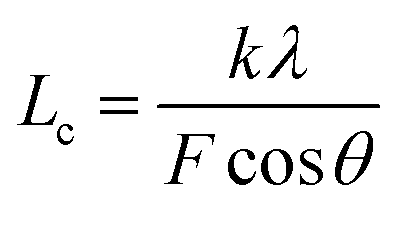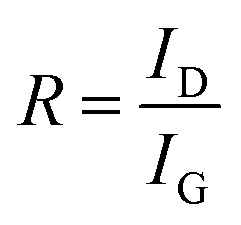DOI:
10.1039/D2RA01786E
(Paper)
RSC Adv., 2022,
12, 13339-13346
New insights into the radial structural differences of polyacrylonitrile fibres during thermal stabilization by the synchronous processing adjustment of time and temperature†
Received
19th March 2022
, Accepted 18th April 2022
First published on 4th May 2022
Abstract
In this study, the synchronous effects of time and temperature on the radial structural differences of polyacrylonitrile (PAN) fibres during thermal stabilization were investigated. For each sample to achieve equal densities (∼1.36 g cm−3), PAN fibres were thermally stabilized for various times between 8–32 min and at corresponding temperatures of 279–252 °C, which was considered to give a synchronous processing adjustment as a time–temperature integral (TTI). Besides, a previously developed mathematic model was utilized to quantitatively evaluate the differences in the radial heterogeneous structures of the stabilized PAN fibres as a function of TTI. It was found that several structural parameters (e.g., the stabilization degrees, the present crystallinities, and the orientation degrees) of PAN chains in the skin regions that mainly determine the fibres' overall performances were dramatically different from those in the core regions. Meanwhile, based on the TTI model, these skin–structure parameters demonstrated a strong correlation with the tensile properties of the resultant carbon fibres. However, while the stabilized PAN fibres had equal densities, their structural parameters, as well as the properties of the resultant carbon fibres, were obviously different.
1. Introduction
Polyacrylonitrile (PAN) fibres have been widely utilized as a type of precursor material for manufacturing carbon fibres (CFs) through a series of processes, including spinning, thermal-oxidative stabilization, carbonization, and surface treatments.1 The structural evolution of PAN fibres during the thermal stabilization process greatly determines the quality and production efficiency of CFs.2 To obtain CFs with better mechanical properties, the industrial processing time during thermal stabilization takes tens of minutes or longer. The current low production efficiency and high costs to manufacture CFs would limit industrial developments in the future. Hence, that the stabilization time has to be rationally reduced is highly necessary.
The mechanical properties (e.g. tensile strength and tensile modulus) of CFs mainly depend on their imperfection level,3,4 and the radial heterogeneity (also called ‘skin–core’ structure)5–7 is considered to be one of the main defects of PAN-based CFs. The formation of a skin–core structure is attributed to the oxygen diffusion along the radial direction of PAN fibres during their thermal stabilization,6,8–10 and the extent of radial oxygen diffusion is a function of the stabilization time.11,12 Due to the stabilized PAN fibres (SFs) having skin–core differences, some structural parameters, like the stabilization degree, aggregation structure, and orientation degree, might thus be different.13 These structural heterogeneities would be eventually inherited by the resultant CFs, which could cause certain losses in the mechanical properties of the CFs.14 Thus, for eliminating the skin–core effects of SFs, some investigations have manifested that adjusting the stabilization time is critical to changing the extent of the skin–core structures of SFs.15,16 When discussing the regulation between the skin–core effects and stabilization time, studies looking at the core region proportion in the cross-sectional images of SFs have shown a downward trend with extending the stabilization time, and have reported that the uneven contrast in the SFs' cross-section gradually disappears.
Besides, some researchers prepared CFs stemming from SFs with different stabilization times, and further measured their mechanical properties, displaying at first an increase and then a decrease trend over the prolonged stabilization time.17 Accordingly, when the stabilization time was shortened, the SFs' inner (core) regions were poorly stabilized due to an insufficient exposure to oxygen.18 These regions could easily evolve into structural defects during the following carbonization process, deteriorating the performances of the CFs. However, when the stabilization time was extended,19,20 excessive amounts of oxygen would be taken in by the SFs' outer (skin) region. Oxygen would then be released by the emission of the small molecular gases during the subsequent carbonization process,21,22 leaving certain defects in the CFs. Thus, further measuring the radial distribution of the structural parameters of SFs during the thermal stabilization process is key to preparing high-performance CFs by scientifically and reasonably reducing the stabilization time.
With the progress of thermal stabilization, the bulk density, which usually serves as a preliminary reference for the stabilization degrees of the SFs, kept increasing. There are some previous studies23–25 that indicated that the preparation of CFs with good mechanical properties generally requires controlling the bulk density within an appropriate range (about 1.36 g cm−3). To achieve SF samples with approximately equal densities, simply controlling the time is irrational, and it needs to be combined with compensation for the temperature adjustments. Thus, we propose a new concept in this area called the ‘time–temperature integral’ (denoted as ‘TTI’), which is employed to evaluate structural differences changing between the skin and core regions of SFs. This is like the time–temperature equivalence of the Williams–Landell–Ferry (WLF) equation, which empirically indicates that physically polymeric performances acquired by maintaining low temperature over a long time could be equally converted into performance at high temperature in a short time. Accordingly, there are some related applications in the gaseous permeabilities of polymeric membranes, the necking behaviours of polyester fibres, and other physical properties of polymer materials.26–29 However, physicochemical reactions during the stabilization process of PAN fibres are highly complex and are simultaneously affected by the stabilization temperature and time. Hence, for the perspective of the processing adjustment, TTI is simply equal to the multiplication of both the Kelvin temperature and time. Essentially, it could allow further figuring out the time effects during thermal stabilization on the SFs' structural evolutions and could provide a practical application basis for reasonably reducing the stabilization time needed to manufacture high-performance CFs.
2. Experimental
2.1 Materials
PAN precursor fibres (Special Acrylic Fibres, 1.21 d/tex, 6000 filaments per tow) were obtained from Courtaulds Ltd. UK. The comonomers of the precursor fibres were mainly made of 92.8 wt% acrylonitrile (AN), 6.0 wt% methyl acrylate (MA), and 1.2 wt% itaconic acid (IA).
2.2 Thermal stabilization and carbonization processes
To obtain a series of SFs samples with equal densities of about 1.36 g cm−3, the duration time, treatment temperature, TTI adjustment values, and the detailed properties were determined and are displayed in Table 1. PAN precursor fibres with a stretching ratio of 1.00 (stretching ratio, λ = v2/v1, where v stands for the velocity of a roller, as shown in Fig. 1) were thermally stabilized in an air or nitrogen atmosphere. The SFs thermally exposed to the air atmosphere were denoted as Air-SFs(G1), Air-SFs(G2), Air-SFs(G3), Air-SFs(G4), and Air-SFs(G5), while the SFs thermally exposed to the nitrogen atmosphere were named as N2-SFs(G1), N2-SFs(G2), N2-SFs(G3), N2-SFs(G4), and N2-SFs(G5).
Table 1 Detailed processing parameters setting during the stabilization process, TTI processing adjustments, and their SFs' densities
| Process code |
Temperature/°C (K) |
Time/min (s) |
TTI adjustments/105 s K |
Density/g cm−3 |
| G1 |
279(552.15) |
8(480) |
2.65 |
1.3634 |
| G2 |
268(541.15) |
14(840) |
4.55 |
1.3635 |
| G3 |
258(531.15) |
20(1200) |
6.37 |
1.3642 |
| G4 |
254(527.15) |
26(1560) |
8.22 |
1.3633 |
| G5 |
252(525.15) |
32(1920) |
10.08 |
1.3644 |
 |
| | Fig. 1 Schematic of the stabilization process and the direct carbonization process under the protection of a nitrogen atmosphere. | |
Subsequently, all of the Air-SFs samples from G1 to G5 had the carbonization treatment carried out under the protection of an inert atmosphere, as shown in Fig. 1. Two ends of the fibres were first fixed on graphite frames with a certain tension, which were then placed inside a straight-tube carbonization furnace to carry out the carbonization treatment according to the program with a heating rate of 5 °C min−1 from room temperature to 1400 °C. Before carbonization, the vacuuming and nitrogen filling of the furnace chamber were repeated three times at least. The sample codes of the resultant CFs were CFs(G1), CFs(G2), CFs(G3), CFs(G4), and CFs(G5), respectively.
2.3 Characterizations
In order to acquire optical micrographs of the SFs, SPI-PON 812, DDSA, NMA, and DMP-30 (a resin embedding kit provided by SPI Supplies Structure Probe, Inc.) were utilized to embed all the SFs samples placed in the vacuum drying chamber at 100 °C for 6 h. An ultra-thin slicer (Leica ARTOS, 3D) equipped with a glass-made cutter was used to prepare the sheet samples with a thickness of ∼500 nm at room temperature (∼23 °C), and an optical microscope was used to observe the morphology of the SFs' cross-sections with the ‘skin–core’ structures. As shown in Fig. 3(a) and (b), obvious skin–core structures of the SFs could be observed. Besides, the rest of the cross-sectional images can be found in the ESI, Fig. S1.† In the present study, we defined the skin proportion (η) to calculate the ratio of the skin regions to that of the whole fibre's cross-section. The quantification is considered as below:| |
 | (2-1) |
where Askin and Acore refer to the area of the skin and core regions, respectively. Image J software was used to determine the skin proportion. For each Air-SFs sample, the results obtained by the average of approximately 15 statistics were plotted as a function of TTI adjustments, as shown in Fig. 2.
 |
| | Fig. 2 Overall skin proportion (η) as a function of TTI adjustment, showing an increasing trend. | |
Additionally, scanning electron microscopy and the energy-dispersive X-ray technique (SEM-EDX, Oxford Instruments) were applied to observe the SFs' cross-sections and to measure the linear distribution of oxygen content along with the radial direction of the SFs.
The densities of the SFs were measured by a gradient density column (LLOYD), filled with a mixed organic solvent of tetrachloromethane and hexamethylene at 23 ± 0.1 °C, and the measurement accuracy was 0.0001 g cm−3. To analyze the stabilization degrees, the results related to the SFs samples that were prepared as KBr pellets were recorded by a PerkinElmer FTIR spectrometer in 32 scans in a combined scan range from 4000 to 400 cm−1 with a resolution of 4 cm−1. The fraction of reacted nitrile groups (FNs) and dehydrogenation index (DHI) were determined by the following formulae:9
| |
 | (2-2) |
| |
 | (2-3) |
where
n is 0.29.
30 In the infrared spectrum, Abs(1590) and Abs(2244) mean the absorbance of –C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif)
N– and –C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif)
N groups, respectively. Besides, Abs(1360) and Abs(1454) refer to the absorbance of –CH
![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif)
and –CH
2– groups,
31 respectively.
A PerkinElmer FTIR spectrometer equipped with a wire-grid polarizer was employed in the characterization of the PAN chain orientations. When using the polarizer, two spectra for each SF sample were obtained by means of both parallel and perpendicular infrared radiations on the sample. The dichroic ratio D and the orientation degree (f) were calculated according to the following formulae:32
| |
 | (2-4) |
| |
R0 = 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cot2α cot2α
| (2-5) |
| |
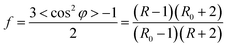 | (2-6) |
where Abs
∥ is the absorbance of the nitrile group with the electric vector of polarized infrared radiation parallel to the fibre axis, while Abs
⊥ is the absorption perpendicular to the fibre axis, and
α is the value of about 70° for uniaxially drawn PAN fibres, indicating the transition-moment angle between the direction of the dipole-moment and PAN-chain axis, and
φ is the angle associated with the molecular segment relative to the stretching direction. The orientation degree of the PAN amorphous chains (
fa) could be accordingly determined by the equation below:
33| | |
f = βfc + (1 − β)fa
| (2-7) |
The XRD patterns were obtained on a D/Max-2500 PC X-ray diffractometer with Cu Kα characterized radiation, which was produced at 40 kV and 40 mA power and a scanning range of 5°–60°. Subsequently, the crystallinities (β) and the crystallite sizes (Lc) of the SFs were calculated by the following formulae:34
| |
 | (2-8) |
| |
 | (2-9) |
where
Ac and
Aa represent the total areas of the pattern peaks of 2
θ = 16.7° and 29.4°, as well as that of 2
θ = 26.5°, respectively. Besides,
λ is the X-ray wavelength (0.1542 nm),
θ is Bragg's angle,
F is the FWHM (Full Width at Half Maximum) value of the diffraction peak, and
k is a constant value of 0.89.
35 Based on Herman's equation, the ordered phase orientation degree (
fc) of the PAN chains could be calculated; however, the results could be also rapidly acquired in the study by using the following empirical equation:
33| |
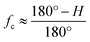 | (2-10) |
To measure the ordered degree of the graphite structures of the CFs, all the samples were analyzed by Raman spectroscopy (inVia Reflex, Rein Shaw, 514 nm laser excitation) in the band range from 100 to 3500 cm−1. The ratio (denoted to R) of the D band intensity (ID) to the G band intensity (IG) was calculated by the following equation:
| |
 | (2-11) |
For testing single-filament mechanical properties, including tensile strength and tensile modulus, the CFs samples were tested on a XQ-1A Fibre Tensile Tester at a pulling rate of 5 mm min−1 and a testing gauge of 25 mm. The final results of the CFs were the average of around 35 tests.
3. Results and discussion
3.1 Methodology
It is recognized that there is an optimal degree of stabilization for producing high-performance CFs. Lower stabilization degrees would result in CFs with deteriorated crystalline structures due to the insufficient formation of the thermally stable cross-linking structures. If the stabilization degrees are too high, the excessive oxygen exposure may also bring about more oxygen-containing structures (chemical groups) in the PAN chains, and the mechanical properties of the resultant CFs could not be better acquired.36–39 Specifically, during the next low-temperature carbonization process, owing to the larger carbon loss with more gaseous emissions of CO, CO2, H2O, O2, HCN, higher molecular weight compounds, miscellaneous tars, etc., the imperfections will be inherited in the final CFs.1 This is also the main reason why SFs with excessive stabilization degrees lead to a relatively poor mechanical performance of the CFs.
In addition, the SFs' density value is a general reflection of the extent of the stabilization reactions, and high-performance CFs are typically obtained when the density of the SFs is around 1.36 g cm−3.23–25 The stabilization degrees are mainly controlled by the processing parameters, such as the time, temperature, and drawing ratio. The densities of the SFs cannot be maintained constant by solely changing the stabilization time. The temperature has to be adjusted accordingly to compensate for the time adjustments. Thus, the synchronous adjustment of the time and temperature during stabilization is used, and a mathematical model to analyze the structural differences in both the skin and core regions was further developed.
The skin regions of SFs are typically considered as the areas with the higher stabilization degrees, while the core regions are not affected by oxygen, having lower stabilization degrees. Besides, we characterized the oxygen content along the fibres' diameter direction using SEM-EDX analysis. As shown in Fig. 3(c) and (d), there are some testing-point positions, namely Air-SFs(G3) points in red, and N2-SFs(G3) points in deep blue, respectively. Other sample images can be seen in Fig. S2 and S3.† The detailed results of the relative oxygen contents are shown in Table 2, and indicate that the outer region of the fibres thermally exposed to oxygen had a higher oxygen content, while that of the core region had a lower relative content. More importantly, the core region of Air-SFs and the entire cross-section of N2-SFs demonstrated similar values of oxygen content. Therefore, we can presumably use the structural parameters of the N2-SFs to represent the parameters of the core region of the Air-SFs.
 |
| | Fig. 3 (a) and (b) Optical images of sample slices of Air-SFs(G3) and N2-SFs(G3), respectively; (c) and (d) SEM images of cross-sections of Air-SFs(G3) and N2-SFs(G3), respectively. | |
Table 2 Detailed results for the relative oxygen contents of Air-SFs(G3) and N2-SFs(G3) diametrically at the five test points
| Position |
Relative oxygen contents of SFs (%) |
| Air-SFs(G3) |
N2-SFs(G3) |
| 1 |
9.84 |
5.95 |
| 2 |
5.92 |
5.73 |
| 3 |
5.97 |
5.99 |
| 4 |
6.07 |
5.81 |
| 5 |
10.06 |
6.07 |
The structural parameters of Air-SFs, including the fraction of reacted nitrile groups (FNs), dehydrogenation index (DHI), crystallinity (β), crystallite size (Lc), and orientation degree (f), were averaged values of both the skin and the core. Meanwhile, the structural parameters of the core of Air-SFs, including FNscore, DHIcore, βcore, Lc,core, and fcore, could be acquired through characterizing the N2-SFs. Thus, the structural parameter of the skin region Air-SFs, including FNsskin, DHIskin, βskin, Lc,skin, and fskin, could be deduced. The detailed formulae are displayed as follows:
| | |
FNs = ηFNsskin + (1 − η)FNscore
| (3-1) |
| | |
DHI = ηDHIskin + (1 − η)DHIcore
| (3-2) |
| | |
β = ηβskin + (1 − η)βcore
| (3-3) |
| | |
Lc = ηLc,skin + (1 − η)Lc,core
| (3-4) |
| | |
f = ηfskin + (1 − η)fcore
| (3-5) |
| | |
fc = ηfc,skin + (1 − η)fc,core
| (3-6) |
| | |
fskin = βskinfc,skin + (1 − βskin)fa,skin
| (3-7) |
Importantly, when an overall structural parameter of the SFs is separated, the structural evolutions in the skin and core regions would perform differently, which may influence the performances of the final CFs. Thus, these specific separated factors should be better measured and controlled in the thermal stabilization to scientifically reduce the duration time and improve the performances of the resultant CFs.
3.2 Integrating the effects of time/temperature on the stabilization/aggregation structures parameters both in the skin and core regions of stabilized PAN fibres
Based on previous studies,9,10,21,22,30 the stabilization degrees and the aggregation structures of SFs were typically measured by several stabilization processing parameters, including the stretching ratio, treatment temperature, and duration time. Due to the ubiquity of the skin–core structure in SFs, the PAN chains' structures in the skin regions were remarkably different from those in the core regions. Utilizing the aforementioned model, two kinds of stabilization degree and aggregate index of the SFs were quantified, which involved the faction of reacted nitrile groups (FNs), dehydrogenation index (DHI), crystallinity (β), and the crystallite size (Lc). Consequently, these structural parameter results that were calculated by eqn (3-1)–(3-4) were highly different even though the SFs' densities were the same.
Fig. 4(a) and (b) show that the stabilization degrees (FNs and DHI) in the skin region were higher than in the core region, which was because, during the thermal stabilization, the fibre's outer layer was first exposed to oxygen, which could subsequently promote cyclization, dehydrogenation and oxidation reactions to facilitate the increase in FNs and DHI.40 When TTI < 6.37 × 105 s K, decreasing trends were observed in response to the stabilization degrees in the skin region, while the trends for the stabilization degrees were converted into upward at a TTI adjustment of 6.37 × 105 s K. Since this may because the skin regions of the SFs were first influenced by the radial diffusion of oxygen, a relatively larger stabilization degree during a short duration time depends on a much higher stabilization temperature.8 Conversely, the main contributor to the increased stabilization degrees was responsible for the prolonged stabilization time.12 On the other hand, when the stabilization time was about 20 min (corresponding to a TTI of 6.37 × 105 s K), the stabilization degrees contributed by the extended stabilization time began to compensate for the decrease caused by the drop in stabilization temperature. However, it was also found that the stabilization degrees in the core region gradually decreased with the increase in TTI. This may be attributed to the PAN chains in the core region being thermally stabilized in the oxygen-deficient environment, and the changes in the stabilization degrees were mainly affected by the temperature.
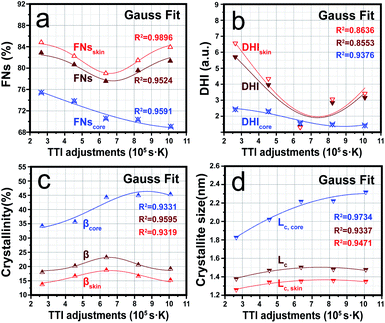 |
| | Fig. 4 Trends in the two kinds of stabilization degrees both in the skin and core regions as a function of TTI: (a) fraction of reacted nitrile groups (FNs), (b) dehydrogenation index (DHI); (c) and (d) crystallinities (β) and crystallite sizes (Lc) of the ‘skin–core’ structures of the SFs with increasing TTI, respectively. | |
Fig. 4(c) and (d) represent a changing plot of the aggregated structures both in the skin and core regions during the thermal stabilization process by the synchronous adjustment of the time and temperature. Due to the fibres' core region not being affected by oxygen diffusion, βcore and Lc,core not only gradually rose with the increase in TTI, but also were larger than βskin and Lc,skin. In addition, βskin and Lc,skin showed a completely different trend compared to that of the core region, which was a trend of initially increasing and then decreasing.
According to some studies, it has been found that some stabilization reactions, including cyclization, dehydrogenation, and oxidation reactions, are initiated at the amorphous PAN chains, and then gradually expand to the ordered region with the progression of stabilization reactions.40 This finally results in crystal PAN chains being destroyed and disordered, becoming new amorphous chain regions that can process other stabilization reactions. Therefore, combined with the stabilization degrees results discussed in the previous section, the evolution trend of the aggregate structures both in the skin and core regions was opposite to that of the stabilization degrees. Furthermore, the extents of the stabilization reactions or the aggregation structures of the SFs were mainly sensitive to the temperature adjustment during the stabilization process. When it comes to a single processing adjustment (time or temperature) to the PAN fibres during thermal stabilization, decreasing the crystallinities and crystallite sizes are the results of an elevated temperature or extended time. Thus, it was found that the stabilization temperature was much more dominant in the downward trend of the stabilization degrees and the upward trend of the aggregation structure evolutions in the core region of the SFs. Importantly, the stabilization degrees and aggregation structures both in the skin and core regions of SFs still showed some greatly different evolutions, even though the densities of the SFs were the same.
3.3 Integrating the effects of time/temperature on the orientation of PAN chains both in the skin and core regions of stabilized PAN fibres
Based on the TTI model, the orientation degrees of PAN chains both in the skin and core regions (fskin and fcore) were examined. Although the density values of each fibre are approximately the same, the overall orientation degree of the chains both in the skin–core regions presented significantly different trends. Fig. 5 plots the orientation degrees in quasicrystals and amorphousness forms (fc and fa) and that of the skin–core regions of SFs (fskin, fcore, fc,skin, fc,core, fa,skin and fa,core) during different TTI adjustment for equal densities.
 |
| | Fig. 5 (a) Overall orientation degrees of Air-SFs and the skin–core regions, (b) orientation degrees in the amorphous Air-SFs and the skin–core regions, (c) orientation degrees in quasicrystals of Air-SFs and the skin–core regions. | |
As the TTI increasingly changed, it could be seen that fa,skin grew slowly, while fc,skin showed an upward changing trend, but its increasing rate was higher than with the former when TTI was less than 6.37 × 105 s K. Subsequently, fc,skin turned to a downward trend with continuing to enhance the TTI, which may be the reason that a certain extent of stabilization reactions, including cyclization, dehydrogenation, and oxidation reactions, which are processed by prolonged duration time, were initiated in the amorphous chains and extended into ordered-chain parts, leading to disorientation in this region. Besides, fcore and fc,core simultaneously showed a similar enhancing trend with increasing TTI, while fa,core did not make a big difference at a relatively low TTI control, but then showed a great climb after the TTI went from 6.37 to 10.08 × 105 s K.
Importantly, it is noteworthy that the stabilization degrees in the core region, FNscore and DHIcore, dropped slightly. In addition, the simultaneous enhancement of βcore and Lc,core could be viewed with the elevated TTI, in which relatively more ordered chains were thus preserved and a relatively high fc,core was maintained. In some regions of the fibres without the diffusion effects of oxygen, a monotonous performance of the structural parameters trend in the core regions was detected with the TTI progressing. This is in contrast to the skin regions that included the effects of oxygen diffusion, whereby the monotonicity was somewhat changed. Generally, combined with the previous discussions, including on the stabilization, aggregation evolutions, the PAN chains' orientation structures in the skin and core regions of the SFs also presented big differences as a function of the TTI.
3.4 Effects of the stabilized PAN fibres' structural differences on the resultant carbon fibres' performances
Some structural characterizations (Raman and WAXD) and mechanical performance tests were applied to reveal the influence of the structural differences of the SFs on the relationship between the CFs' structures and their mechanical properties. For visible excitation testing, the typical features in the Raman spectra of carbon fibres or other carbon-like materials are the D band (lying at ∼1360 cm−1) and the G band (lying at ∼1580 cm−1), which are mainly resonations of sp3-carbons (or defected graphite) and sp2-carbons (or ordered graphite), respectively.41
As shown in Fig. 6(a) and (b), Raman spectra of the resultant CFs were recorded and quantified by the ratio (eqn (2-11)) of the D band intensity (ID) to the G band intensity (IG), which can reflect the graphitic structure, stacking order, and crystal lateral thickness of the final CFs.42,43 Consequently, it was found that with an elevated TTI, the results for the DG ratio showed the trend of at first a decrease and then an increase. This indicated that the final CFs with relatively more ordered structures should be at the lowest DG ratio value when TTI was 6.37 × 105 s K, and the mechanical properties may be accordingly better. WAXD curves and the quantified results, and the graphite stack size of the crystal plane (002), can be observed in Fig. 6(c) and (d), respectively. Obvious diffraction peaks of (002) could be found in the spectra of the CFs, and the calculated Lc results displayed two trends during the TTI control: (i) a plain plot keeping low during the high stabilization temperature–short time treatment (when TTI < 6.37 × 105 s K); (ii) starting with the highest value when the TTI reached 6.37 × 105 s K, it was converted into a slowly dropping trend with the elevated TTI.
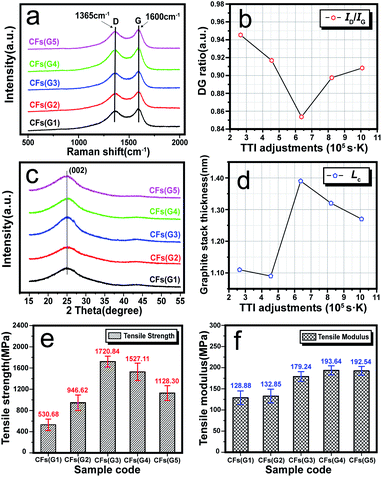 |
| | Fig. 6 (a) Raman spectra of the resultant CF samples, including CFs(G1–G5), (b) ID/IG trend of the resultant CF samples with elevated TTI adjustments, (c) XRD curves of the resultant CF samples, (d) graphitic stacking thickness examined by the diffraction peak denoted to the (002) crystal plane, of 2θ ≈ 17° in the XRD pattern of the CF samples; (e) and (f) tensile strength and tensile modulus of the resultant CF samples, respectively, which stemmed from the PAN precursor thermally exposed to an air atmosphere. | |
After carbonization of the SFs, the mechanical properties of the CFs were measured, and the trends with increasing TTI were plotted in Fig. 6(e) and (f), such as the tensile strength and tensile modulus. Similarly, the tensile strength also tended to increase first and then decreased with the maximum value (1720.84 MPa) at the TTI of 6.37 × 105 s K. Accordingly, this may be due to what the proper stabilization degrees of the SFs are for retaining a maximum fc,skin value of the SFs. This could cause a nice performance of the final CFs in terms of the tensile strength. However, the tensile modulus of the CFs was enhanced slightly with the elevated TTI. There may be positively correlated to some reasons: (a) the upward trend of skin proportion of the SFs with increasing TTI control, as well as (b) the rising-up of fa,skin for the formation of more graphitizable structures inside the skin regions, which may provide a greater extent of tensile modulus after carbonization treatment.
Through a series of reasonable adjustments of the TTI, the structural parameters both in the skin and core regions of the SFs during the thermal stabilization process, involving the stabilization degrees, aggregation structures, and orientation degrees, etc., could be better clarified. Also, it could imply that although the PAN fibre samples were thermally stabilized with equal densities of ∼1.36 g cm−3, there were massive differences among the SFs' structural parameters, and each resultant CFs sample performed in conformity with the graphitic structures and the mechanical properties (tensile strength and tensile modulus) with increasing the TTI control. This means that a suitable stabilization processing in the future needs to be utilized for obtaining preferred structures and the performances of industrial CF products.
4. Conclusions
In summary, through synchronous adjustments of the stabilization time and temperature, PAN fibres were prepared with equal densities of around 1.36 g cm−3. The synchronous effects on the radial structural differences of the PAN fibres were comprehensively investigated by introducing an integrating framework, called the time–temperature integral (TTI). Consequently, remarkable structural differences were observed in both the skin and core regions, and the skin–structure evolutions were the determining factors that dominantly influenced the overall performances of the SFs. When the TTI adjustment was 6.37 × 105 s K, the stabilization degrees in the skin regions reached a minimum, and the orientation in the quasicrystals of the skin regions was the highest, which facilitated the aggregate-structure integration, and simultaneously maximized the crystallinity and crystalline size. Therefore, during the TTI adjustment during the stabilization process, a strong correlation between the skin–structure evolutions and performances of the resultant CFs was observed. Furthermore, when prepared by the SFs with these skin–structure features, CFs with a relatively preferred graphitic stacking thickness could be obtained, which made the mechanical properties better. However, this was in contrast to the structural evolutions in the core regions, where there were insignificant effects on the performances of the CFs. Admittedly, this could help the understanding of why, even when the densities of the SFs were identical, the structures and the properties of the resultant CFs showed different results.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research is supported by the Natural Science Foundation of Jiangsu Province (Grant No. BK2019021292), the special fund of Jiangsu Province for the Transformation of Scientific and Technological Achievements (BA2019043), and the Jiangsu Provincial Key Research and Development Program (BE2021014-2).
References
- E. Frank, L. M. Steudle, D. Ingildeev, J. M. Spörl and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2014, 53, 5298 CrossRef PubMed.
- P. Wang, J. Liu, Z. Yue and R. Li, Carbon, 1992, 30, 120 CrossRef.
- J. Liu, Z. Yue and H. Fong, Small, 2009, 5, 542 Search PubMed.
- H. Okuda, R. J. Young, F. Tanaka, J. Watanabe and T. Okabe, Carbon, 2016, 107, 481 CrossRef.
- G. K. Layden, Carbon, 1972, 10, 63 CrossRef.
- S. Nunna, M. Naebe, N. Hameed, B. L. Fox and C. Creighton, Polym. Degrad. Stab., 2017, 136, 30 CrossRef.
- M. J. Yu, C. G. Wang, Y. J. Bai, Y. Xu and B. Zhu, J. Appl. Polym. Sci., 2008, 107, 1945 Search PubMed.
- S. Nunna, C. Creighton, N. Hameed, M. Naebe, L. C. Henderson, M. Setty and B. L. Fox, Polym. Test., 2017, 59, 211 CrossRef.
- S. Nunna, M. Naebe, N. Hameed, C. Creighton, S. Naghashian, M. J. Jennings, S. Atkiss, M. Setty and B. L. Fox, Polym. Degrad. Stab., 2016, 125, 114 CrossRef.
- W. Watt and W. Johnson, Nature, 1975, 257, 212 CrossRef.
- L. Audouin, V. Langlois, J. Verdu and J. C. M. de Bruijn, J. Mater. Sci., 1994, 29, 583 CrossRef.
- Y. Ge, Z. Fu, M. Zhang and H. Zhang, J. Appl. Polym. Sci., 2021, 138, 49603 CrossRef CAS.
- L. Chen, J. Liu, J. Chen, X. Li, Z. Zhao and X. Wang, Polymer, 2020, 210, 123043 CrossRef CAS.
- X. Liu, C. Zhu, J. Guo, Q. Liu, H. Dong, Y. Gu, R. Liu, N. Zhao, Z. Zhang and J. Xu, Mater. Lett., 2014, 128, 420 Search PubMed.
- Y. Hou, T. Sun, H. Wang and D. Wu, J. Appl. Polym. Sci., 2008, 108, 3996 CrossRef.
- T. H. Ko, H. Y. Ting and C. H. Lin, J. Appl. Polym. Sci., 1988, 35, 640 Search PubMed.
- H. Ogawa and K. Saito, Carbon, 1995, 33, 788 CrossRef.
- J. Wang, L. Hu, C. Yang, W. Zhao and Y. Lu, RSC Adv., 2016, 6, 73411 Search PubMed.
- T. H. Ko, J. Appl. Polym. Sci., 1991, 42, 1957 CrossRef.
- P. H. Wang, Z. R. Yue, R. Y. Li and J. Liu, J. Appl. Polym. Sci., 1995, 56, 300 Search PubMed.
- E. Fitzer, W. Frohs and M. Heine, Carbon, 1986, 24, 395 CrossRef.
- B. Szepcsik and B. Pukánszky, Thermochim. Acta, 2019, 671, 208 CrossRef.
- J. Liu, S. Xiao, Z. Shen, L. Xu, L. Zhang and J. Peng, Polym. Degrad. Stab., 2018, 150, 91 Search PubMed.
- İ. Karacan and G. Erdoğan, Polym. Eng. Sci., 2011, 52, 952 Search PubMed.
- T. H. Ko and C. H. Li, Polym. Compos., 1995, 16, 232 Search PubMed.
- G. E. Gaides and A. J. Mchugh, Polymer, 1989, 30, 2123 CrossRef.
- S. M. Allen, M. Fujin, V. Stannett, H. B. Hopfenberg and J. L. Williams, J. Membr. Sci., 1977, 2, 164 CrossRef.
- S. Kobayashi, Fiber, 1970, 26, 559 Search PubMed.
- J. M. Guenet, H. S. Jeon, C. Khatri, S. K. Jha, N. P. Balsara, M. M. Green, A. Brulet and A. Thierry, Macromolecules, 1997, 30, 4596 CrossRef.
- G. L. Collins, N. W. Thomas and G. E. Williams, Carbon, 1988, 26, 679 CrossRef.
- I. Karacan and G. Erdogan, Fibers Polym., 2012, 13, 302 Search PubMed.
- S. Dalton, F. Heatley and P. M. Budd, Polymer, 1999, 40, 5543 CrossRef.
- E. N. Sabet, P. Nourpanah and S. Arbab, Polymer, 2016, 90, 146 CrossRef.
- G. Hinrichsen, J. Polym. Sci., Part C: Polym. Symp., 1972, 38, 314 Search PubMed.
- J. B. Nichols, J. Appl. Phys., 1954, 25, 847 Search PubMed.
- M. Jing, C. Guo, Y. Bai, B. Zhu and Y. Wang, Polym. Bull., 2007, 58, 551 CrossRef.
- T. Ko, H. Ting and C. Lin, J. Appl. Polym. Sci., 1988, 35, 640 Search PubMed.
- İ. Karacan and G. Erdoğan, Fibers Polym., 2012, 13, 863 Search PubMed.
- L. M. Manocha and O. P. Bahl, Fibre Sci. Technol., 1980, 13, 212 Search PubMed.
- L. Chen, Z. Shen, J. Liu, J. Liang and X. Wang, RSC Adv., 2020, 10, 6361 Search PubMed.
- A. C. Ferrari, Solid State Commun., 2007, 143, 57 CrossRef.
- J. Lu, W. Li, H. Kang, L. Feng, J. Xu and R. Liu, Polym. Test., 2020, 81, 106267 CrossRef CAS.
- K. Kong, L. Deng, I. A. Kinloch, R. J. Young and S. J. Eichhorn, J. Mater. Sci., 2012, 47, 5410 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ac,
Jing Chen‡a,
Zhigang Shen*c,
Jie Liuab and
Xiaoxu Wang*ab
ac,
Jing Chen‡a,
Zhigang Shen*c,
Jie Liuab and
Xiaoxu Wang*ab