DOI:
10.1039/D1RA08878E
(Paper)
RSC Adv., 2022,
12, 13347-13351
Reverse atom transfer radical polymerization of dimethyl itaconate initiated by a new azo initiator: AIBME†
Received
6th December 2021
, Accepted 4th April 2022
First published on 4th May 2022
Abstract
Reverse atom transfer radical polymerization (RATRP) was used to synthesize poly(dimethyl itaconate) (PDMI) using an AIBME/CuBr2/dNbpy system. The number average molecular weight (Mn) of PDMI was as high as Mn = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, the monomer conversion rate reached up to 70%, and the dispersity remained low (Đ = 1.06–1.38). The first-order kinetics of PDMI are discussed in detail. The AIBME initiator had a higher initiation efficiency than the AIBN initiator. As the ratio of initiator (AIBME) to catalyst (CuBr2) decreased, the Mn and Đ of PDMI decreased. At 60 °C and 80 °C, the Mn of PDMI was much higher than the theoretical number average (Mn,th), and the Đ of PDMI broadened with the conversion rate. At 100 °C, the Đ of PDMI remained low, and the Mn of PDMI was closer to the Mn,th. As the ratio of monomer (DMI) to initiator (AIBME) increased, the Mn of PDMI changed little over time. These phenomena could be explained by the influence of the initiator and catalyst on polymerization kinetics.
000 g mol−1, the monomer conversion rate reached up to 70%, and the dispersity remained low (Đ = 1.06–1.38). The first-order kinetics of PDMI are discussed in detail. The AIBME initiator had a higher initiation efficiency than the AIBN initiator. As the ratio of initiator (AIBME) to catalyst (CuBr2) decreased, the Mn and Đ of PDMI decreased. At 60 °C and 80 °C, the Mn of PDMI was much higher than the theoretical number average (Mn,th), and the Đ of PDMI broadened with the conversion rate. At 100 °C, the Đ of PDMI remained low, and the Mn of PDMI was closer to the Mn,th. As the ratio of monomer (DMI) to initiator (AIBME) increased, the Mn of PDMI changed little over time. These phenomena could be explained by the influence of the initiator and catalyst on polymerization kinetics.
Introduction
With the consumption of fossil resources, humans must face the emerging resource crisis, energy crisis and environmental issues. As a substitute for fossil resources, sustainable bio-based resources have become a research “hotspot”. As an important bio-based resource, itaconic acid can be obtained from citric acid.1 Itaconic acid is produced on a large scale via biological fermentation of sugars by Aspergillus terreus.2 Itaconic acid can be used to prepare elastomers, plastics, coatings and adhesives.3–9 It can be esterified with alkyl alcohols with different side-group lengths (e.g., methanol, ethanol and butanol) to synthesize the dialkyl itaconate.10 These alkyl alcohols are derived from wood chips, corn and other biological raw materials.11–14 Itaconates are similar in structure to acrylic monomers derived from petrochemical products and they have double bonds, so they are considered essential resources for sustainable development. The polymerization rate of itaconates is much lower than that of acrylic monomers because itaconates have two asymmetric large side groups with significant steric hindrance. The steric hindrance of monomers has a negative effect on living polymerization.15–17 The free-radical polymerization (FRP) kinetics and thermal properties of itaconates have been studied.18–21 Besides, we have used diisoamyl itaconate and isoprene as monomers to synthesize a bio-based crosslinkable poly(diisoamyl itaconate-co-isoprene) (PDII) elastomer through redox-initiated emulsion polymerization. The number average molecular weight (Mn) and glass transition temperature of the PDII elastomer were 352000 g mol−1 and −39.5 °C, respectively. The mechanical properties of the PDII elastomer can be comparable to or even better than those of traditional synthetic rubbers.5 However, the polymerization of itaconates based on traditional FRP has resulted in uncontrollable Mn and dispersity (Đ) due to chain transfer. The emergence of reversible deactivation radical polymerization (RDRP) has solved these shortcomings. Controlled radical polymerizations can be used to synthesize polymers with new topological structures and polymers with different components, and to link functional groups in different parts of polymers or various compounds.22 Controlled radical polymerization of itaconates has been studied. For example, the polymerization characteristics of dimethyl itaconate (DMI) were studied through atom transfer radical polymerization (ATRP). The polymerization of DMI using ATRP exhibited a linear dependence of monomer consumption versus time up to ∼50% conversion.15 Poly(N-phenylitaconimide) (polyPhII) was prepared by continuous activator regeneration atom transfer radical polymerization with Mn = 11900 g mol−1 and high Đ (1.52).23 Derivatives of itaconic acid have been synthesized to well-defined bio-based acrylic thermoplastic elastomers by reversible addition–fragmentation chain-transfer (RAFT) polymerizations.24 Poly(itaconate) was synthesized by organocatalyzed living radical polymerization with a monomer conversion rate of 62%, the Mn of the poly(itaconate) was as high as 20000 g mol−1, and the Đ of the poly(itaconate) was between 1.28 and 1.46.25 However, the polymerization of itaconates based on RDRP has problems, such as a long reaction time and high Đ (>1.2). ATRP has been commercialized in the USA, Europe, and Japan, and has important commercial value.26 Therefore, studying itaconates using ATRP is important. RATRP was developed based on normal ATRP systems. The RATRP system adopts a high-price copper complex (CuII-X2/L) and radical initiator (I2). The mechanism is shown in Scheme 1.26–29 The primary role of the ligand (L) in ATRP is to dissolve the transition-metal salt in the organic medium and adjust the redox potential of the metal center to obtain the appropriate reactivity and kinetics of atom transfer.30 Dimethyl 2,2′-azobis(2-methyl propionate) (AIBME) is an essential azo free-radical initiator in the polymerization reaction, and its structure is shown in Fig. 1(A). AIBME has a low self-accelerating decomposition temperature. Compared with other azo initiators, AIBME has moderate initiation activity, which results in a controlled polymerization reaction. AIBME can initiate the polymerization of monomers at low temperatures. In addition, AIBME does not contain cyano groups, and its decomposition products are non-toxic.31 The polymerization of DMI through RATRP initiated by AIBME has not been reported. In the present study, RATRP was used to synthesize PDMI, in which the AIBME was employed as a new azo initiator. By using 4,4′-dinonyl-2,2′-bipyridyl(dNbpy) ligand, RATRP can be carried out under homogeneous conditions. The kinetics of RATRP of DMI are discussed using the system AIBME/CuBr2/dNbpy.
 |
| | Scheme 1 Mechanism of RATRP. Reproduced with permission from ref. 22. | |
 |
| | Fig. 1 Molecular structure of (A) AIBME and (B) AIBN. | |
Results and discussion
Effect of AIBME![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :CuBr2 ratio on RATRP of DMI
:CuBr2 ratio on RATRP of DMI
A catalyst is the critical factor in controlling an active polymerization reaction. Homogeneous polymerization of DMI was carried out at 60 °C. Fig. S1 and 2† show the 1H nuclear magnetic resonance (NMR) spectrum of DMI and PDMI, respectively, and indicated the synthesis of PDMI. From Table 1, the polymerization reaction of DMI could occur in the absence of a catalyst, and PDMI had high Đ (1.45). These results demonstrated that polymerization was out-of-control in the absence of a catalyst. Eqn (1) shows how the Đ (= Mw/Mn) of polymers prepared by ATRP, in the absence of chain-termination and chain-transfer reactions, relates to the concentration of dormant species (Pn-X) and deactivator (CuII-X2), the rate constants of propagation (kp) and deactivation (kdeact), the number-average degree of polymerization (DPn) and monomer conversion. The DPn of the produced polymers is determined by the initial concentration ratio of the monomer:initiator. Thus, the Đ of PDMI can also be decreased by increasing the concentration of the deactivator CuBr2.27,29,32 From Table 1, as the ratio of [AIBME]:[CuBr2] decreases, the Đ of PDMI decreases. Especially when the [AIBME]:[CuBr2] is 1:1, the Đ of PDMI reaches 1.09. Through Fig. 2(A), we compared the first-order linear kinetic plots of PDMI under different CuBr2 concentrations. We found that when [AIBME]:[CuBr2] = 1:1, the first-order kinetic plot of PDMI was linear, indicating that the concentration of growing radicals was constant. When [AIBME]:[CuBr2] = 1:0.5, deviation occurred because the concentration of CuBr2 was not sufficient to deactivate the propagating radicals, and the deactivation rate (Kdeact) was less than the activation rate (Kact), which resulted in termination of molecular-chain coupling.23,25 Fig. 2(B) shows the Mn and Đ as the conversion rate changes. When [AIBME]:[CuBr2] = 1:0.5 and [AIBME]:[CuBr2] = 1:1, the apparent initiator efficiency of PDMI could reach up to 91.6% (sample 4 in Table S1†) and 81.9% (sample 6 in Table S2†), respectively. However, according to Tables S1 and 2,† the polymerization of PDMI showed low apparent initiator efficiency (f) overall. f was calculated according to eqn (2), where Mn,th and Mn,GPC represent the theoretical number average molecular weight and the number average molecular weight based on GPC, respectively.27,33 Mn,th was calculated according to eqn (3), where [M0] and [I0] represent the initial concentrations of the monomer and initiator, respectively, and [Mw] is the molecular weight of the monomer.33 The slow decomposition of the initiator AIBME at 60 °C can explain this phenomenon. Then, the influence of the reaction temperature was analyzed.
Table 1 Characterization data of the homogeneous polymerization of DMI at 60 °C
| Samplea |
DMI/AIBME/CuBr2/dNbpy |
Time/h |
Conv.b/% |
Mn,thc |
Mn,GPCd |
ĐGPCe |
| [DMI] = 18 mmol, 25% cyclohexanone by volume. Conversion based on 1H NMR spectroscopy. Mn,th is the theoretical number average molecular weight based on eqn (3). Mn,GPC based on GPC. Đ = Mw/Mn is the dispersity based on GPC. |
| 1 |
200:1:0:0 |
5 |
91.1 |
14400 |
19800 |
1.45 |
| 2 |
200:1:0.1:0.2 |
12 |
86.5 |
13700 |
10100 |
1.32 |
| 3 |
200:1:0.5:1 |
12 |
23.3 |
3700 |
6600 |
1.15 |
| 4 |
200:1:1:2 |
12 |
10.9 |
1700 |
4600 |
1.09 |
 |
| | Fig. 2 (A) First-order kinetic plot. (B) Dependence of the number average molecular weight (Mn) and dispersity (Đ) with conversion. Under the following conditions: at 60 °C, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 200:1:0.5:1, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 200:1:1:2, [DMI] = 54 mmol, 25% cyclohexanone by volume. | |
Effect of the reaction temperature on RATRP of DMI
The reaction temperature is an important factor for azo-initiator efficiency. The higher the temperature, the higher the initiation rate.34 Studies have shown that temperature has a significant influence on RATRP reaction kinetics.27 When [DMI]:[AIBEM]:[CuBr2]:[dNbpy] = 200:1:1:2, the polymerization is carried at 60 °C, 80 °C and 100 °C, respectively. Fig. 3(A) is a graph showing the first-order linear kinetics of PDMI at different temperatures. The kinetic plot of homogeneous RATRP of PDMI showed significant curvature at 80 °C and 100 °C. Besides, according to Tables S2, 3 and 4,† the conversion rate at 80 °C and 100 °C was higher than that at 60 °C at the beginning of the polymerization. After 24 h of polymerization, the conversion rate at 80 °C and 100 °C was lower than that at 60 °C. These observations can be explained. At 80 °C and 100 °C, the AIBME initiator may be decomposed rapidly into free radicals, so the activation rate is greater than the deactivation rate and the initial conversion rate increases sharply. However, with increasing time, the growing PDMI chain at 80 °C and 100 °C is deactivated rapidly by CuBr2 to form a dormant species, resulting in a slow increase in conversion rate. Fig. 3(B) shows the Mn and Đ as the conversion rate changes. The Mn of PDMI at 60 °C is higher than Mn,th, indicating that the apparent initiator efficiency is relatively low. Besides, the Đ of PDMI broadened significantly with conversion at 60 °C, whereas the Đ of PDMI remained low at 80 °C and 100 °C. At 60 °C, the initiator decomposed slowly, and thus the radical quasi-steady-state could not be established, which caused the possible termination reactions. According to Table S4,† the apparent initiator efficiency of PDMI at 100 °C was high (>80%). However, the Mn barely increased at 100 °C, possibly because the propagating radicals generated at 100 °C were deactivated rapidly by CuBr2 to form a dormant species, resulting in a low polymerization rate. Another reason is that chain transfer occurs in the polymerization reaction at 100 °C, leading to a possible termination reaction.27| |
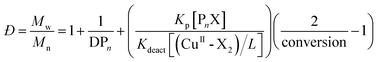 | (1) |
| | |
f = (Mn,th/Mn,GPC) × 100%
| (2) |
| |
 | (3) |
 |
| | Fig. 3 (A) First-order kinetic plot. (B) Dependence of the number average molecular weight (Mn) and dispersity (Đ) with conversion. Under the following conditions: varying temperature, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 200:1:1:2, [DMI] = 54 mmol, 25% cyclohexanone by volume. | |
Effect of the DMI:AIBME ratio on RATRP of DMI
According to eqn (3), increasing the ratio of monomer:initiator can theoretically increase the Mn.27,33 Fig. 4(A) is the first-order linear kinetic plot of PDMI. The kinetic plot of homogeneous RATRP of PDMI also showed significant curvature. Fig. 4(B) shows the Mn and Đ as the conversion rate changes: the increase in the monomer (DMI) to initiator (AIBME) ratio is not aligned with the increase in the Mn. Besides, the Mn of PDMI is lower than the Mn,th: this may be due to the steric hindrance of the two side groups of DMI monomer. The steric hindrance of the DMI monomer leads to a slow reaction rate. Đ remains at a narrow level of 1.09–1.22 (Tables S4–S7†), indicating a rapid and dynamic exchange between the active chain and dormant chain end.15
 |
| | Fig. 4 (A) First-order kinetic plot. (B) Dependence of the number average molecular weight (Mn) and dispersity (Đ) with conversion. Under the following conditions: at 100 °C, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 100:1:1:2, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 200:1:1:2, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 300:1:1:2, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 400:1:1:2, [AIBME] = 0.27 mmol, 25% cyclohexanone by volume. | |
Effect of the initiator on RATRP of DMI
Finally, we compared the kinetic characteristics of the two initiators AIBME (Fig. 1(A)) and AIBN (Fig. 1(B)). Fig. 5(A) is the first-order linear kinetic plot of PDMI. As shown in Fig. 5(B), the Đ of both AIBME and AIBN systems remained at a low level. However, there was a long induction period in the AIBN initiator system, and the conversion rate of the AIBME system was nearly twice that of the AIBN system (Tables S4 and 8†). Besides, the AIBN system showed low apparent initiator efficiency (<35%) (Table S8†) because the AIBN-initiator activity is lower than that of AIBME.31
 |
| | Fig. 5 (A) First-order kinetic plot. (B) Dependence of the number average molecular weight (Mn) and dispersity (Đ) with conversion. Under the following conditions: varying initiator, at 100 °C, [DMI]:[AIBME]:[CuBr2]:[dNbpy] = 200:1:1:2, [DMI]:[AIBN]:[CuBr2]:[dNbpy] = 200:1:1:2, [DMI] = 54 mmol, 25% cyclohexanone by volume. | |
Conclusions
Well-defined PDMI was synthesized by RATRP with a new azo initiator: AIBME. The latter had a higher initiation activity than that of the traditional azo initiator AIBN. Compared with the RATRP system with AIBN, the RATRP system with AIBME could: (i) greatly improve the conversion rate of the DMI monomer; (ii) be used to synthesize PDMI with low Đ in a shorter time and at a lower temperature. However, the Mn and conversion of the PDMI barely increased with increasing time under a high temperature and catalyst concentration. Besides, as the ratio of monomer (DMI) to initiator (AIBME) increased, the Mn of PDMI changed little over time. At high temperature and catalyst concentration, the growing PDMI chain could be deactivated by CuBr2 to form a dormant species, resulting in a slow increase. The steric hindrance of the DMI monomer led to a low reaction rate. The use of the AIBME initiator shows great potential.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (51988102, 51503010) and the National Key Research and Development Program of China (2017YFB0306903).
Notes and references
- H. Fouilloux and C. M. Thomas, Macromol. Rapid Commun., 2021, 42, 2000530 CrossRef CAS PubMed.
- M. Zhao, X. Lu, H. Zong, J. Li and B. Zhuge, Biotechnol. Lett., 2018, 40, 455–464 CrossRef CAS.
- J. Cunha da Cruz, A. Machado de Castro and E. F. Camporese Sérvulo, 3 Biotech, 2018, 8, 138 CrossRef PubMed.
- H. Hajian and M. Wan, Curr. Res. J. Biol. Sci., 2015, 7, 37–42 CrossRef CAS.
- R. Wang, J. Ma, X. Zhou, Z. Wang, H. Kang, L. Zhang, K.-c. Hua and J. Kulig, Macromolecules, 2012, 45, 6830–6839 CrossRef CAS.
- H. Kang, M. Li, Z. Tang, J. Xue, X. Hu, L. Zhang and B. Guo, J. Mater. Chem. B, 2014, 2, 7877–7886 RSC.
- X. Hu, H. Kang, Y. Li, M. Li, R. Wang, R. Xu, H. Qiao and L. Zhang, Polym. Chem., 2015, 6, 8112–8123 RSC.
- H. Qiao, R. Wang, H. Yao, X. Zhou, W. Lei, X. Hu and L. Zhang, Polym. Chem., 2015, 6, 6140–6151 RSC.
- X. Zhou, H. Ji, G.-H. Hu, R. Wang and L. Zhang, Polym. Chem., 2019, 10, 6131–6144 RSC.
- D. Boschert, A. Schneider-Chaabane, A. Himmelsbach, A. Eickenscheidt and K. Lienkamp, Chem.–Eur. J., 2018, 24, 8217–8227 CrossRef CAS PubMed.
- C. Redmond Molly, L. Valentine David and L. Sessions Alex, Appl. Environ. Microbiol., 2010, 76, 6412–6422 CrossRef CAS PubMed.
- M. Yu, Y. Zhang, I. C. Tang and S.-T. Yang, Metab. Eng., 2011, 13, 373–382 CrossRef CAS PubMed.
- A. ElMekawy, L. Diels, H. De Wever and D. Pant, Environ. Sci. Technol., 2013, 47, 9014–9027 CrossRef CAS.
- H. B. Aditiya, T. M. I. Mahlia, W. T. Chong, H. Nur and A. H. Sebayang, Renewable Sustainable Energy Rev., 2016, 66, 631–653 CrossRef CAS.
- M. Fernández-García, M. Fernández-Sanz, J. L. de la Fuente and E. L. Madruga, Macromol. Chem. Phys., 2001, 202, 1213–1218 CrossRef.
- Z. Szablan, A. A. Toy, T. P. Davis, X. Hao, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2432–2443 CrossRef CAS.
- Z. Szablan, A. A. Toy, A. Terrenoire, T. P. Davis, M. H. Stenzel, A. H. E. Müller and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3692–3710 CrossRef CAS.
- E. L. Madruga and M. Fernández-García, Polymer, 1994, 35, 4437–4442 CrossRef CAS.
- M. Fernández-García, E. L. Madruga and R. Cuervo-Rodriguez, Polymer, 1996, 37, 263–268 CrossRef.
- M. Fernández-García and E. L. Madruga, Polymer, 1997, 38, 1367–1371 CrossRef.
- T. Hirano, R. Takeyoshi, M. Seno and T. Sato, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2415–2426 CrossRef CAS.
- K. Matyjaszewski and T. P. Davis, in Handbook of Radical Polymerization, DOI: DOI:10.1002/0471220450.ch16, 2002, pp. 895–900.
- S. Okada and K. Matyjaszewski, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 822–827 CrossRef CAS.
- K. Satoh, D.-H. Lee, K. Nagai and M. Kamigaito, Macromol. Rapid Commun., 2014, 35, 161–167 CrossRef CAS PubMed.
- K. Hu, J. Sarkar, J. Zheng, Y. H. M. Lim and A. Goto, Macromol. Rapid Commun., 2020, 41, 2000075 CrossRef CAS PubMed.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- J. Xia and K. Matyjaszewski, Macromolecules, 1999, 32, 5199–5202 CrossRef CAS.
- P. Krys and K. Matyjaszewski, Eur. Polym. J., 2017, 89, 482–523 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromol. Theory Simul., 2008, 17, 359–375 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- Y. Li, Y. Zhang and X. Z. Wang, J. Chem. Eng. Data, 2020, 65, 1411–1424 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572–7573 CrossRef CAS.
- M. Buback, B. Huckestein, F.-D. Kuchta, G. T. Russell and E. Schmid, Macromol. Chem. Phys., 1994, 195, 2117–2140 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
HaiJun Ji,
Hui Yang,
Jie Yu,
Jiaqi Wang,
Liqun Zhang
,
HaiJun Ji,
Hui Yang,
Jie Yu,
Jiaqi Wang,
Liqun Zhang






