
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy and application of graphene oxide in the synthesis of 2,3-disubstituted quinolines via a Povarov multicomponent reaction and subsequent oxidation†
Samantha Caputo‡
 a,
Alessandro Kovtun‡b,
Francesco Brunocde,
Enrico Raveracdef,
Chiara Lambruschinia,
Manuela Meluccib and
Lisa Moni*a
a,
Alessandro Kovtun‡b,
Francesco Brunocde,
Enrico Raveracdef,
Chiara Lambruschinia,
Manuela Meluccib and
Lisa Moni*a
aDepartment of Chemistry and Industrial Chemistry, University of Genoa, Via Dodecaneso 31, 16146 GENOVA, Italy. E-mail: lisa.moni@unige.it
bConsiglio Nazionale delle Ricerche, Istituto per la Sintesi Organica e la Fotoreattività (CNR-ISOF), Via Gobetti 101, 40129 BOLOGNA, Italy
cMagnetic Resonance Center (CERM), University of Florence, Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy
dDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy
eConsorzio Interuniversitario Risonanze Magnetiche di Metalloproteine (CIRMMP), Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy
fFlorence Data -scienze, University of Florence, Italy
First published on 26th May 2022
Abstract
The carbocatalyzed synthesis of 2,3-disubstituted quinolines is disclosed. This process involved a three-component Povarov reaction of anilines, aldehydes and electron-enriched enol ethers, which gave the substrate for the subsequent oxidation. Graphene oxide (GO) was exploited as a heterogeneous, metal-free and sustainable catalyst for both transformations. The multicomponent reaction proceeded under simple and mild reaction conditions, exhibited good functional group tolerance, and could be easily scaled up to the gram level. A selection of tetrahydroquinolines obtained was subsequently aromatized to quinolines. The multistep synthesis could also be performed as a one-pot procedure. Investigation of the real active sites of GO was carried out by performing control experiments and a by full characterization of the carbon material by X-ray photoelectron spectroscopy (XPS) and solid-state nuclear magnetic resonance (ssNMR).
Introduction
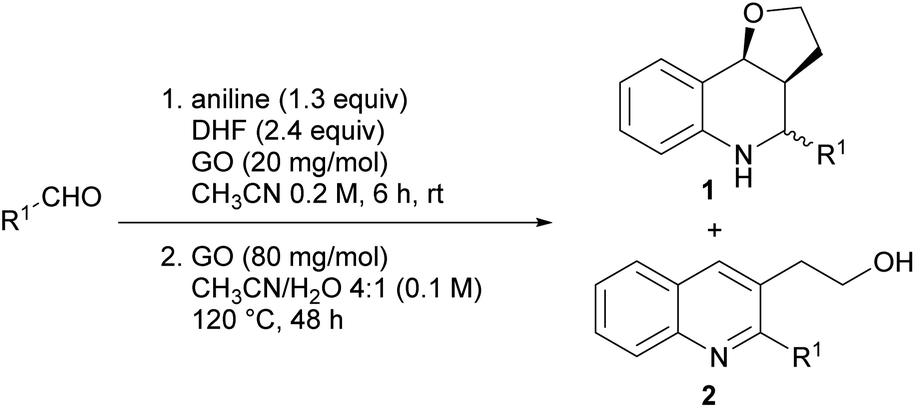
The need to develop new sustainable synthetic processes is becoming more and more compelling,1,2 and the reduction of the environmental footprint of chemical processing represents a strategic geopolitical asset as well.3 One approach to increase the sustainability of chemical processing is the development of synthetic strategies with a limited number of steps and with improved operational simplicity (e.g. reducing the unit operations).4 In this context “multi-bond forming processes”, such as domino (or cascade) reactions and multicomponent reactions (MCRs), are particularly promising.5 MCRs are one-pot reactions where three or more reagents are combined in the same reaction vessel, to obtain a product that contains the largest possible number of atoms of the reagents.6–9 Recently, the range of possible multicomponent reactions has been greatly expanded by using photocatalysis and electrocatalysis.10–13 When MCRs are included in a one-pot multistep process the advantages multiply, saving time and energy (step and operational efficiency) and reducing waste, therefore approaching the concept of an ideal eco-friendly synthesis.14,15Recently carbon nanomaterials, with particular reference to graphene oxide (GO), have shown to exhibit interesting catalytic activity in organic transformations,16–18 such as C–C bond formation,19,20 C–N and C–O oxidation,21–23 thiol oxidation24 and different MCRs.25–32
GO is a π-conjugated planar material characterized by the following structure and active sites:16,18,33 a few nm large aromatic Csp2-domains surrounded by oxygen functional groups (e.g. alcohols, epoxides and carboxylic acids) as well as free radicals, which are due to internal structural defects. Because of this multifunctional character, GO can be considered as a multicatalytic system, intrinsically versed towards one-pot multistep syntheses. Nevertheless, GO-assisted one-pot multistep reactions have been seldom explored,34 at least using organo- or metal-functionalized GO composites.35 Building on our experience in the development of new synthetic methodologies using MCRs36–44 and in one-pot multistep processes,45,46 and inspired by the potential of MCRs involving the employment of an external oxidant,47 we decided to study the GO-promoted Povarov multicomponent reaction between anilines, aldehydes and 2,3-dihydrofuran (DHF) that acts as dienophile to give tetrahydroquinolines (THQs) 1, and their subsequent oxidation to 2,3 disubstituted quinolines 2 (Scheme 1).
 | ||
| Scheme 1 Syntheses of 2,3-disubstituted quinolines 2 via the Povarov multicomponent reaction and subsequent oxidation. | ||
In this context, the dual capacity of GO to serve both as an acid and an oxidant should favour the in situ formation of imine, its activation in the aza-Diels–Alder process and the subsequent oxidation of the THQ adduct into the corresponding quinoline.
THQs48 and quinolines49,50 are privileged heteroaromatic scaffolds, largely present in the structures of bioactive compounds and natural products, with many applications in different chemical domains, such as organic synthesis or material science.
Even though the Povarov reaction has been extensively investigated for the synthesis of THQs, some drawbacks persist: strongly acidic conditions, use of noxious and expensive catalysts, formation of mixtures of products, unsatisfactory yields, limits on the scope (i.e. aliphatic aldehydes), and longer reaction times.51
In the literature, the routes to 2,3-disubstituted quinolines via a Povarov multicomponent reaction and subsequent oxidation are still relatively underexplored, presenting either a limited scope52,53 or requiring multistep procedure (Scheme 1).54 In 2018, Meléndez Gómez reported a simple and efficient one-pot synthesis of quinolines 2; however the scope in that case was restricted to 2,3-di(hydroxyalkyl)quinolines.55
Herein, we report the first GO-promoted one-pot multistep synthesis of substituted quinolines and the investigation of the real active sites of GO using X-ray photoelectron spectroscopy (XPS) and solid-state nuclear magnetic resonance (ssNMR).
Results and discussion
The Povarov reaction was initially optimized in acetonitrile using aniline, p-chlorobenzaldehyde and 2,3-dihydrofuran (DHF) in the presence of 20 mg of GO for 1 mmol of aldehyde.After the reaction mixture was stirred at room temperature for 24 h, the furo-[3,2-c]tetrahydroquinoline 1a was isolated in a 56% yield as a mixture of endo- and exo-isomers (Table 1, entry 1).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent (M) | Dienophile (equiv.) | Temperature | GO (mg mmol−1) | Time (h) | endo/exo ratioa | Yield of 1ab |
| a endo/exo ratio was calculated after purification by column chromatography.b Isolated yield.c The reaction was performed with 1.5 equiv. of the dienophile; after 2 h, an additional amount of 1.5 equiv. was added and the mixture was stirred further for 2 h.d Reaction was performed in the presence of MnCl2 4H2O (0.12 mg mmol−1): the amount of the catalyst was decided based on the amount of Mn contained on GO (about 5000 ppm, see ESI for details).e reaction was performed in the presence of reduced graphene oxide (rGO). | |||||||
| 1 | CH3CN (0.4) | 1.5 | rt | 20 | 24 | 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 :20 |
56% |
| 2 | CH2Cl2 (0.4) | 1.5 | rt | 20 | 24 | — | — |
| 3 | Toluene (0.4) | 1.5 | rt | 20 | 24 | — | Trace |
| 4 | Pentane (0.4) | 1.5 | rt | 20 | 24 | — | 18% |
| 5 | EtOAc (0.4) | 1.5 | rt | 20 | 24 | 80:20 |
22% |
| 6 | 1,4-Dioxane (0.4) | 1.5 | rt | 20 | 24 | 80:20 |
13% |
| 7 | 2-Me-THF (0.4) | 1.5 | rt | 20 | 24 | — | 20% |
| 8 | DMF (0.4) | 1.5 | rt | 20 | 48 | 60:40 |
36% |
| 9 | iPrOH (0.4) | 1.5 | rt | 20 | 24 | 90:10 |
43% |
| 10 | tBuOH (0.4) | 1.5 | rt | 20 | 24 | 70:30 |
48% |
| 11 | H2O (0.4) | 1.5 | rt | 20 | 24 | — | 20% |
| 12 | CH3CN (0.4) | 1.5 | 60 °C | 20 | 24 | 80:20 |
27% |
| 13 | CH3CN (0.4) | 1.5 | 10 °C | 20 | 24 | — | — |
| 14 | CH3CN (0.4) | 1.5 | rt | 40 | 4 | 80:20 |
57% |
| 15 | CH3CN (0.8) | 1.5 | rt | 20 | 4 | 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :20 :20 |
60% |
| 16 | CH3CN (0.8) | 1.5 | rt | 10 | 4 | 80:20 |
42% |
| 17 | CH3CN (0.8) | 3 | rt | 20 | 4 | 75:25 |
62% |
| 18 | CH3CN (0.8) | 1.5 + 1.5c | rt | 20 | 4 | 8020 |
62% |
| 19 | CH3CN (0.8) | 1.5 | rt | — | 4 | — | <5% |
| 20d | CH3CN (0.8) | 1.5 | rt | — | 4 | 80:20 |
20% |
| 21e | CH3CN (0.2) | 1.5 | rt | 20 (rGO) | 6 | 75:25 |
21% |
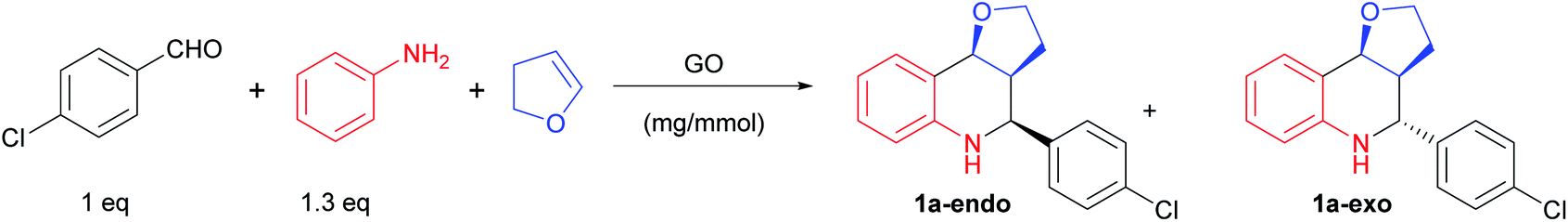
Starting from this result, different reaction conditions were explored (Table 1). The effect of different solvents was first evaluated. Apolar solvents, such as pentane, toluene, dichloromethane (entries 2–5), or ethers (entries 6,7) afforded very low conversions and poor yields. Better results were obtained with protic solvents, such as iPrOH (entry 9) and tBuOH (entry 10), although the concomitant formation of the corresponding acetal decreased the yield (the corresponding di-isopropyl acetal was isolated in a 25% yield using iPrOH). In DMF (entry 8), the reaction turned out to be very slow: based on the literature data,56 we suppose a role of DMF in stabilizing the imine and deactivating the GO surface by covalent bonding. Having assessed that the best results were obtained by using CH3CN as solvent, we investigated other reaction parameters, such as the temperature (entries 12 and 13), concentration (entry 15), and amount of catalyst and dienophile (entries 14–18). We found that the best conditions in terms of yield, atom economy and environmental impact, were the following: CH3CN 0.8 mol dm−3, in the presence of 20 mg mmol−1 of GO, 1.5 equiv. of DHF for 4 h at room temperature. The stereochemistry of the products was determined by the combined use of the coupling constant and literature data.57
Proofs of a genuine GO catalysis were gained via dedicated control experiments. The background reaction was not productive (entry 19), while the potential co-catalysis promoted by the metal contamination of GO (i.e. manganese(II)) was ruled out via a designed experiment with MnCl2 4H2O (entry 20).58 Using reduced GO (rGO) as a carbocatalyst, the desired product was obtained in a poor yield, proving the important role of the oxygenated groups (entry 21).
In order to evaluate the versatility of the synthetic protocol, we moved on to establish the scope, with varying the aldehyde, aniline and dienophile, under the optimized reaction conditions (Scheme 2).
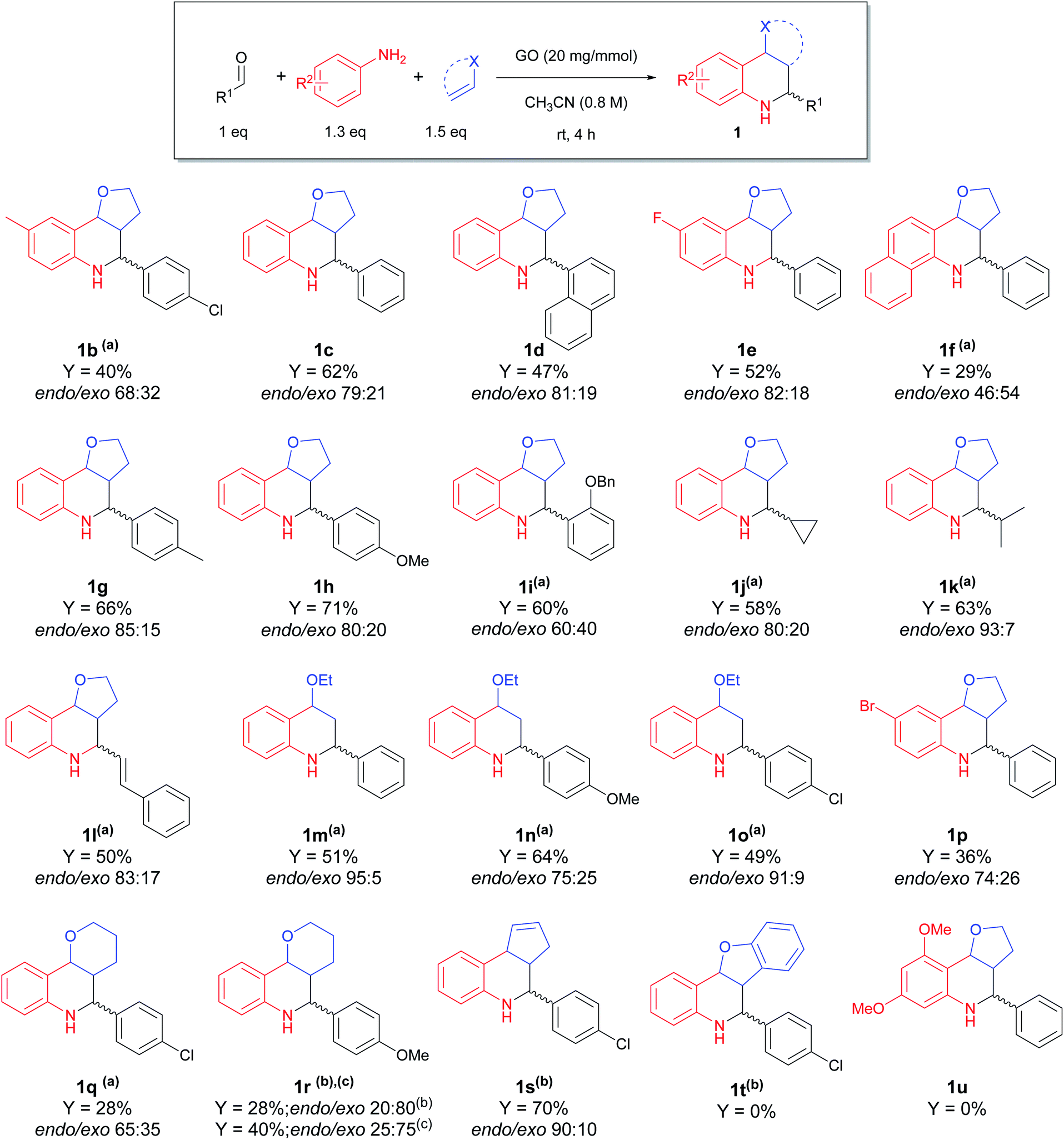 | ||
| Scheme 2 Scope of the carbocatalyzed Povarov reaction. Different reaction conditions: (a) rt, 24–48 h; (b) 60 °C, 24–48 h; (c) CH3CN/H2O = 4:1, 60 °C, 24–48 h. | ||
Good results were obtained when performing the reaction with DHF as dienophile: interestingly, good yields were observed employing aliphatic (1j and 1k) and α,β-unsaturated (1l) aldehydes, substrates rarely used in the Povarov reaction;54,59,60 on the other hand, substituted anilines (1b, 1f and 1p) and 2,3-dihydropyran (DHP) as dienophile (1q and 1r) gave lower yields. To improve the conversion of the less reactive substrates, we slightly changed the reaction conditions, applying longer time and/or higher reaction temperature, affording the products in acceptable yields and with endo as the major isomer. For compound 1r, where surprisingly the inversion of diastereoselectivity was observed, we tried to improve the yield by using a mixture of CH3CN/H2O as 4:1,61 showing an increase in the yield from 28% to 40%.
Different behaviour was observed by using cyclopentadiene as dienophile: even though the reaction a required longer time (24 h) and higher temperature (60 °C), due to the lower reactivity of the dienophile (4 equiv.), the corresponding THQ 1s was obtained with a 70% yield and high diastereoselectivity. Ethyl vinyl ether (1m–o) gave good yields, even though it required a longer reaction time.
Reaction with benzaldehyde, 3,5-dimethoxyaniline and DHF afforded a complex mixture of different products and the corresponding tetrahydroquinoline 1u was not observed.
Employing benzofuran as dienophile, no reaction occurred, despite heating at 60 °C for 24 h to promote the reactivity.
To assess the efficiency of the catalyst and verify its recyclability, we performed a scale-up experiment using 7.5 mmol of p-chlorobenzaldehyde (Scheme 3). In this case, a notable 66% isolated yield of 1a was obtained under slightly different conditions (CH3CN 0.2 mol dm−3, 2.5 equiv. of DHF, 24 h).
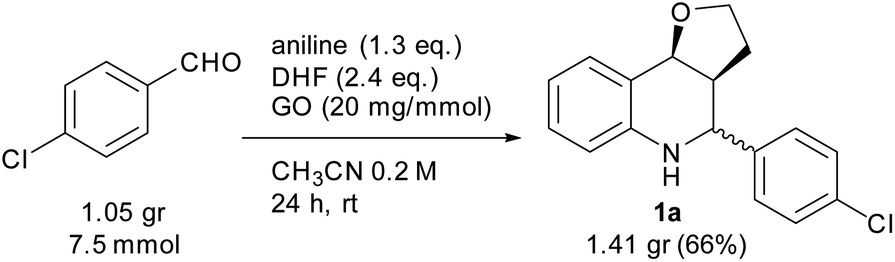 | ||
| Scheme 3 Scale-up experiment. | ||
At the end of the experiment, the solvent was removed by evaporation and the product was recovered by dissolving the mixture in dichloromethane/ethyl acetate followed by centrifugation at 7800 rpm for 5 min. The catalyst was washed by repeating the process 10 times to remove all traces of organic compounds. The recovered catalyst was washed again with water and methanol, and subsequently reused in consecutive reactions. A slight deactivation of the GO catalyst was noticed after the first run, while a significant decrease was noted from the 4th run. The above results indicate that the GO catalyst showed moderate recyclability within three runs (Fig. 1).
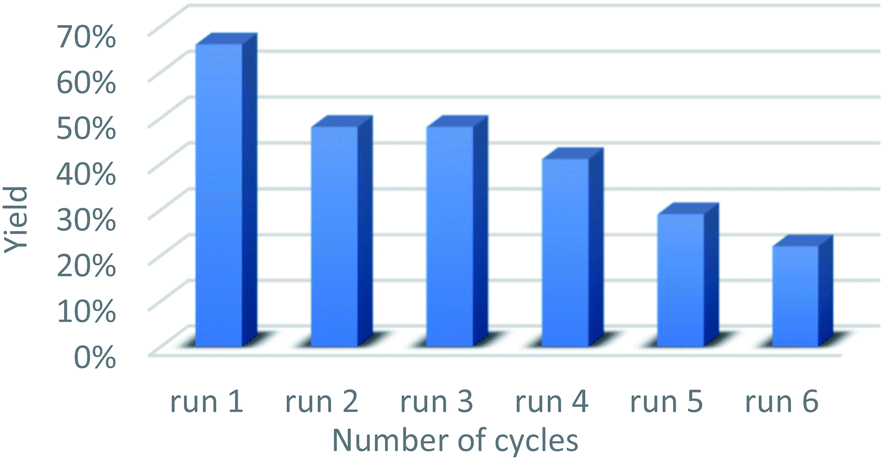 | ||
| Fig. 1 Recyclability experiment for the carbocatalyzed Povarov reaction. | ||
After the recyclability experiments, the recovered GO after the first and the last run were analyzed by XPS and ssNMR. XPS showed a significant decrease in the epoxide and hydroxide groups during the reaction (Fig. 2). The recyclability experiments were repeated with other batches of GO (one produced by Graphenea, and one by Abalonyx, which contained a much lower amount of Mn), giving similar results in terms of the yields and XPS analyses (see ESI for details†). The XPS survey spectra of GO in the reaction conditions (control sample of GO in CH3CN at room temperature for 24 h) and GO after multiple reactions (see ESI for details†) were mainly composed of O 1s and C 1s signals; the presence of N 1s, Si 2p, S 2p, Mn 2p and Cl 2p signals were found at lower intensities. The binding energies and relative atomic concentrations of all the samples are reported in the ESI†.
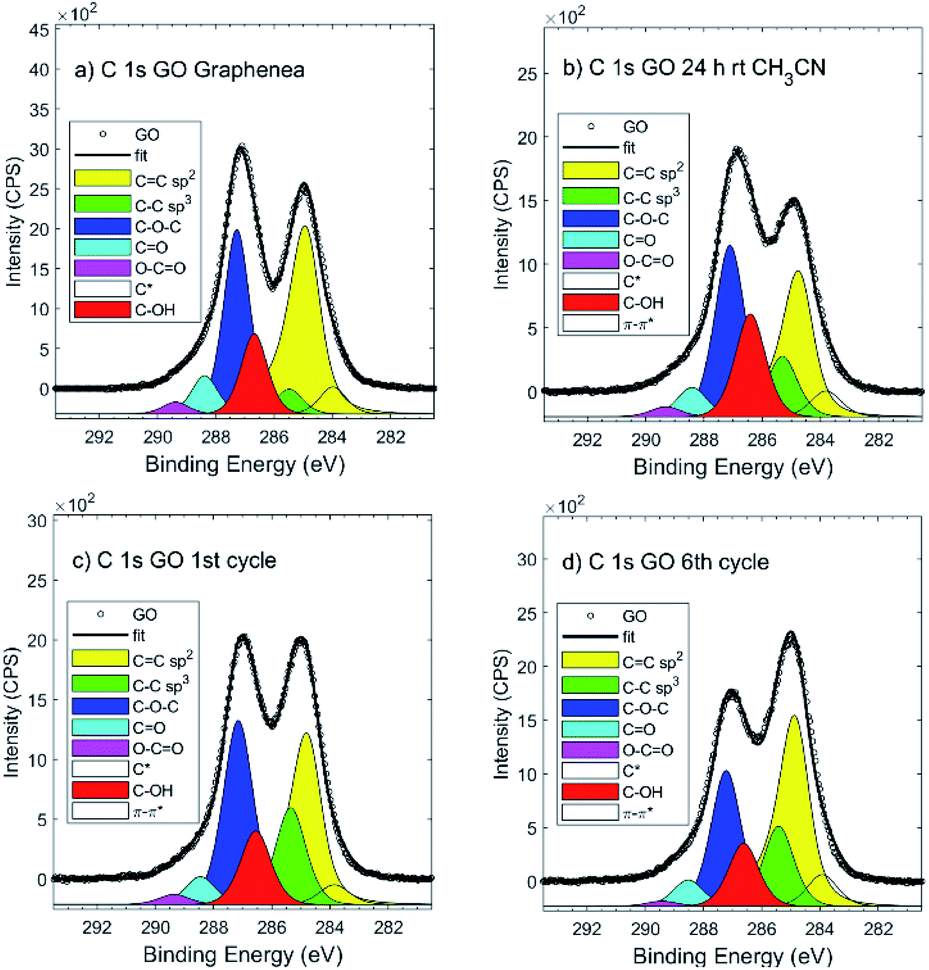 | ||
| Fig. 2 XPS C 1s signal of: (a) commercial Graphenea GO; (b) GO after CH3CN at room temperature for 24 h; (c) GO after the 1st cycle; (d) GO after the 6th cycle. | ||
The XPS C 1s signal analysis showed a significant decrease in C–O groups content and a concomitant increase in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C Sp2 and C–C Sp3 regions. However, the efficiency of the reaction might not be directly related to the overall oxidation degree of the material, but rather to the increase in the amounts of nitrogen (N) and chlorine (Cl).
C Sp2 and C–C Sp3 regions. However, the efficiency of the reaction might not be directly related to the overall oxidation degree of the material, but rather to the increase in the amounts of nitrogen (N) and chlorine (Cl).
In particular, N was so far present in both GO Abalonyx and GO Graphenea, about 0.3–0.4% in the control samples. After the first and last reactions, the amount of N proportionally grew, and the efficiency seemed to decrease as a function of N (Fig. 3a). The Cl showed a less evident behaviour: the initial amount in GO Graphenea was negligible, but in GO Abalonyx, it was initially present in a significant amount (0.5%). If we consider only the trend in GO Graphenea, the amount of Cl grew after the first and the last reactions (Fig. 3b). Since the increase in the amount of nitrogen was higher than the increase measured for chlorine, the presence of these two elements seemed to be associated with the interaction of the aldehyde and aniline with the GO surface, rather than with the final product.
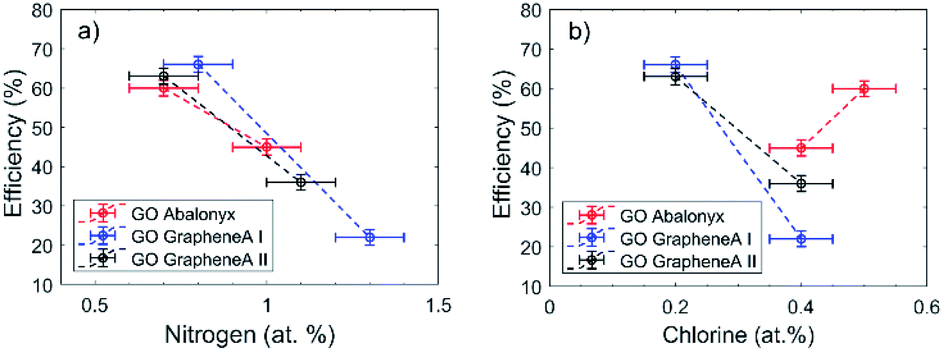 | ||
| Fig. 3 (a) Nitrogen vs. reaction efficiency and (b) chlorine vs. reaction efficiency. | ||
The simplest interpretation of the N and Cl trends is that the p-chlorobenzaldehyde and aniline are bound to the GO surface, blocking the active sites and preventing further Povarov reaction. Obviously, the presence of these molecules on GO affected the O/C decrease measured.
In order to clarify the nature of the interaction between the organic molecules and GO, we treated the recovered GO under acidic conditions,62 and we repeated the XPS analysis, finding no significant variation in GO's chemical structure: the O/C ratio slightly decreased, but N and Cl amounts were identical to in the untreated sample. Acid treatment apparently was not able to regenerate the GO as previously reported,61 probably due to the presence of grafted reagents on the active sites. Moreover, we carried out the Povarov reaction using the GO recovered after acidic treatment, but it gave 1a in only a 17% yield.
To investigate further the GO functionalization after the Povarov reaction, characterization by NMR spectroscopy was performed. The 13C ssNMR spectra showed peaks corresponding to aromatic C-sp2 at 130 ppm, hydroxyl groups at 70 ppm, and epoxy at 60 ppm; carboxyl was present in lower amounts.63 From the quantitative 13C ssNMR spectra obtained by direct excitation (see Fig. S7 and S8 in the ESI†), a decrease in C–OH and C–O–C groups was observed; in fact, the epoxy ring opening is one of the most common mechanism for covalent grafting, as reported in the literature through XPS and NMR,64 but in the present study it could be excluded: the C–O–C/C–OH ratio obtained by ssNMR was close to 2 for all the samples. The overall reduction of GO after the Povarov reaction, observed by XPS, could be confirmed also by ssNMR. The signal for the aliphatic carbon (Csp3) appeared at 31 ppm in both the direct excitation and cross-polarization spectra of GO after the reaction (Fig. 4).
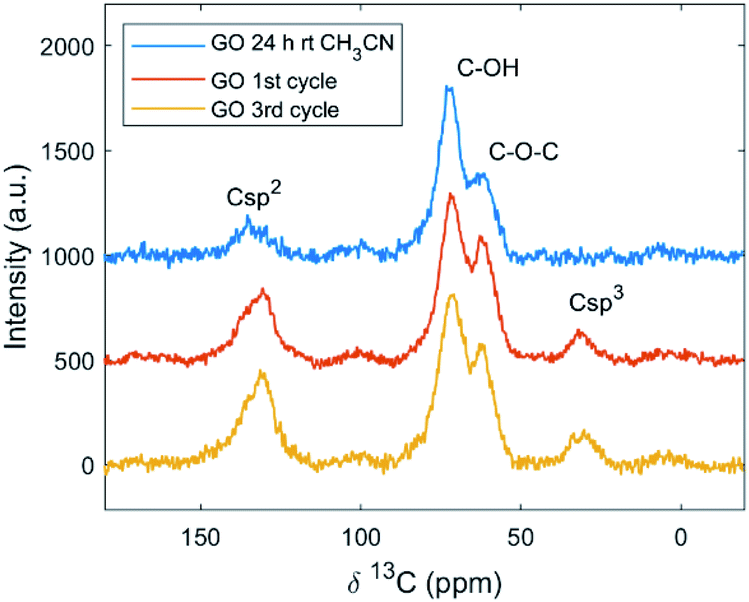 | ||
| Fig. 4 CP 1H–13C ssNMR signals of commercial Abalonyx GO after CH3CN at room temperature for 24 h; GO after the 1st cycle; GO after the 3rd cycle. | ||
The 1H–13C wPMLG-HETCOR spectra65–67 (Fig. 5) presented a correlation between the signal of 13C at 31 ppm and a signal in the 1–2 ppm region in 1H, further proof of the formation of aliphatic carbons on GO surfaces. In the literature, there are a few reports of examples of saturated C–H bond signals in the 30 ppm region, such as the NaOH-promoted Suzuki cross-coupling reaction of phenylboronic acid68 or the methylene groups in the covalent grafting of amines.64 The relative amount of (Csp3) obtained from the direct excitation 13C NMR spectra (14% ca) seemed to be independent from the GO type and number of cycles.
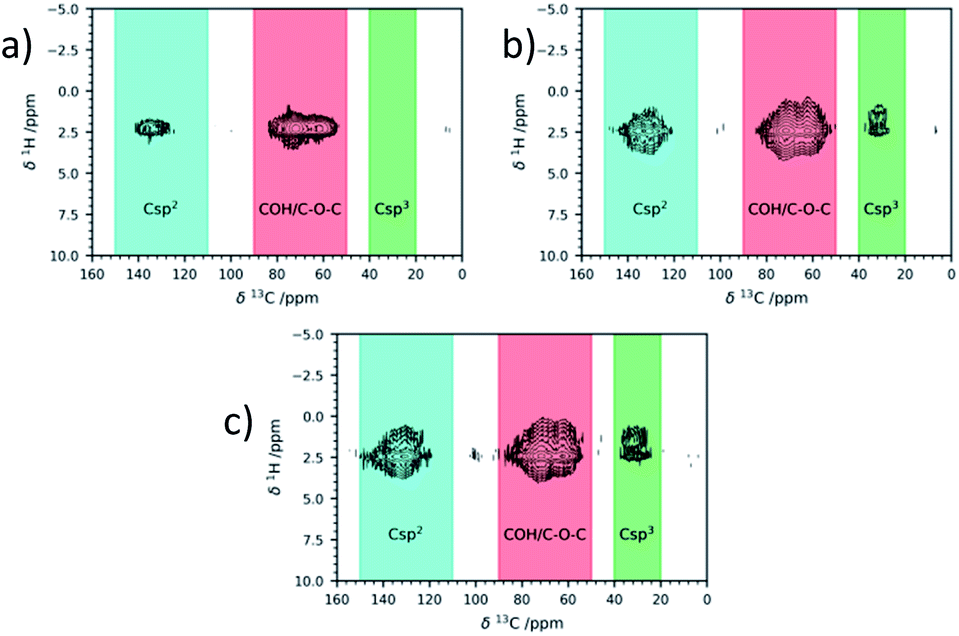 | ||
| Fig. 5 1H–13C correlation ssNMR signals of commercial Abalonyx GO after CH3CN at room temperature for 24 h (a); GO after the 1st cycle (b); GO after the 3rd cycle (c). | ||
Summarising, XPS and NMR analysis showed that: i) GO after the Povarov reaction was significantly reduced, (ii) no epoxy ring opening was observed, and iii) p-chlorobenzaldehyde and aniline could be covalently grafted, as well as dihydrofuran, while iv) the presence of aliphatic Csp3 may suggest that a fraction of reaction sites was inactivated after the reaction.
Although the mechanism of the Povarov reaction has not yet been certainly established, a step-ways manner is generally proposed rather than a concerted one:69,70 The in situ-formed imine, activated by BrØnsted acid (BA) or Lewis acid (LA) catalysts, undergoes nucleophilic attack from the dienophile, with the formation of a cationic intermediate, which evolves into the final THQ after a quick intramolecular Friedel–Crafts reaction. Actually, GO presents both BA and LA properties. Indeed, while the BA character of GO has been well established, recently, the Lewis acidity, due to the basal plane epoxide ring opening, was found to be crucial for its catalytic activity involving multicomponent reactions.62 Therefore, based on our experimental findings and on the previous literature, a possible mechanism is proposed and depicted in Scheme 4. Initially, the BA groups on GO promote the protonation of the aldehyde and, after nucleophilic attack by aniline, the dehydration step, give imine I. GO can donate a proton to the imine I, forming the reactive iminium species II, which undergoes reaction with DHF, following the generally accepted mechanism of the Povarov reaction, to generate 1a. Another possibility is that GO might act as a Lewis acidic carbenium catalyst,71 interacting with imine to give complex III; the electron-rich olefin, such as DHF, attacks complex III to form intermediate IV, which undergoes an intramolecular cyclization to produce complex V. Then, the detachment of the product with subsequent aromatization provides THQ 1a.
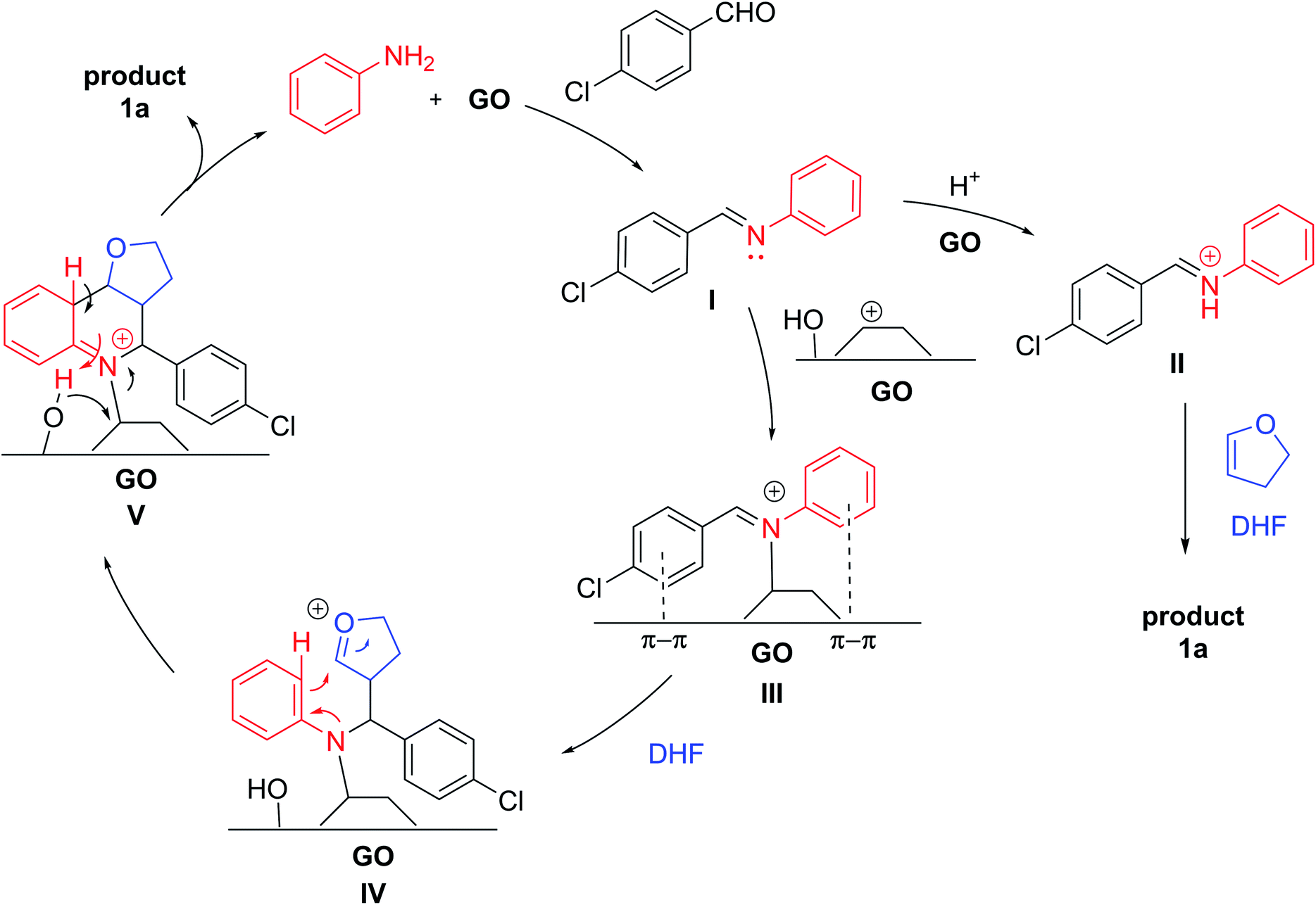 | ||
| Scheme 4 Proposed catalytic activities of GO in the Povarov reaction. | ||
Based on our experimental results, the decrease in efficiency was related to the partial passivation of the surface of the material due to the covalent bonds with organic substrates. Although a direct covalent interaction between GO and the Povarov product cannot be excluded (for instance, the covalent bond with intermediate V), the reagents may interact with the surface independently. Actually, aniline is able to chemically interact with the GO surface, opening an epoxy group and/or reacting with the Cα of a hydroxyl group.72 Eventually, alcohols or amino-alcohol moieties grafted on GO should interact with the aldehyde in an unproductive pathway with partial deactivation of the carbon material. Moreover, the appearance of new peaks at 31 ppm in the 13C-NMR ssNMR spectra suggested the interaction of DHF as well. This molecule can undergo GO-promoted acid-hydration, to afford the corresponding hemiacetal, which can covalently interact with the GO surface, increasing the passivation.
Then, we proceeded to investigate the carbocatalized oxidation of the Povarov products to give substituted quinolines. GO has been largely exploited for the oxidation of organic compounds,22,23,73,74 although very few examples have appeared recently in the literature regarding the oxidation of heterocycles.75
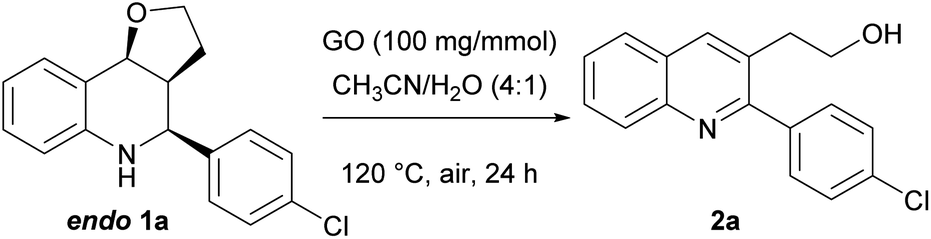
Aiming to develop a one-pot procedure, we initially treated THQ 1a (as a diastereoisomeric mixture) in conditions similar to those used for the Povarov reaction, except for the temperature, which was increased to 110 °C. Contrary to what is reported in the literature with the most commonly used oxidant 2,3-dichloro-5,6-dicyano-1,4-benzoquinoneone (DDQ),54 where a mixture of different products is generally obtained, the reaction provided the 2,3-disubstituted quinoline 2a as a single product, even though it proceeded very slowly, affording a low yield after 4 days (Y = 28%) (Table 2, entry 1). Product 2a was the result of an elimination–oxidation process, generally promoted by acidic conditions.76,77 In addition, in our case, based on the ratio of endo/exo isomers of the starting material recovered, only the endo isomer appeared to have reacted, while the exo isomer remained untouched.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst (mg mmol−1) | Solvent (M) | Time (h) | Temperature | Molar ratio (1a/2a)a | Yield exo 1a | Yield 2ab |
| a Conversion was calculated after purification by column chromatography.b Isolated yield.c Reaction was performed starting from a mixture endo/exo of 80:20.d Reaction performed starting from pure endo.e Reaction was performed in the presence of Na2CO3 (40 mg mmol−1). |
|||||||
| 1c | GO (20) | CH3CN (0.2) | 96 | 110 °C | 50:50 |
— | 28% |
| 2d | GO (50) | CH3CN (0.2) | 48 | 120 °C | 35:65 |
— | 56% |
| 3d,e | GO (50) | CH3CN (0.2) | 48 | 120 °C | 55:45 |
— | 44% |
| 4d | GO (50) | CH3CN (0.1) | 96 | 120 °C | 20:80 |
— | 49% |
| 5d | GO (50) | CH3CN (0.1) | 96 | 90 °C | 40:60 |
— | 45% |
| 6d | GO (50) | CH3CN/H2O 4:1 (0.2) |
48 | 120 °C | 15:85 |
— | 63% |
| 7d | GO (100) | CH3CN (0.1) | 48 | 120 °C | 5:95 |
— | 88% |
| 8d | GO (100) | CH3CN/H2O 4:1 (0.1) |
24 | 120 °C | 0:100 |
— | 92% |
| 9d | GO (100) | CH3CN/H2O 4:1 (0.1) |
4 | 120 °C (MW) | 20:80 |
— | 71% |
| 10d | GO (100) | CH3CN/H2O 2:1 (0.1) |
3 | 120 °C (MW) | 10:90 |
— | 88% |
| 11c | GO (100) | CH3CN/H2O 4:1 (0.1) |
24 | 120 °C | 15:85 |
13% | 73% |

To simplify the optimization, at first, we decided to study the oxidation reaction starting from the pure endo (easily accessible by trituration of the mixture). Then, the reaction was carried out with a slight increase in the temperature and catalyst, leading to a better, albeit still unsatisfactory, conversion and yield (entry 2). Based on the work of Zang,75 we repeated the oxidation in the presence of a substoichiometric amount of Na2CO3, suggesting that the base does not have a role in the process (entry 3). Lower temperatures or longer times did not produce significant improvements, while the use of a mixture of CH3CN/H2O as 4:1 afforded a great conversion of 1a endo into quinoline 2a (Y = 63%, entry 6). Furthermore, the increase in GO to 100 mg mmol−1 (entry 7) was explored, while performing the reaction in CH3CN 0.1 M to allow a better dispersion of the catalyst in the solvent, giving almost a complete oxidation of 1a endo (Y = 88%). The best reaction conditions were found to be the use of a mixture of CH3CN/H2O as 4:1 (0.1 M) and 100 mg mmol−1 of GO; indeed, complete conversion of THQ endo into quinoline was observed, affording the desired adduct in a 92% isolated yield (entry 8).
To shorten the reaction time, we tried to perform the oxidation process under microwave (MW) irradiation (entries 9 and 10). This provided an outstanding conversion and time reduction, and quinoline was obtained in an 88% isolated yield.
Finally, we tried the oxidation of a diastereoisomeric mixture of 1a (endo/exo = 80:20) under the optimal reaction conditions, obtaining a complete conversion of 1a endo into quinoline 2a (73%) and partial recovery of 1a exo (13%) (entry 11).
To explain the mechanism of the oxidation and its diastereoselectivity, we performed several experiments (Scheme 5). Since the oxidation–elimination product is generally formed under acidic conditions by elimination and subsequent aromatization by the air, we were interested to verify if GO was only effective as an acid promoter just in the first step (Scheme 5a, pathway A). With this aim, N-substituted THQ 3 was synthetized through a reductive amination reaction and reacted under the optimized conditions, recovering the unaltered starting material (Scheme 5b). This result suggests that elimination might not be the first step, or, in any case, the GO does not only mediate the elimination step. However, could GO catalyze first the C–N oxidation, and the intermediate then be aromatized by the elimination process (Scheme 5a, pathway B)? To investigate this possibility, THQ 1s, where the elimination is prevented by the lack of oxygen, was treated under the optimized conditions, but also in this case only the starting material was recovered (Scheme 5c).
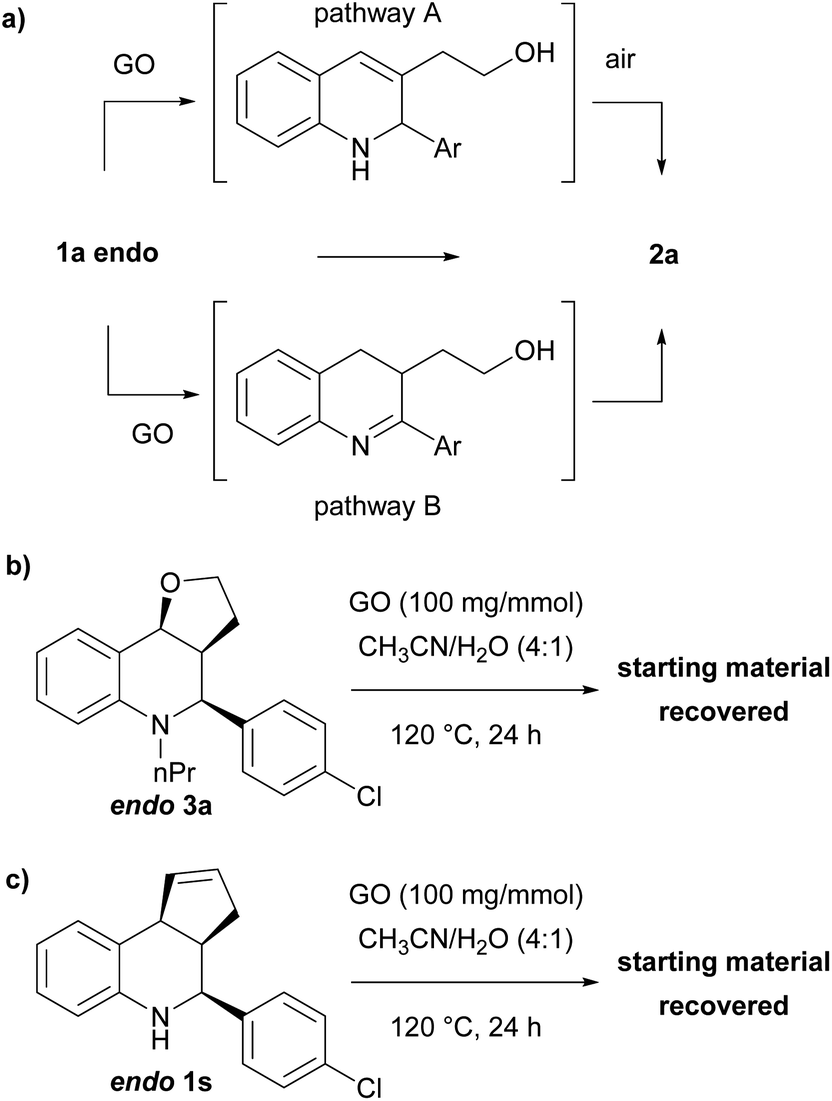 | ||
| Scheme 5 Investigation of the oxidation mechanism and control experiments. | ||
Based on these experimental results, we suggest that the process is concerted and that the interaction between THQ and GO surface is expected to be more favourable for the endo isomer where both hydrogens have a cis relationship, so the steric hindrance of the aryl group does not prevent the simultaneous interaction between the THQ core and the oxygen of the tetrahydrofuran moiety.
Moreover, other control experiments were performed to investigate the catalytic activity and the role of GO and oxygen in the process (Table 3). The potential co-catalysis promoted by metal contaminations of GO was ruled out by performing the reaction with MnCl2 4H2O (entry 2): actually, no reaction occurred; as well as by heating THQ 1a without GO as a control experiment (entry 1).
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield |
| a MnCl2 4H2O was used instead of GO considering that GO contains 5000 ppm of Mn by ICP-OES analysis, the control experiment was performed by using the equivalent amount of Mn(II) (see ESI for details). | ||
| 1 | No GO | 11% |
| 2 | MnCl2·4H2O (0.12 mg mmol−1)a | — |
| 3 | GO (100 mg mmol−1) under Ar | 53% |
| 4 | rGO (100 mg mmol−1) | 53% |
| 5 | rGO (100 mg mmol−1) under Ar | 21% |
The carbocatalytic oxidation carried out under an argon atmosphere (entry 3) brought a low conversion with a 53% isolated yield, demonstrating that oxygen is important, although its absence does not completely suppress the reaction. Furthermore, when reduced graphene oxide (rGO) was employed, the same decrease in yield was noted (entry 4).
These results suggested that both oxygen and hydroxyl and epoxy groups on GO play a role during oxidation. As a further demonstration of this, the yield dramatically decreased when the reaction was carried out with the concomitant presence of rGO and an argon atmosphere (entry 5). Based on the experimental evidence and literature data,21 we deduce that GO serves both as an oxidant, thanks to the hydroxyl and epoxy groups, and as a catalyst in the aerobic oxidation of the substrate. Further proof of this dual mechanism was obtained from the recyclability experiments. The reusability of GO was investigated following the same protocol used for the MCR. Even though the XPS analysis disclosed a significant reduction in the carbon material after 3 runs, the yield dropped only up to 50%, similar to what happened when using rGO (see ESI for details†). The magnitude of the GO reduction was significantly higher than for GO after the Povarov reaction: after the first oxidation cycle the O/C dropped from 0.28 ± 0.02 for the control sample to 0.12 ± 0.01, as well as all the C–O groups decreased, as revealed by C 1s XPS analysis: the epoxide and hydroxide contracted from 22.4% and 6.6% to 5.2% and 3.1%, respectively.
Finally, an oxidation using GO recovered from the 3rd run of the Povarov reaction was performed, obtaining 2a in a high yield (81%). This result suggests that the two processes might involve different functional groups on the GO surface, making the one-pot process feasible.
Next, the scope of the optimized protocol was investigated for a variety of tetrahydroquinolines (Scheme 6). The reaction proceeded smoothly, and complete consumption of the endo isomer was observed almost in each case. Quinolines bearing different aromatic substituents at position 2 were obtained in excellent yields. Interesting results were obtained even with compounds bearing alkyl or unsaturated substituents, allowing the isolation of products 2j, 2k and 2l in good to excellent yields.
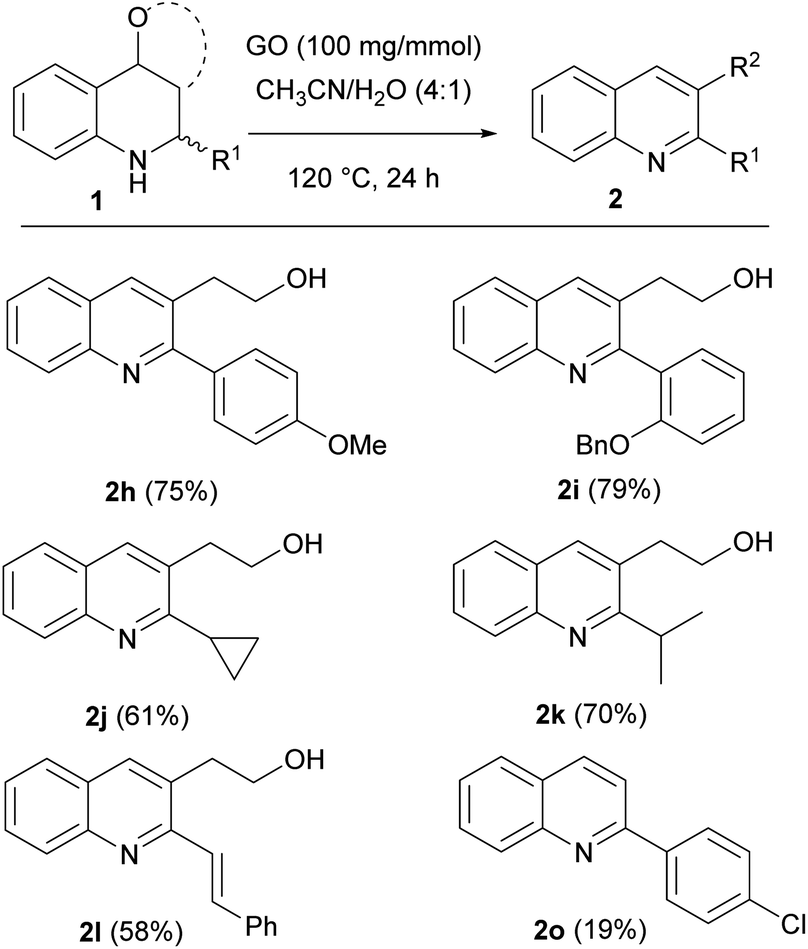 | ||
| Scheme 6 Scope of the oxidation of THQs in the presence of GO. | ||
Unfortunately, THQ 1o gave the corresponding quinoline 2o in a poor yield due to the extended degradation.
Finally, having demonstrated the efficiency of this protocol, we investigated the one-pot procedure leading to 2, without isolating the Povarov product 1 (Table 4). After a short optimization (see ESI†), we found the best results employing an increased dilution in the Povarov step. Then, as soon as the multicomponent reaction was complete, acetonitrile/water solution and fresh GO were directly added and the mixture was heated at 120 °C for 48 h.
Although a complete conversion of THQs has never been observed, the final quinolines were isolated in good yields, if compared with the previously reported two-step procedure. In two cases (Table 4, entries 1 and 3), the one-pot sequence was superior; whereas for compounds 2h and 2j (entries 2 and 4), the two-step sequence seemed to be more efficient. Interestingly, we were also able to isolate the not-reacted THQ as a mixture of diastereoisomer with exo as the major one, which confirmed its lower reactivity in the oxidation step.
In addition, we also wanted to demonstrate that the presence of an additional functional group on the aromatic ring (2i) may be exploited for further derivatizations. As polycycle-fused quinolines are typical privileged structures, widely distributed in natural products and medicinal chemistry,78–81 we studied the conversion of compound 2i into the benzo-oxepin-fused quinoline 4 by hydrogenolysis of the benzyl protecting group, followed by a Mitsunobu reaction with tbutyl azodicarboxylate (TBAD) and triphenylphosphine.82
The desired product was isolated after column chromatography in excellent yield (Scheme 7). Interestingly, this simple protocol allowed the synthesis of one of the rare examples of an oxepine-containing polycyclic-fused quinoline system.83–86
 | ||
| Scheme 7 Synthesis of oxepin-annulated quinolines 4. | ||
Material and methods
General procedure for the povarov reaction
A screw-capped vial was equipped with a magnetic stirring bar and charged with GO (20 mg mmol−1) and CH3CN (0.8 M). The resulting suspension was sonicated for 2 min, using an ultrasonic bath. The appropriate aldehyde (1 equiv.), aniline (1.3 equiv.) and dienophile (1.5 equiv.) were added to the mixture and the sealed reaction vial was stirred at room temperature (rt) or 60 °C for 4–48 h. After completion of the reaction, the mixture was filtered through a Celite cake, washing with DCM/EtOAc 1:1 (15 mL). The filtrate was evaporated to dryness in vacuo and the residue was purified by flash column chromatography (SiO2), eluting with PE/EtOAc or PE/acetone or PE/Et2O/DCM, to give the THQ as a mixture endo/exo isomers. The diastereomeric ratio was determined by 1H NMR, by integration.
General procedure for the synthesis of 2,3-substituted quinolines
A screw-capped vial was equipped with a magnetic stirring bar and charged with GO (100 mg mmol−1) and CH3CN/H2O (4:1, 0.1 M). The resulting suspension was sonicated for 2 min, using an ultrasonic bath. The appropriate THQ (1 equiv.) was added and the mixture was stirred at 120 °C for 24 h. After completion of the reaction, the mixture was filtered through a Celite cake, washing with DCM/EtOAc 1:1 (15 mL) and MeOH (5 mL). The filtrate was evaporated to dryness and the residue was purified by flash column chromatography (SiO2), eluting with PE/acetone or PE/EtOAc.
General procedure for the one-pot synthesis of 2,3-substituted quinolines
A screw-capped vial was equipped with a magnetic stirring bar and charged with GO (20 mg mmol−1) and CH3CN (0.2 M). The resulting suspension was sonicated for 2 min, using an ultrasonic bath. The appropriate aldehyde (1 equiv.), aniline (1.3 equiv.) and 2,3-dihydrofuran (2.5 equiv.) were added to the mixture. The sealed reaction vial was stirred at rt for 6–24 h. After completion of the reaction, GO (80 mg mmol−1) and CH3CN/H2O (to achieve a mixture of 4:1 0.1 M) were added and the resulting suspension was stirred at 120 °C for 48 h. Then, the reaction mixture was filtered through a Celite cake, washing with DCM/EtOAc 1:1 (15 mL) and MeOH (5 mL). The filtrate was evaporated to dryness and the residue was purified by flash column chromatography (SiO2), eluting with PE/acetone.
Structural characterization of GO
X-Ray photoelectron spectroscopy (XPS) and solid-state nuclear magnetic resonance analyses (ssNMR) were performed on both commercial GO Graphenea and Abalonyx; after catalysis, the modification of GO was compared with the control sample, consisting of a GO suspension in CH3CN at rt for 24 h. The GO after the oxidation reaction was compared with GO suspension in CH3CN:H20 of 4:1 at 120 °C for 48 h. XPS and ssNMR were performed on fresh tablets obtained from a vacuum oven (50 °C at 10 mbar for 1 h)-dried GO, which was compressed for 2 min at 100 bar by stainless steel discs.
XPS spectra were acquired by a hemispherical analyser (Phoibos 100, Specs, Germany) using a non-monochromatic Mg Kα excitation set at 125 W (XR50, Specs, Germany). Survey and high resolution spectra was acquired in fixed analyser transmission mode (FAT) on a large area of ca 7 × 3 mm2 and overall energy resolution of 0.9 eV measured on freshly sputtered silver (Ag 3d). The spectrometer was calibrated to the Au 4f7/2 peak at 84.0 eV. Static charging effects were corrected for by calibrating all the spectra to C 1s 285.0 eV. Deconvolutions were performed using CasaXPS software after Shirley background subtraction. C 1s was fitted as described in the literature,87 using the asymmetric line-shape for aromatic C-sp2 and symmetric line-shapes (pseudo-voigt) for the C–O defects. The binding energies of the synthetic components of C 1s were: CC sp2 at 284.4 eV, CC* sp2 at 283.6 eV, C–C sp3 285.0 eV, C–OH at 286.2 eV, C–O–C at 286.9 eV, CO at 288.2 eV and O–CO at 289.1 eV. The O/C ratio was obtained from the O 1s and C 1s area ration and from C 1s fitting according to the stoichiometric ratios of C–O groups.
Solid-state NMR experiments were recorded on a Bruker Avance II spectrometer operating at 700 MHz 1H Larmor frequency (16.4 T), corresponding to 176 MHz 13C Larmor frequency. The spectrometer was equipped with a 3.2 mm BVT MAS probehead in double resonance mode. The magic angle spinning (MAS) frequency of the sample was set to 11.111 kHz. In the 1D direct excitation 13C-NMR spectra, the excitation pulse duration was set to 3.0 μs, corresponding to a 56° flip angle, the spectral window was 625 ppm, and the interscan delays were optimized to 1.9 s to make the experiment quantitative. For the 2D {1H}-13C HETCOR experiments, cross-polarization was achieved by matching the k = 1 Hartmann–Hahn condition. The 90° pulse duration on 13C and 1H were set to 4.8 us and 3.3 us, respectively. The spectral windows for 1H and 13C were 20 and 250 ppm, respectively. During the 1H magnetization evolution under the chemical shift in the indirect dimension, the PMLG decoupling sequence was used to suppress 1H–1H dipolar couplings. In these experiments, the interscan delay was set to 1 s.
The {1H}-13C HETCOR spectra were coprocessed for denoising as recently proposed by some of us,88 and processed applying a square-sine window function on both time dimensions. The presence of paramagnetic impurities originating from Mn ions is particularly critical for the spectrum acquisition of pristine GO produced by Graphenea as they present a relatively high Mn residual, in accordance with the results published by Panich.89 Hummers derived commercial GOs (Abalonyx and Graphenea) were produced by using also permanganate and the cleaning procedures may leave a small fraction of Mn: 0.15 ± 0.03 at% by XPS and 0.5% by ICP-OES in Graphenea and below 0.03%, the sensitivity limit for XPS, in Abalonyx. GO Abalonyx presents less experimental problems and, luckily, the control sample of GO Graphenea presented a significantly lower amount of Mn with respect to the pristine sample. On the other side, the reduced graphene oxide, rGO, presented also paramagnetic properties due to the high C-sp2 content (above 70% of C atoms).
Conclusions
In conclusion, we demonstrated that commercial graphene oxide can promote the 3C Povarov reaction between aldehydes, anilines and electron-enriched enol ethers. Even though GO has been already used as a heterogeneous catalyst for MCRs, very few studies regarding the real active sites, when using multiple reagents, have appeared in the literature. With this aim, control experiments and a full characterization of the carbon material by XPS and ssNMR were carried out, revealing that GO undergoes a significant reduction and covalent chemical modifications upon interaction with small organic molecules, which lead to a partial inactivation. Furthermore, we demonstrated that GO can promote the subsequent oxidation of some Povarov substrates to yield 2,3-disubstituted quinolines. These can also be performed as a one-pot procedure. The present method is versatile and allows the preparation of a library of THQs and quinolines under easy conditions and without the need for an inert atmosphere. GO is a bench-stable and easy-to-handle catalyst and this is an important advantage with respect to the Lewis acids typically used to promote Povarov MRCs. Studies are in progress to find other useful applications of GO in one-pot multistep syntheses involving MCRs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge PRIN-2017 project 2017W8KNZW. This work benefited from access (ITA002) to CERM/CIRMMP, Italy centre of Instruct-ERIC, a Ladmak ESFRI project, for the ssNMR measurements and support. S. C. and L. M. acknowledge Prof. M. Grotti, F. Soggia and W. Sgroi for determination of Mn in GO. A. K. and M. M. acknowledge GrapheneCore3 881603 —Graphene Flagship project for financial support. A. K. thanks Dr D. Jones for the enlightening discussions about the XPS interpretation of GO. F. B. acknowledges the Italian Ministry of Education, University and Research (MIUR) and European Social Fund (ESF) for the PON R&I 2014–2020 program, actions IV.4 ‘Doctorates and research contracts on Innovation topics’.Notes and references
- P. T. Anastas and J. B. Zimmerman, The United Nations sustainability goals: How can sustainable chemistry contribute?, Curr. Opin. Green Sustain., 2018, 13, 150–153 CrossRef
.
- S. Axon and D. James, The UN Sustainable Development Goals: How can sustainable chemistry contribute? A view from the chemical industry, Curr. Opin. Green Sustain., 2018, 13, 140–145 CrossRef
- https://ec.europa.eu/environment/strategy/chemicals-strategy_en.
- L. Jiang and W. Yi, in Sustainable Organic Synthesis: Tools and Strategies, The Royal Society of Chemistry, 2022, 10.1039/9781839164842-00472, pp. 472–487
- N. J. Green and M. S. Sherburn, Multi-Bond Forming Processes in Efficient Synthesis, Aust. J. Chem., 2013, 66, 267 CrossRef CAS
- J. Zhu and H. Bienaymé, Multicomponent reactions, John Wiley and Sons, 2005 Search PubMed
- H. Bienaymé, C. Hulme, G. Oddon and P. Schmitt, Maximizing Synthetic Efficiency: Multi-Component Transformations Lead the Way, Chem. Eur. J., 2000, 6, 3321–3329 CrossRef
- S. Zhi, X. Ma and W. Zhang, Consecutive multicomponent reactions for the synthesis of complex molecules, Org. Biomol. Chem., 2019, 17, 7632–7650 RSC
- Q.-W. Gui, B.-B. Wang, S. Zhu, F.-L. Li, M.-X. Zhu, M. Yi, J.-L. Yu, Z.-L. Wu and W.-M. He, Four-component synthesis of 3-aminomethylated imidazoheterocycles in EtOH under catalyst-free, oxidant-free and mild conditions, Green Chem., 2021, 23, 4430–4434 RSC
- Z. Pan, X. Yang, B. Chen, S. Shi, T. Liu, X. Xiao, L. Shen, L. Lou and Y. Ma, Employing Visible-Light Photoredox Catalysis in Multicomponent–Multicatalyst Reactions: One-Pot Synthesis of Spiroquinazolin-2-(thi)ones, J. Org. Chem., 2022, 87, 3596–3604 CrossRef PubMed
- Y. Wu, J.-Y. Chen, J. Ning, X. Jiang, J. Deng, Y. Deng, R. Xu and W.-M. He, Electrochemical multicomponent synthesis of 4-selanylpyrazoles under catalyst- and chemical-oxidant-free conditions, Green Chem., 2021, 23, 3950–3954 RSC
- P. Capurro, C. Lambruschini, P. Lova, L. Moni and A. Basso, Into the Blue: Ketene Multicomponent Reactions under Visible Light, J. Org. Chem., 2021, 86, 5845–5851 CrossRef CAS PubMed
- G. A. Coppola, S. Pillitteri, E. V. Van der Eycken, S.-L. You and U. K. Sharma, Multicomponent reactions and photo/electrochemistry join forces: atom economy meets energy efficiency, Chem. Soc. Rev., 2022, 51, 2313–2382 RSC
- L. Banfi, C. Lambruschini, L. Moni and R. Riva, in Green Synthetic Processes and Procedures, The Royal Society of Chemistry, 2019, pp. 115–140, 10.1039/9781788016131-00115
- R. C. Cioc, E. Ruijter and R. V. A. Orru, Multicomponent reactions: advanced tools for sustainable organic synthesis, Green Chem., 2014, 16, 2958–2975 RSC
- C. K. Chua and M. Pumera, Carbocatalysis: the state of “metal-free” catalysis, Chemistry, 2015, 21, 12550–12562 CrossRef CAS PubMed
- L. Lombardi and M. Bandini, Graphene Oxide as a Mediator in Organic Synthesis: a Mechanistic Focus, Angew. Chem., Int. Ed. Engl., 2020, 59, 20767–20778 CrossRef CAS PubMed
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Harnessing the chemistry of graphene oxide, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC
- J. Fang, Z. Peng, Y. Yang, J. Wang, J. Guo and H. Gong, Graphene-Oxide-Promoted Direct Dehydrogenative Coupling Reaction of Aromatics, Asian J. Org. Chem., 2018, 7, 355–358 CrossRef CAS
- H. Wu, C. Qiu, Z. Zhang, B. Zhang, S. Zhang, Y. Xu, H. Zhou, C. Su and K. P. Loh, Graphene-Oxide-Catalyzed Cross-Dehydrogenative Coupling of Oxindoles with Arenes and Thiophenols, Adv. Synth. Catal., 2019, 362, 789–794 CrossRef
- H. He, Z. Li, K. Li, G. Lei, X. Guan, G. Zhang, F. Zhang, X. Fan, W. Peng and Y. Li, Bifunctional Graphene-Based Metal-Free Catalysts for Oxidative Coupling of Amines, ACS Appl. Mater. Interfaces, 2019, 11, 31844–31850 CrossRef CAS PubMed
- C. Su, M. Acik, K. Takai, J. Lu, S. J. Hao, Y. Zheng, P. Wu, Q. Bao, T. Enoki, Y. J. Chabal and K. P. Loh, Probing the catalytic activity of porous graphene oxide and the origin of this behaviour, Nat. Commun., 2012, 3, 1298 CrossRef PubMed
- D. R. Dreyer, H. P. Jia and C. W. Bielawski, Graphene oxide: a convenient carbocatalyst for facilitating oxidation and hydration reactions, Angew. Chem., Int. Ed. Engl., 2010, 49, 6813–6816 CAS
- D. R. Dreyer, H. P. Jia, A. D. Todd, J. Geng and C. W. Bielawski, Graphite oxide: a selective and highly efficient oxidant of thiols and sulfides, Org. Biomol. Chem., 2011, 9, 7292–7295 RSC
- M. Bavadi and K. Niknam, Synthesis of functionalized dihydro-2-oxopyrroles using graphene oxide as heterogeneous catalyst, Mol. Diversity, 2018, 22, 561–573 CrossRef CAS PubMed
- K. B. Dhopte, R. S. Zambare, A. V. Patwardhan and P. R. Nemade, Role of Degree of Oxidation of Graphene Oxide on Biginelli Reaction Kinetics, ChemistrySelect, 2017, 2, 10997–11006 CrossRef CAS
- A. Gupta, R. Kaur, D. Singh and K. K. Kapoor, Graphene oxide: a carbocatalyst for the one-pot multicomponent synthesis of highly functionalized tetrahydropyridines, Tetrahedron Lett., 2017, 58, 2583–2587 CrossRef CAS
- A. Gupta, D. Kour, V. K. Gupta and K. K. Kapoor, Graphene oxide mediated solvent-free three component reaction for the synthesis of 1-amidoalkyl-2-naphthols and 1,2-dihydro-1-arylnaphth[1,2-e][1,3]oxazin-3-ones, Tetrahedron Lett., 2016, 57, 4869–4872 CrossRef CAS
- N. Kausar, P. Mukherjee and A. R. Das, Practical carbocatalysis by graphene oxide nanosheets in aqueous medium towards the synthesis of diversified dibenzo[1,4]diazepine scaffolds, RSC Adv., 2016, 6, 88904–88910 RSC
- S. Kundu and B. Basu, Graphene oxide (GO)-catalyzed multi-component reactions: green synthesis of library of pharmacophore 3-sulfenylimidazo[1,2-a]pyridines, RSC Adv., 2015, 5, 50178–50185 RSC
- A. Sengupta, C. Su, C. Bao, C. T. Nai and K. P. Loh, Graphene Oxide and Its Functionalized Derivatives as Carbocatalysts in the Multicomponent Strecker Reaction of Ketones, ChemCatChem, 2014, 6, 2507–2511 CrossRef CAS
- O. Mohammadi, M. Golestanzadeh and M. Abdouss, Recent advances in organic reactions catalyzed by graphene oxide and sulfonated graphene as heterogeneous nanocatalysts: a review, New J. Chem., 2017, 41, 11471–11497 RSC
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Carbocatalysis by graphene-based materials, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, Graphite Oxide as an Auto-Tandem Oxidation-Hydration-Aldol Coupling Catalyst, Adv. Synth. Catal., 2011, 353, 528–532 CrossRef CAS
- F. Zhang, H. Jiang, X. Wu, Z. Mao and H. Li, Organoamine-Functionalized Graphene Oxide as a Bifunctional Carbocatalyst with Remarkable Acceleration in a One-Pot Multistep Reaction, ACS Appl. Mater. Interfaces, 2015, 7, 1669–1677 CrossRef CAS PubMed
- G. Vitali Forconesi, L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Synthesis of Polyoxygenated Heterocycles by Diastereoselective Functionalization of a Bio-Based Chiral Aldehyde Exploiting the Passerini Reaction, Molecules, 2020, 25(14), 3227 CrossRef PubMed
- P. Capurro, L. Moni, A. Galatini, C. Mang and A. Basso, Multi-Gram Synthesis of Enantiopure 1,5-Disubstituted Tetrazoles Via Ugi-Azide 3-Component Reaction, Molecules, 2018, 23, 2758 CrossRef PubMed
- A. Pinna, A. Basso, C. Lambruschini, L. Moni, R. Riva, V. Rocca and L. Banfi, Stereodivergent access to all four stereoisomers of chiral tetrahydrobenzo[f][1,4]oxazepines, through highly diastereoselective multicomponent Ugi–Joullié reaction, RSC Adv., 2020, 10, 965–972 RSC
- F. Ibba, P. Capurro, S. Garbarino, M. Anselmo, L. Moni and A. Basso, Photoinduced Multicomponent Synthesis of alpha-Silyloxy Acrylamides, an Unexplored Class of Silyl Enol Ethers, Org. Lett., 2018, 20, 1098–1101 CrossRef CAS PubMed
- L. Moni, F. De Moliner, S. Garbarino, J. Saupe, C. Mang and A. Basso, Exploitation of the Ugi 5-Center-4-Component Reaction (U-5C-4CR) for the Generation of Diverse Libraries of Polycyclic (Spiro)Compounds, Front. Chem., 2018, 6, 369 CrossRef PubMed
- S. Caputo, L. Banfi, A. Basso, A. Galatini, L. Moni, R. Riva and C. Lambruschini, Diversity-Oriented Synthesis of Various Enantiopure Heterocycles by Coupling Organocatalysis with Multicomponent Reactions, Eur. J. Org. Chem., 2017, 2017, 6619–6628 CrossRef CAS
- C. Lambruschini, D. Galante, L. Moni, F. Ferraro, G. Gancia, R. Riva, A. Traverso, L. Banfi and C. D'Arrigo, Multicomponent, fragment-based synthesis of polyphenol-containing peptidomimetics and their inhibiting activity on beta-amyloid oligomerization, Org. Biomol. Chem., 2017, 15, 9331–9351 RSC
- M. Spallarossa, L. Banfi, A. Basso, L. Moni and R. Riva, Access to Polycyclic Alkaloid-Like Structures by Coupling the Passerini and Ugi Reactions with Two Sequential Metal-Catalyzed Cyclizations, Adv. Synth. Catal., 2016, 358, 2940–2948 CrossRef CAS
- L. Moni, L. Banfi, A. Basso, L. Carcone, M. Rasparini and R. Riva, Ugi and Passerini reactions of biocatalytically derived chiral aldehydes: application to the synthesis of bicyclic pyrrolidines and of antiviral agent telaprevir, J. Org. Chem., 2015, 80, 3411–3428 CrossRef CAS PubMed
- L. Moni, C. F. Gers-Panther, M. Anselmo, T. J. Muller and R. Riva, Highly Convergent Synthesis of Intensively Blue Emissive Furo[2,3-c]isoquinolines by a Palladium-Catalyzed Cyclization Cascade of Unsaturated Ugi Products, Chemistry, 2016, 22, 2020–2031 CrossRef CAS PubMed
- L. Moni, M. Denissen, G. Valentini, T. J. Muller and R. Riva, Diversity-oriented synthesis of intensively blue emissive 3-hydroxyisoquinolines by sequential Ugi four-component reaction/reductive Heck cyclization, Chemistry, 2015, 21, 753–762 CrossRef CAS PubMed
- A. Basso, L. Moni, L. Banfi and R. Riva, External-Oxidant-Based Multicomponent Reactions, Synthesis, 2016, 48, 4050–4059 CrossRef
- I. Muthukrishnan, V. Sridharan and J. C. Menendez, For recent review see: Progress in the Chemistry of Tetrahydroquinolines, Chem. Rev., 2019, 119, 5057–5191 CrossRef CAS PubMed
- J. B. Bharate, R. A. Vishwakarma and S. B. Bharate, Metal-free domino one-pot protocols for quinoline synthesis, RSC Adv., 2015, 5, 42020–42053 RSC
- G. A. Ramann and B. J. Cowen, Recent Advances in Metal-Free Quinoline Synthesis, Molecules, 2016, 21(8), 986 CrossRef PubMed
- V. V. Kouznetsov, Recent synthetic developments in a powerful imino Diels–Alder reaction (Povarov reaction): application to the synthesis of N-polyheterocycles and related alkaloids, Tetrahedron, 2009, 65, 2721–2750 CrossRef CAS
- B. Crousse, J.-P. Bégué and D. Bonnet-Delpon, Synthesis of 2-CF3-Tetrahydroquinoline and Quinoline Derivatives from CF3-N-Aryl-aldimine, J. Org. Chem., 2000, 65, 5009–5013 CrossRef CAS PubMed
- V. P. Zaitsev, N. M. Mikhailova, D. N. Orlova, E. V. Nikitina, E. V. Boltukhina and F. I. Zubkov, Synthesis and oxidation of 2-furyl-4-R-substituted and furo[3,2-c]-condensed 1,2,3,4-tetrahydro-1,10-phenanthrolines and quinolines, Chem. Heterocycl. Compd., 2009, 45, 308–316 CrossRef CAS
- V. Sridharan, C. Avendaño and J. Carlos Menéndez, Convenient, two-step synthesis of 2-styrylquinolines: an application of the CAN-catalyzed vinylogous type-II Povarov reaction, Tetrahedron, 2009, 65, 2087–2096 CrossRef CAS
- C. M. Meléndez Gómez, M. Marsiglia, R. Escarsena, E. del Olmo and V. V. Kouznetsov, Synthesis of 2,3-di(ω-hydroxyalkyl)quinolines from anilines and cyclic enols using sequential cycloaddition/aromatization reactions, Tetrahedron Lett., 2018, 59, 22–25 CrossRef
- R. Ramírez-Jiménez, M. Franco, E. Rodrigo, R. Sainz, R. Ferritto, A. M. Lamsabhi, J. L. Aceña and M. B. Cid, Unexpected reactivity of graphene oxide with DBU and DMF, J. Mater. Chem. A, 2018, 6, 12637–12646 RSC
- B. H. S. T. da Silva, N. L. Marana, A. C. Mafud and L. C. da Silva-Filho, A theoretical and experimental study to unequivocal structural assignment of tetrahydroquinoline derivatives, Struct. Chem., 2013, 25, 327–337 CrossRef
- The quantity of catalyst was decided based on the amount of Mn contained on GO (about 6000 ppm, see ESI for details†).
- B. Crousse, J. Legros, M. Ourévitch and D. Bonnet-Delpon, Facile Synthesis of Tetrahydroquinolines and Julolidines through Multicomponent Reaction, Synlett, 2006, 2006, 1899–1902 CrossRef
- A. Olmos, J. Sommer and P. Pale, Scandium(III) Zeolites as New Heterogeneous Catalysts: [4 + 2]Cyclocondensation of in situ Generated Aryl Imines with Alkenes, Chem. Eur. J., 2011, 17, 1907–1914 CrossRef CAS PubMed
- L. Lombardi, D. Bellini, A. Bottoni, M. Calvaresi, M. Monari, A. Kovtun, V. Palermo, M. Melucci and M. Bandini, Allylic and Allenylic Dearomatization of Indoles Promoted by Graphene Oxide by Covalent Grafting Activation Mode, Chem. Eur. J., 2020, 26, 10427–10432 CrossRef CAS PubMed
- V. D. Ebajo, C. R. L. Santos, G. V. Alea, Y. A. Lin and C.-H. Chen, Regenerable Acidity of Graphene Oxide in Promoting Multicomponent Organic Synthesis, Sci. Rep., 2019, 9, 15579 CrossRef PubMed
- L. B. Casabianca, M. A. Shaibat, W. W. Cai, S. Park, R. Piner, R. S. Ruoff and Y. Ishii, NMR-Based Structural Modeling of Graphite Oxide Using Multidimensional 13C Solid-State NMR and ab Initio Chemical Shift Calculations, J. Am. Chem. Soc., 2010, 132, 5672–5676 CrossRef CAS PubMed
- I. A. Vacchi, C. Spinato, J. Raya, A. Bianco and C. Ménard-Moyon, Chemical reactivity of graphene oxide towards amines elucidated by solid-state NMR, Nanoscale, 2016, 8, 13714–13721 RSC
- M. Leskes, R. S. Thakur, P. K. Madhu, N. D. Kurur and S. Vega, Bimodal Floquet description of heteronuclear dipolar decoupling in solid-state nuclear magnetic resonance, J. Chem. Phys., 2007, 127, 024501 CrossRef PubMed
- E. Vinogradov, P. K. Madhu and S. Vega, Proton spectroscopy in solid state nuclear magnetic resonance with windowed phase modulated Lee–Goldburg decoupling sequences, Chem. Phys. Lett., 2002, 354, 193–202 CrossRef CAS
- E. Vinogradov, P. K. Madhu and S. Vega, High-resolution proton solid-state NMR spectroscopy by phase-modulated Lee–Goldburg experiment, Chem. Phys. Lett., 1999, 314, 443–450 CrossRef CAS
- Y. Li, H. Chen, L. Y. Voo, J. Ji, G. Zhang, G. Zhang, F. Zhang and X. Fan, Synthesis of partially hydrogenated graphene and brominated graphene, J. Mater. Chem., 2012, 22, 15021–15024 RSC
- L. R. Domingo, M. J. Aurell, J. A. Sáez and S. M. Mekelleche, Understanding the mechanism of the Povarov reaction. A DFT study, RSC Adv., 2014, 4, 25268 RSC
- M. Ríos-Gutiérrez, H. Layeb and L. R. Domingo, A DFT study of the mechanism of Brønsted acid catalysed Povarov reactions, Tetrahedron, 2015, 71, 9339–9345 CrossRef
- Y. Huang, C. Qiu, Z. Li, W. Feng, H. Gan, J. Liu and K. Guo, Tritylium Cation as Low Loading Lewis Acidic Organocatalyst in Povarov Reactions, ACS Sustainable Chem. Eng., 2016, 4, 47–52 CrossRef CAS
- K. Spyrou, M. Calvaresi, E. K. Diamanti, T. Tsoufis, D. Gournis, P. Rudolf and F. Zerbetto, Graphite Oxide and Aromatic Amines: Size Matters, Adv. Funct. Mater., 2015, 25, 263–269 CrossRef CAS
- H.-P. Jia, D. R. Dreyer and C. W. Bielawski, C–H oxidation using graphite oxide, Tetrahedron, 2011, 67, 4431–4434 CrossRef CAS
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, Graphene Oxide: A Convenient Metal-Free Carbocatalyst for Facilitating Aerobic Oxidation of 5-Hydroxymethylfurfural into 2, 5-Diformylfuran, ACS Catal., 2015, 5, 5636–5646 CrossRef CAS
- J. Zhang, S. Chen, F. Chen, W. Xu, G.-J. Deng and H. Gong, Dehydrogenation of Nitrogen Heterocycles Using Graphene Oxide as a Versatile Metal-Free Catalyst under Air, Adv. Synth. Catal., 2017, 359, 2358–2363 CrossRef CAS
- E. Vicente-Garcia, F. Catti, R. Ramon and R. Lavilla, Unsaturated lactams: new inputs for povarov-type multicomponent reactions, Org. Lett., 2010, 12, 860–863 CrossRef CAS PubMed
- A. A. Kudale, J. Kendall, D. O. Miller, J. L. Collins and G. J. Bodwell, Povarov reactions involving 3-aminocoumarins: synthesis of 1,2,3,4-tetrahydropyrido[2,3-c]coumarins and pyrido[2,3-c]coumarins, J. Org. Chem., 2008, 73, 8437–8447 CrossRef CAS PubMed
- S. Chakrabarty, M. S. Croft, M. G. Marko and G. Moyna, Synthesis and evaluation as potential anticancer agents of novel tetracyclic indenoquinoline derivatives, Bioorg. Med. Chem., 2013, 21, 1143–1149 CrossRef CAS PubMed
- M. R. Jorshari, M. Mamaghani and P. Jahanshahi, Synthesis, delivery, and molecular docking of fused quinolines as inhibitor of Hepatitis A virus 3C proteinase, Sci. Rep., 2021, 11, 18970 CrossRef CAS PubMed
- M. C. Al-Matarneh, R.-M. Amărandi, I. I. Mangalagiu and R. Danac, Synthesis and Biological Screening of New Cyano-Substituted Pyrrole Fused (Iso)Quinoline Derivatives, Molecules, 2021, 26, 2066 CrossRef CAS PubMed
- R. Gattu, P. R. Bagdi, R. S. Basha and A. T. Khan, Camphorsulfonic Acid Catalyzed One-Pot Three-Component Reaction for the Synthesis of Fused Quinoline and Benzoquinoline Derivatives, J. Org. Chem., 2017, 82, 12416–12429 CrossRef CAS PubMed
- L. Moni, L. Banfi, A. Basso, A. Brambilla and R. Riva, Diversity-oriented synthesis of dihydrobenzoxazepinones by coupling the Ugi multicomponent reaction with a Mitsunobu cyclization, Beilstein J. Org. Chem., 2014, 10, 209–212 CrossRef PubMed
- W. Gao, C. Nie, L. Xu, B. Zhao and Y. Li, Synthesis of novel oxepine-containing polycyclic-fused quinoline systems, Heterocycl. Commun., 2014, 20(3), 161 CAS
- P. Langer, S. Rotzoll and H. Görls, Regioselective Synthesis of Oxepin- and Oxocin-Annulated Quinolines by Combined ‘Claisen-Rearrangement/Olefin-Metathesis' Reactions, Synthesis, 2008, 45–52 CrossRef
- K. Mahesh, K. Ravi, P. K. Rathod and P. Leelavathi, Convenient synthesis of quinoline-fused triazolo-azepine/oxepine derivatives through Pd-catalyzed C–H functionalisation of triazoles, New J. Chem., 2020, 44, 2367–2373 RSC
- Y. Li, G. Lin and W. Gao, Synthesis of Polycyclic-fused Heterocycles Combining Both Quinoline Ring and Benzoxepine Framework in A Single Molecule, Polycyclic Aromat. Compd., 2012, 32, 469–486 CrossRef CAS
- A. Kovtun, D. Jones, S. Dell'Elce, E. Treossi, A. Liscio and V. Palermo, Accurate chemical analysis of oxygenated graphene-based materials using X-ray photoelectron spectroscopy, Carbon, 2019, 143, 268–275 CrossRef CAS
- F. Bruno, R. Francischello, G. Bellomo, L. Gigli, A. Flori, L. Menichetti, L. Tenori, C. Luchinat and E. Ravera, Multivariate Curve Resolution for 2D Solid-State NMR spectra, Anal. Chem., 2020, 92, 4451–4458 CrossRef CAS PubMed
- A. M. Panich, A. I. Shames and N. A. Sergeev, Paramagnetic Impurities in Graphene Oxide, Appl. Magn. Reson., 2012, 44, 107–116 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01752k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |