of ammonium persulfate and dicyandiamide. Compared to g-C3N4, the bandgap of the oxygen-doped g-C3N4 increased from 2.70 to 2.75 eV, and its CB decreased from −0.60 to −0.81 V. The more negative CB position indicates more powerful reduction ability, and hence the photocatalytic performance will increase significantly.
3.2 Copolymerization of g-C3N4
The photocatalytic reactivity of g-C3N4 is attributed to its π-conjugated framework because of its sp2-hybridization system. However, the aromatic π system of g-C3N4 in its pristine form generally encounters intrinsic drawbacks, namely, the rapid recombination of charge carriers, inadequate absorption and utilization of sunlight and low surface areas as a result of the defective polymerization of its organic framework, considerably restricting its photochemical functions.93–95 Thus, employing the merits of the polymeric π-conjugated system, it is envisaged that the delocalization of the π electrons can be further extended by modifying and anchoring the existing molecular structure of g-C3N4 with other structure-matching aromatic groups or organic additives during the copolymerization process to modulate its intrinsic properties.96–100 The copolymerization approach is regarded as typical molecular doping to adjust the conventional π systems, electronic properties, optical absorption, band structure and importantly photocatalytic performance of g-C3N4.101–105
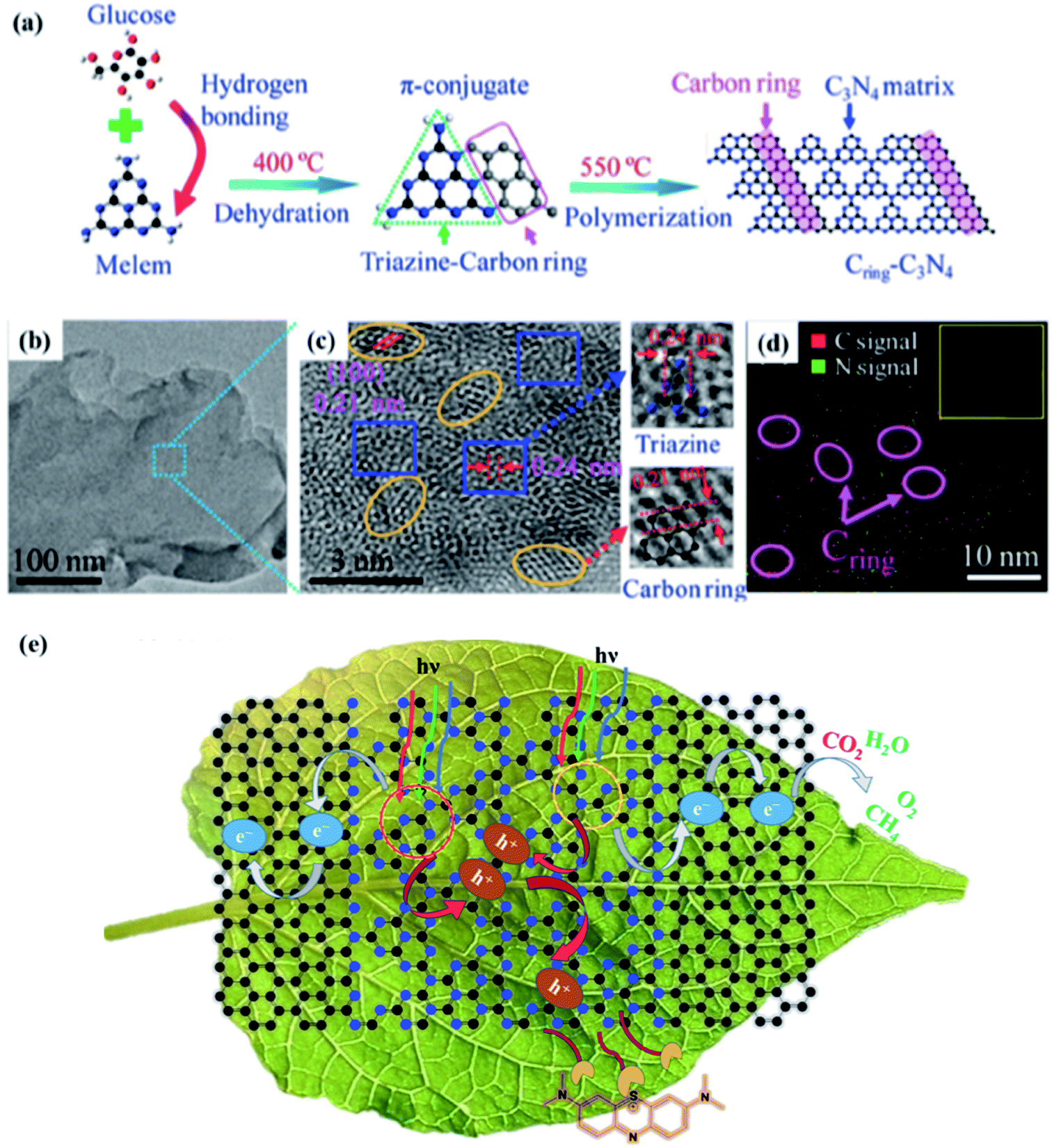
Che and co-workers106a investigated a thermal-conjugate in-plane polymerization approach using glucose as the starting material to integrate Cring into the triazine units of g-C3N4. The synthetic route for (Cring)-C3N4 including the dehydration and polymerization process is shown in FIG. 3A. The successful fusion of Cring in the C3N4 matrix was confirmed by the TEM and HRTRM analysis (Fig. 3b and c). The lattice fringe in orange ellipses (Fig. 3c) is 0.21 nm, in accordance with the (100) plane of graphite. The enrichment of C (elliptic region) can be observed in Fig. 3d, further suggesting the presence of Cring. In addition, the integration of Cring in the matrix of g-C3N4 can significantly broaden its visible-light absorption, facilitate charge carrier separation, and thus promote its photo-catalytic overall water splitting (OWS) activity. The Cring-C3N4 photocatalyst accelerated the surface reaction kinetics and enhanced the separation of photocarriers in the photoconversion of CO2 to CH4 and adsorption and photodegradation of methylene blue106b (Fig. 3e).
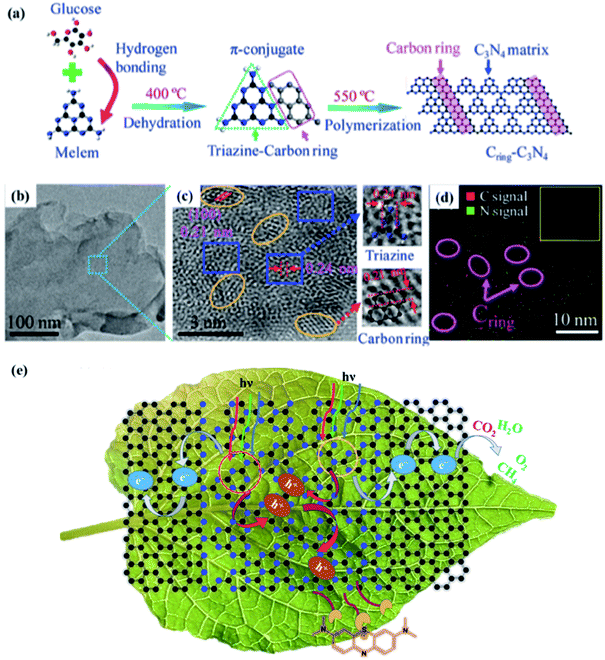 |
| | Fig. 3 (a) Synthetic procedure for Cring-g-C3N4. (b) TEM image and (c) HRTEM image of Cring-g-C3N4. The elliptic and rectangular areas represent a carbon ring and g-C3N4 matrix, respectively. (d) EDS mapping of Cring-g-C3N4. The inset image is the corresponding mapping area. Adapted with permission from ref. 106a. Copyright 2017, the American Chemical Society. (e) Photoconversion of CO2 to CH4 and the adsorption and photodegradation of methylene blue. Adapted with permission from ref. 106b. Copyright 2021, the American Chemical Society. | |
It is expected that the fusion of conjugated aromatic groups in g-C3N4 units is a favourable route to regulate the electronic features of g-C3N4 to furnish materials with desirable physical and chemical properties, consequently delivering highly efficient photocatalysts. The current research on the copolymerization of co-monomer-modified g-C3N4 is expected to offer a new path for the sustainable conversion of solar energy due to its fascinating novel properties.
3.3 Crystalline g-C3N4
g-C3N4 derived via direct polymerization exhibits an amorphous or hypocrystalline structure due to the incomplete polymerization of the precursors, resulting a relatively poor photocatalytic performance. Due to kinetic hindrance problem in the solid-state route, the formation of highly crystalline g-C3N4 is a major challenge. However, the insertion of suitable metal salts can enhance the mass-transfer efficiency and improve the crystallinity, thus enabling a new route to synthesize highly crystalline g-C3N4. Bhunia and co-workers107 demonstrated that highly crystalline g-C3N4 (PTI-0.13) can be synthesized via the polymerization of melamine, 2,4,6- triaminopyrimidine, and a mixture of LiCl and KCl in air at 550 °C.
3.4 Surface-engineered g-C3N4
Surface engineering of the interface of g-C3N4 can enhance the amount of adsorption sites by increasing the absorption of protons and inhibit carrier recombination, ultimately resulting in an improved photocatalytic performance. Yu and co-workers108 proposed a surface engineering strategy to gradually dope graphitic carbon rings in g-C3N4. By changing the amount of 1,3,5-cyclohexanetriol, graphitic carbon rings were incorporated at different depths in g-C3N4. Gradual surface doping furnished g-C3N4 with suitable electronic band structures and built-in electric fields, resulting in accelerated carrier separation and improved photocatalytic performance. In another case, Wang's group109 fabricated proton- and –OH-modified g-C3N4 spheres and evaluated the effects of the introduction of protons and –OH on the photocatalytic degradation and HER activity. The results suggested that proton functionalization has a lower influence on the photocatalytic hydrogen process, while –OH can dramatically enhance the photocatalytic hydrogen production rate.
3.5 Single-atom engineering of g-C3N4
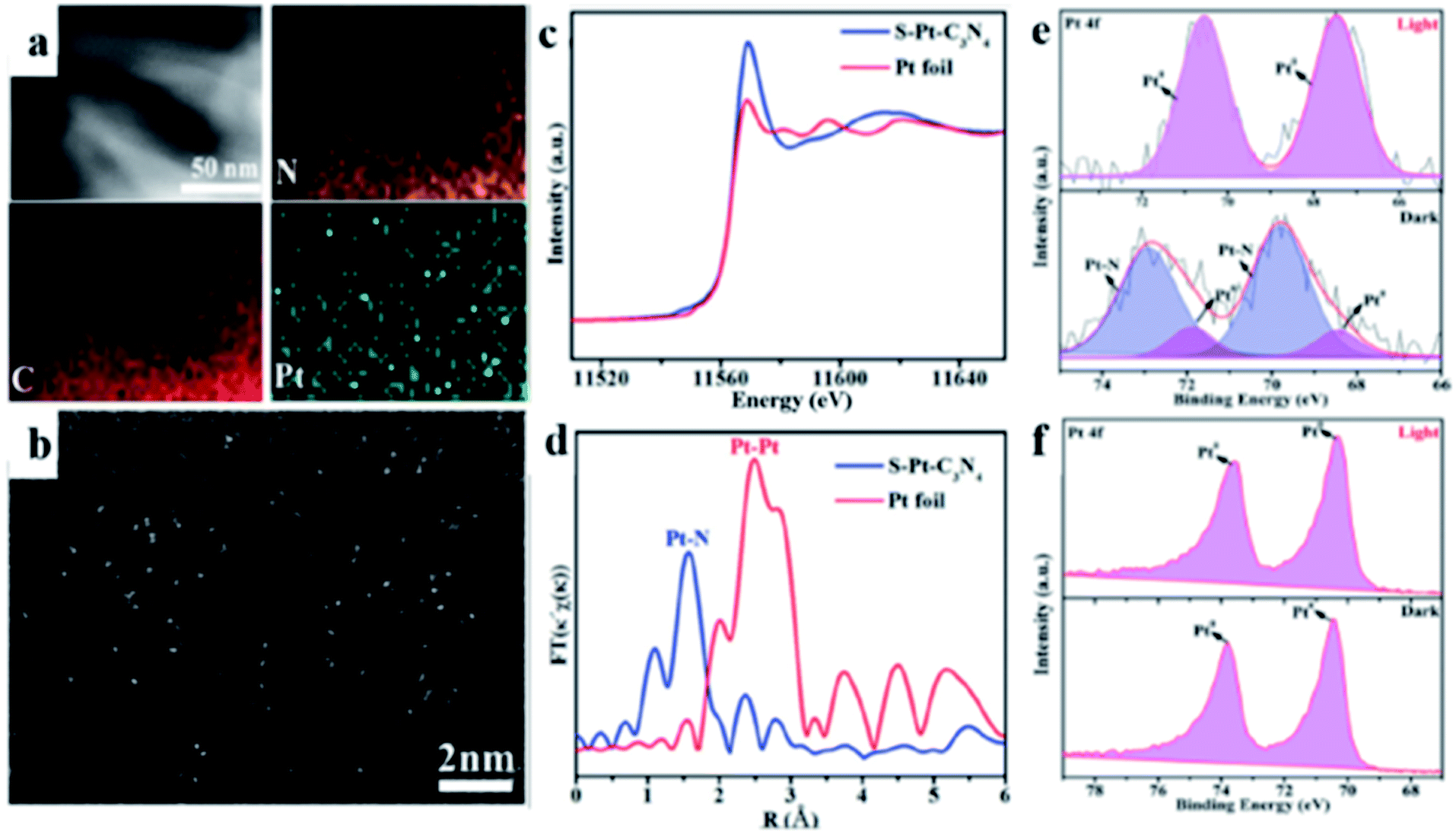
In the conversion of solar energy, single-atom photocatalysts are of tremendous importance due to their maximum utilization of metal active sites, high catalytic activity, and selectivity. Accordingly, g-C3N4 is an excellent support for single-metal atoms (SMAs) because the lone electron pairs present in the N atoms can coordinate with SMAs. To date, various SMAs such as Pt,110–112 Ni,113 and Cu114,115 have been investigated for the photocatalytic HER. Zhou and co-workers110 demonstrated a low-temperature in situ photocatalytic reduction approach to synthesize a high-density Pt single atom (PtSA/g-C3N4). Zhang and co-workers112 selected single-atom Pt/C3N4 nanorods (S–Pt–C3N4) as a model material and employed synchronous illumination X-ray photoelectron spectroscopy to monitor the bond evolution process (Fig. 4).
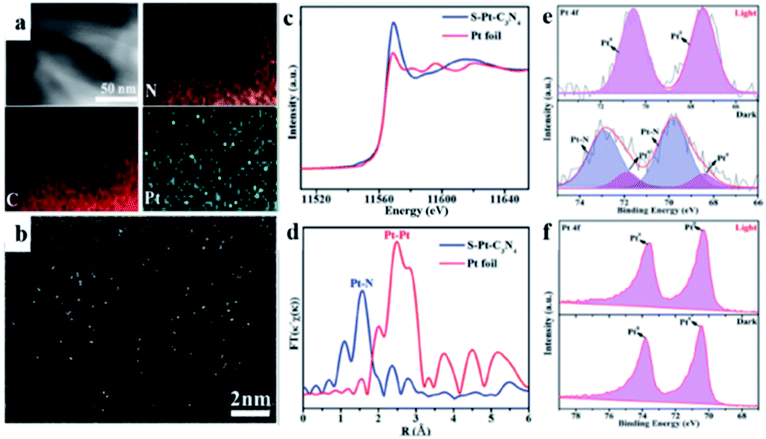 |
| | Fig. 4 (a) Elemental mapping and (b) HAADF-STEM images of the S–Pt–C3N4 sample. (c) K-edge XANES spectra of the S–Pt–C3N4 catalyst and Pt foil. (d) Fourier transform Pt L3-edge EXAFS spectra for S–Pt–C3N4 and Pt foil without phase correction. SI-XPS Pt 4f spectra of (e) S–Pt– C3N4 and (f) M-Pt-C3N4. Adapted with permission from ref. 112. Copyright 2020, John Wiley & Sons, Inc. | |
4 g-C3N4-based heterojunction photocatalysts
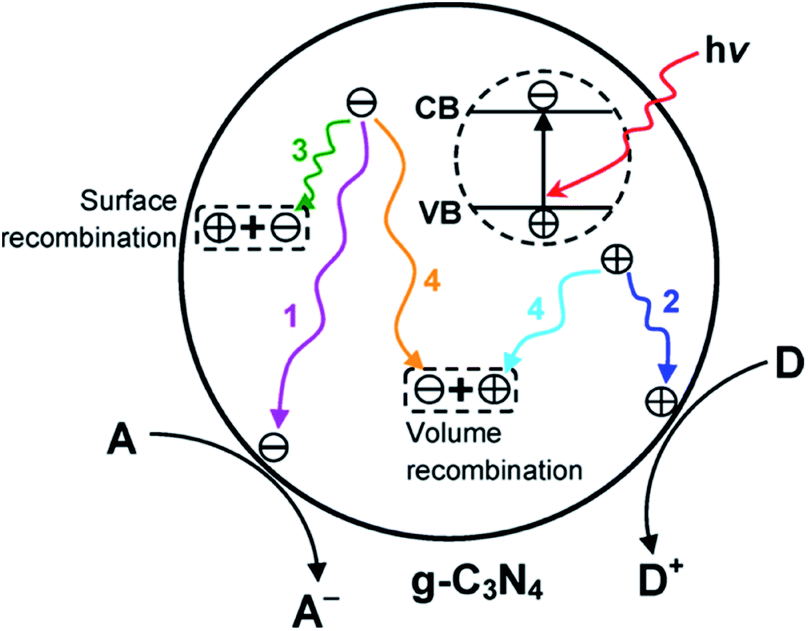
g-C3N4 exhibits a bandgap energy, also known as the void region, covering the top of the filled VB to the bottom of the vacant CB. Initially, the absorption of a photon with energy equal to or greater than the bandgap results in the excitation of an electron from the VB to CB, leaving behind an empty state, which constitutes a positive hole.116,117 Once spatially separated, the charge carriers in the excited states, which then migrate to the surface of g-C3N4, are responsible for initiating the reduction and oxidation processes for the photocatalytic conversion of reactant molecules (Fig. 5).
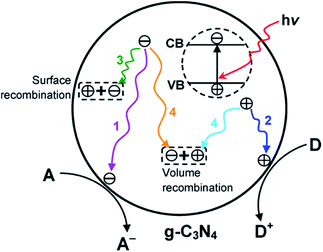 |
| | Fig. 5 Schematic illustration of photoexcited electron–hole pairs in g-C3N4 with possible decay pathways. A and D denote electron acceptor and electron donor, respectively. Adapted with permission from ref. 31. | |
The location of the reactive sites may be either on the g-C3N4 surface, where the photoexcitation occurs, or across the interface of another semiconductor or cocatalyst.118 In this case, the main role of g-C3N4 is to absorb light, produce electron–hole pairs and transfer them to its surface or to a cocatalyst. Indeed, g-C3N4 is chemically active only when the photo-induced electron–hole pair is consumed simultaneously before recombination occurs in a fraction of a nanosecond.119–121
The excitation of the bandgap upon the absorption of light and the formation of electron–hole pairs can also be rapidly accompanied by a few deexcitation pathways (Fig. 5). It is noteworthy that the successful migration of charge carriers to the acceptor molecules, which results in reduction and oxidation processes (pathways 1 and 2, respectively), also competes with the recombination processes.122 For example, the recombination process can take place via two different paths, as follows: (1) on the surface of the particle depicted in pathway 3 (surface recombination) and (2) in the bulk of g-C3N4 illustrated in pathway 4 (volume recombination). These recombination processes are disadvantageous to the photocatalytic efficiency. When the recombination process occurs, the excited electrons revert to the VB, dissipating the energy as heat without reacting with the adsorbed species on the surface of g-C3N4.123 Therefore, various strategies are employed to prevent the recombination of charge carriers in g-C3N4 and upgrade its photocatalytic performance.
The construction of heterojunctions is another feasible approach to enhance the properties of g-C3N4. Especially, its charge separation efficiencies can be dramatically accelerated via the construction of heterojunctions. According to the component, the heterojunction can be classified into five types, i.e., type II, Z-scheme, S-scheme, g-C3N4/metal, and g-C3N4/carbon heterojunctions.
4.1 g-C3N4-based type II heterojunction
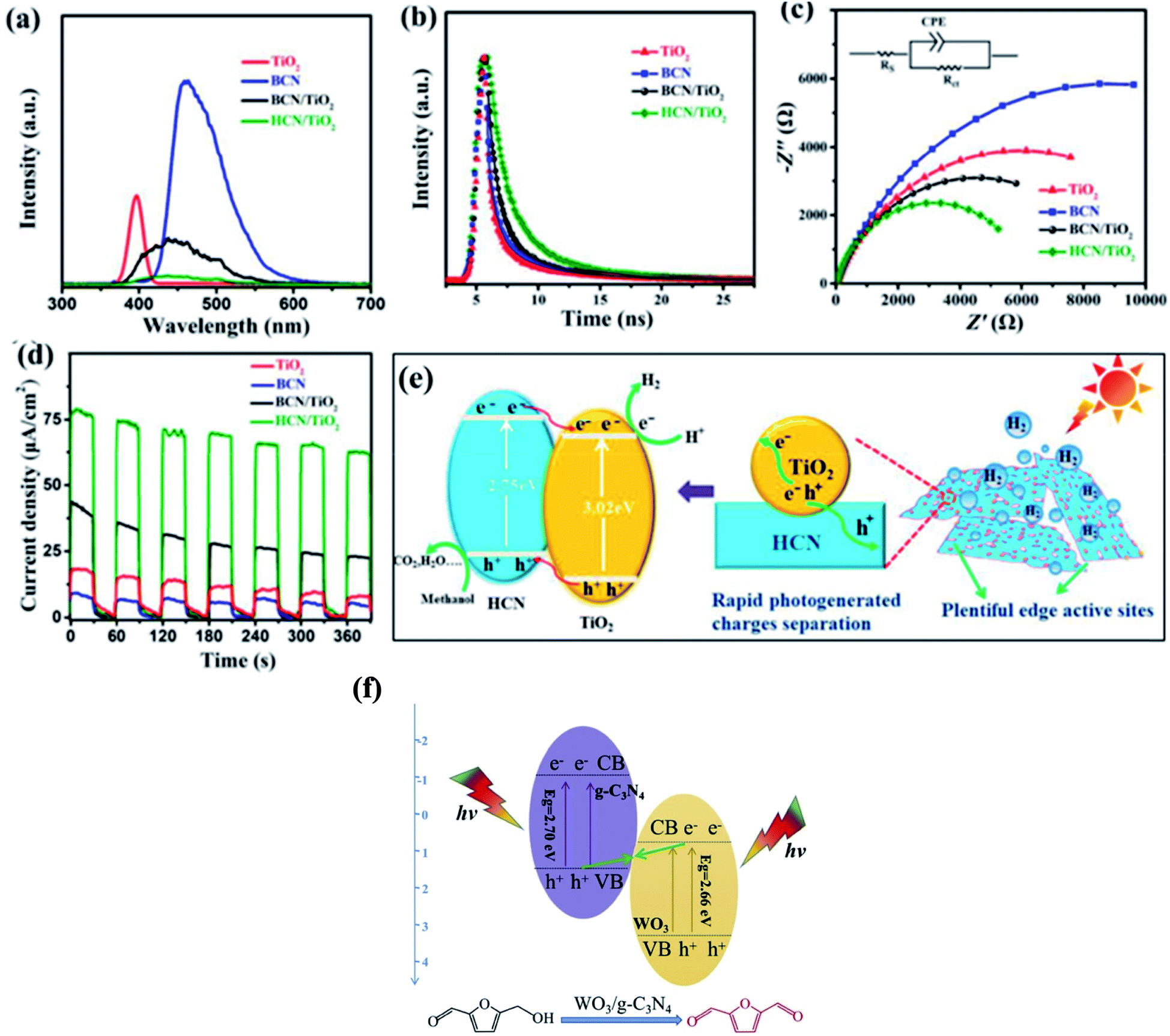
A g-C3N4-based type II heterojunction can be formed with TiO2,124 ZnO,125 WO3,126 CdS,127 ZnIn2S4,128 BiVO4,129 Ag3PO4,130 and many others. Xiao and co-workers124a proposed an in situ exfoliation and conversion strategy to synthesize a holey C3N4 nanosheet/TiO2 nanocrystal heterojunction (Fig. 6a–e). In type-II heterojunctions, the CB and VB of semiconductor A are higher than that of semiconductor B and their different chemical potentials show band bending at the junction. Type-II heterojunctions are less effective given that they provide low reduction and oxidation potentials, and the transfer of charges is difficult due to electrostatic repulsion between similar charges124b and their quantum efficiency has been found to be low. For the type-II heterostructure, reduction and oxidation reactions occur at semiconductor B with lower reduction ability, while semiconductor A has low oxidation ability. Consequently, the redox potential of type-II heterojunctions significantly decreases. The electrons in the CB of semiconductor B and holes in the VB of semiconductor A migrate owing to the electrostatic repulsion between the hole–hole and electron–electron pairs. A novel type of structure named Z-scheme, which has a new charge migration mechanism, has been developed to overcome the drawbacks of type-II heterojunctions.
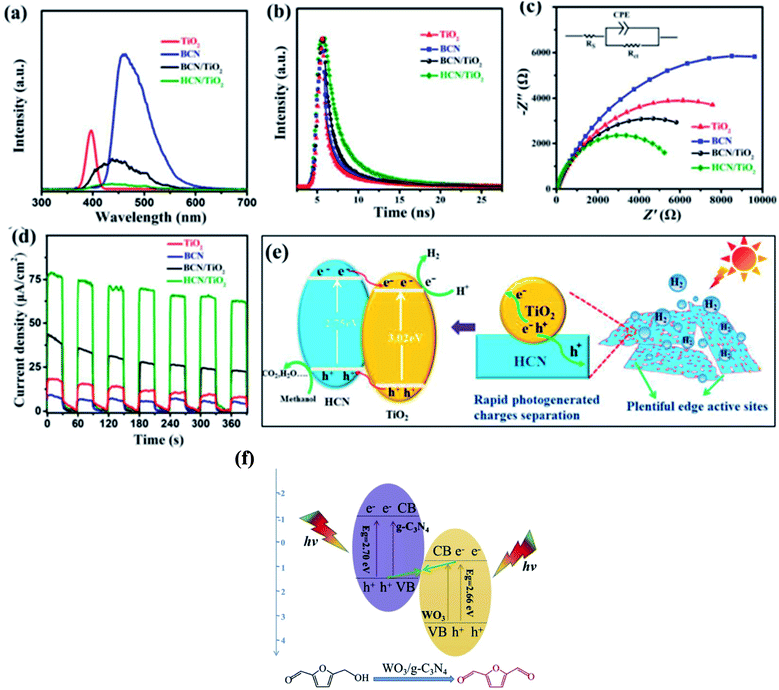 |
| | Fig. 6 (a) PL spectra (360 nm excitation), (b) time-resolved fluorescence spectra, (c) Nyquist plots from electrochemical impedance spectroscopy, and (d) transient photocurrent response of TiO2, g-C3N4, g-C3N4/TiO2, and holey C3N4/TiO2. (e) Proposed synergistic effect of surface reactions and charge separation in holey C3N4/TiO2. Adapted with permission from ref. 124a. Copyright 2020, Elsevier. Z-scheme mechanism for the photocatalytic oxidation of 5-HMF to DFF over WO3/g-C3N4. Adapted with permission from ref. 132b. Copyright 2019, Elsevier. | |
4.2 g-C3N4-based Z-scheme heterojunction
In the traditional type II heterojunctions, electrons tend to inject in the CB with mild reduction ability, while holes tend to inject in the VB with weak oxidation ability because of the thermodynamic effect. Therefore, after their separation and transfer, both the reduction and oxidation ability would be weakened. Accordingly, the exploration of heterojunctions that simultaneously promote carrier separation and maintain the oxidation and reduction ability is of practical importance. In principle, the transport direction of the electrons and holes of green plants are opposite to the traditional type II heterojunction, which is named Z-scheme heterojunction. Z-scheme systems can be divided into three types, i.e., traditional Z-scheme, all-solid-state Z-scheme, and direct Z-scheme. Their detailed differences can be seen in ref. 131a The Z-scheme photocatalytic system, as a new type of heterostructure, can improve the separation of electron–hole pairs and enhance the redox ability compared with the corresponding pristine materials, resulting in wide opportunities in various application areas, especially in terms of tremendous potential in improving the photocatalytic reaction activity.124b,131b–d However, the Z-scheme has many problems associated with electron mediators such as their complex fabrication process, high reverse reaction, and shielding outcome by shuttle redox mediators.124b Taking an all-solid-state Z-scheme material as an example, Pan et al.132a engineered a g-C3N4/RGO/Fe2O3 Z-scheme system with RGO as an electron shuttle. In comparison with g-C3N4, g-C3N4/RGO/Fe2O3 exhibited a dramatically enhanced photocurrent, suggesting an improvement in the charge carrier separation due to the incorporation of RGO. In addition, the lower PL intensity of g-C3N4/RGO/Fe2O3 suggested the inhibited recombination of carriers. Therefore, g-C3N4/RGO/Fe2O3 exhibited efficient solar light-induced OWS activity. Wu et al.132b utilised a WO3/g-C3N4 composite, which was prepared via a directly calcination method with ammonium tungstate hydrate and melamine as raw materials, for the photocatalytic oxidation of 5-hydroxymethylfurfural (5-HMF) to 2,5-diformylfuran (DFF) (Fig. 6f).
4.3 g-C3N4-based S-scheme heterojunction
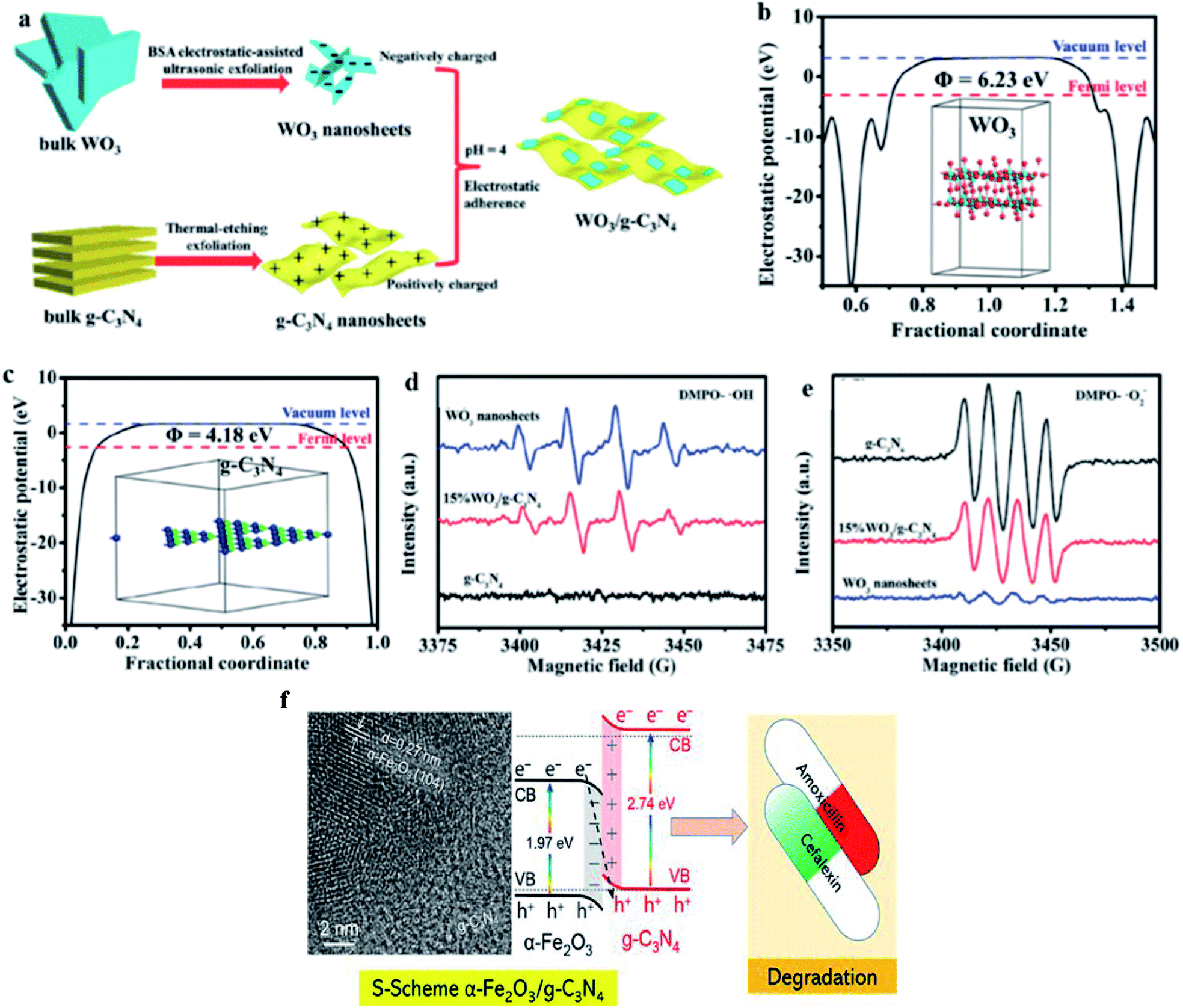
Yu's group proposed the S-scheme heterojunction because of the great fundamental problems of type II heterojunctions and the complexity and misunderstanding of Z-scheme photocatalysts.131a Similar to the type II heterojunction, the S- scheme system contains an oxidation photocatalyst and a reduction photocatalyst with staggered band structures. However, the charge transfer route of the S-scheme system is completely different from the type II heterojunction.133a The major advantages of S-scheme photocatalysts are the reservation of photogenerated holes and electrons in the VB and CB of the oxidation and reduction photocatalysts. The simultaneous recombination of useless electrons and holes generated by the internal electrical field imparts strong redox ability.124b As discussed before, in type II heterojunctions, the redox abilities in both the CB and VB are weakened. In contrast, the strong redox potential is maintained in the S-scheme heterojunction due to the powerful photoinduced electrons and holes accumulated in the CB of the reduction photocatalyst and VB of the oxidation photocatalyst, respectively, with the recombination of the extra carriers. The disadvantages and limitations of S-scheme photocatalysts include the fact that the scope of their application is mainly applicable to powder photocatalysts and they do not apply to photoelectrochemistry and solar cells with external circuits. The two selected semiconductors constructing S-scheme photocatalysts are mainly n-type semiconductors and should have suitable band structures and obvious Fermi level differences.133b For example, Fu et al.134a employed an electrostatic self-assembly method (Fig. 7a–e) to design an ultrathin WO3/g-C3N4 S-scheme heterogeneous composite. Cao et al.134b described that S-scheme Fe2O3/g-C3N4 photocatalysts can degrade commercial cefalexin and amoxicillin (20 mg L−1) under visible light, with five- and nine-times higher performances than that of g-C3N4, respectively. Furthermore, the detailed evidence to propose S-scheme Fe2O3/g-C3N4 heterojunctions and comparison of their photocatalytic performance in the degradation of antibiotics were also included in this protocol (Fig. 7f).
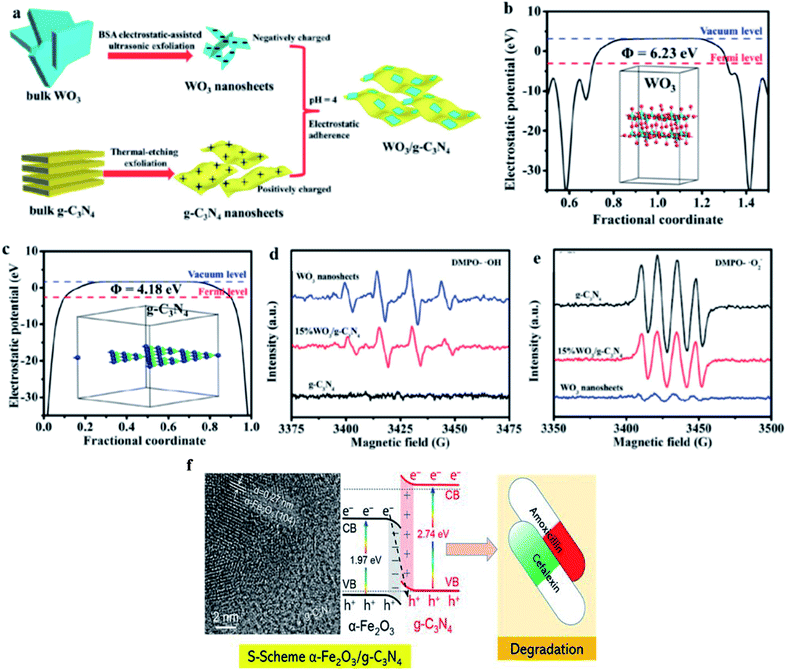 |
| | Fig. 7 (a) Procedure for the formation of 2D/2D WO3/g-C3N4 heterojunctions by electrostatic interaction. Electrostatic potentials of (b) WO3 (001) surface and (c) g-C3N4 (001) surface. Insets show the structural models of the materials for DFT calculation. EPR spectra of (d) DMPO− ˙OH in aqueous and (e) DMPO−˙O2− in methanol dispersion in the presence of g-C3N4, WO3 nanosheets and 15% WO3/g-C3N4. Adapted with permission from ref. 134. Copyright 2018, Elsevier. S-scheme α-Fe2O3/g-C3N4 nanocomposite as heterojunction photocatalyst for the degradation of antibiotics. Adapted with permission from ref. 134b. Copyright 2022, the American Chemical Society. | |
4.4 g-C3N4/metal heterojunction
Many studies have demonstrated that the formation of a g-C3N4/metal heterojunction can effectively enhance the activity of g-C3N4. The mechanism of g-C3N4/metal is different from g-C3N4. When a metal contacts a semiconductor, an energy barrier difference is produced at the interface due to their different work functions and this energy barrier is denoted as the Mott–Schottky energy barrier. The Mott–Schottky energy barrier can greatly promote the electron transfer, and hence promote charge-carrier separation. Specifically, noble metals such as Ag135–138 and Au139,140 exhibit surface plasmon resonance effects upon visible-light irradiation, which can dramatically promote the light absorption and near field enhancement. However, the incorporation of noble metals increases the expense, and thus the exploration of green and low-cost systems is of significant importance.
4.5 g-C3N4/carbon
In g-C3N4/carbon heterojunction systems, carbon is regarded as a promising component due to its merits of being metal free and conductive, excellent physico-chemical stability, low cost, and good light absorption ability.141 The commonly used carbon materials, including graphene,142 graphyne,143–145 CNT,146–151 C60,152 and carbon dots,153–157 can significantly enhance the absorption ability and promote charge separation. Xiang and co-workers158 synthesized graphene/g-C3N4 composites via the pyrolysis of melamine, graphite oxide, and hydrazine hydrate. The obtained sample exhibited dramatically enhanced photocatalytic hydrogen evolution activity in comparison with graphene and g-C3N4. After the incorporation of graphene, the photogenerated electrons of g-C3N4 can transfer to the surface of graphene, and hence inhibit the recombination of carriers of g-C3N4 and promote the photocatalytic water-splitting reaction.
In summary, the above-mentioned studies provide a new pathway for the design of g-C3N4-based hybrids for elevated charge transfer in the target photocatalytic reactions. This can undoubtedly promote the smart design of g-C3N4-based nanomaterials. It is expected that the novel fabrication procedures can lead to huge application potential in the other layered photocatalysts and multicomponent hybrid photocatalysts.
5 Synthetic applications of g-C3N4-based photocatalysts
5.1 g-C3N4-catalyzed reactions of quinoxalin-2(1H)-ones with N-aryl glycines under visible light
Yu et al.159 reported the visible light-induced heterogeneous g- C3N4-catalyzed decarboxylative reaction of the low-cost and easily available quinoxalin-2(1H)-ones with N-aryl glycines as starting materials (Scheme 1). The simplicity of this synthesis and excellent regioselectivity make this protocol a promising tool for building molecules with diverse skeletons and discovering bioactive compounds.
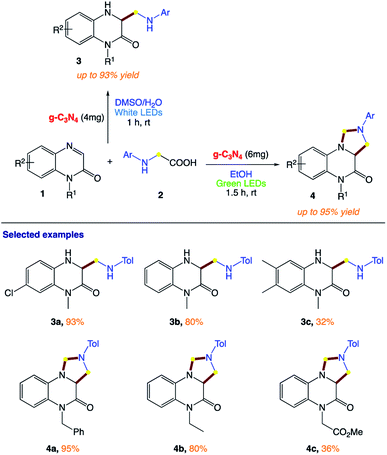 |
| | Scheme 1 g-C3N4-catalyzed reactions of quinoxalin-2(1H)-ones with N-aryl glycines under visible light. | |
The authors proposed a tentative mechanism for these divergent transformations (Scheme 2). Upon irradiation with visible light, the g-C3N4 semiconductor is excited to generate electrons and holes, which play important roles in organic transformations, acting as single-electron acceptors and donors, respectively. Initially, radical intermediate A is formed through a photooxidation process by releasing a CO2 molecule and proton. Then, radical A is added to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of 1 to afford intermediate C. Then, intermediate C undergoes a single-electron photo-reduction to generate anion D. Then intermediate D can be converted into product 3 after protonation. Alternatively, the oxidation of radical A affords iminium species B. The combination of iminium species B and anion D gives intermediate E, followed by intramolecular cyclization to produce product 4 by releasing ArNH2.
N bond of 1 to afford intermediate C. Then, intermediate C undergoes a single-electron photo-reduction to generate anion D. Then intermediate D can be converted into product 3 after protonation. Alternatively, the oxidation of radical A affords iminium species B. The combination of iminium species B and anion D gives intermediate E, followed by intramolecular cyclization to produce product 4 by releasing ArNH2.
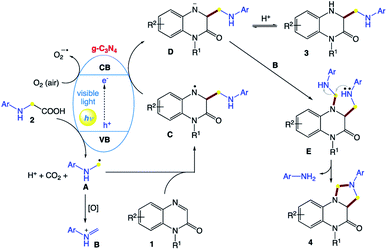 |
| | Scheme 2 Proposed reaction mechanism for g-C3N4-catalyzed reactions of quinoxalin-2(1H)-ones with N-aryl glycines under visible light. | |
5.2 Synthesis of thiocyanated heterocycles
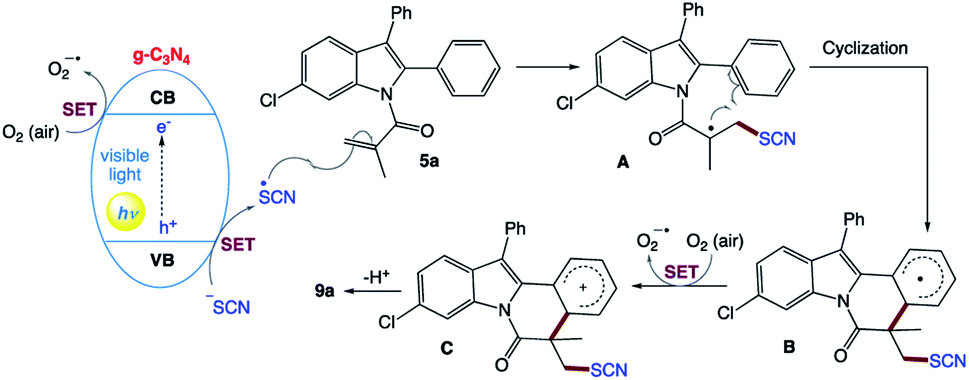
B. Yu and co-workers160 reported a metal-free photocatalytic strategy for the preparation of thiocyanated heterocycles from the inexpensive NH4SCN using carbon nitride (g-C3N4) as a general heterogeneous catalyst in a green solvent under an air atmosphere and irradiation from a blue LED (Scheme 3). This general method showed advantages such as having a metal-free procedure and an external-oxidant-free procedure, utilizing a recyclable catalyst and a green solvent, wide applicability, applied at room temperature, and easy to scale up.
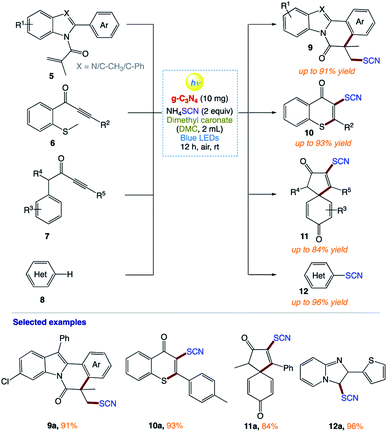 |
| | Scheme 3 Synthesis of thiocyanated heterocycles. | |
They proposed the mechanism for this visible light-promoted reaction, which was catalyzed by g-C3N4 (Scheme 4). Firstly, under the irradiation of visible light, g-C3N4 absorbs photons and generates holes in its valence band (VB) and electrons in its conduction band (CB). Then, the holes generated in the VB (EVB = +1.2 V vs. SCE) obtain electrons from NH4SCN via single-electron transfer (SET) to generate the SCN radical. Simultaneously, the electrons in the CB are transferred to O2 (air) through SET to generate O2˙−. Then, the SCN radical is added to the CC bonds of 5a to generate radical intermediate A, which is then subjected to intramolecular cyclization to obtain radical intermediate B. Furthermore, B is oxidized by O2 (air) to form carbocation C through an SET process. Finally, C is transformed into the target product 9a by deprotonation.
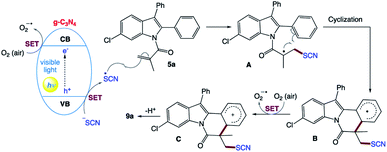 |
| | Scheme 4 Proposed reaction mechanism for the synthesis of thiocyanated heterocycles. | |
5.3 Selective oxidative cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C bonds in aryl olefins
C bonds in aryl olefins
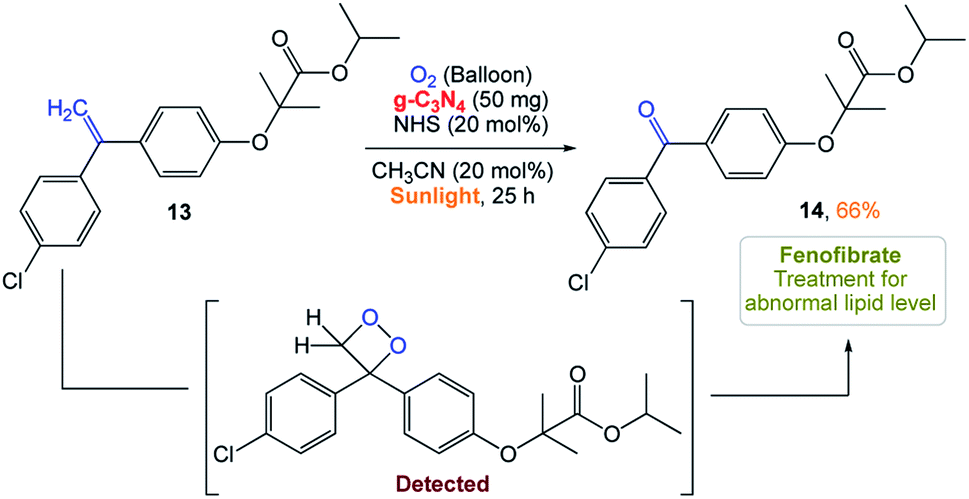
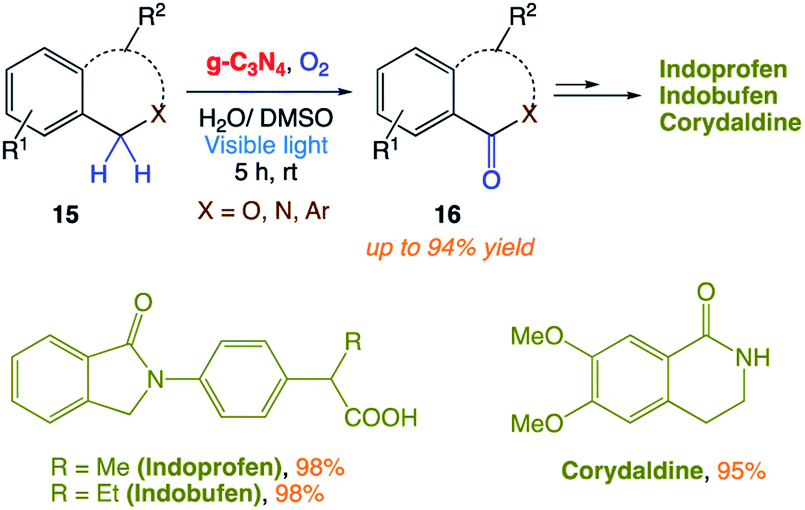
S. Das and co-workers161 reported the use of polymeric carbon nitride (PCN) as a photocatalyst for the selective cleavage of CC bonds in aryl olefins. This method was selective, scalable in the presence of sunlight and the photocatalyst was easily recyclable. Excellent substrate scope with high selectivity is the main advantage of this recyclable catalyst. In addition, they could perform late-stage CC bond cleavage in aryl olefins and degradation of complex molecules to achieve highly valuable products and pharmaceuticals (Scheme 5).
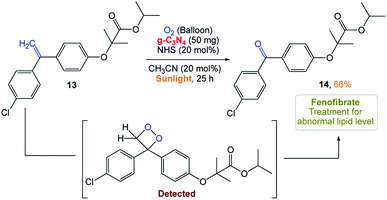 |
| | Scheme 5 Metal-free heterogeneous g-C3N4 photocatalyst for the selective oxidative cleavage of CC bonds in aryl olefins by directly harvesting solar energy. | |
5.4 Aerobic benzylic C–H oxygenations
Y. Cai and co-workers162 reported a metal-free heterogeneous photocatalytic system for highly efficient benzylic C–H oxygenations using oxygen as an oxidant. This visible light-mediated oxidation reaction utilized graphitic carbon nitride (g-C3N4) as a recyclable, nontoxic and low cost photocatalyst. The mild reaction conditions allowed the generation of synthetically and biologically valuable materials from readily accessible alkyl aromatic precursors in good yields (Scheme 6).
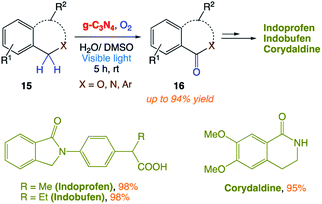 |
| | Scheme 6 g-C3N4-based heterogeneous photocatalyst for visible light-mediated aerobic benzylic C–H oxygenation. | |
The probable reaction mechanism was proposed by the authors (Scheme 7). Due to the structural and electronic properties of g-C3N4 with a band gap of 2.7 eV, visible-light excitation leads to the efficient separation of photogenerated electron hole pairs. The photoelectrons (CBM of g-C3N4 located at −0.88 V vs. RHE) can activate O2 to the superoxide radical anion O2˙− (reduction potential at −0.56 V vs. SCE). The photogenerated hole of g-C3N4 further undergoes single-electron transfer (SET) with the substrate including isochromans, benzylarenes and N-acyl-derived tetrahydroisoquinolines or isoindolines to afford benzylic radical A. Regarding the substrate N-aryl-derived tetrahydroisoquinolines or isoindolines, SET leads to the formation of the corresponding nitrogen radical cation B, followed by base-promoted deprotonation to generate benzylic radical C. Furthermore, A and C react with the peroxide radical ˙OOH from superoxide anions to generate intermediates E and F. Finally, they transform into the products with the simultaneous formation of water as the side product.
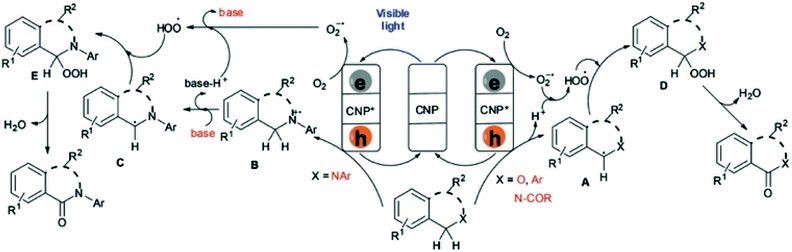 |
| | Scheme 7 Proposed reaction mechanism for g-C3N4-based heterogeneous photocatalyst for visible light-mediated aerobic benzylic C–H oxygenations. Adapted with permission from ref. 162. | |
5.5 Aerobic cyclization of thiobenzanilides
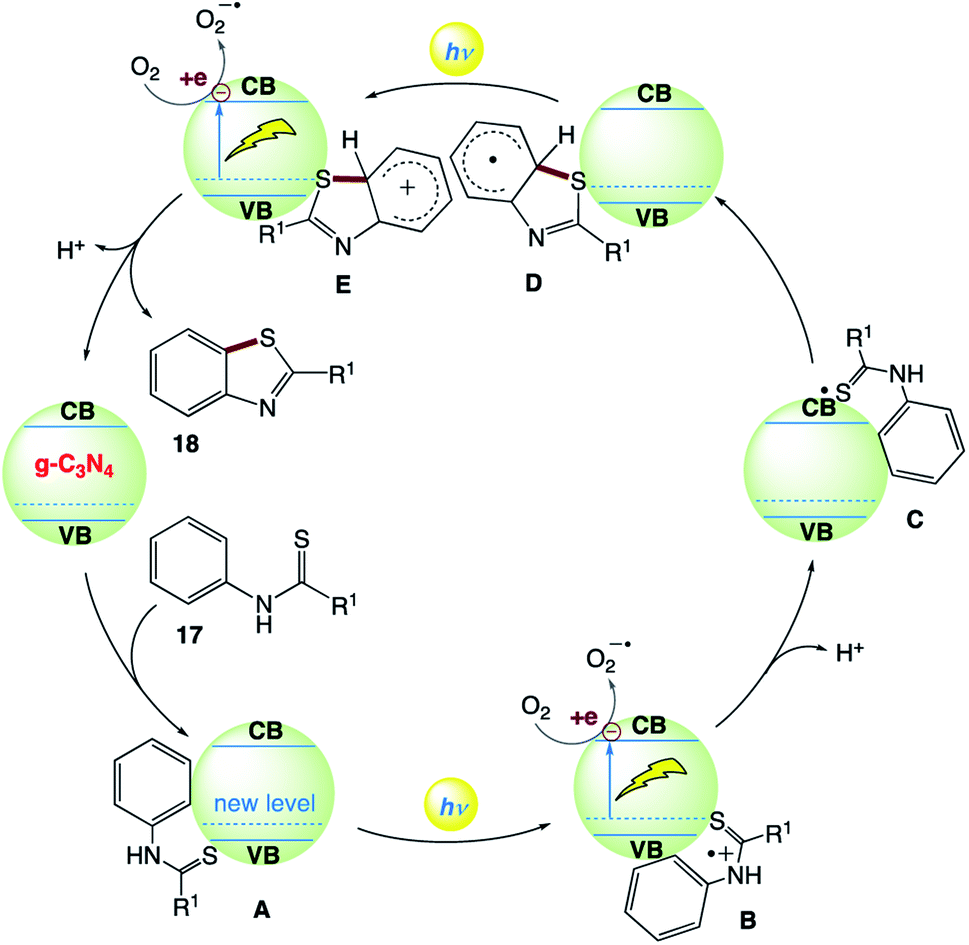
C. Y. Zhou and co-workers163 reported the use of a metal-free heterogeneous photocatalyst for the synthesis of benzothiazoles via intramolecular C–H functionalization/C–S bond formation of thiobenzanilides from benzothioamide 17 by the inexpensive graphitic carbon nitride (g-C3N4) under visible light irradiation. This reaction provides access to a broad range of 2-substituted benzothiazoles 18 in high yields under an air atmosphere at room temperature (Scheme 8).
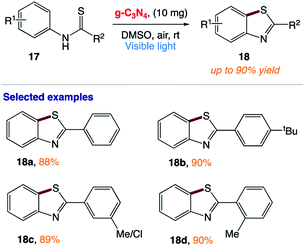 |
| | Scheme 8 Visible-light carbon nitride-catalyzed aerobic cyclization of thiobenzanilides under ambient conditions. | |
The plausible reaction mechanism was proposed by the authors (Scheme 9). Initially, N-substituted benzothioamide 17 adsorbs on the surface of g-C3N4 to form complex A with the formation of a new donor level above the valence band of g-C3N4. Visible-light irradiation of A drives the promotion of an electron to the conduction band of g-C3N4 with the generation of radical cation B. The photogenerated electron quenching by O2 and loss of a proton from B produce radical C. The sulfur-centered radical of C intramolecularly attacks the benzene ring to form aryl radical D, which releases an electron to the conduction band of g-C3N4 to generate cation E under visible-light irradiation. Then, E undergoes deprotonation and rearomatization to provide the final product 18.
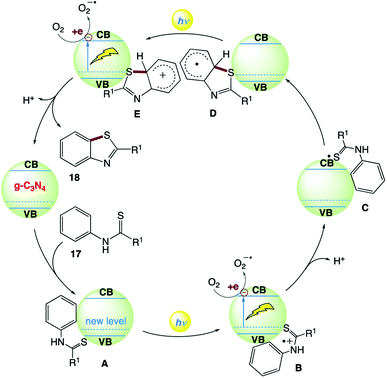 |
| | Scheme 9 Proposed reaction mechanism for visible-light carbon nitride-catalyzed aerobic cyclization of thiobenzanilides under ambient air conditions. | |
5.6 Graphitic carbon nitride for α-aminoalkyl radical additions, allylations, and heteroarylations
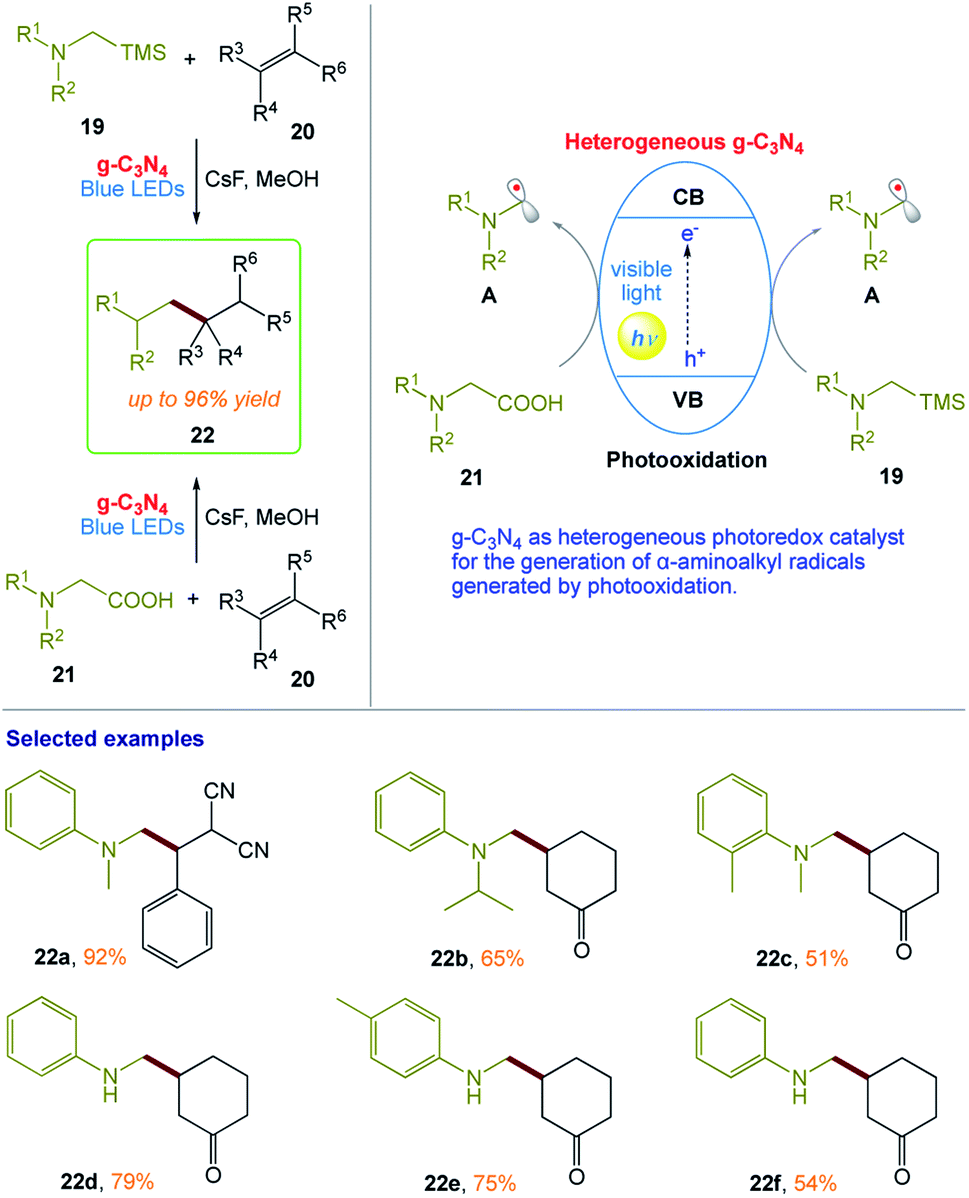
M. Rueping and co-workers164 reported a protocol for the photooxidative activation of α-silylamines and α-amino acids for desilylative and decarboxylative additions, allylations, and heteroarylations in the presence of graphitic carbon nitride (g-C3N4). This procedure has broad scope and provides the desired products in high yields (Scheme 10). In this protocol, the heterogeneous nature of the reaction system enables the recovery and reuse of the catalyst without loss in its reactivity. The reaction can also be conducted in a continuous flow fashion, even on the gram-scale using a simple and reusable continuous-flow photoreactor, which illustrates the practicability of this heterogeneous photocatalysis protocol.
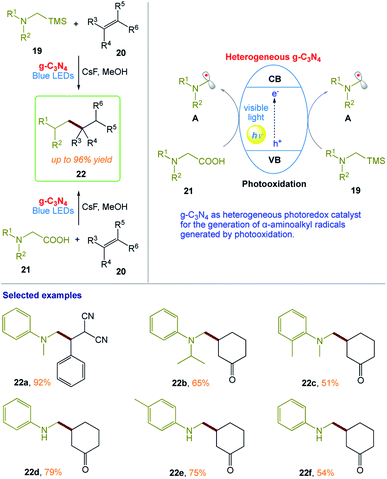 |
| | Scheme 10 Heterogeneous visible-light photoredox catalysis with graphitic carbon nitride for α-aminoalkyl radical additions, allylations, and heteroarylations. | |
5.7 Meerwein hydration reaction of alkenes
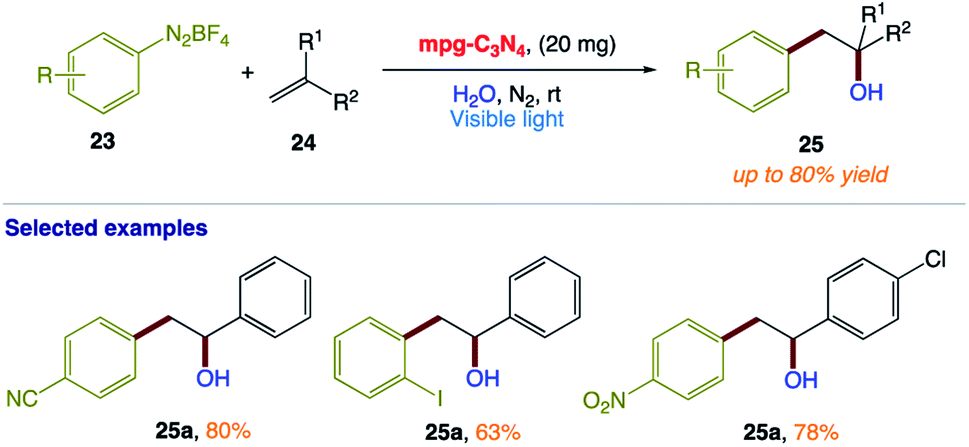
T. Niu and co-workers165 reported a green and efficient visible light-induced Meerwein hydration reaction of alkenes in aqueous medium using mpg-C3N4 as a recyclable photocatalyst. This protocol provides a direct approach for the preparation of racemic alcohols via a free radical mechanism (Scheme 11). In this reaction, water acts as both a solvent and reagent without any additives or co-solvents.
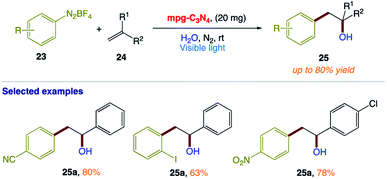 |
| | Scheme 11 Heterogeneous visible-light-induced Meerwein hydration reaction of alkenes in water using mpg-C3N4 as a recyclable photocatalyst. | |
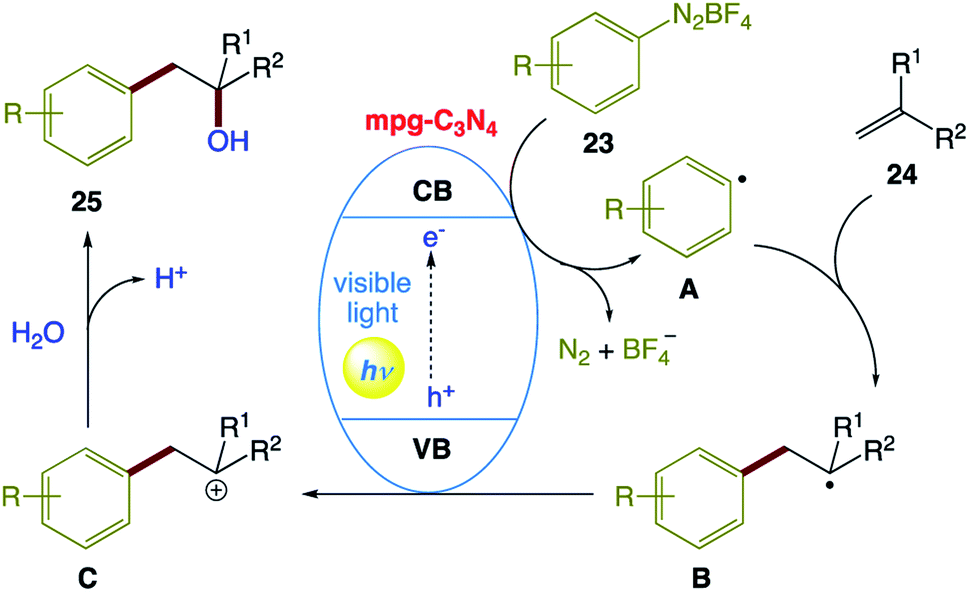
The plausible reaction mechanism proposed by the authors (Scheme 12). Initially, mpg-C3N4 is excited upon irradiation with visible light, which undergoes single-electron transfer (SET) to generate aryl radical A from diazonium salt 23. Adding A to alkene 24 generates the corresponding benzyl radical B, which is oxidized by the valence band of mpg-C3N4 to form intermediate C. Then, H2O as a nucleophilic reagent attacks intermediate C, followed by dehydrogenation to afford the corresponding product 25.
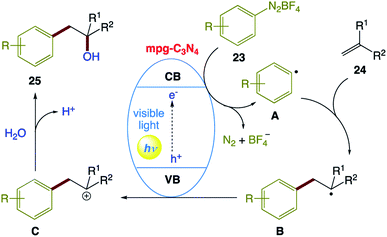 |
| | Scheme 12 Proposed reaction mechanism for visible-light-induced Meerwein hydration reaction of alkenes in water using mpg-C3N4 as a recyclable photocatalyst. | |
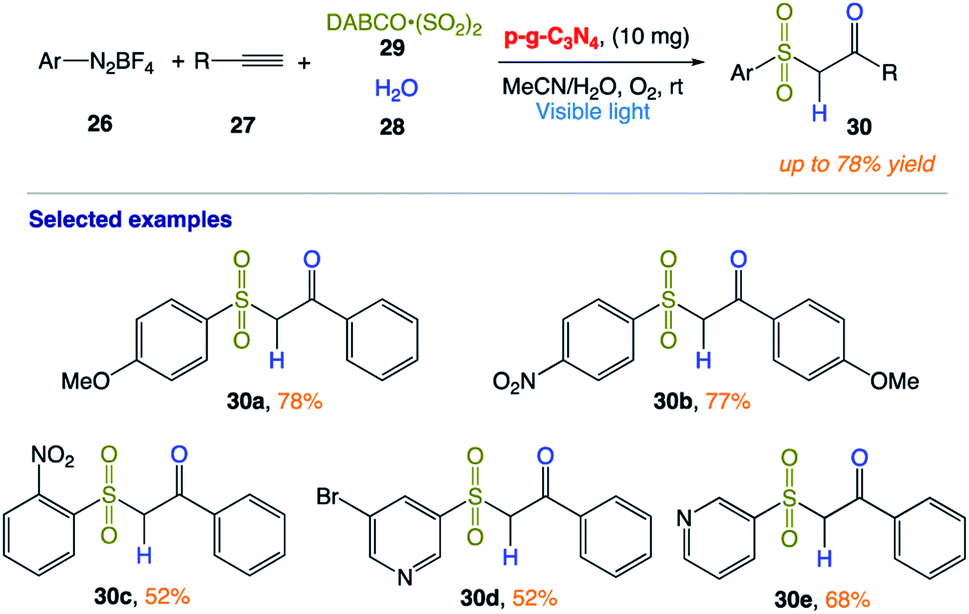
5.8 Multicomponent hydrosulfonylation of alkynes
T. Niu and co-workers166 reported the synthesis of β-ketosulfones 30 via the facile and efficient graphitic carbon nitride (p-g-C3N4)-photocatalyzed hydrosulfonylation of alkynes with the insertion of sulfur dioxide under aerobic conditions at room temperature. In this protocol, isotope experiments confirmed that the oxygen atom of the products comes from H2O, while the O2 plays an important role in the reaction by quenching the DABCO radical cation (Scheme 13).
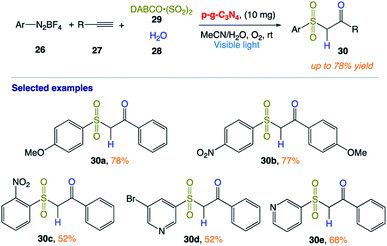 |
| | Scheme 13 Heterogeneous carbon nitride photocatalyst multicomponent hydrosulfonylation of alkynes to access β-keto sulfones with the insertion of sulfur dioxide in aerobic aqueous medium. | |
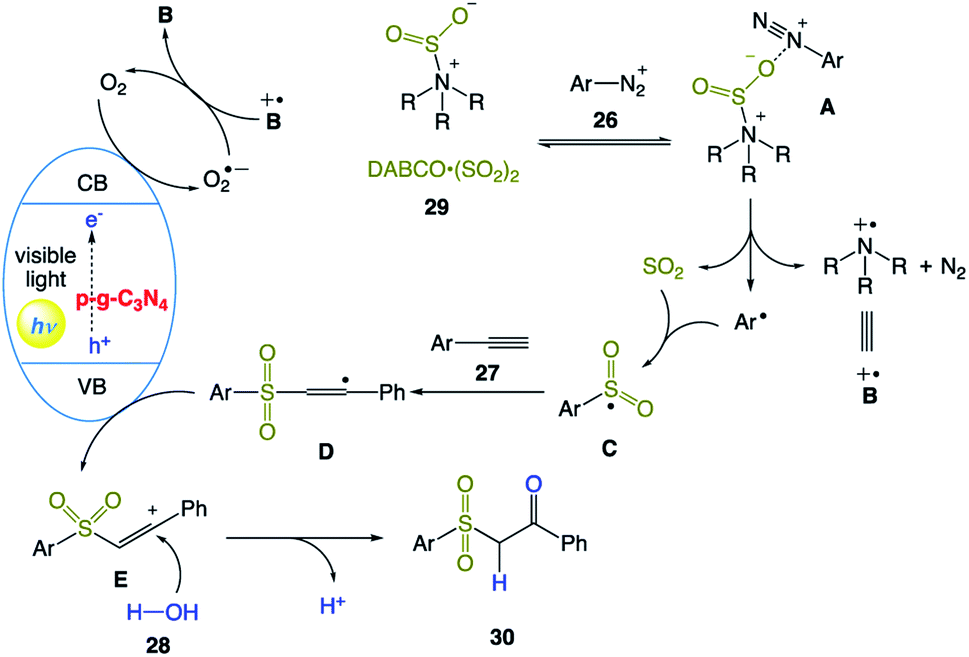
The plausible mechanism was proposed by the authors (Scheme 14). Initially, aryldiazonium cation 26 with DABCO·(SO2)2 29 provides complex A through electrostatic interactions. Subsequent homolytic cleavage of an N–S bond and single-electron transfer generate an aryl radical, sulfur dioxide, and radical cation intermediate B˙+. Then, the aryl radical reacts with sulfur dioxide to give sulfonyl radical C. Meanwhile, electrons and holes are produced upon the excitation of p-g-C3N4 under visible light irradiation. Consequently, an electron reduces molecular oxygen to produce a superoxide radical, which is quenched immediately with DABCO radical cation B˙+. Simultaneously, the obtained sulfonyl radical C reacts with alkynes 27 to form intermediate D. Subsequent SET oxidation of intermediate D forms alkenyl carbocation E, which undergoes hydrolysis the to produce the desired β-keto sulfones 30.
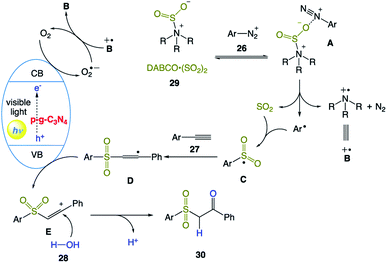 |
| | Scheme 14 Proposed reaction mechanism for heterogeneous carbon nitride photocatalyst multicomponent hydrosulfonylation of alkynes to access β-keto sulfones with the insertion of sulfur dioxide in aerobic aqueous medium. | |
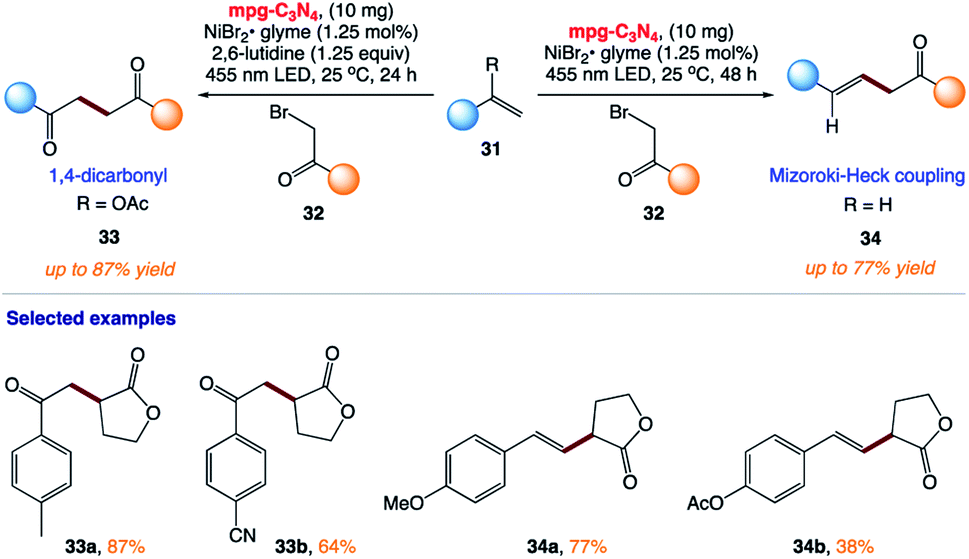
5.9 Mizoroki–Heck-type reactions and synthesis of 1,4-dicarbonyl compounds
B. König and co-workers167 reported the synthesis of 1,4-dicarbonyl compounds and substituted alkenes (Mizoroki–Heck type coupling) starting from secondary and tertiary alkyl halides and vinyl acetate or styrene derivatives using visible-light photocatalysis (Scheme 15). This protocol uses mesoporous graphitic carbon nitride (mpg-CN) as a heterogeneous organic semiconductor photocatalyst and Ni(II) salts as Lewis acid catalysts.
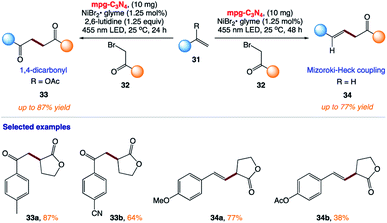 |
| | Scheme 15 Mizoroki–Heck type coupling and synthesis of 1,4-dicarbonyl compounds using mpg-C3N4 organic semiconductor visible light photo-redox catalysis. | |
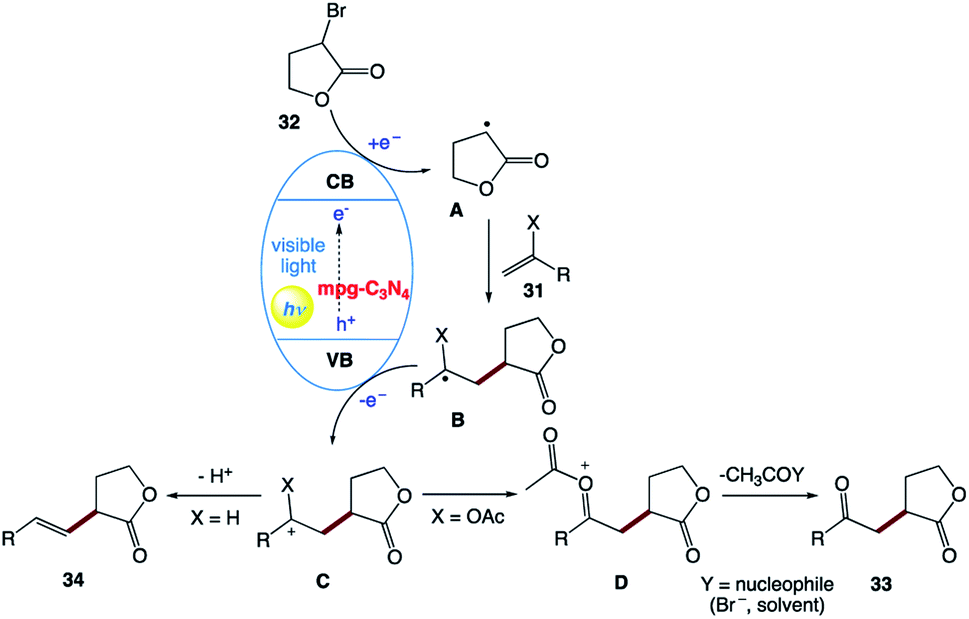
The plausible mechanism was proposed by the authors (Scheme 16). The exact role of the nickel(II) salt is not clear; however, the authors speculated that the nickel salt acts as a Lewis acid to coordinate with the ester group of alkyl bromide 32, and thereby lowers the reduction potential of alkyl bromide 32. Alternatively, it may be possible that it can activate the double bond by coordinating with olefin 31 given that the formation of the product was observed in the presence of other Lewis acids. However, reactions involving non-activated alkyl bromides did not yield any product, which ruled out the typical cross-coupling mechanism. Based on these observations, they believed that mpg-CN under photochemical illumination generates two-dimensional surface redox centers as electron–hole pairs. The photogenerated electron effectively reduces alkyl bromide 32 and generates alkyl radical A. Then, alkyl radical A adds to the double bond of vinyl acetate B and the resulting radical is oxidized by the photogenerated hole, delivering carbocation C. Successive loss of the acetyl group presumably supported by the nucleophiles present in the reaction medium such as bromide anions or the solvent yields the corresponding 1,4-dicarbonyl compounds 33, whereas proton loss in the case of olefins 34 results in a Mizoroki–Heck type cross-coupling product.
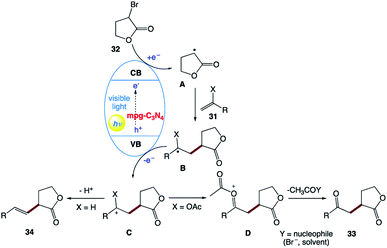 |
| | Scheme 16 Proposed reaction mechanism for the reaction involving the mpg-C3N4 semiconductor. | |
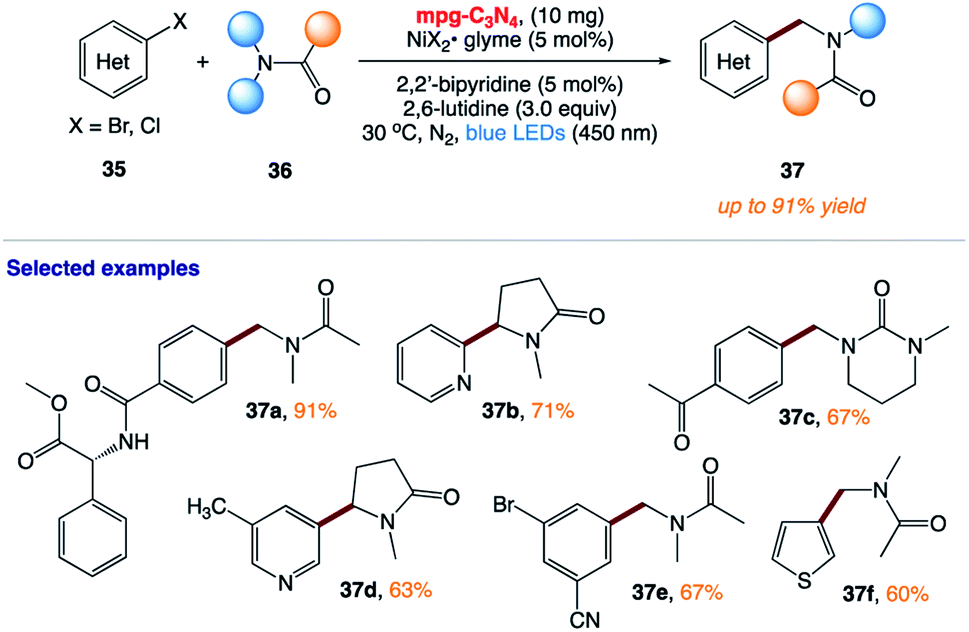
5.10 Photocatalytic (het)arylation of C(sp3)–H bonds with carbon nitride
B. König and co-workers168 reported the use of mesoporous graphitic carbon nitride (mpg-CN) as a heterogeneous organic semiconductor photocatalyst for the direct arylation of C(sp3)–H bonds in combination with nickel catalysis (Scheme 17). This protocol is based on a durable organic semiconductor mpg-CN photocatalyst and a simple nickel catalyst, providing a powerful alternative to conventional homogeneous photoredox catalysts.
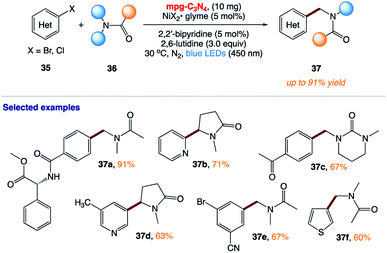 |
| | Scheme 17 Photocatalytic (het)arylation of C(sp3)–H bonds with carbon nitride. | |
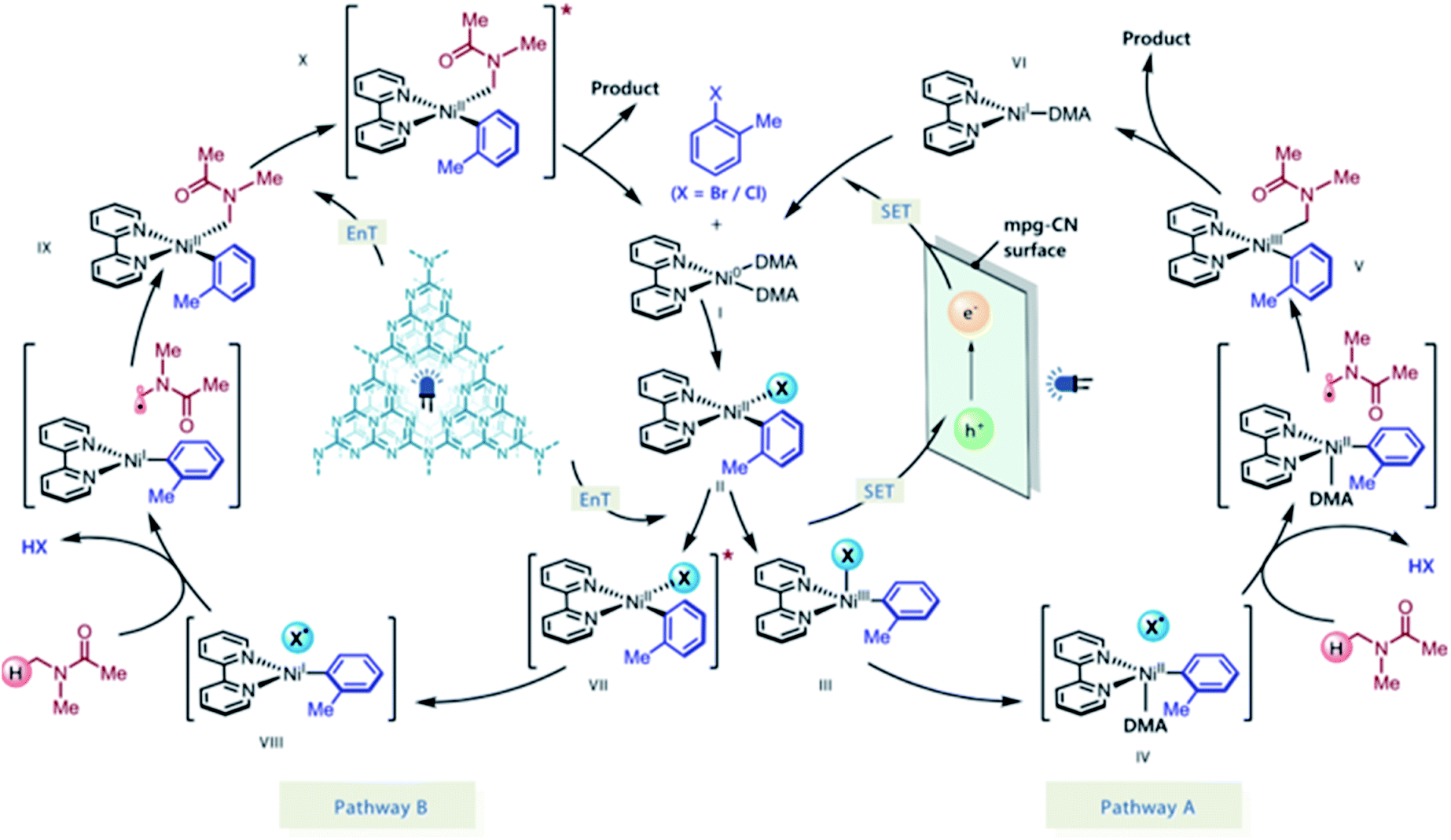
The authors postulated two plausible mechanistic scenarios (Scheme 18). Initially, the Ni(0) complex I undergoes oxidative addition with an aryl halide, delivering Ni(II) oxidative addition complex II. Concurrently, light absorption by the mpg-CN semiconductor photocatalyst triggers charge separation, producing two-dimensional surface redox centers as electron hole pairs. In pathway A, SET oxidation of complex II by the photogenerated hole (VBM located at +1.2 V vs. SCE, E1/2 (NiII/NiIII) = +0.85 V vs. SCE) affords species III, which may undergo Ni(III)-X homolysis to give a halogen radical and Ni(II) species IV. The resulting halogen radical can rapidly abstract a hydrogen atom from DMA (H–Br BDE ∼88 kcal mol−1, H–Cl BDE ∼102 kcal mol−1, and α-amino C–H BDE ∼89–94 kcal mol−1), which immediately recombines with species IV to form V. Subsequently, the reductive elimination of V results in the desired product and Ni(I) species VI. Finally, reduction of VI by the electron located on the semiconductor surface (CBM located at −1.5 V vs. SCE, E1/2 (NiI/Ni0) = −1.42 V vs. SCE) regenerates Ni(0) and completes the catalytic cycle. In pathway B, mpg-CN serves as a light-absorbing antenna undergoing an energy transfer process (EnT) (singlet–triplet band gap of ca. 0.39 eV) to produce electronically excited Ni(II) species VII. Homolysis of the Ni(II)–X bond and HAT followed by a rebound of the resulting carbon-centered radical with VIII generates Ni(II) species IX. Reductive elimination from the electronically excited species X, promoted by EnT with mpg-CN, provides the final product and regenerates the Ni(0) species, thus completing the catalytic cycle.
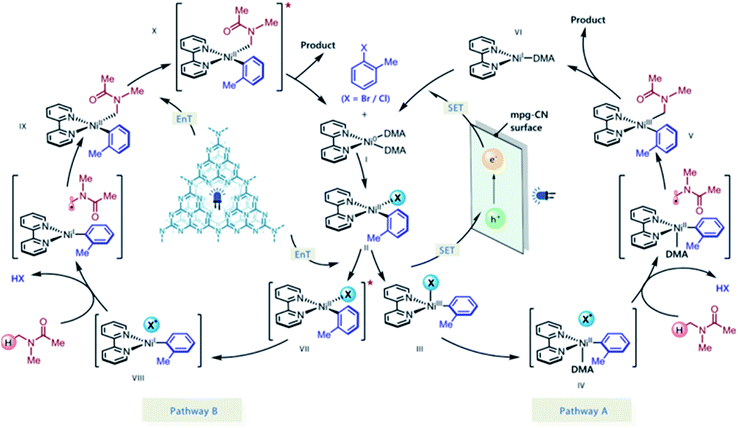 |
| | Scheme 18 Proposed reaction mechanism for mpg-CN/nickel photocatalytic C(sp3)–H arylation. Adapted with permission from ref. 168. | |
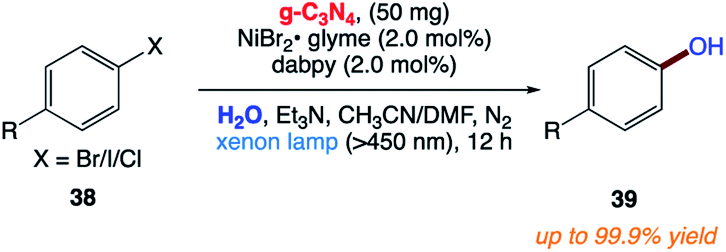
5.11 Synthesis of phenols from aryl halides
F. Zhang and co-workers169 reported the synthesis of phenols 39 via the facile and efficient hydroxylation reaction of aryl halides 38 with water through the merger of a heterogeneous organic semiconductor graphitic carbon nitride (g-C3N4) photocatalyst and a homogeneous nickel(II) bipyridine organometallic catalyst under visible-light irradiation at room temperature (Scheme 19).
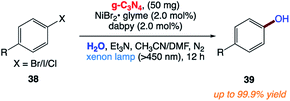 |
| | Scheme 19 Visible light-catalytic hydroxylation of aryl halides with water to phenols by carbon nitride and nickel complex cooperative catalysis. | |
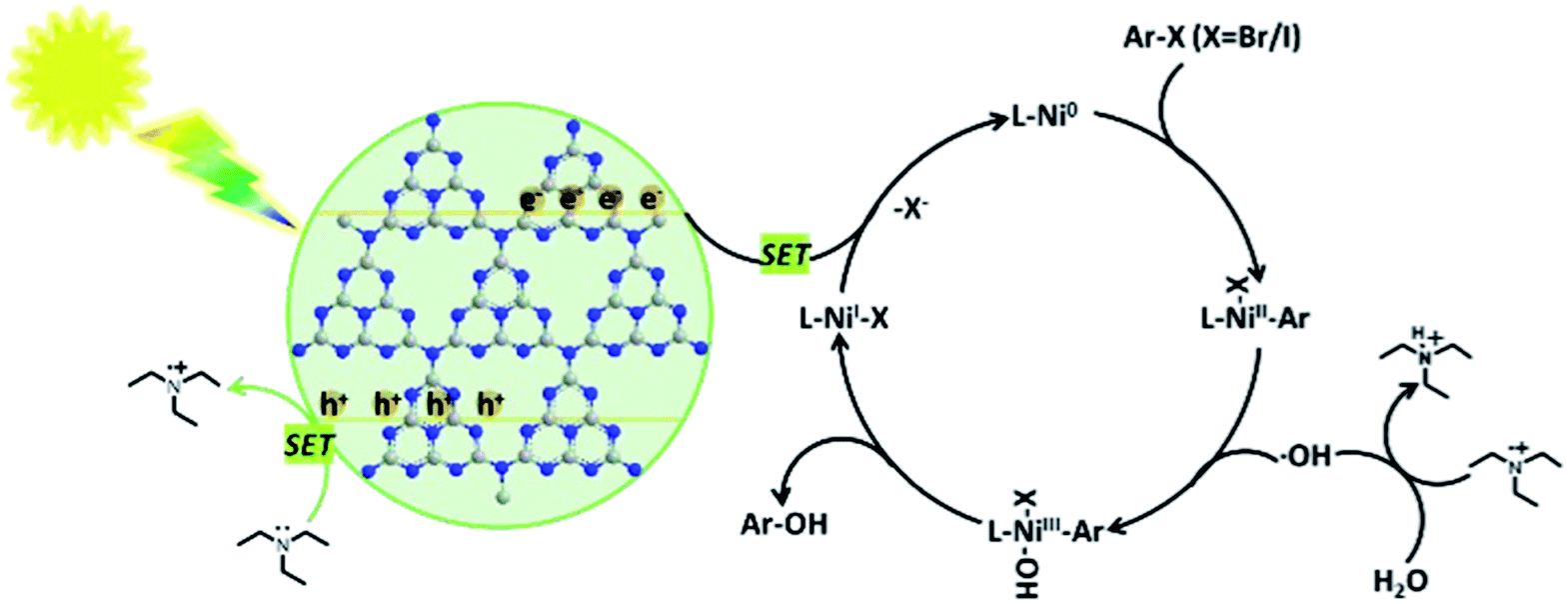
The reaction mechanism was proposed by the authors (Scheme 20). In the proposed mechanism for the Nidabpy/g-C3N4 dual catalytic system, g-C3N4 acts as a photocatalyst and Et3N provides electrons. After visible light illumination, g-C3N4 is excited to enable the effective separation of the photogenerated electron–hole pairs. The photogenerated holes can cause the oxidation of Et3N to form a cationic radical. Then, the Et3N radical undergoes a hydrogen atom transfer process with water to generate s hydroxyl radical. At this stage, the concurrent oxidative addition of aromatic halides to Ni(0) species should deliver the Ni(II) species, which would be trapped by the hydroxyl radical to form an Ni(III) organometallic adduct. Thereafter, reductive elimination would deliver the desired phenols product and Ni(I) species. Finally, the photogenerated electron by semiconductor excitation is utilized for the reduction of the Ni(I) species to Ni(0) species via single-electron transfer. Then, concurrent oxidative addition of aromatic halides to Ni(0) species delivers the Ni(II) species to complete the Ni catalytic cycle.
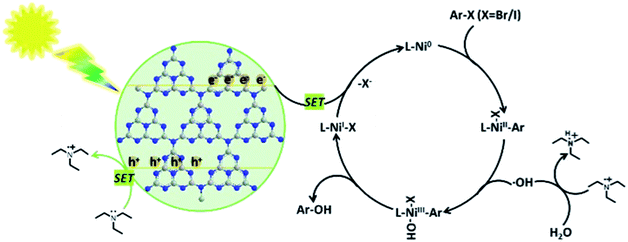 |
| | Scheme 20 Proposed mechanism of Nidabpy/g-C3N4-catalyzed visible-light-driven hydroxylation of aromatic halides with water. Adapted with permission from ref. 169. | |
5.12 Sonogashira coupling synthesis of ynones
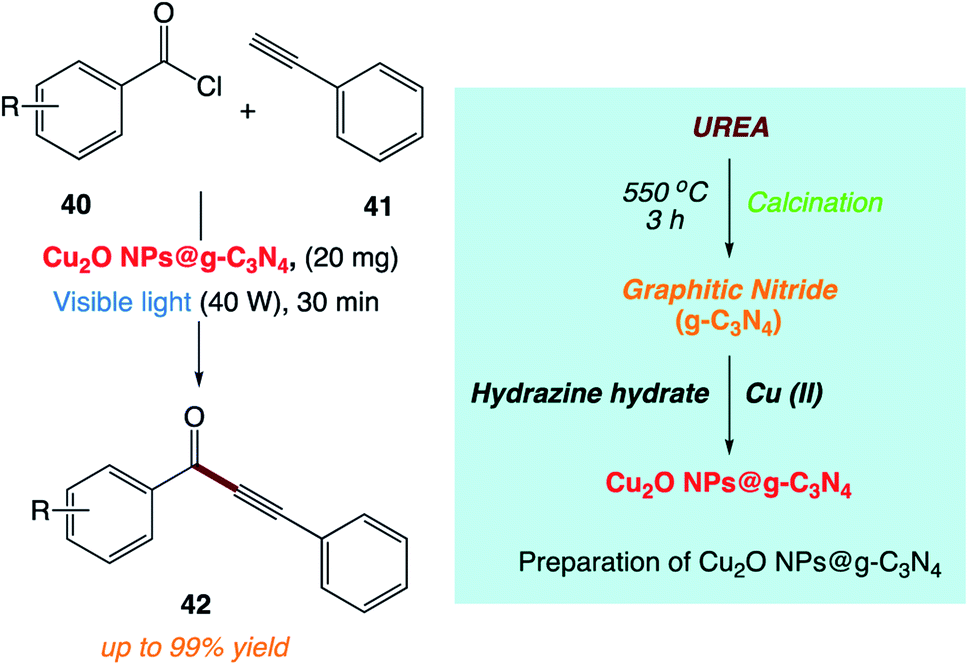
H. Kaur and co-workers170 reported the use of copper nanoparticles supported on graphitic carbon nitride (Cu2O NPs@ g-C3N4) as a photoactive catalyst for the synthesis of ynones under mild and sustainable conditions (Scheme 21). This methodology is simple and efficient and allows the reaction to progress more efficiently under visible light at room temperature.
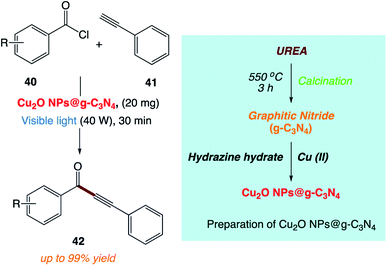 |
| | Scheme 21 Sonogashira coupling between benzoyl chloride and phenyl acetylene catalyzed by Cu2O NPs@g-C3N4. | |
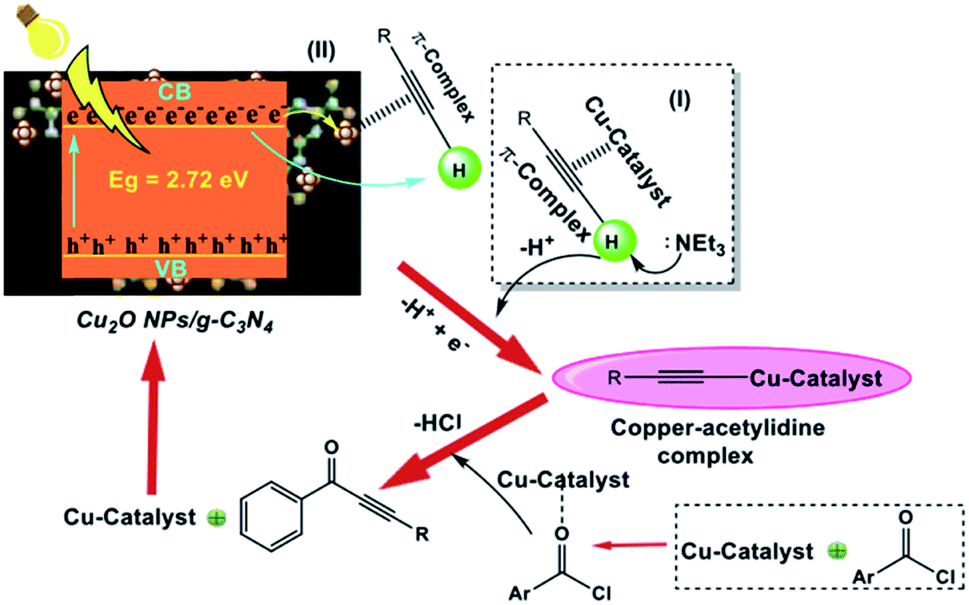
The authors proposed a plausible reaction mechanism for Cu2O NPs/g-C3N4 (Scheme 22). In this mechanism, they proposed that the reaction proceeds via the Csp-H activation of alkyne molecules under visible light irradiation given that Cu2O NP/g-C3N4 has the ability to promote the formation of the copper acetylide complex more effectively. The copper acetylide complex reacts with the preformed iminium ion to form a 2-pyridinyl propargyl amine intermediate. Subsequently, the cycloisomerization of the intermediate affords the desired product.
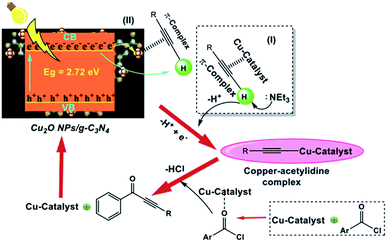 |
| | Scheme 22 Proposed reaction mechanism for the synthesis of ynones catalyzed by Cu2O NP/g-C3N4 catalyst. Adapted with permission from ref. 170. | |
5.13 Anaerobic oxidation of alcohols to ketones
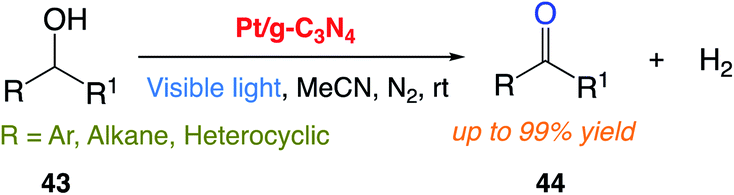
J. Liu and co-workers171 reported the use of a platinum nanocluster/graphitic carbon nitride (Pt/g-C3N4) composite solid catalyst with photocatalytic anaerobic oxidation function for the highly active and selective transformation of alcohols 43 to ketones 44. The desired products were successfully obtained in good to excellent yield from various functionalized alcohols at room temperature (Scheme 23).
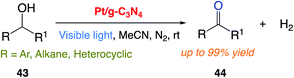 |
| | Scheme 23 . Photocatalytic dehydrogenation of alcohols under anaerobic conditions. | |
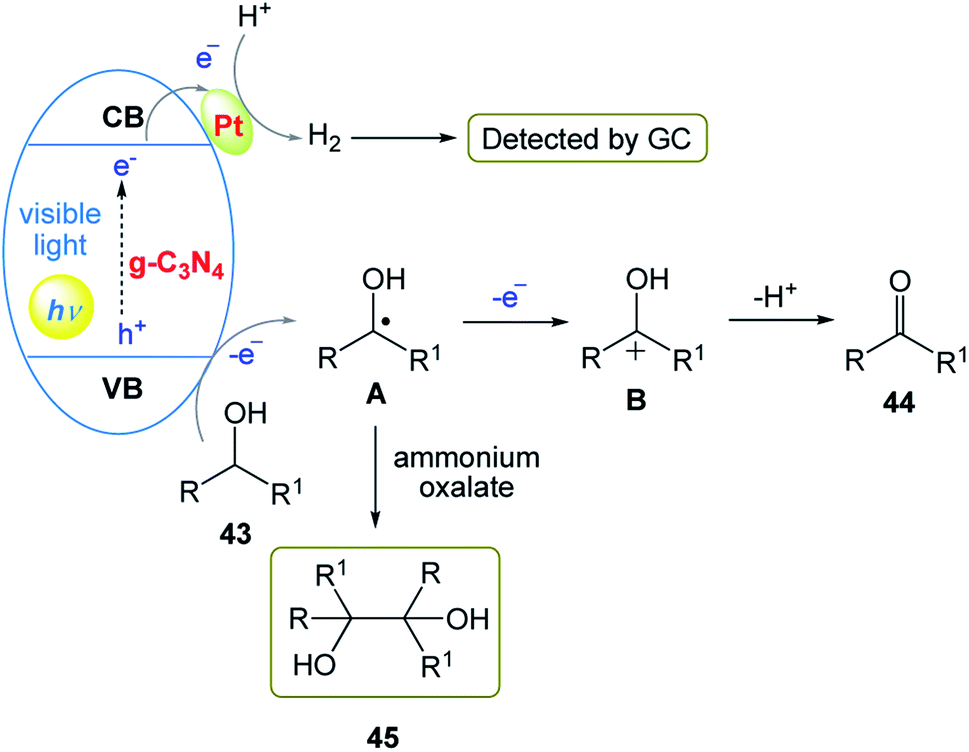
The authors proposed a possible two-electron oxidation mechanism for the photocatalytic anaerobic dehydrogenation of alcohols (Scheme 24). In this mechanism, firstly, holes (h+) on the valence band (VB) and electrons (e−) on the conduction band (CB) are generated under the irradiation of solar light. Then, the Pt-mediated h+ extracts hydrogen from substrate 43 to release α-alcohol radicals. Subsequently, the α-alcohol radical intermediate is oxidized to afford the corresponding product 44. Simultaneously, the electrons in the CB band transfer to the Pt nanoclusters through the Schottky-junction to drive the formation of H2 molecules. Furthermore, both byproduct 45 and H2 provide evidence for the reaction mechanism for the photocatalytic anaerobic dehydrogenation of alcohols.
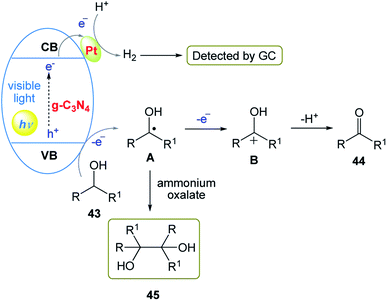 |
| | Scheme 24 Proposed two-electron oxidation mechanism of photocatalytic anaerobic dehydrogenation of alcohols initiated by Pt/g-C3N4. | |
5.14 Photocatalytic decarboxylative alkyl/acyl radical addition and reductive dimerization of para-quinone methides
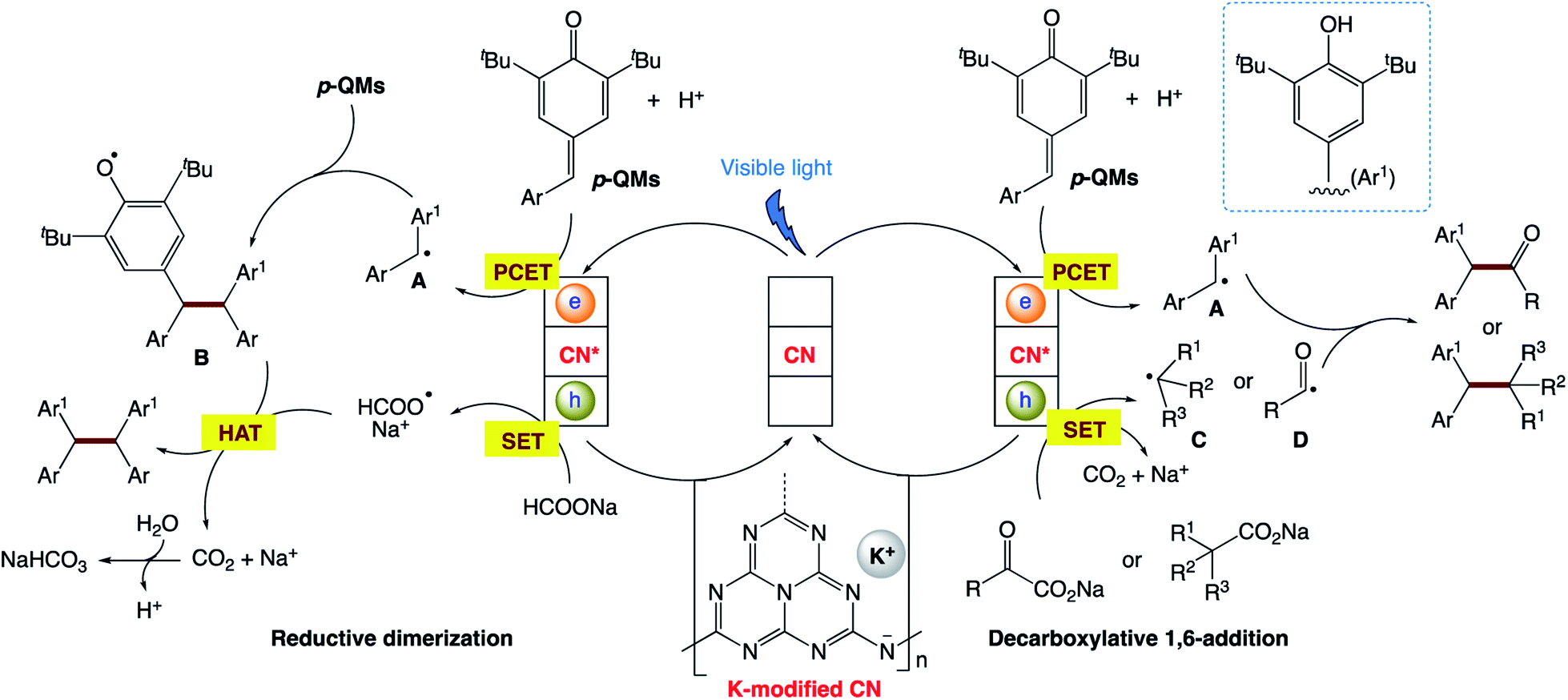
Y. Cai and co-workers172 reported a modified carbon nitride-based heterogeneous photocatalytic system for both the decarboxylative addition and reductive dimerization of para-quinone methides (Scheme 25). The potassium-intercalated carbon nitride (CN–K) was facilely prepared via the direct KCl-induced structure remodeling of bulk g-C3N4, exhibiting remarkable catalytic activity. The heterogeneous nature and mild reaction conditions of this protocol allow good catalyst recyclability, broad substrate scope, scale-up in a continuous flow, and applications for the target synthesis.
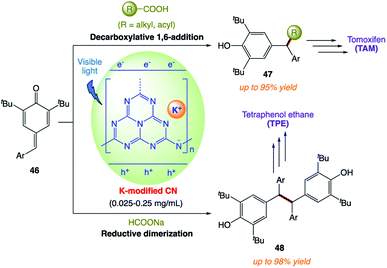 |
| | Scheme 25 Potassium-modified carbon nitride for heterogeneous photocatalytic decarboxylative alkyl/acyl radical addition and reductive dimerization of para-quinone methides. | |
The authors proposed the mechanism (Scheme 26), in which, firstly, visible-light absorption by CN–K leads to efficient charge separation, yielding two-dimensional surface redox centers as electron–hole pairs. The photo-electrons reduce p-QMs to the corresponding diaryl methyl radical A via the PCET process. For the reductive dimerization reaction, radical 1,6-addition of A to p-QMs generates radical B. The photogenerated hole of CN–K with a valence band potential of +0.84 eV undergoes single-electron oxidation with HCOONa to form HCOO˙, followed by hydrogen atom transfer (HAT) to B, affording the target dimerization products. For decarboxylative 1,6-addition reaction, the photogenerated hole of CN–K undergoes single-electron transfer (SET) with carboxylic acid anions (+0.91 to +1.25 V vs. SCE for benzylic acid anions) to deliver the corresponding alkyl radical C or acyl radical D. The direct coupling of radical A with radical C or D affords the target 1,6-addition adducts.
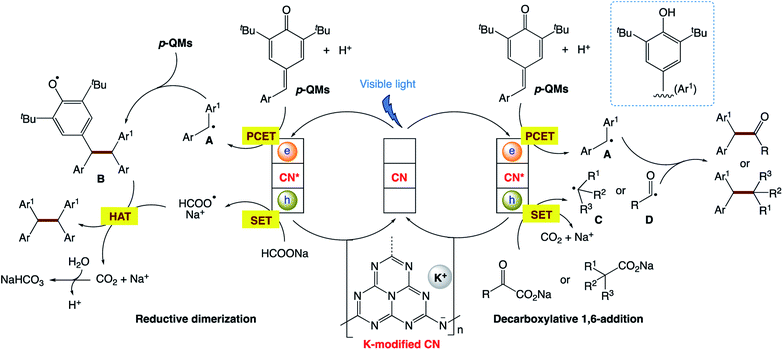 |
| | Scheme 26 Proposed reaction mechanism for potassium-modified carbon nitride for heterogeneous photocatalytic decarboxylative alkyl/acyl radical addition and reductive dimerization of para-quinone methides. | |
6 Conclusions and perspectives
In the field of photocatalysis, g-C3N4-based photocatalytic materials are considered one of the best candidates in terms of their properties, preparation, and applications. g-C3N4-based photocatalysts have significant advantages and simple strategies can be used to change the properties of these photocatalysts, while their overall structure composition remains unchanged. The outstanding properties of g-C3N4-based photocatalysts make them the preferred form of visible light photocatalysts for a variety of applications compared to other photocatalysts. This review includes the application of g-C3N4 via numerous synthetic strategies in an expanded manner. The application of g-C3N4 in chemical functionalization via the covalent or noncovalent approach using a wide range of chemical modifiers has been proven to be a useful strategy to amplify and introduce new properties in this photocatalyst. The properties and utilization of g-C3N4 will give researchers some new insights, ideas, and momentum, thus enhancing the production of clean energy and their application in efficient visible light-driven catalysts in sustainable development to next-level perspectives and encourage researchers for the future development in this research field. Although significant efforts have already been devoted to the modification of g-C3N4 materials and optimizing their photocatalytic activity, the potential of g-C3N4 materials is yet to be fully exploited. In summary, we believe that with the great efforts of researchers in synthesis, characterization, and mechanistic understanding, we can finally obtain an ideal photocatalyst that achieves the aim of utilizing g-C3N4 to carry out various chemical transformations.
Conflicts of interest
“There are no conflicts to declare”.
References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Y. Zheng, Z. M. Pan and X. C. Wang, Chin. J. Catal., 2013, 34, 524–535 CrossRef CAS.
- J. V. Liebig, Ann. Pharm., 1834, 10, 1–47 CrossRef.
- H. Huang, S. Yang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2014, 26, 5160–5165 CrossRef CAS.
- F. Goettmann, A. Fischer, M. Antonietti and A. Thomas, Chem. Commun., 2006, 4530–4532 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- Y. F. Sun, S. Gao, F. C. Lei, C. Xiao and Y. Xie, Acc. Chem. Res., 2015, 48(3), 712–721 CrossRef.
-
(a) J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121–4126 CrossRef CAS PubMed;
(b) J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
-
(a) E. Wirnhier, M. Dçblinger, D. Gunzelmann, J. Senker, B. V. Lotsch and W. Schnick, Chem.–Eur. J., 2011, 17, 3213–3221 CrossRef CAS;
(b) M. Xu, L. Han and S. J. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 12533–12540 CrossRef CAS.
-
(a) G. Liao, C. Li, X. Li and B. Fang, Cell Rep. Phys. Sci., 2021, 2, 100355 CrossRef CAS;
(b) G. Liao, C. Li, S. Y. Liu, B. Fang and H. Yang, Trends Chem., 2022, 4, 111–127 CrossRef CAS;
(c) Y. Liu, G. Xu, D. Ma, Z. Li, Z. Yan, A. Xu, W. Zhong and B. Fang, J. Cleaner Prod., 2021, 328, 129745 CrossRef CAS;
(d) Y. Liu, S. Shen, Z. Li, D. Ma, G. Xu and B. Fang, Mater. Charact., 2021, 174, 111031 CrossRef CAS;
(e) G. Liao, Y. Gong, L. Zhang, H. Gao, G. J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC;
(f) G. Liao, X. Tao and B. Fang, Matter, 2022, 5, 377–379 CrossRef CAS;
(g) P. Shandilya, P. Mandyal, V. Kumar and M. Sillanpää, Sep. Purif. Technol., 2022, 281, 119825 CrossRef CAS;
(h) A. Sudhaik, P. Raizada, P. Shandilya and P. Singh, J. Environ. Chem. Eng., 2018, 6(4), 3874–3883 CrossRef CAS;
(i) A. Sudhaik, P. Raizada, P. Shandilya, D. Y. Jeong, J. H. Lim and P. Singh, J. Ind. Eng. Chem., 2018, 67, 28–51 CrossRef CAS;
(j) P. Raizada, A. Sudhaik, P. Singh, P. Shandilya, V. K. Gupta, A. Hosseini-Bandegharaei and S. Agrawal, J. Photochem. Photobiol., A, 2019, 374, 22–35 CrossRef CAS;
(k) P. Raizada, A. Sudhaik, P. Singh, P. Shandilya, P. Thakur and H. Jung, Arabian J. Chem., 2020, 13(1), 3196–3209 CrossRef CAS;
(l) P. Shandilya, A. Guleria and B. Fang, J. Environ. Chem. Eng., 2021, 9(6), 106461 CrossRef CAS;
(m) A. Guleria, R. Sharma, A. Singh, N. K. Upadhyay and P. Shandilya, J. Water Process. Eng., 2021, 43, 102305 CrossRef;
(n) P. Shandilya, S. Sambyal, R. Sharma, A. Kumar and D. V. Vo, Mater. Res. Found., 2021, 112, 121–161 Search PubMed;
(o) P. Shandilya, P. Raizada and P. Singh, water Pollut. Rem., 2021, 119–146 Search PubMed;
(p) R. Sharma, G. G. Arizaga, A. K. Saini and P. Shandilya, Sustainable Mater. Technol., 2021, 29, e00319 CrossRef CAS;
(q) A. Guleria, R. Sharma and P. Shandilya, Adv. Mater., 2021, 100, 113–160 CAS;
(r) P. Raizada, A. Sudhaik, P. Singh, P. Shandilya, A. K. Saini, V. K. Gupta, J. H. Lim and H. Jung, Sep. Purif. Technol., 2019, 212, 887–900 CrossRef CAS;
(s) A. Kumar, P. Shandilya, D. V. Vo, G. Sharma, M. Naushad, P. Dhiman and F. J. Stadler, Environ. Chem. Lett., 2022, 20, 597–632 CrossRef CAS;
(t) P. Shandilya, R. Sharma, R. K. Arya, A. Kumar, D. V. Vo and G. Sharma, Int. J. Hydrogen Energy, 2021 DOI:10.1016/j.ijhydene.2021.08.190;
(u) P. Huang, J. Huang, J. Li, T. D. Pham, L. Zhang, J. He, G. W. Brudvig, N. A. Deskins, A. I. Frenkel and G. Li, J. Phys. Chem. C, 2022, 126(20), 8596–8604, DOI:10.1021/acs.jpcc.2c01216;
(v) C. Wang, W. Shi, K. Zhu, X. Luan and P. Yang, Langmuir, 2022, 38(18), 5934–5942 CrossRef CAS PubMed.
-
(a) V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88(1), 59 CrossRef CAS;
(b) V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2015, 88(3), 227 CrossRef CAS;
(c) V. Srivastava, P. K. Singh and P. P. Singh, Asian J. Chem., 2016, 28(10), 2159 CrossRef CAS;
(d) V. Srivastava, P. K. Singh and P. P. Singh, Croat. Chem. Acta, 2017, 90(3), 435 CrossRef CAS;
(e) V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377 RSC;
(f) V. Srivastava, P. K. Singh, S. Kanaujia and P. P. Singh, New J. Chem., 2018, 42, 688 RSC;
(g) P. K. Singh, P. P. Singh and V. Srivastava, Croat. Chem. Acta, 2018, 91(3), 383 CrossRef;
(h) V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 40 CrossRef CAS;
(i) V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 1333 CrossRef CAS;
(j) V. Srivastava, P. K. Singh and P. P. Singh, Tetrahedron Lett., 2019, 60, 151041 CrossRef CAS;
(k) V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, RSC Adv., 2020, 10, 20046 RSC;
(l) V. Srivastava, P. K. Singh and P. P. Singh, Rev. Roum. Chim., 2020, 65(3), 221 CrossRef;
(m) A. Srivastava, P. K. Singh, A. Ali, P. P. Singh and V. Srivastava, RSC Adv., 2020, 10, 39495 RSC;
(n) V. Srivastava, P. K. Singh, A. Srivastava and P. P. Singh, RSC Adv., 2021, 11, 14251–14259 RSC;
(o) V. Srivastava and P. P. Singh, Org. Biomol. Chem., 2021, 19, 313–321 RSC;
(p) V. Srivastava, P. K. Singh, A. Srivastava, S. Sinha and P. P. Singh, Photochem, 2021, 1, 237–246 CrossRef;
(q) P. P. Singh, P. K. Singh, M. Z. Beg, A. Kashyap and V. Srivastava, Synth. Commun., 2021, 51(20), 3033–3058 CrossRef CAS;
(r) V. Srivastava, P. K. Singh, S. Tivari and P. P. Singh, Org. Chem. Front., 2022, 9, 1485 RSC;
(s) V. Srivastava, P. K. Singh and P. P. Singh, J. Photochem. Photobiol., C, 2022, 50, 100488 CrossRef CAS.
- J. Liu, H. Wang, Z. P. Chen, H. Moehwald, S. Fiechter, R. van de Krol, L. Wen, L. Jiang and M. Antonietti, Adv. Mater., 2015, 27, 712–718 CrossRef CAS PubMed.
- Z. Xu, L. Guan, H. Li, J. Sun, Z. Ying, J. Wu and N. Xu, J. Phys. Chem. C, 2015, 119, 29062–29070 CrossRef CAS.
- Y. Cui, Z. Ding, Z. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef CAS.
- Z. Zhang, K. Leinenweber, M. Bauer, L. A. J. Garvie, P. F. McMillan and G. H. Wolf, J. Am. Chem. Soc., 2001, 123, 7788–7796 CrossRef CAS PubMed.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- G. Dong, Y. Zhang, Q. Pan and J. Qiu, J. Photochem. Photobiol., C, 2014, 20, 33–50 CrossRef CAS.
- Q. Han, N. Chen, J. Zhang and L. Qu, Mater. Horiz., 2017, 4, 832–850 RSC.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CrossRef CAS.
- J. Liang, Y. Zheng, J. Chen, J. Liu, D. Hulicova-Jurcakova, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 3892–3896 CrossRef CAS PubMed.
- H. Ji, F. Chang, X. Hu, W. Qin and J. Shen, Chem. Eng. J., 2013, 218, 183–190 CrossRef CAS.
- Q. Han, B. Wang, Y. Zhao, C. Hu and L. Qu, Angew. Chem., Int. Ed., 2015, 54, 11433–11437 CrossRef CAS PubMed.
- F. Dong, Z. Wang, Y. Sun, W. K. Ho and H. Zhang, J. Colloid Interface Sci., 2013, 401, 70–79 CrossRef CAS PubMed.
- Y. Zou, B. Yang, Y. Liu, Y. Ren, J. Ma, X. Zhou, X. Cheng and Y. Deng, Adv. Funct. Mater., 2018, 28, 1806214 CrossRef.
- G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083–8091 RSC.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Cora, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- B. Zhu, P. Xia, W. Ho and J. Yu, Appl. Surf. Sci., 2015, 344, 188–195 CrossRef CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176, 44–52 Search PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- A. Zambon, J. M. Mouesca, C. Gheorghiu, P. A. Bayle, J. Pecaut, M. Claeys-Bruno, S. Gambarelli and L. Dubois, Chem. Sci., 2016, 7, 945–950 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- D. Hollmann, M. Karnahl, S. Tschierlei, K. Kailasam, M. Schneider, J. Radnik, K. Grabow, U. Bentrup, H. Junge and M. Beller, et al., Chem. Mater., 2014, 26, 1727–1733 CrossRef CAS.
- T. Sano, S. Tsutsui, K. Koike, T. Hirakawa, Y. Teramoto, N. Negishi and K. Takeuchi, J. Mater. Chem. A, 2013, 1, 6489–6496 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. J. Cui, Z. X. Ding, P. Liu, M. Antonietti, X. Z. Fu and X. C. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- A. Li, W. Zhu, C. Li, T. Wang and J. Gong, Chem. Soc. Rev., 2019, 48, 1874–1907 RSC.
-
(a) Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC;
(b) F. He, Z. Wang, Y. Li, S. Peng and B. Liu, Appl. Catal., B, 2020, 269, 118828 CrossRef CAS.
- J. Wang, Z. Wang and Z. Zhu, Appl. Catal., B, 2017, 204, 577–583 CrossRef CAS.
- A. Bandyopadhyay, D. Ghosh, N. M. Kaley and S. K. Pati, J. Phys. Chem. C, 2017, 121(3), 1982–1989 CrossRef CAS.
- X. Lu, L. Gai, D. Cui, Q. Wang, X. Zhao and X. Tao, Mater. Lett., 2007, 61(21), 4255–4258 CrossRef CAS.
- J. Wang, L. Zhang, F. Long, W. Wang, Y. Gu, S. Mo, Z. Zou and Z. Fu, RSC Adv., 2016, 6, 23272–23278 RSC.
- X. Bai, L. Wang, R. Zong and Y. Zhu, J. Phys. Chem. C, 2013, 117(19), 9952–9961 CrossRef CAS.
- B. Z. Desalegn, H. S. Jadhav and J. G. Seo, ChemCatChem, 2019, 11(12), 2870–2878 CrossRef CAS.
- Y. Zeng, X. Liu, C. Liu, L. Wang, Y. Xia, S. Zhang, S. Luo and Y. Pei, Appl. Catal., B, 2018, 224, 1–9 CrossRef CAS.
- J. Liu and B. Cheng, Appl. Surf. Sci., 2018, 430, 348–354 CrossRef CAS.
- Z. Mo, X. Zhu, Z. Jiang, Y. Song, D. Liu, H. Li, X. Yang, Y. She, Y. Lei, S. Yuan, H. Li, L. Song, Q. Yan and H. Xu, Appl. Catal., B, 2019, 256, 117854 CrossRef CAS.
- J. Wu, N. Li, X. H. Zhang, H. B. Fang, Y. Z. Zheng and X. Tao, Appl. Catal., B, 2018, 226, 61–70 CrossRef CAS.
- M. Yin, F. Jia, C. Wu, P. Zheng, Y. Fan and Z. Li, Mater. Sci. Eng., B, 2017, 223, 35–42 CrossRef CAS.
- Q. Liu, J. Shen, X. Yu, X. Yang, W. Liu, J. Yang, H. Tang, H. Xu, H. Li, Y. Li and J. Xu, Appl. Catal., B, 2019, 248, 84–94 CrossRef CAS.
- T. Y. Ma, Y. Tang, S. Dai and S. Z. Qiao, Small, 2014, 10(12), 2382–2389 CrossRef CAS PubMed.
- J. Li, H. Hao, J. Zhou, W. Li, N. Lei and L. Guo, Appl. Surf. Sci., 2017, 422, 626–637 CrossRef CAS.
- L. Ma, G. Wang, C. Jiang, H. Bao and Q. Xu, Appl. Surf. Sci., 2018, 430, 263–272 CrossRef CAS.
- Z. Tong, D. Yang, X. Zhao, J. Shi, F. Ding, X. Zou and Z. Jiang, Chem. Eng. J., 2018, 337, 312–321 CrossRef CAS.
- S. Kang, X. Li, C. Yin, J. Wang, M. S. Aslam, H. Qi, Y. Cao, J. Jin and L. Cui, J. Colloid Interface Sci., 2019, 554, 269–277 CrossRef CAS PubMed.
- Z. Fang, Y. Hong, D. Li, B. Luo, B. Mao and W. Shi, ACS Appl. Mater. Interfaces, 2018, 10(24), 20521–20529 CrossRef CAS PubMed.
- Y. Li, Z. Ruan, Y. He, J. Li, K. Li, Y. Jiang, X. Xu, Y. Yuan and K. Lin, Appl. Catal., B, 2018, 236, 64–75 CrossRef CAS.
- S. Obregoń, A. Vaźquez, M. A. Ruíz-Go&macute;ez and V. Rodríguez- Gonzaĺez, Appl. Surf. Sci., 2019, 488, 205–212 CrossRef.
- X. Chen, R. Shi, Q. Chen, Z. Zhang, W. Jiang, Y. Zhu and T. Zhang, Nano Energy, 2019, 59, 644–650 CrossRef CAS.
-
(a) Y. Cai, Y. Tang, L. Fan, Q. Lefebvre, H. Hou and M. Rueping, ACS Catal., 2018, 8, 9471–9476 CrossRef CAS;
(b) A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS PubMed;
(c) I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed;
(d) Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed;
(e) P. Geng, Y. Tang, G. Pan, W. Wang, J. Hu and Y. Cai, Green Chem., 2019, 21, 6116–6122 RSC;
(f) K. S. Lakhi, D. H. Park, K. Al-Bahily, W. Cha, B. Viswanathan, J. H. Choy and A. Vinu, Chem. Soc. Rev., 2017, 46, 72–101 RSC;
(g) J.-Q. Di, M. Zhang, Y.-X. Chen, J.-X. Wang, S.-S. Geng, J.-Q. Tang and Z.-H. Zhang, Green Chem., 2021, 23, 1041–1049 RSC;
(h) S.-J. Chen, T.-F. Niu and B.-Q. Ni, Chin. J. Synth. Chem., 2021, 29, 562–569 Search PubMed;
(i) T. Niu, S. Chen, M. Hong, T. Zhang, J. Chen, X. Dong and B. Ni, Green Chem., 2020, 22, 5042–5049 RSC;
(j) B. Ni, B. Zhang, J. Han, B. Peng, Y. Shan and T. Niu, Org. Lett., 2020, 22, 670–674 CrossRef CAS PubMed.
- J. Liu, W. Li, L. Duan, X. Li, L. Ji, Z. Geng, K. Huang, L. Lu, L. Zhou and Z. Liu, et al., Nano Lett., 2015, 15, 5137–5142 CrossRef CAS PubMed.
- S. Guo, Y. Zhu, Y. Yan, Y. Min, J. Fan and Q. Xu, Appl. Catal., B, 2016, 185, 315–321 CrossRef CAS.
- Y. Bu and Z. Chen, Electrochim. Acta, 2014, 144, 42–49 CrossRef CAS.
- G. Dong, Z. Ai and L. Zhang, RSC Adv., 2014, 4, 5553–5560 RSC.
- X. She, L. Liu, H. Ji, Z. Mo, Y. Li, L. Huang, D. Du, H. Xu and H. Li, Appl. Catal., B, 2016, 187, 144–153 CrossRef CAS.
- G. Dong, K. Zhao and L. Zhang, Chem. Commun., 2012, 48, 6178–6180 RSC.
- P. Zhang, X. Li, C. Shao and Y. Liu, J. Mater. Chem. A, 2015, 3, 3281–3284 RSC.
- L. Zhang, X. Chen, J. Guan, Y. Jiang, T. Hou and X. Mu, Res. Bull., 2013, 48, 3485–3491 CrossRef CAS.
- D. H. Lan, H. T. Wang, L. Chen, C. T. Au and S. F. Yin, Carbon, 2016, 100, 81–89 CrossRef CAS.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- C. Xu, Q. Han, Y. Zhao, L. Wang, Y. Li and L. Qu, J. Mater. Chem. A, 2015, 3, 1841–1846 RSC.
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CrossRef CAS.
- S. Lin, X. Ye, X. Gao and J. Huang, J. Mol. Catal. A: Chem., 2015, 406, 137–144 CrossRef CAS.
- C. Lu, R. Chen, X. Wu, M. Fan, Y. Liu, Z. Le, S. Jiang and S. Song, Appl. Surf. Sci., 2016, 360(Part B), 1016–1022 CrossRef CAS.
- F. Raziq, Y. Qu, X. Zhang, M. Humayun, J. Wu, A. Zada, H. Yu, X. Sun and L. Jing, J. Phys. Chem. C, 2016, 120, 98–107 CrossRef CAS.
- H. Pan, H. Zhang, H. Liu and L. Chen, Solid State Commun., 2015, 203, 35–40 CrossRef CAS.
-
(a) Y. Wang, H. Li, J. Yao, X. Wang and M. Antonietti, Chem. Sci., 2011, 2, 446–450 RSC;
(b) Z. Lin and X. Wang, Angew. Chem., Int. Ed., 2013, 52, 1735–1738 CrossRef CAS PubMed.
- H. Li, Y. Liu, X. Gao, C. Fu and X. Wang, ChemSusChem, 2015, 8, 1189–1196 CrossRef CAS PubMed.
- G. Zhang, M. Zhang, X. Ye, X. Qiu, S. Lin and X. Wang, Adv. Mater., 2014, 26, 805–809 CrossRef CAS PubMed.
- Q. Han, C. Hu, F. Zhao, Z. Zhang, N. Chen and L. Qu, J. Mater. Chem. A, 2015, 3, 4612–4619 RSC.
- Y. Wang, Y. Di, M. Antonietti, H. Li, X. Chen and X. Wang, Chem. Mater., 2010, 22(18), 5119–5121 CrossRef CAS.
- Z. Lin and X. Wang, ChemSusChem, 2014, 7(6), 1547–1550 CrossRef CAS PubMed.
- Y. C. Lu, J. Chen, A. J. Wang, N. Bao, J. J. Feng, W. Wang and L. Shao, J. Mater. Chem. C, 2015, 3, 73–78 RSC.
- H. Ma, Y. Li, S. Li and N. Liu, Appl. Surf. Sci., 2015, 357(Part A), 131–138 CrossRef CAS.
- Y. Jiang, Z. Sun, C. Tang, Y. Zhou, L. Zeng and L. Huang, Appl. Catal., B, 2019, 240, 30–38 CrossRef CAS.
- S. Zhang, J. Li, X. Wang, Y. Huang, M. Zeng and J. Xu, J. Mater. Chem. A, 2015, 3, 10119–10126 RSC.
- J. Zhang, M. Zhang, R. Q. Sun and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145–10149 CrossRef CAS PubMed.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008–3014 CrossRef CAS.
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. Zou, ACS Catal., 2013, 3, 912–919 CrossRef CAS.
- G. Zhang and X. Wang, J. Catal., 2013, 307, 246–253 CrossRef CAS.
- X. Fan, L. Zhang, R. Cheng, M. Wang, M. Li, Y. Zhou and J. Shi, ACS Catal., 2015, 5, 5008–5015 CrossRef CAS.
- X. Ye, Y. Cui and X. Wang, ChemSusChem, 2014, 7, 738–742 CrossRef CAS PubMed.
- J. Zhang, X. An, N. Lin, W. Wu, L. Wang, Z. Li, R. Wang, Y. Wang, J. Liu and M. Wu, Carbon, 2016, 100, 450–455 CrossRef CAS.
- W. Ho, Z. Zhang, W. Lin, S. Huang, X. Zhang, X. Wang and Y. Huang, ACS Appl. Mater. Interfaces, 2015, 7(9), 5497–5505 CrossRef CAS PubMed.
- S. Chen, C. Wang, B. R. Bunes, Y. Li, C. Wang and L. Zang, Appl. Catal., A, 2015, 498, 63–68 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- Y. Guo, S. Chu, S. Yan, Y. Wang and Z. Zou, Chem. Commun., 2010, 46, 7325–7327 RSC.
- M. Z. Rahman, R. J. Ran, Y. Tang, M. Jaroniec and S. Qiao, J. Mater. Chem. A, 2016, 4, 2445–2452 RSC.
-
(a) W. Che, W. Cheng, T. Yao, F. Tang, W. Liu, H. Su, Y. Huang, Q. Liu, J. Liu, F. Hu, Z. Pan, Z. Sun and S. Wei, J. Am. Chem. Soc., 2017, 139(8), 3021–3026 CrossRef CAS PubMed;
(b) H. Ashrafi, M. Akhond, M. Zare and G. Absalan, Ind. Eng.
Chem. Res., 2021, 60, 3893–3906 CrossRef CAS.
- M. K. Bhunia, K. Yamauchi and K. Takanabe, Angew. Chem., Int. Ed., 2014, 53(41), 11001–11005 CrossRef CAS PubMed.
- Y. Yu, W. Yan, X. Wang, P. Li, W. Gao, H. Zou, S. Wu and K. Ding, Adv. Mater., 2018, 30(9), 1705060 CrossRef PubMed.
- S. Wang, H. Zhao, X. Zhao, J. Zhang, Z. Ao, P. Dong, F. He, H. Wu, X. Xu, L. Shi, C. Zhao, S. Wang and H. Sun, Chem. Eng. J., 2020, 381, 122593 CrossRef CAS.
- P. Zhou, F. Lv, N. Li, Y. Zhang, Z. Mu, Y. Tang, J. Lai, Y. Chao, M. Luo, F. Lin, J. Zhou, D. Su and S. Guo, Nano Energy, 2019, 56, 127–137 CrossRef CAS.
- Z. Zeng, Y. Su, X. Quan, W. Choi, G. Zhang, N. Liu, B. Kim, S. Chen, H. Yu and S. Zhang, Nano Energy, 2020, 69, 104409 CrossRef CAS.
- L. Zhang, R. Long, Y. Zhang, D. Duan, Y. Xiong, Y. Zhang and Y. Bi, Angew. Chem., Int. Ed., 2020, 59(15), 6224–6229 CrossRef CAS PubMed.
- X. Jin, R. Wang, L. Zhang, R. Si, M. Shen, M. Wang, J. Tian and J. Shi, Angew. Chem., Int. Ed., 2020, 59(17), 6827–6831 CrossRef CAS PubMed.
- G. Wang, T. Zhang, W. Yu, R. Si, Y. Liu and Z. Zhao, ACS Catal., 2020, 10(10), 5715–5722 CrossRef CAS.
- X. Xiao, Y. Gao, L. Zhang, J. Zhang, Q. Zhang, Q. Li, H. Bao, J. Zhou, S. Miao, N. Chen, J. Wang, B. Jiang, C. Tian and H. Fu, Adv. Mater., 2020, 32(33), 2003082 CrossRef CAS PubMed.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Jr Balkus, ACS Catal., 2012, 2, 949–956 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. Lett., 2012, 3, 663–672 CrossRef CAS PubMed.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Jr Balkus, ACS Catal., 2012, 2, 949–956 CrossRef CAS.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS.
- K. Zhou, Y. Zhu, X. Yang, X. Jiang and C. Li, New J. Chem., 2011, 35, 353–359 RSC.
- A. Furube, T. Asahi, H. Masuhara, H. Yamashita and M. Anpo, J. Phys. Chem. B, 1999, 103, 3120–3127 CrossRef CAS.
- B. Liu and E. S. Aydil, Chem. Commun., 2011, 47, 9507–9509 RSC.
-
(a) Y. Xiao, S. Guo, G. Tian, B. Jiang, Z. Ren, C. Tian, W. Li and H. Fu, Sci. Bull., 2021, 66(3), 275–283 CrossRef CAS;
(b) P. Shandilya, S. Sambyal, R. Sharma, P. Mandyal and B. Fang, J. Hazard. Mater., 2022, 428, 128218 CrossRef CAS PubMed.
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Dalton Trans., 2012, 41, 6756–6763 RSC.
- K. Katsumata, R. Motoyoshi, N. Matsushita and K. Okada, J. Hazard. Mater., 2013, 260, 475–482 CrossRef CAS PubMed.
- F. Jiang, T. Yan, H. Chen, A. Sun, C. Xu and X. Wang, Appl. Surf. Sci., 2014, 295, 164–172 CrossRef CAS.
- Y. Qin, H. Li, J. Lu, Y. Feng, F. Meng, C. Ma, Y. Yan and M. Meng, Appl. Catal., B, 2020, 277, 119254 CrossRef CAS.
- J. Cheng, X. Yan, Q. Mo, B. Liu, J. Wang, X. Yang and L. Li, Ceram. Int., 2017, 43(1), 301–307 CrossRef CAS.
- L. Liu, Y. Qi, J. Lu, S. Lin, W. An, Y. Liang and W. Cui, Appl. Catal., B, 2016, 183, 133–141 CrossRef CAS.
-
(a) Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6(7), 1543–1559 CrossRef CAS;
(b) C. Chen, X. Xie, B. Anasori, A. Sarycheva, T. Makaryan, M. Zhao, P. Urbankowski, L. Miao, J. Jiang and Y. Gogotsi, Angew. Chem., Int. Ed., 2018, 57, 1846–1850 CrossRef CAS PubMed;
(c) P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed;
(d) Y. L. Wang, Y. Tian, Z. L. Lang, W. Guan and L. K. Yan, J. Mater. Chem. A, 2018, 6, 21056–21063 RSC.
-
(a) Z. Pan, G. Zhang and X. Wang, Angew. Chem., Int. Ed., 2019, 58(21), 7102–7106 CrossRef CAS PubMed;
(b) H. Zhang, F. Feng, Y. Zhu, Y. Wu and T. Wu, J. Photochem. Photobiol., A, 2019, 371, 1–9 CrossRef CAS.
-
(a) X. Li, B. Kang, F. Dong, Z. Zhang, X. Luo, L. Han, J. Huang, Z. Feng, Z. Chen, J. Xu, B. Peng and Z. L. Wang, Nano Energy, 2021, 81, 105671 CrossRef CAS;
(b) Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CrossRef CAS.
-
(a) J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS;
(b) V. V. Pham, T. K. Truong, L. V. Hai, H. P. P. La, H. T. Nguyen, V. Q. Lam, H. D. Tong, T. Q. Nguyen, A. Sabbah, K. H. Chen, S. J. You and T. M. Cao, ACS Appl. Nano Mater., 2022, 5(3), 4506–4514 CrossRef CAS.
- W. Zhao, Y. Guo, S. Wang, H. He, C. Sun and S. Yang, Appl. Catal., B, 2015, 165, 335–343 CrossRef CAS.
- P. Christopher, H. Xin and S. Linic, Nat. Chem., 2011, 3(6), 467–472 CrossRef CAS PubMed.
- C. Wan, L. Zhou, L. Sun, L. Xu, D.-g. Cheng, F. Chen, X. Zhan and Y. Yang, Chem. Eng. J., 2020, 396, 125229 CrossRef CAS.
- F. He, S. Wang, H. Zhao, Y. Wang, J. Zhang, Q. Yan, P. Dong, Z. Tai, L. Chen, Y. Wang and C. Zhao, Appl. Surf. Sci., 2019, 485, 70–80 CrossRef CAS.
- L. Zhang, N. Ding, L. Lou, K. Iwasaki, H. Wu, Y. Luo, D. Li, K. Nakata, A. Fujishima and Q. Meng, Adv. Funct. Mater., 2019, 29(3), 1806774 CrossRef.
- A. Zada, M. Humayun, F. Raziq, X. Zhang, Y. Qu, L. Bai, C. Qin, L. Jing and H. Fu, Adv. Energy Mater., 2016, 6(21), 1601190 CrossRef.
- W. Wang, J. C. Yu, D. Xia, P. K. Wong and Y. Li, Environ. Sci. Technol., 2013, 47(15), 8724–8732 CrossRef CAS PubMed.
- J. Wan, C. Pu, R. Wang, E. Liu, X. Du, X. Bai, J. Fan and X. Hu, Int. J. Hydrogen Energy, 2018, 43(14), 7007–7019 CrossRef CAS.
- Q. Xu, B. Zhu, B. Cheng, J. Yu, M. Zhou and W. Ho, Appl. Catal., B, 2019, 255, 117770 CrossRef CAS.
- Y. Y. Han, X. L. Lu, S. F. Tang, X. P. Yin, Z. W. Wei and T. B. Lu, Adv. Energy Mater., 2018, 8(16), 1702992 CrossRef.
- Y. Li, H. Zhang, P. Liu, D. Wang, Y. Li and H. Zhao, Small, 2013, 9(19), 3336–3344 CAS.
- F. Ding, Z. Zhao, D. Yang, X. Zhao, Y. Chen and Z. Jiang, Ind. Eng. Chem. Res., 2019, 58(9), 3679–3687 CrossRef CAS.
- L. Song, X. Kang and S. Zhang, Int. J. Energy Res., 2018, 42(4), 1649–1656 CrossRef CAS.
- K. C. Christoforidis, Z. Syrgiannis, V. La Parola, T. Montini, C. Petit, E. Stathatos, R. Godin, J. R. Durrant, M. Prato and P. Fornasiero, Nano Energy, 2018, 50, 468–478 CrossRef CAS.
- Y. Chen, J. Li, Z. Hong, B. Shen, B. Lin and B. Gao, Phys. Chem. Chem. Phys., 2014, 16(17), 8106–8113 RSC.
- Y. Xu, H. Xu, L. Wang, J. Yan, H. Li, Y. Song, L. Huang and G. Cai, Dalton Trans., 2013, 42(21), 7604–7613 RSC.
- H. Zhao, S. Wang, F. He, J. Zhang, L. Chen, P. Dong, Z. Tai, Y. Wang, H. Gao and C. Zhao, Carbon, 2019, 150, 340–348 CrossRef CAS.
- X. Bai, L. Wang, Y. Wang, W. Yao and Y. Zhu, Appl. Catal., B, 2014, 152–153, 262–270 CrossRef CAS.
- K. Wang, X. Wang, H. Pan, Y. Liu, S. Xu and S. Cao, Int. J. Hydrogen Energy, 2018, 43(1), 91–99 CrossRef CAS.
- D. Qu, J. Liu, X. Miao, M. Han, H. Zhang, Z. Cui, S. Sun, Z. Kang, H. Fan and Z. Sun, Appl. Catal., B, 2018, 227, 418–424 CrossRef CAS.
- F. Wang, P. Chen, Y. Feng, Z. Xie, Y. Liu, Y. Su, Q. Zhang, Y. Wang, K. Yao, W. Lv and G. Liu, Appl. Catal., B, 2017, 207, 103–113 CrossRef CAS.
- W. J. Ong, L. K. Putri, Y. C. Tan, L. L. Tan, N. Li, Y. H. Ng, X. Wen and S. P. Chai, Nano Res., 2017, 10(5), 1673–1696 CrossRef CAS.
- Q. Liu, T. Chen, Y. Guo, Z. Zhang and X. Fang, Appl. Catal., B, 2016, 193, 248–258 CrossRef CAS.
- Q. Xiang, J. Yu and M. Jaroniec, J. Phys. Chem.C, 2011, 115(15), 7355–7363 CrossRef CAS.
- Y. F. Si, X. L. Chen, X. Y. Fu, K. Sun, X. Song, L. B. Qu and B. Yu, ACS Sustainable Chem. Eng., 2020, 8(29), 10740–10746 CAS.
- F. L. Zeng, H. L. Zhu, X. L. Chen, L. B. Qu and B. Yu, Green Chem., 2021, 23, 3677 RSC.
- Y. Zhang, N. Hatami, N. S. Lange, E. Ronge, W. Schilling, C. Joossc and S. Das, Green Chem., 2020, 22, 4516–4522 RSC.
- P. Geng, Y. Tang, G. Pan, W. Wang, J. Hu and Y. Cai, Green Chem., 2019, 21, 6116 RSC.
- J. Bai, S. Yan, Z. Zhang, Z. Guo and C. Y. Zhou, Org. Lett., 2021, 23, 4843–4848 CrossRef CAS PubMed.
- Y. Cai, Y. Tang, L. Fan, Q. Lefebvre, H. Hou and M. Rueping, ACS Catal., 2018, 8, 9471–9476 CrossRef CAS.
- J. Wang, L. Xue, M. Hong, B. Ni and T. Niu, Green Chem., 2020, 22, 411–416 RSC.
- B. Ni, B. Zhang, J. Han, B. Peng, Y. Shan and T. Niu, Org. Lett., 2020, 22(2), 670–674 CrossRef CAS PubMed.
- J. Khamrai, S. Das, A. Savateev, M. Antonietti and B. König, Green Chem., 2021, 23, 2017 RSC.
- S. Das, K. Murugesan, G. J. V. Rodríguez, J. Kaur, J. P. Barham, A. Savateev, M. Antonietti and B. König, ACS Catal., 2021, 11, 1593–1603 CrossRef CAS.
- K. Wang, M. Tong, Y. Yang, B. Zhang, H. Liu, H. Li and F. Zhang, Green Chem., 2020, 22, 7417–7423 RSC.
- A. S. Sharma, V. S. Sharma and H. Kaur, ACS Appl. Nano Mater., 2020, 3, 1191–1202 CrossRef CAS.
- D. Sun, P. Li, X. Wang, Y. Wang, J. Wang, Y. Wang, Y. Lu, L. Duan, S. Sarina, H. Zhub and J. Liu, Chem. Commun., 2020, 56, 11847–11850 RSC.
- Q. Yang, G. Pan, J. Wei, W. Wang, Y. Tang and Y. Cai, ACS Sustainable Chem. Eng., 2021, 9, 2367–2377 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Vishal Srivastava
a and
Vishal Srivastava