
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-driven oxidation of CH4 to C1 chemicals catalysed by an organometallic Ru complex with O2†
Tatsuya Nakanoa,
Tsukasa Abe b,
Takahiro Matsumoto*acd,
Kento Kimuraa,
Genta Nakamuraa,
Shinya Hayamie,
Yoshihito Shiota*b,
Kazunari Yoshizawa*bc and
Seiji Ogo*ac
b,
Takahiro Matsumoto*acd,
Kento Kimuraa,
Genta Nakamuraa,
Shinya Hayamie,
Yoshihito Shiota*b,
Kazunari Yoshizawa*bc and
Seiji Ogo*ac
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: matsumoto.takahiro.236@m.kyushu-u.ac.jp
bInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
cInternational Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka, 819-0395, Japan
dPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), Kawaguchi 332-0012, Japan
eGraduate School of Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
First published on 28th April 2022
Abstract
CH4 conversion is one of the most challenging chemical reactions due to its inertness in terms of physical and chemical properties. We have achieved photo-induced C–H bond breaking of CH4 and successive C–O bond formation to form CH3OH concomitant with HCHO by an organometallic Ru complex with O2.
CH4 is one of the most promising resources of energy and materials because it has high affinity with renewable energy, and has become capable of being easily and abundantly obtained in biomethane form from biomass by means of recent technological developments.1,2 In order to use CH4 in industrial processes instead of naphtha, innovative and useful transformation methods are now strongly demanded. However, its inertness in view of its physical and chemical properties makes CH4 one of the most unreactive molecules.3 To date, there have been three type of catalysts, i.e., enzymatic, heterogeneous and homogeneous, found for the direct oxidation of CH4 to CH3OH with O2 as an oxidant.4–7 Soluble and particulate methane monooxygenases (sMMOs and pMMOs) are well-known enzymes that oxidise CH4 to CH3OH with O2 under ambient conditions. Their active sites are constructed from Fe and Cu centres for sMMOs and pMMOs, respectively.4 They cleave the unreactive C–H bond of CH4 with subsequent C–O bond formation, proposed to be promoted by the Fe and Cu oxido species. By mimicking the active-site structure of sMMOs and pMMOs, heterogeneous catalysts5,6 and homogeneous Cu catalysts7 have been developed to catalyse aerobic CH4 oxidation to CH3OH. The heterogeneous zeolite catalysts need high temperature, and the homogeneous Cu catalysts need H2O2 as a reductant for the catalytic reaction. These catalysts may possess metal oxido cores that can promote C–H bond activation like MMOs. In addition to the MMO-inspired catalysts, a homogeneous inorganic compound of ClO2 works as a light-triggered oxidizing reagent to convert CH4 to CH3OH and HCOOH with O2 in a non-catalytic system.8 Homogeneous organometallic complexes other than CH4-to-CH3OH catalysts have also been designed for the conversion of CH4 to various significant compounds, capitalizing on the flexible and designable tuning of the ligand environment surrounding the metal centre(s).9 Recently, various heterogeneous catalysts have also been developed for CH4 conversion.6 While many efforts have been made to date for catalytic CH4 conversion, the direct catalytic conversion of CH4 to C1 chemicals of CH3OH and HCHO by a homogeneous organometallic catalyst with light irradiation has not yet been reported. Here, we report aerobic CH4 oxidation to CH3OH and HCHO catalysed by a homogeneous Ru complex in water with input of light energy. Most photocatalysts of organometallic complexes have been developed for redox reactions that mean single electron transfer between metal complexes and external electron donors/acceptors.10 Recently, charge transfers, such as ligand-to-metal or metal-to-ligand originating from organometallic complexes, have been utilized for chemical reactions such as material transformations apart from single-electron transfer reactions.9g,11 This advanced method should expand the possibility of photo-induced organometallic catalysis. We have developed a novel photo-driven C–H activating catalyst by means of charge transfer derived from a homogenous Ru complex.
A water-soluble and oxygen-sensitive RuII complex, [RuII(η5-C5Me5)(H2O)3]+ (1), was oxygenated by O2 in H2O to rapidly generate oxidised species like a bis(μ-oxido) Ru2IV species, [Ru2IV(η5-C5Me5)2(μ-O)2]2+ (2) (Fig. 1). This species was not formed in CH3CN but formed in H2O, which was likely to be caused by stabilization of the Ru2IV(μ-O)2 core in a polar environment. Density functional theory (DFT) calculations indicated that 2 was stabilized in H2O but destabilized under vacuum conditions relative to the corresponding starting RuII triaqua complex 1 (Fig. S1†). The oxygenated species 2 is stable in H2O at ambient temperature unlike bis(μ-oxido) Fe2 species that are generally unstable at ambient temperature.12 Its stability gave us a chance to irradiate 2 with light to form a highly active excited state.
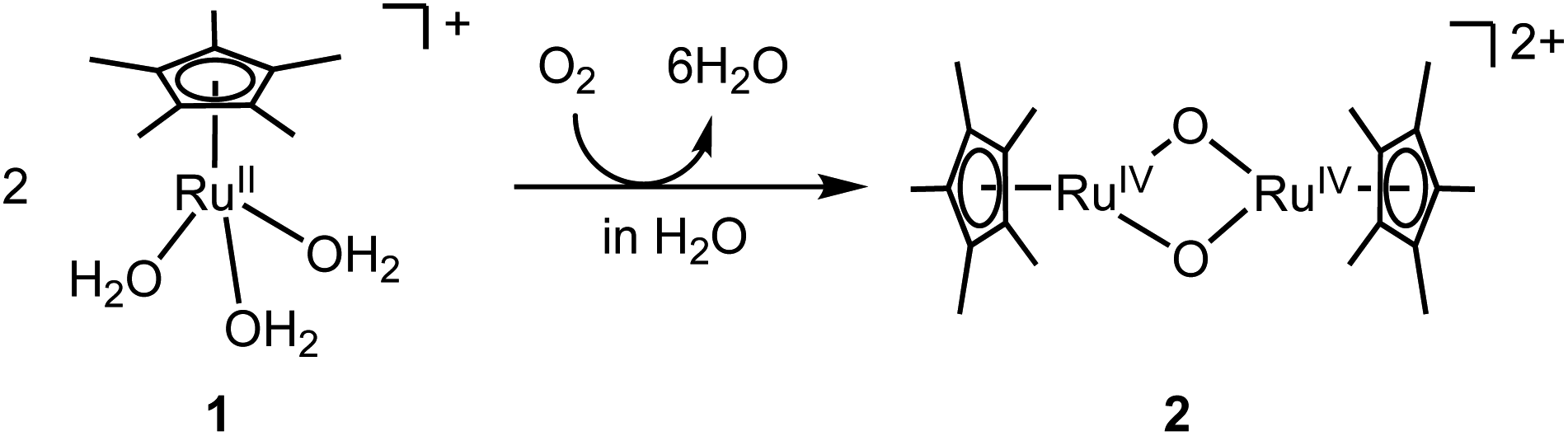 | ||
| Fig. 1 Synthesis of bis(μ-oxido) Ru2IV complex 2 from oxygenation of mononuclear RuII triaqua complex 1 in H2O. | ||
The structure of 2 was estimated by electrospray ionization-mass spectrometry (ESI-MS) (Fig. S2 and S3†) and DFT calculations (Fig. 2). The positive-ion ESI mass spectrum of 2 in H2O shows a prominent signal at m/z 521.9 that corresponds to [2 + OH]+, and a characteristic isotopic distribution that matches well with the calculated isotopic distribution (Fig. S2a–c†). It can be strongly suggested that complex 2 bears oxido ligands by isotope-labelling experiments using O2 in H218O and 18O2 in H2O during oxygenation of 1. The positive-ion ESI mass spectrum obtained from the reaction of 1 with O2 in H218O shows a prominent signal at m/z 527.9 that corresponds to [18O-labeled 2 + 18OH]+ (Fig. S2d†), while the positive-ion ESI mass spectrum obtained from the reaction of 1 with 18O2 in H2O shows a prominent signal at m/z 521.9 that corresponds to [2 + OH]+ (Fig. S2e†). These labelling experiments clearly reveal the presence of water-exchangeable ligands in 2 (Fig. S3†), which means that the oxido ligands should be coordinated to the RuIV centre. It is well known that oxido ligand(s) coordinating to metal centre(s) can be easily exchanged by external water.13 The high-valent metal centre is likely to bind an oxido ligand rather than a hydroxido ligand because such an oxido ligand has little ability to accept a proton to form a hydroxido ligand, caused by the Lewis basicity of the oxido ligand necessarily being lowered by delocalizing the electron density of the electron-rich oxido ligand toward the electron-deficient high-valent metal centre.12,14
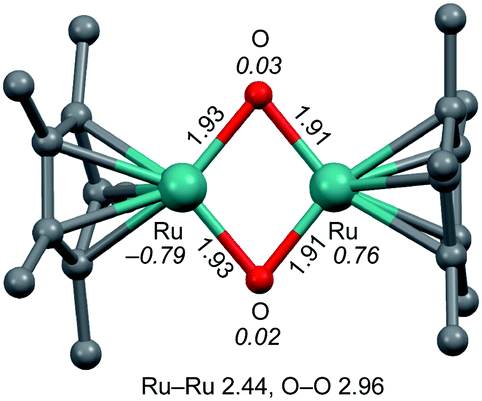 | ||
| Fig. 2 Optimized structure of bis(μ-oxido) Ru2IV complex 2. The structure of 2 in the ground singlet state was optimized by DFT calculations. Units are in Å. The italicized values represent the spin densities of the Ru and O atoms. H atoms are omitted for clarity. | ||
DFT calculations indicated that the optimized structure of 2 contains a bis(μ-oxido) Ru2IV core rather than a (μ-peroxido) Ru2III core, as shown in Fig. 2. Multinuclear Ru (hydr)oxido complexes have been reported, which are structurally similar to the Ru2(μ-O)2 centre of 2.15 The electron-donating η5-C5Me5 ligand allows the dinuclear Ru centre to access high-valent oxidation states of IV and the steric hindrance of the methyl groups of the η5-C5Me5 ligand creates a small cavity around the Ru atoms for the arrangement of only two oxido ligands. The Ru2IV(μ-O)2 structure seems to be characteristic of this ligand environment system. Changing ligand environments with respect to electronic effects and steric hindrance can provide various Rux2{μ-O(Hy)}z structures (x = III–VI, y = 0–1, and z = 1–3).15 The distances of the two Ru centres and the two O atoms in 2 were calculated to be 2.44 and 2.96 Å (Fig. 2), respectively, which correspond to the interacting dinuclear Ru centres and the cleavage of the O–O bond. Spin density analysis indicates each RuIV centre has S = 1 and an interatomic interaction of two RuIV centres with the antiferromagnetic exchange interaction results in S = 0 in the ground state of 2 (1R, Table S1†), which is consistent with the experimental observation with a superconducting quantum interference device (SQUID) that the bis(μ-oxido) Ru2IV complex 2 is diamagnetic. On the basis of experimental and DFT results, the bis(μ-oxido) Ru2IV species 2 can be generated from four-electron reduction of O2 by two RuII centres via O–O bond breaking.
An ultraviolet-visible (UV-vis) spectral change from 1 to 2 by oxygenation in H2O shows a decrease in absorption bands around 330 nm (ε = 900 M−1 cm−1) and 400 nm (ε = 1300 M−1 cm−1) derived from 1 and an increase in a broad band around 290 nm (ε = 3800 M−1 cm−1) derived from 2 (Fig. S4†). Since the characteristic absorption band of 2 is observed in the UV region, we irradiated 2 with UV light for excitation. Time-dependent (TD)-DFT calculations are consistent with the experimental UV-vis spectra of 1 and 2 (Fig. S5†). The TD-DFT calculations of 2 show an absorption band at 263 nm, assigned to the charge transfer from the ground singlet state to the excited triplet state (Fig. S5b†). While the oxido ligands in 2 show little radical character with a spin density of almost zero (1R, Table S1†), the oxido ligands in the excited triplet state are capable of showing a radical character (3R*, Table S1†), described below in detail. This radical character must originate in the abstraction of an H atom from CH4 in the initiation step.
Following spectroscopic, mass-spectrometric and DFT analyses of 2, we investigated its photo-induced oxidation of CH4 in H2O. An aqueous solution of 2 under a CH4/O2 atmosphere (partial pressures of CH4 and O2 = 4 and 2 MPa, respectively) was irradiated by UV light (250–385 nm, 15 mW cm−2) for 5 h. Subsequently, the resulting aqueous solution was analysed by gas chromatography-mass spectrometry (GC-MS) after removing Ru complex(es) by passage through a silica gel column. CH3OH and HCHO were observed by GC-MS analysis (Fig. S6†), with their retention times and fragment patterns clearly corresponding to those of authentic CH3OH and HCHO. No HCOOH was observed by GC-MS. Control experiments were conducted without 2, UV light, CH4, or O2, all showing no product formation. When visible light (385–740 nm) was used instead of UV light, no reaction occurred. We determined the TONs of CH3OH and HCHO as 1.1 and 3.0, respectively; thus, the total TON was estimated to be 4.1. Considering that CH3OH was formed by 2-electron oxidation of CH4 with 2-electron oxidant 2 and CH3OH was 2-electron oxidized to form HCHO by 2, we calculated the TONs as follows: (mol of CH3OH)/(mol of 2) for CH3OH and (mol of HCHO) × 2/(mol of 2) for HCHO. Although the order of these TON values is the same as those of trinuclear Cu oxide systems that catalysed CH4 oxidation to CH3OH by O2 using H2O2 as reductant (TON = 1.4 or ∼6),7 our system needs only O2. We also determined the yields of CH3OH and HCHO based on CH4 to be 0.12 and 0.17%, respectively. We confirmed that photo-induced CH3OH oxidation yielded HCHO with 2 under the same conditions as the photo-induced CH4 oxidation. No HCOOH was also detected in the CH3OH oxidation. In order to confirm the origin of the oxygen atom of CH3OH, we conducted an isotope labelling experiment of photo-induced oxidation of CH4 by 2 with 16O2 in H218O. No 18O-incorporated methanol (CH318OH) was formed, but CH316OH was observed. This result indicates that in the process of C–H bond activation of CH4, coupling of a CH3 radical with O2 occurs prior to OH rebound to the CH3 radical. After the C–H bond cleavage of CH4, DFT calculations indicate that the interaction of the CH3 radical with the OH ligand coordinating to the Ru centre is energetically higher than a transition state corresponding to the release of a CH3 radical from the (μ-hydroxido)(μ-oxido) Ru2III,IV core (Fig. S7†). The insights, benefitting from the reports of activation of weaker C–H bonds in hydrocarbons rather than CH4 by metal oxido species without light irradiation, also permit us to propose H atom abstraction from the C–H bond of CH4.12,14,16
We followed the reaction of bis(μ-oxido) Ru2IV species 2 with CH4 and O2 under light irradiation by ESI-MS (Fig. S8†). The ESI-MS results indicate that the main signal derived from 2 decreased as a signal at m/z 371.1, assignable to a tetramethylfulvene-coordinating RuII complex [RuII(tetramethylfulvene)(η5-C5Me5)]+, and unidentified signals increased. The formation of tetramethylfulvene complex indicates that the methyl group of η5-C5Me5 was oxidized.17
Photo-driven oxidation of C2H6 by using 2 with O2 (partial pressures of C2H6 and O2 = 2 and 1 MPa, respectively) also occurs as in the case with CH4 oxidation. The products of C2H5OH and CH3CHO were observed by GC-MS (Fig. S9†), where their TONs were determined to be 0.31 and 0.46, respectively, based on the same calculation protocol as for CH4 oxidation. The total TON was calculated as 0.77. A trace amount of CH3COOH was observed in the C2H6 oxidation. The TON of C2H6 oxidation is slightly lower than that of the CH4 oxidation, which suggests that the oxidation reaction with 2 is relevant to the molecular size of the external substrate. The ten methyl groups of two η5-C5Me5 ligands seem to protect the bis(μ-oxido) Ru2IV core, which allows a smaller molecule to access the active bis(μ-oxido) centre.
DFT calculations indicate that photo-excitation of 2 is required to cause H atom abstraction from CH4 (Fig. 3), which is consistent with the experimental result that 2 shows no reactivity toward CH4 without light irradiation. Fig. 3 shows the computed energy surfaces for the C–H bond activation by 2 in the open-shell singlet and triplet states. To obtain reaction coordinates of the C–H bond dissociation, we performed intrinsic reaction coordinate (IRC) calculations in the ground state. The potential energy surfaces of the excited states were obtained by a single-point calculation using the TD-DFT method along the reaction coordinate. The reactions involve the interaction of the oxido ligand with the H atom, followed by H atom abstraction from CH4 to generate a CH3 radical with the (μ-hydroxido)(μ-oxido) Ru2III,IV species. Calculated activation energies for the C–H bond cleavage of CH4 by the catalyst are 33.4 kcal mol−1 in the ground state S0 and 19.6 kcal mol−1 in the triplet excited state T27. These results lead us to conclude that CH4 activation with 2 is likely to occur in the transition state in the potential energy surface of the excited state.
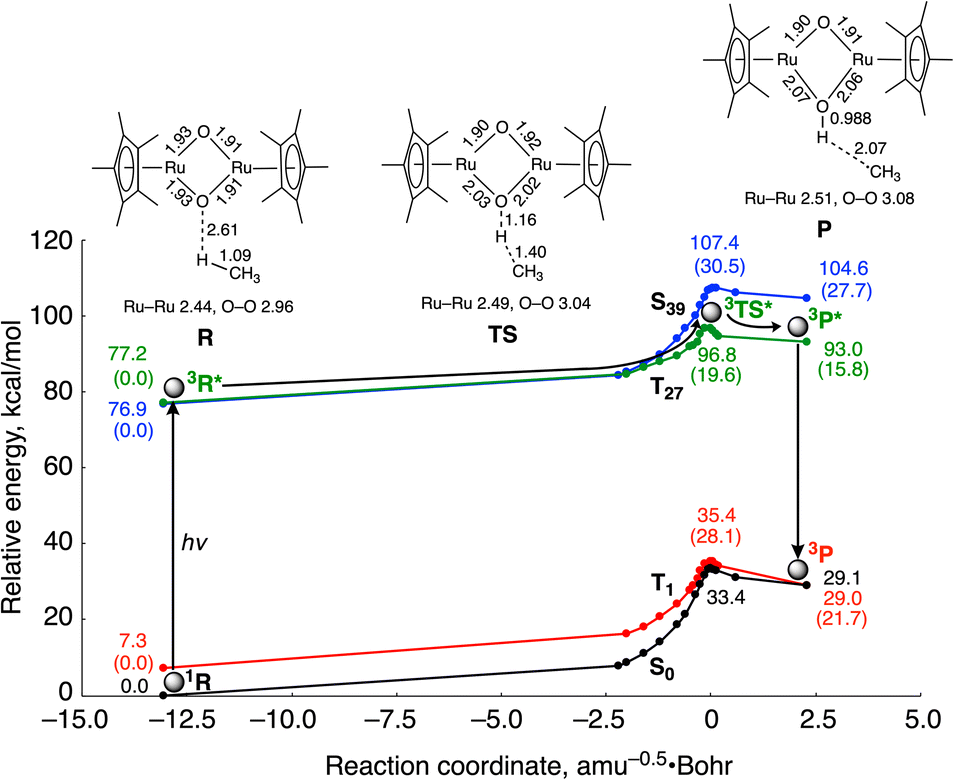 | ||
| Fig. 3 Computed energy surfaces for the C–H bond activation of CH4 by 2 in the ground state S0 and the three excited states T1, T27 and S39. R: reactant complex, TS: transition state and P: product complex. The values in parentheses are relative energies from R in each state. Distances and energies are given in units of Å and kcal mol−1, respectively. | ||
Spin density analysis shows the oxido ligands of the reactant complex in the ground singlet state 1R have little radical character (O1: 0.01; O2: 0.03), while those in the excited triplet state 3R* have a more radical character (O1: 0.23; O2: 0.24) (Table S1†). The increase in the spin densities of the μ-oxido ligands in the bis(μ-oxido)dicopper complexes enhances the reactivity for H atom abstraction from CH4.18 Therefore, it is considered that the increase in the spin densities in the μ-oxido moieties diminishes the activation energy of H atom abstraction from CH4. We considered this the reason why the spin densities in the μ-oxido moieties increase by irradiation with UV light. Since the two unpaired electrons in the Ru centres are antiferromagnetically coupled in the ground singlet state of 2, the delocalized electrons of the μ-oxido moieties are cancelled. In contrast, UV light irradiation induces metal-to-metal charge transfer (MMCT) (Fig. S5†) to cause the spin inversion of an unpaired electron in the Ru centre. Therefore, the delocalized electrons of the μ-oxido moieties are enhanced, resulting in the radical character of the μ-oxido moieties. In the transition state (TS), the C atom of the CH3 radical increases in radical character with H atom migration, while the spin density of the Ru centre decreases. Thus, O–H bond formation and CH3 radical formation occur simultaneously.
On the basis of experimental analyses and DFT calculations, we propose a reaction mechanism of photo-induced CH4 oxidation by the Ru complex with O2 (Fig. 4). The bis(μ-oxido) Ru2IV species 2 is excited by UV light to generate the excited species 3. The highly active excited species 3 is able to abstract an H atom from CH4 to afford (μ-hydroxido)(μ-oxido) Ru2III,IV species 4 with the CH3 radical. The CH3 radical reacts with O2 to form a CH3OO radical, which can be coupled intermolecularly to generate a CH3OOOOCH3 species. This releases O2 to form a CH3O radical,19 which abstracts an H atom from 4 to afford CH3OH together with regeneration of 2. Based on thermodynamic energy calculations for CH4 oxidation to CH3OH with 2 in the ground state in H2O at standard temperature (eqn (1)–(4), the energies are corrected by zero-point vibrational energies and Gibbs free energies at 298.15 K), the process of H atom abstraction from CH4 to a CH3 radical is an endergonic reaction (ΔG = 28.0 kcal mol−1), although the processes of CH3 radical with O2 (ΔG = −27.0 kcal mol−1) and CH3O radical with 2 (ΔG = −24.9 kcal mol−1) are exergonic reactions. The overall reaction of CH4 with O2 to CH3OH is exergonic (ΔG = −23.9 kcal mol−1). Because only the first step of H atom abstraction by 2 needs external energy, we must input light energy into this system.
| CH4 + 2 = CH3˙ + 4 − 28.0 kcal | (1) |
| CH3˙ + ½ O2 = CH3O˙ + 27.0 kcal | (2) |
| CH3O˙ + 4 = CH3OH + 2 + 24.9 kcal | (3) |
| CH4 + ½ O2 = CH3OH + 23.9 kcal | (4) |
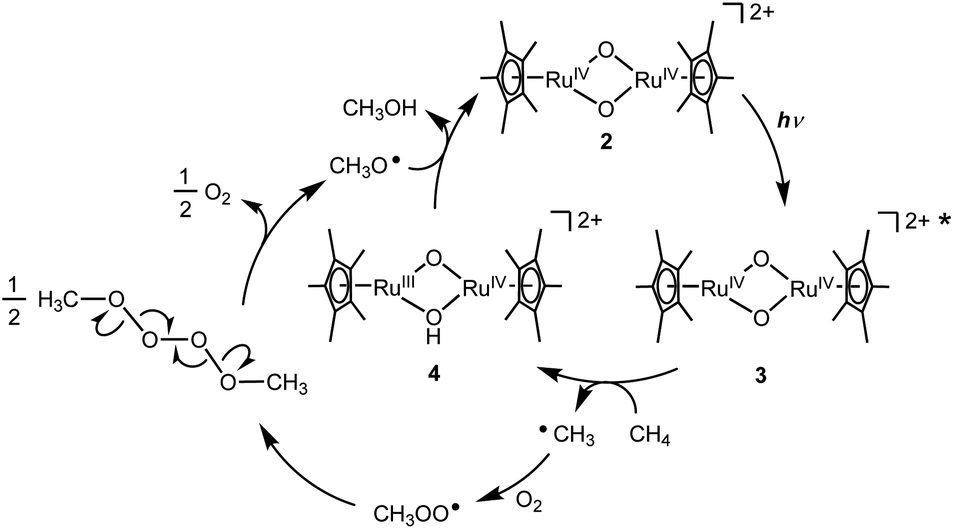 | ||
| Fig. 4 A proposed mechanism for the photo-induced oxidation of CH4 to CH3OH by Ru complex with O2 in H2O. | ||
In conclusion, we have succeeded in the photo-induced conversion of CH4 to C1 chemicals of CH3OH and HCHO catalysed by the water-soluble bis(μ-oxido) Ru2IV complex with O2. This is the first case of catalytic oxidation of CH4 to CH3OH and HCHO with a homogeneous catalyst by using only O2. The light-triggered radical character of the oxido ligands enables the activation of the unreactive C–H bond of CH4, as evidenced by experimental results and DFT calculations. We think it will be possible to apply such a photo-excited metal complex to the activation of various unactivated molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from JST PRESTO Grant Number JPMJPR17S9, JST CREST Grant Numbers JPMJCR15P5 and JPMJCR18R2, Japan and the World Premier International Research Center Initiative (WPI), Japan. The computations were mainly carried out using the computer facilities at the Research Institute for Information Technology, Kyushu University.References
- R. Singh, P. K. Mishra, N. Srivastava, A. Shrivastav and K. R. Srivastava, in Bioenergy Research: Evaluating Strategies for Commercialization and Sustainability, ed., N. Srivastava and M. Srivastava, Wiley-VCH, Weinheim, 2021, pp. 245–254 Search PubMed.
- G. D. Saratale, J. Damaraja, S. Shobana, R. G. Saratale, S. Periyasamy, G. Zhen and G. Kumar, in Green Energy to Sustainability: Strategies for Global Industries, ed., A. A. Vertès, N. Qureshi, H. P. Blaschek and H. Yukawa, Wiley-VCH, Weinheim, 2020, pp. 447–459 Search PubMed.
- (a) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–9520 CrossRef CAS PubMed; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 471, 507–514 CrossRef PubMed; (c) Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes, ed., A. E. Shilov and G. B. Shul’pin, Kluwer Academic Publishers, Dordrecht, 2000 Search PubMed; (d) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS; (e) R. H. Crabtree, Chem. Rev., 1995, 95, 987–1007 CrossRef CAS.
- (a) R. Banerjee, J. C. Jones and J. D. Lipscomb, Annu. Rev. Biochem., 2019, 88, 409–431 CrossRef CAS PubMed; (b) M. O. Ross and A. C. Rosenzweig, J. Biol. Inorg Chem., 2017, 22, 307–319 CrossRef CAS PubMed; (c) V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574–8621 CrossRef CAS PubMed; (d) C. E. Tinberg and S. J. Lippard, Acc. Chem. Res., 2011, 44, 280–288 CrossRef CAS PubMed.
- (a) S. L. Scott, Science, 2021, 373, 277–278 CrossRef CAS PubMed; (b) D. Kiani, S. Sourav, Y. Tang, J. Baltrusaitis and I. E. Wachs, Chem. Soc. Rev., 2021, 50, 1251–1268 RSC; (c) J. Meyet, A. P. van Bavel, A. D. Horton, J. A. van Bokhoven and C. Copéret, Catal. Sci. Technol., 2021, 11, 5484–5490 RSC; (d) M. H. Mahyuddin, Y. Shiota and K. Yoshizawa, Catal. Sci. Technol., 2019, 9, 1744–1768 RSC; (e) V. L. Sushkevich, D. Palagin, M. Ranocchiari and J. A. van Bokhoven, Science, 2017, 356, 523–527 CrossRef CAS PubMed; (f) B. E. R. Snyder, P. Vanelderen, M. L. Bols, S. D. Hallaert, L. H. Böttger, L. Ungur, K. Pierloot, R. A. Schoonheydt, B. F. Sels and E. I. Solomon, Nature, 2016, 536, 317–321 CrossRef CAS PubMed.
- (a) M. Yovanovich, A. J. da Silva, R. F. B. de Souza, V. Ussui, A. O. Neto and D. R. R. Lazar, Int. J. Electrochem. Sci., 2021, 16, 210735 CrossRef CAS; (b) X. Cai, S. Fang and Y. H. Hu, J. Mater. Chem. A, 2021, 9, 10796–10802 RSC; (c) N. Feng, H. Lin, H. Song, L. Yang, D. Tang, F. Deng and J. Ye, Nat. Commun., 2021, 12, 4652 CrossRef CAS PubMed; (d) B. Li, X. Song, S. Feng, Q. Yuan, M. Jiang, L. Yan and Y. Ding, Appl. Catal., B, 2021, 293, 120208 CrossRef CAS; (e) H. Song, X. Meng, S. Wang, W. Zhou, X. Wang, T. Kako and J. Ye, J. Am. Chem. Soc., 2019, 141, 20507–20515 CrossRef CAS PubMed.
- (a) E. Moharreri, T. Jafari, D. Rathnayake, H. Khanna, C.-H. Kuo, S. L. Suib and P. Nandi, Sci. Rep., 2021, 11, 19175 CrossRef CAS PubMed; (b) S. I. Chan, Y.-J. Lu, P. Nagababu, S. Maji, M.-C. Hung, M. M. Lee, I.-J. Hsu, P. D. Minh, J. C.-H. Lai, K. Y. Ng, S. Ramalingam, S. S.-F. Yu and M. K. Chan, Angew. Chem., Int. Ed., 2013, 52, 3731–3735 CrossRef CAS PubMed.
- K. Ohkubo and K. Hirose, Angew. Chem., Int. Ed., 2018, 57, 2126–2129 CrossRef CAS PubMed.
- (a) A. E. Shilov and G. B. Shul’pin, Russ. Chem. Rev., 1987, 56, 442–464 CrossRef; (b) R. A. Periana, Science, 1998, 280, 560–564 CrossRef CAS PubMed; (c) Y. Yamada, K. Morita, N. Mihara, K. Igawa, K. Tomooka and K. Tanaka, New J. Chem., 2019, 43, 11477–11482 RSC; (d) A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC; (e) A. K. Cook, S. D. Schimler, A. J. Matzger and M. S. Sanford, Science, 2016, 351, 1421–1424 CrossRef CAS PubMed; (f) K. T. Smith, S. Berritt, M. González-Moreiras, S. Ahn, M. R. Smith III, M.-H. Baik and D. J. Mindiola, Science, 2016, 351, 1424–1427 CrossRef CAS PubMed; (g) A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- (a) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- (a) Y. Ueda, Y. Masuda, T. Iwai, K. Imaeda, H. Takeuchi, K. Ueno, M. Gao, J. Hasegawa and M. Sawamura, J. Am. Chem. Soc., 2022, 144, 2218–2224 CrossRef CAS PubMed; (b) Y. Park, L. Tian, S. Kim, T. P. Pabst, J. Kim, G. D. Scholes and P. J. Chirik, JACS Au, 2022, 2, 407–418 CrossRef CAS PubMed; (c) Y. Park, S. Kim, L. Tian, H. Zhong, G. D. Scholes and P. J. Chirik, Nat. Chem., 2021, 13, 969–976 CrossRef CAS PubMed.
- (a) L. Que, Jr., Acc. Chem. Res., 2007, 40, 493–500 CrossRef PubMed; (b) M. Costas, J.-U. Rohde, A. Stubna, R. Y. N. Ho, L. Quaroni, E. Münck and L. Que, Jr., J. Am. Chem. Soc., 2001, 123, 12931–12932 CrossRef CAS PubMed; (c) L. Que, Jr. and W. B. Tolman, Angew. Chem., Int. Ed., 2002, 41, 1114–1137 CrossRef.
- (a) R. Tagore, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 2193–2203 CrossRef CAS PubMed; (b) R. Tagore, H. Chen, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2006, 128, 9457–9465 CrossRef CAS PubMed; (c) K. Chen, M. Costas and L. Que, Jr., J. Chem. Soc., Dalton Trans., 2002, 672–679 RSC.
- (a) J. A. Halfen, S. Mahapatra, E. C. Wilkinson, S. Kaderli, V. G. Young, Jr., L. Que, Jr., A. D. Zuberbühler and W. B. Tolman, Science, 1996, 271, 1397–1400 CrossRef CAS PubMed; (b) S. Mahapatra, J. A. Halfen and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 11575–11586 CrossRef CAS; (c) S. Mahapatra, J. A. Halfen, E. C. Wilkinson, G. Pan, X. Wang, V. G. Young, Jr., C. J. Cramer, L. Que, Jr. and W. B. Tolman, J. Am. Chem. Soc., 1996, 118, 11555–11574 CrossRef CAS; (d) B. M. T. Lam, J. A. Halfen, V. G. Young, Jr., J. R. Hagadorn, P. L. Holland, A. Lledós, L. Cucurull-Sánchez, J. J. Novoa, S. Alvarez and W. B. Tolman, Inorg. Chem., 2000, 39, 4059–4072 CrossRef CAS PubMed.
- (a) T. Suzuki, Y. Suzuki, T. Kawamoto, R. Miyamoto, S. Nanbu and H. Nagao, Inorg. Chem., 2016, 55, 6830–6832 CrossRef CAS PubMed; (b) A. C. Dengel, A. M. EI-Hendawy, W. P. Griffith, C. A. O′Mahoney and D. J. Williams, J. Chem. Soc., Dalton Trans., 1990, 737–742 RSC; (c) P. Neubold, B. S. P. C. D. Vedova, K. Wieghardt, B. Nuber and J. Weiss, Inorg. Chem., 1990, 29, 3355–3363 CrossRef CAS; (d) J. M. Power, K. Evertz, L. Henling, R. Marsh, W. P. Schaefer, J. A. Labinger and J. E. Bercaw, Inorg. Chem., 1990, 29, 5058–5065 CrossRef CAS; (e) P. Neubold, B. S. P. C. D. Vedova, K. Wieghardt, B. Nuber and J. Weiss, Angew. Chem., Int. Ed., 1989, 28, 763–765 CrossRef; (f) R. P. Tooze, G. Wilkinson, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1986, 2711–2720 RSC.
- (a) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622–8648 CrossRef CAS PubMed; (b) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060–1081 CrossRef CAS PubMed; (c) H. Kotani, H. Shimomura, K. Ikeda, T. Ishizuka, Y. Shiota, K. Yoshizawa and T. Kojima, J. Am. Chem. Soc., 2020, 142, 16982–16989 CrossRef CAS PubMed; (d) J. Kaizer, E. J. Klinker, N. Y. Oh, J.-U. Rohde, W. J. Song, A. Stubna, J. Kim, E. Münck, W. Nam and L. Que, Jr., J. Am. Chem. Soc., 2004, 126, 472–473 CrossRef CAS PubMed.
- (a) L. Fan, M. L. Turner, M. B. Hursthouse, K. M. A. Malik, O. V. Gusev and P. M. Maitlis, J. Am. Chem. Soc., 1994, 116, 385–386 CrossRef CAS; (b) M. E. N. Clemente, P. J. Saavedra, M. C. Vásquez, M. A. Paz-Sandoval, A. M. Arif and R. D. Ernst, Organometallics, 2002, 21, 592–605 CrossRef; (c) C. Gemel, K. Mereiter, R. Schmid and K. Kirchner, Organometallics, 1997, 16, 5601–5603 CrossRef CAS.
- Y. Shiota and K. Yoshizawa, Inorg. Chem., 2009, 48, 838–845 CrossRef CAS PubMed.
- Y. Hori, T. Abe, Y. Shiota and K. Yoshizawa, Bull. Chem. Soc. Jpn., 2019, 92, 1840–1846 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01772e |
| This journal is © The Royal Society of Chemistry 2022 |