
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStepwise copolymerization of polybenzimidazole for a low dielectric constant and ultrahigh heat resistance†
Xianzhu Zhong a,
Aniruddha Nagac,
Jiabei Zhoua,
Kenji Takadaa,
Fitri Adila Amat Yusofb,
Tetsu Mitsumatab,
Kenji Oqmhulaa,
Kenta Hongoa,
Ryo Maezonoa and
Tatsuo Kaneko*a
a,
Aniruddha Nagac,
Jiabei Zhoua,
Kenji Takadaa,
Fitri Adila Amat Yusofb,
Tetsu Mitsumatab,
Kenji Oqmhulaa,
Kenta Hongoa,
Ryo Maezonoa and
Tatsuo Kaneko*a
aGraduate School of Advanced Science and Technology, Energy and Environment Area, Japan Advanced Institute of Science and Technology (JAIST), 1-1 Asahidai, Nomi, Ishikawa 923-1292, Japan. E-mail: kaneko@jaist.ac.jp
bDepartment of Materials Science & Technology, Faculty of Engineering, Niigata University, Ikarashi, Nishi-ku, Niigata, 950-2181, Japan
cSchool of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
First published on 19th April 2022
Abstract
Bio-based polymer materials having great potential due to the depletion of fossil-fuel resources have been applied as single-use and medicinal materials but their low thermomechanical resistance have limited wider applications. Here, ultrahigh thermoresistant bio-based terpolymers with a low dielectric constant, comprising polybenzimidazole and poly(benzoxazole-random-aramid), were prepared by a method involving stepwise polycondensation of three monomers, 3,4-diaminobenzoic acid for benzimidazoles, 3-amino-4-hydroxylbenzoic acid for benzoxazoles, and 4-aminobenzoic acid for aramids. For optimized monomer compositions, the obtained terpolymers exhibited dielectric constants lower than 3, and a 10% mass loss at approximately 760 °C which is a temperature higher than that for any other polymer material reported so far. The high thermal degradation temperatures of the prepared terpolymers were a result of the high interaction enthalpies of hydrogen bonding between imidazole rings in the polymer chains, which were obtained from density functional theory calculations using trimer models. Furthermore, the applicability of the prepared terpolymers as a wire-coating material for a simple motor insulation was demonstrated, indicating that it has significant potential to be used as a thermostable material with a low dielectric constant (k).
1. Introduction
Bio-based polymer materials developed from natural resources are sustainable and reliable, and therefore an ideal alternative to petroleum-based polymer materials.1–5 Bio-based materials, such as poly(lactic acid) (PLA) and polyhydroxybutyrates (PHB), have been developed and used for various applications.3,6,7 However, as most bio-based polymer materials have a low thermal resistance and poor electric function, they cannot be applied as engineering plastics that need to function in extreme conditions, such as in aerospace and electric vehicles, and high-power engines.8–10Bio-based polybenzimidazole (PBI), which exhibits excellent thermal resistance, has been previously synthesized from 3,4-diaminobenzoic acid (DABA) via the Smiles rearrangement of the hydroxyl group of fermented 3-amino-4-hydroxybenzoic acid (AHBA).11,12 In addition, poly(benzimidazole-co-amide) (PBI-co-PA) was synthesized by introducing amide groups to increase the processability of the polymer. Interestingly, it was found that the thermal decomposition temperature of PBI-co-PA increased with the incorporation of a small amount of PA (up to 15 mol%), in spite of the low degradation temperature of the PA units. Another PBI copolymer, poly(benzimidazole-co-benzoxazole) (PBI-co-PBO) was synthesized through the copolymerization of DABA with AHBA. This copolymer exhibited excellent low dielectric constant (low-k) performance; however, its thermal stability and processability were not optimized.13 Therefore, the development of a PBI derivative as a processable polymer material with outstanding thermal resistance and low-k performance remains a significant challenge.9,14–17
In this work, we design a series of PBI derivatives possessing improved thermal, mechanical and dielectric properties, by incorporating aramid and benzoxazole components into PBI. Among these derivatives, a terpolymer of appropriate composition exhibits a significant low k, as well as an ultrahigh thermodegradation temperature that is much higher than those of conventional polymers, including PBI and PBO.
2. Experimental section
2.1. Materials
AHBA (purity: >97%), DABA (purity: 98%), 4-aminobenzoic acid (PABA) (purity: >99%), and sodium hydroxide (NaOH) (purity: >98%) were purchased from TCI (Tokyo Chemical Industry). Poly(phosphoric acid) (PPA) (purity: 85%) was obtained from Sigma-Aldrich. Methane sulfonic acid (MSA) (purity: >98%) and trifluoroacetic acid (TFA) (purity: >98%) were supplied by Wako pure chemical Industries, Ltd. pH test paper, supplied by (Macherey-Nagel GmbH & Co. KG, Düren, Germany). Copper wire were purchased from SENKO Co., Ltd. All the solvents and reagents in this research were used as received without any further processing or purification.2.2. Syntheses
2.3. Measurements
Fourier transform infrared (FT-IR) spectra of terpolymers were recorded in a PerkinElmer Spectrum with a diamond-attenuated total reflection (ATR) accessory. The wavenumber range was set as 4000 to 400 cm−1. Solid-state 13C NMR CP/TOSS (Total Suppression of Spinning Sidebands) spectra of the terpolymer were recorded with a Bruker Advance III spectrometer operating at 125 MHz. Terpolymer samples were filled into 7 mm diameter zirconia rotor with a Kel-F cap and then spun at 8 kHz. The contact time and the period between successive accumulations were set as 2 s and 5 s respectively. The total number of the scans was set as 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
The viscosity of terpolymer was measured through a viscometer SIBATA 026300-3. Conc. sulfuric acid (H2SO4) was used as a solvent. Thermo-gravimetric analysis (TGA) curves of the terpolymers were recorded using a HITACHI STA7200. In a platinum crucible, the samples (5 mg) were placed and heated under a nitrogen atmosphere to 1000 °C with a heating rate of 10 °C min−1. The 5% mass loss temperature (Td5) and 10% mass loss temperature (Td10) of the samples were taken as indices of the thermal decomposition temperatures.
The stress–stain curves of the terpolymers were recorded via tensile mode mechanical tests using an Instron-3365 mechanical tester instrument at room temperature. Samples were shaped into a rectangular film with a length of 40 mm, width of 7 mm and a thickness of 15 μm. The elongation speed was set as 0.4 mm min−1.
The electrical resistivity of the terpolymer films was measured using a digital megohmmeter (DSM-8104 HIOKI) at 25 °C. External electromagnetic noise was shielded by a Faraday cage during the measurement. Through conductive rubber electrodes with a guard electrode, two terminal methods were adopted to apply 1 kV DC electric voltage to the film which had dimensions of 40 × 40 mm. The formula ρv = (S/t) × Rm was used to calculate the volume resistivity ρv (Ω cm), where S is the area of the electrode, t represents the thickness of the film.
The crystallinity was investigated using an X-ray diffractometer (SmartLab; Rigaku Corp., Akishima, Japan). Wide-angle X-ray diffraction (WAXD) patterns was checked from the facade of the film with a graphite-monochromatized Cu Kα radiation beam generated at 100 mA and 40 kV.
The measurement of dielectric constant of the terpolymer films were carried out in an LCR meter (HIOKI IM3536) at 20 °C with a frequency of 1 MHz. The measurement was carried out by two terminals method with an applied electric potential of 1.0 V. The film was sandwiched between two electrodes with a constant pressure of 86 kPa and the sample holder was set in a Faraday cage. The relative dielectric constant (kr) was calculated via the formula of kr = Cd/k0A, where C is the capacitance, d is thickness of film, A is the cross-sectional area of film and k0 is the dielectric constant in vacuum (=8.854 × 10−12 F m−1).
The coating experiment was performed using the copper wire with a diameter of 0.6 mm. The assembled mini motor is powered by 2 batteries of 1.5 V. In the experiment of heat resistance of PBI–PBO–PA coating, two copper wires coated with terpolymer were connected with a sample holder made of strainless-steel in a parallel circuit. An electrical potential of 10.0 V was applied to the circuit using a dc power supply (PWR200MH KIKUSUI). Both copper wires were approximately 8.0 cm long, therefore the electric current flowed could be same for both wires. The electric current flowed in the main circuit was measured to be 16.0 A.
2.4. Theoretical calculations
To understand thermal stability of the terpolymer, we made several trimer models and evaluated their interaction enthalpies of the H-bonds between molecular chains theoretically, following our previous study:13 All the DFT simulations were carried out using Gaussian 16.18 To properly describe the charge transfer relevant to the H-bonds, CAM-B3LYP was chosen as the exchange-correlation functional,19 associating with the cc-pVQZ basis set.20 A basis-set superposition error was reduced by applying the counterpoise correction to optimize each of the geometries.213. Results and dicussions
3.1. Syntheses
Terpolymers were synthesized in bulk by a one-pot polymerization method. Although the polymerization mixture became viscous and the reaction appeared successful, casting of the resulting polymers did not produce self-standing films, as can be seen from the photograph in the inset of Scheme 1. If the macromolecular backbone is too rigid, processing becomes difficult in some cases. Considering the good processability of PBI, as shown in previous reports,9,13 it was hypothesized that the continuous structure of benzimidazole units might be effective for film preparation.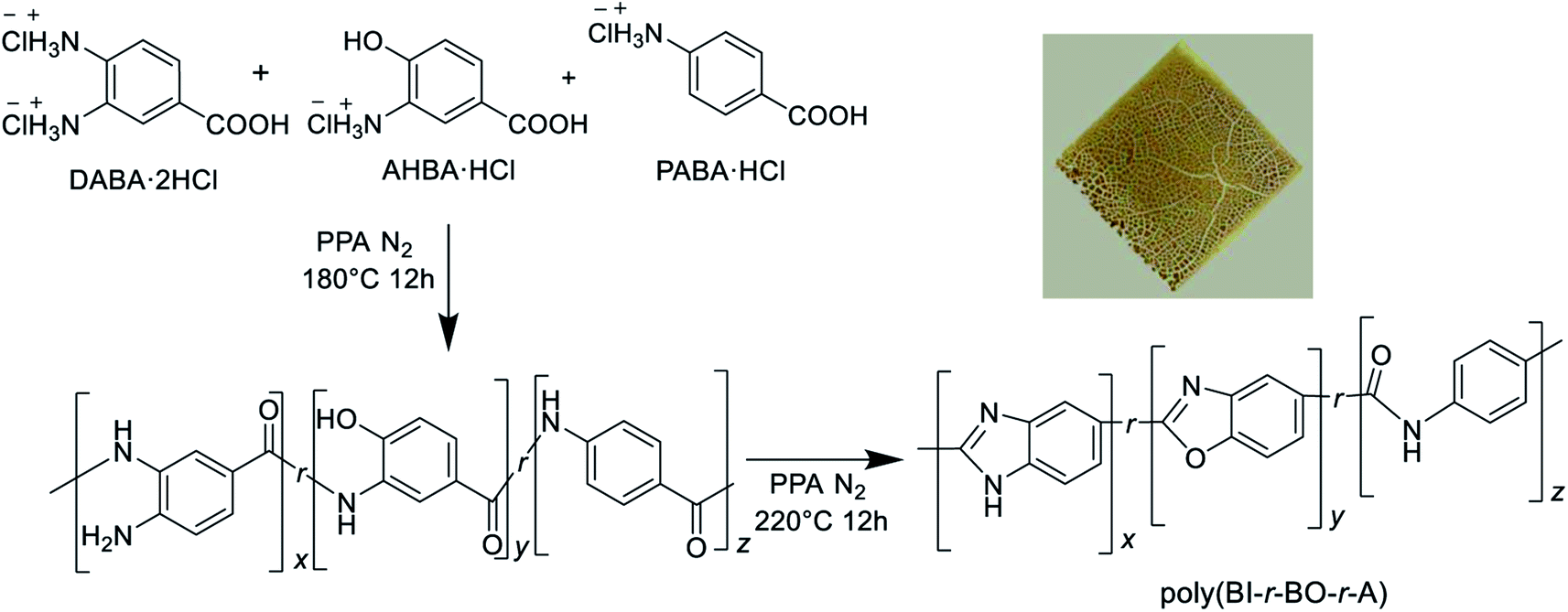 | ||
| Scheme 1 Synthetic pathway of terpolymers from three aminobenzoic acid derivatives through simple polymerization. Inset: picture of cast film. | ||
Therefore, a stepwise method was adopted for the synthesis; at first, the polybenzimidazoles were synthesized for short-range polymerization; then, the monomers for PBO and PA were added and successively polymerized to produce poly(BI-b-BO-A), as shown in Scheme 2.
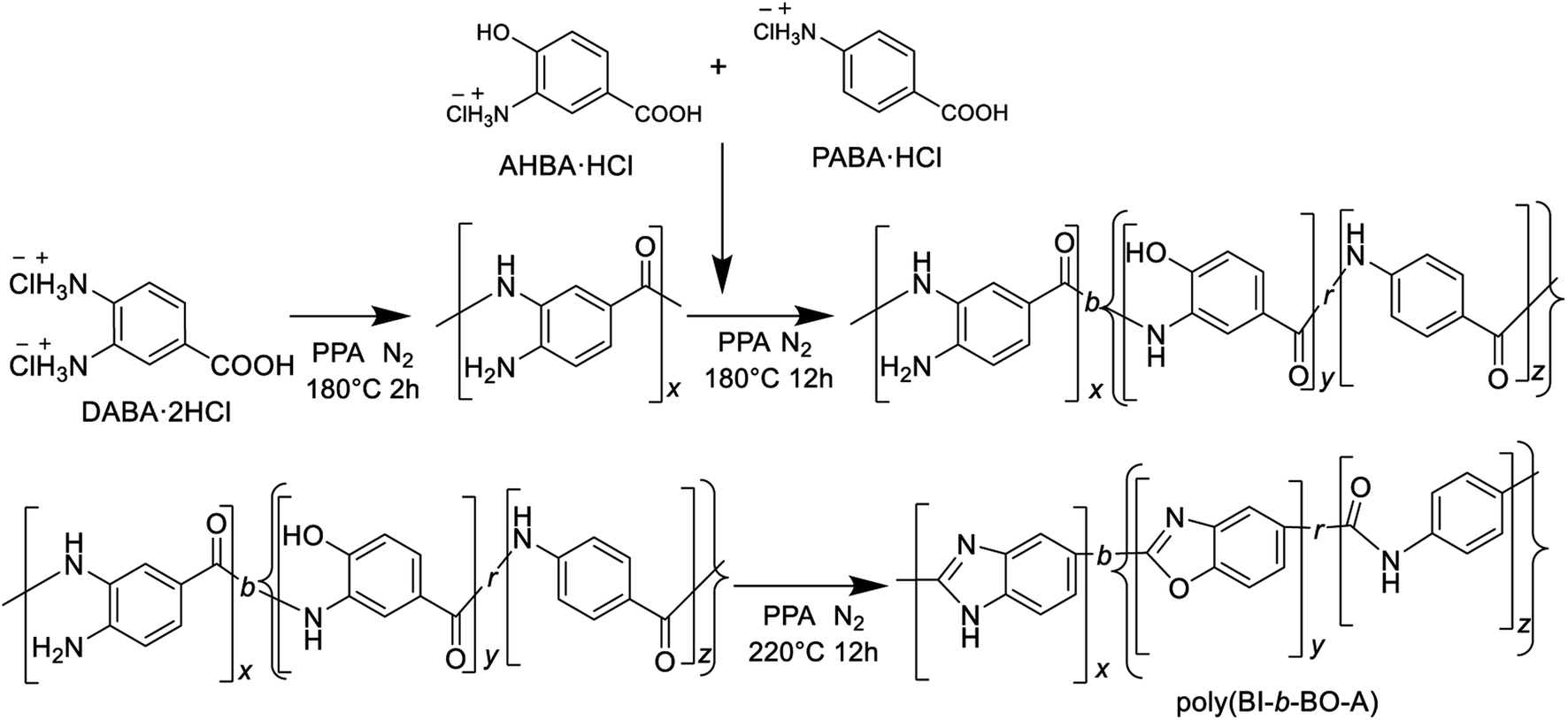 | ||
| Scheme 2 Synthetic pathway of P(BI-b-BO-A) from three aminobenzoic acid derivatives through stepwise polymerization. | ||
The self-standing film was successfully fabricated, and the film was further dried at 80 °C for 12 h (Fig. 1a). The film was pliable enough to be folded into the shape of an origami airplane (Fig. 1b); after unfolding, it did not tear or break (Fig. 1c).
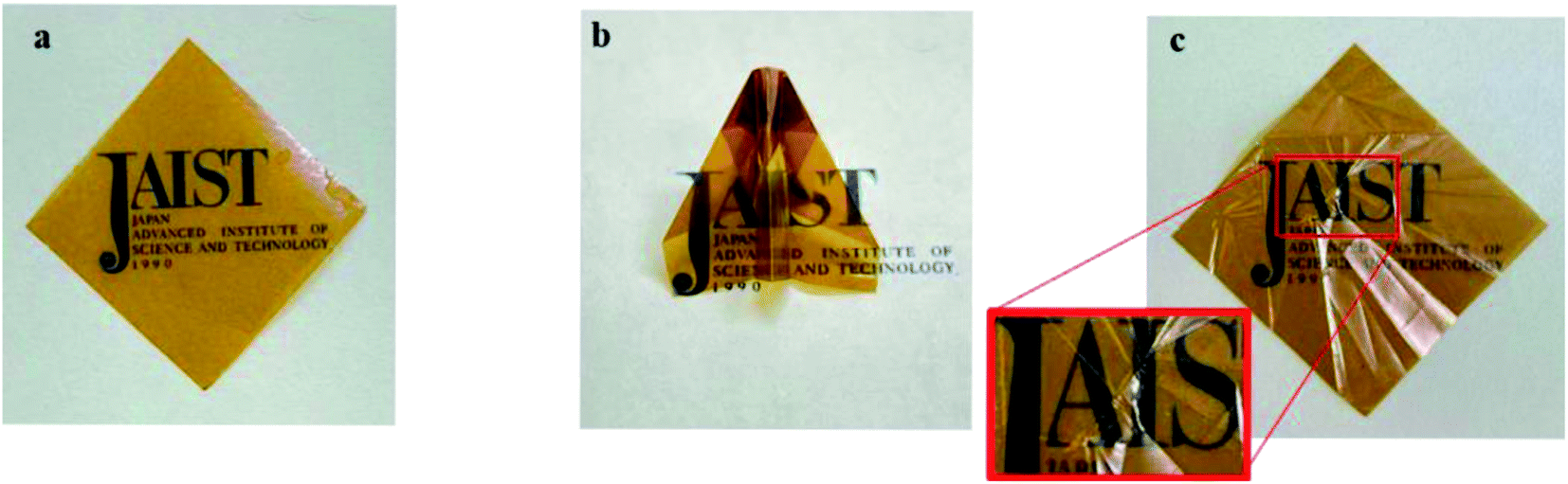 | ||
| Fig. 1 Images of the solution casted film of terpolymer. (a) Pristine, (b) origami-folded, and (c) unfolded films. | ||
The structures of the polymers were confirmed by Fourier Transform-Infrared (FT-IR) and 13C solid-state Nuclear Magnetic Resonance (NMR) spectroscopies. In the FT-IR spectra (Fig. S1†), for terpolymer in all the ratios, distinct C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC, and C–N absorption peaks were observed at approximately 1690 cm−1, 1590 cm−1 and 1380 cm−1 respectively. Besides, the CO absorption peaks gradually showed at approximately 1745 cm−1 and became stronger as PA ratio increasing from 0 to 30%, but the C–O peaks became weaker and vanished ultimately because of the decreasing composition of PBO. In the solid-state NMR spectrum (Fig. 2), six distinct signals in the range of 100 to 180 ppm and some very broad signals between 190 and 220 ppm were detected. The signal at 167 ppm was assigned to oxazole carbon (marked as 5), and the signal at 160 ppm contained the imidazole carbon (marked as 1) among others. Very broad signals in the range of 190 to 220 ppm were assigned to amide carbonyl carbons that connect BI and BO to A. The other signals were assigned to the benzene carbons. The close resemblance between the solid-state NMR spectra of the polymers synthesized via one-pot and stepwise polymerization, suggested that they had analogous structures.
N, CC, and C–N absorption peaks were observed at approximately 1690 cm−1, 1590 cm−1 and 1380 cm−1 respectively. Besides, the CO absorption peaks gradually showed at approximately 1745 cm−1 and became stronger as PA ratio increasing from 0 to 30%, but the C–O peaks became weaker and vanished ultimately because of the decreasing composition of PBO. In the solid-state NMR spectrum (Fig. 2), six distinct signals in the range of 100 to 180 ppm and some very broad signals between 190 and 220 ppm were detected. The signal at 167 ppm was assigned to oxazole carbon (marked as 5), and the signal at 160 ppm contained the imidazole carbon (marked as 1) among others. Very broad signals in the range of 190 to 220 ppm were assigned to amide carbonyl carbons that connect BI and BO to A. The other signals were assigned to the benzene carbons. The close resemblance between the solid-state NMR spectra of the polymers synthesized via one-pot and stepwise polymerization, suggested that they had analogous structures.
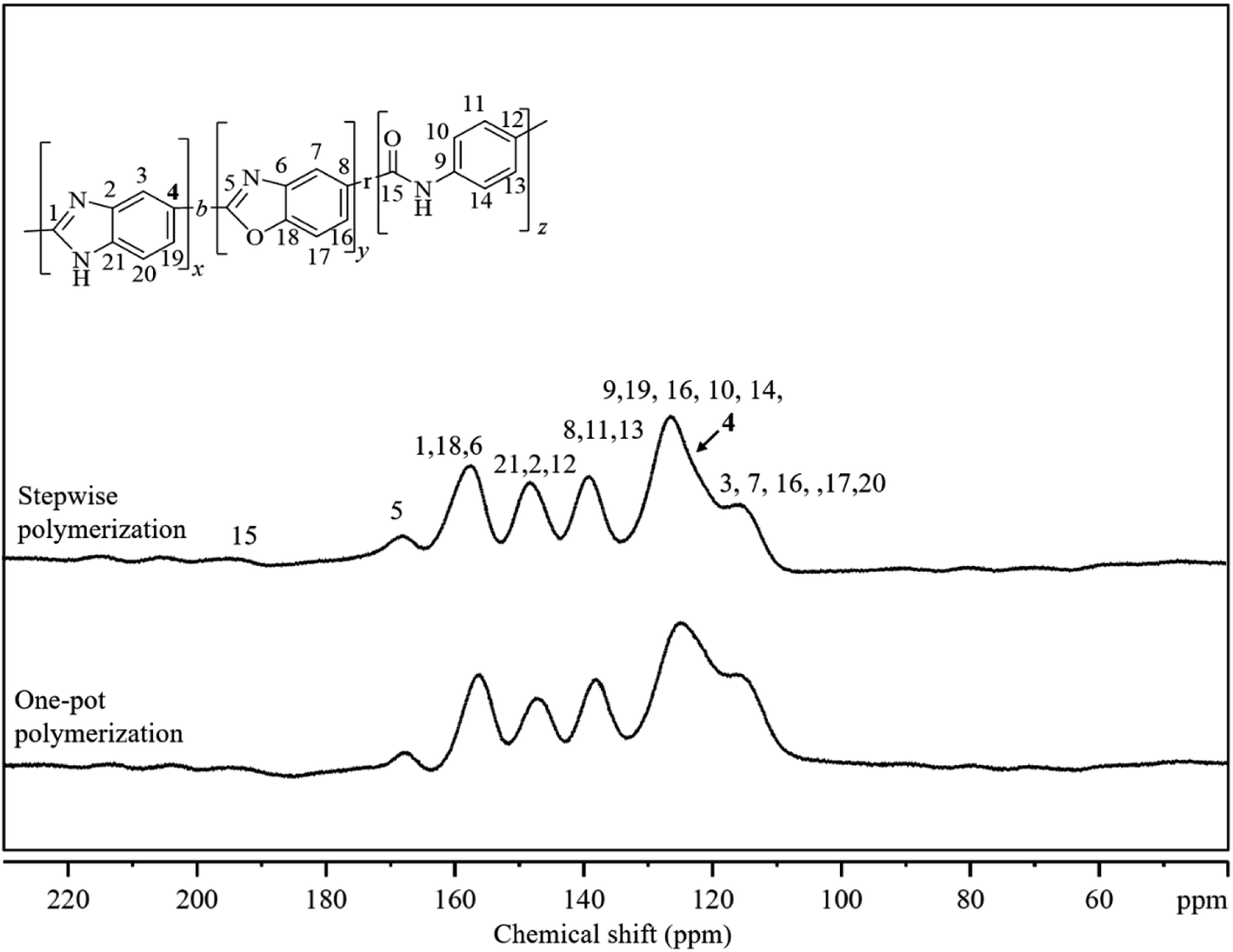 | ||
| Fig. 2 Solid-state 13C CP/TOSS NMR spectra of terpolymers in 70–21–9 (mol%) prepared by a one-pot polymerization and a stepwise polymerization method. | ||
The NMR signal from the carbon marked as 4 (C4), is expected to be sensitive to the presence of neighboring units such as BI, BO, and A. Therefore, the broadness of this signal for the polymer prepared by simple polymerization can be attributed to the presence of all the three neighboring units around C4. In contrast, the sharper C4 signal detected for the polymer prepared by stepwise polymerization can be explained by the PBI block formation.
The molecular weight of the polymers could not be evaluated by chromatography owing to their poor solubility in popular solvents. However, the inherent viscosities (ηinh) of the polymers were evaluated in a solution of concentrated sulfuric acid to compare their degrees of polymerization, as described in Table 1.
| Polymera composition (%) | Td5b (°C) | Td10b (°C) | σc (MPa) | γc (%) | Ec (GPa) | ηinh1 (ηinh2)d (dL g−1) |
|---|---|---|---|---|---|---|
| a Terpolymers in varying compositions of PBO and PA, in which PBO varied from 30% to 0 while PA composition varied from 0 to 30%. PBI composition were fixed at 70%.b Thermal property indices, measured by TGA at nitrogen atmosphere. 5% and 10% weight loss thermal decomposition temperatures (Td5 and Td10).c Mechanical properties σ, γ, and E measured by stress–strain tensile test refer to tensile strength at break, strain at beak, and Young's modulus, respectively.d ηinh1 refers to inherent viscosity of terpolymer synthesized by stepwise polymerization, while ηinh2 shown in parentheses refers to inherent viscosity of terpolymer synthesized via one-pot polymerization. | ||||||
| 70–30–0 | 638 | 703 | 80 ± 1.5 | 2.3 ± 0.1 | 3.48 | 1.89 (0.84) |
| 70–27–3 | 634 | 701 | 76 ± 3.2 | 3.2 ± 0.2 | 2.38 | 1.75 (0.78) |
| 70–21–9 | 624 | 707 | 76 ± 1.0 | 5.1 ± 0.1 | 1.49 | 1.65 (0.77) |
| 70–15–15 | 637 | 716 | 74 ± 1.4 | 6.7 ± 0.1 | 1.23 | 1.38 (0.78) |
| 70–9–21 | 658 | 736 | 72 ± 1.2 | 8.6 ± 0.1 | 0.84 | 1.30 (0.75) |
| 70–3–27 | 675 | 758 | 63 ± 1.7 | 10.3 ± 0.1 | 0.61 | 1.28 (0.74) |
| 70–0–30 | 686 | 763 | 48 ± 1.6 | 12.0 ± 0.2 | 0.40 | 1.24 (0.64) |
The inherent viscosities of terpolymers synthesized by stepwise polymerization (ηinh1) ranged from 1.24 to 1.89 dL g−1, which are much higher than those of the terpolymers synthesized by simple polymerization (ηinh2: 0.64–0.84 dL g−1). DABA possesses a high polymerizability owing to the reactive functional groups of o-diamine. The higher values of ηinh1 compared to ηinh2 are responsible for the formation of the pliable film from P(BI-b-BO-A) (Fig. 1). In addition, ηinh decreases with a decrease in the amount of PBO (and increase in PA) for both types of terpolymers. This can be attributed to the higher amine reactivity due to the presence of the o-hydroxy groups.
3.2. Thermal properties
The thermal stabilities of the terpolymers synthesized by stepwise polymerization were estimated using thermogravimetric analysis (TGA) under a nitrogen atmosphere. According to the thermograms obtained (Fig. 3a), none of the terpolymers were found to exhibit any significant loss of mass up to ∼520 °C; mass loss started occurring at approximately 550 °C. Significant mass losses were observed with an increase in temperatures up to 1000 °C. Interestingly, the completion of thermal degradation was not detected below 1000 °C, and the residue yields were still higher than 69 wt%. The thermal decomposition temperatures Td5 and Td10 for P(BI-b-BO-A) of all compositions have been listed in Table 1 and plotted against the PA compositions in Fig. 3b. Both Td5 and Td10 were almost constant for low mole percentages of PA. However, beyond a critical PA composition of 9 mol%, they gradually increased with increasing mol% of PA. Terpolymers having 30 mol% of PA showed the highest Td5 and Td10 values of 686 °C and 763 °C, respectively. This can be attributed to the occurrence of inter-chain hydrogen bonds between the imidazoles and primary amines, which increase in number with the increase in relative mol% of PA compared to benzoxazoles. Overall, the terpolymers were found to exhibit ultrahigh thermal stability, and a Td10 of ∼760 °C, which is the highest value reported so far for plastic films, and is comparable to the melting points of light metals such as aluminum and magnesium.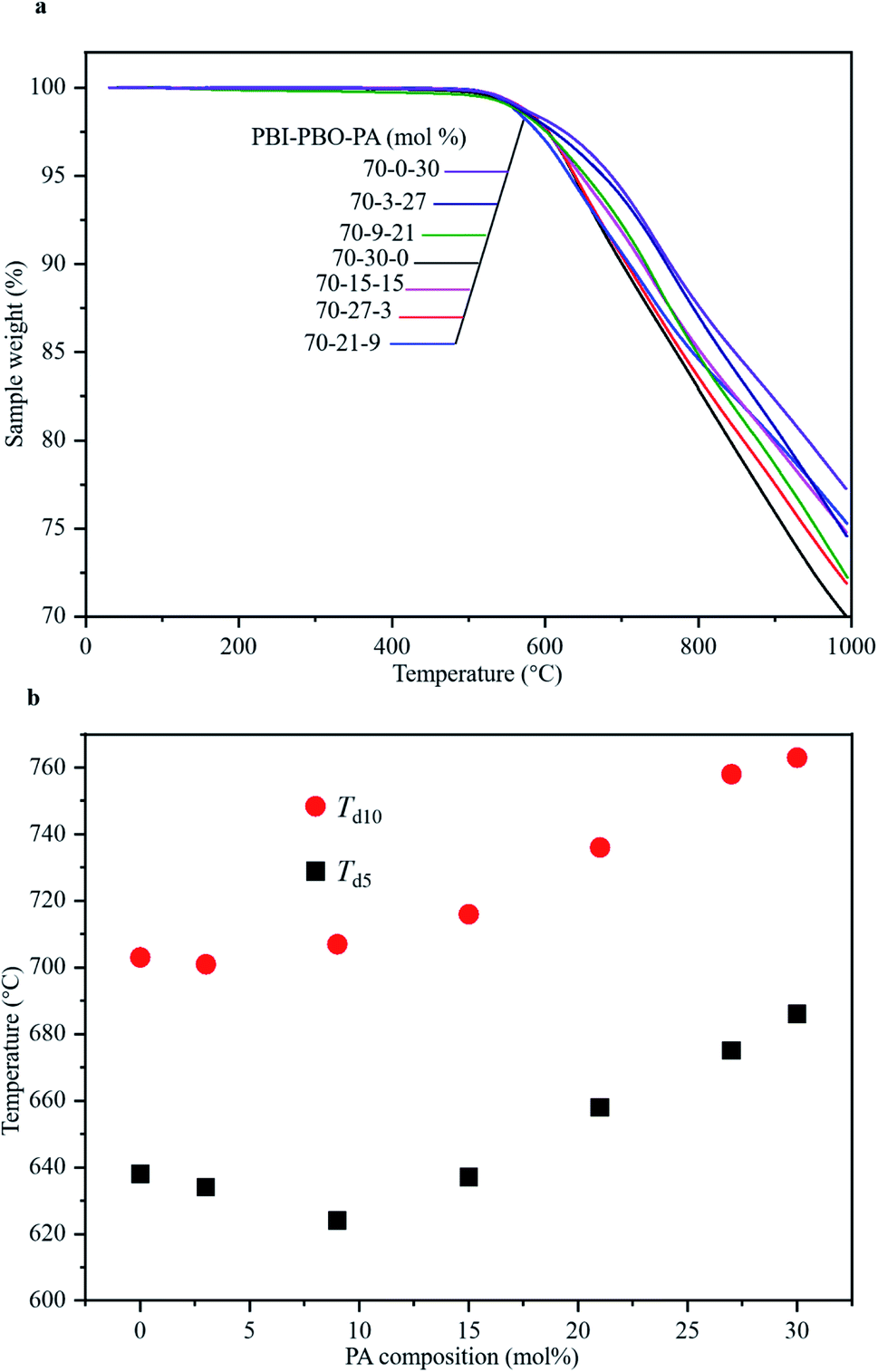 | ||
| Fig. 3 Thermal characterization of terpolymers. (a) TGA curves of terpolymers with various compositions. (b) Td5 and Td10 of terpolymers of different compositions. | ||
The burning characteristics of the terpolymers were investigated using a combustion test, in which a terpolymer film with a composition of 70%–21%–9% was taken as the representative terpolymer and a polyethylene film was used as the reference. A visual of the entire burning process can be found in Video S1.† Unlike that for the polyethylene film, no obvious flame was visible during the burning of the terpolymer film (Fig. 4). In addition, the ignition of the terpolymer film was quenched immediately after removal from the fire, and no dripping of the material was observed during the entire test, suggesting that the terpolymer film is extremely flame-retardant.
 | ||
| Fig. 4 Burning photographs depicting the burning terpolymer (left) and polyethylene (right). | ||
To elucidate the mechanism of thermal stability of the terpolymer, the interaction enthalpy of the hydrogen bonds (H-bonds) between its molecular chains was investigated via density functional theory (DFT) calculations. Several trimer models comprising three units of 2-phenylbenzimidazole (2BI), 2-phenylbenzoxazole (2BO), and benzanilide (BA) were used to simulate the polymer chains. Since PBI had the highest composition (70 mol%) in our terpolymers, the interaction enthalpy of the hydrogen bonding between imidazole N–H (model 1) and imidazole N (model 2) were calculated in detail. The results have been presented in Table 2. The interaction enthalpy for H-bonding between the two BI primary models was calculated to be −13.32 kcal mol−1, which was higher than that calculated for any other combination of BI, BO and BA primary models (entries 1–5). The results indicate that the H-bonding between two imidazole rings is the strongest of the five combinations. Next, the models were extended to trimers with BI as the central unit. In a previous work from our group, the H-bonding between two models of BI-BI-BI was found to be very weak (with an interaction enthalpy of −4.77 kcal mol−1), presumably due to the lowered reactivity of both N–H and –N by resonance stabilization in the long π-conjugation (entry 6).13 However, the incorporation of BA in either one of the two trimer models was found to increase the H-bond interaction enthalpies (entries 7-10). In the present study, it was observed that the H-bonding was strengthened by the incorporation of BO and BA on both sides of the central BI. The interaction enthalpy between the two trimer models of BA-BI-BA (entry 11) was estimated to be −14.13 kcal mol−1, which is slightly higher than that of entry 1, owing to the limitation of resonance stabilization, and the possible induction effects due to the substitution with BA. For BO-BI-BO and BO-BI-BA, the H-bonding enthalpies were calculated to be −13.57 kcal mol−1 and −13.80 kcal mol−1, respectively. The absolute values of the H-bonding interaction enthalpies were found to increase with increase in the amount of incorporated PA, indicating an elevation in the interaction enthalpy of the H-bond due to the incorporation of PA into PBI chain, thereby increasing the thermal stability.
| Entry | Monomer 1 (H-donor) | Monomer 2 (H-acceptor) | Interaction enthalpy (kcal mol−1) |
|---|---|---|---|
| a H-bonding enthalpy calculated via DFT theory.b H-bonding enthalpy calculated via DFT theory in previous work. | |||
| 1b | BI | BI | −13.32 |
| 2b | BI | BO | −9.23 |
| 3b | BI | BA | −10.24 |
| 4b | BA | BO | −9.23 |
| 5b | BA | BA | −6.85 |
| 6a | BI-BI-BI | BI-BI-BI | −4.77 |
| 7a | BA-BI-BI | BI-BI-BI | −10.58 |
| 8a | BI-BI-BI | BA-BI-BI | −10.01 |
| 9a | BI-BI-BA | BI-BI-BI | −12.46 |
| 10a | BI-BI-BI | BI-BI-BA | −12.68 |
| 11b | BA-BI-BA | BA-BI-BA | −14.13 |
| 12b | BO-BI-BO | BO-BI-BO | −13.57 |
| 13b | BO-BI-BA | BO-BI-BA | −13.80 |
3.3. Mechanical properties
The mechanical properties of the terpolymer films synthesized by stepwise polymerization were investigated using the stress–strain analysis (Fig. 5a). The tensile strength at break (σ), strain at break (ε), and Young's modulus (E) were evaluated therefrom. The stress–strain curves showed a consistent increase of stress with strain up until the breakage point, corresponding to the maximum permissible strain (strain at break or ε, Table 1). The values of the mechanical properties showed regular variations with the molar compositions of PA (Fig. 5b). With the increase in mol% of PA, the value of strain at break increased from 2.3 to 12.0%, while the tensile strength decreased from 80 to 48 MPa, and the Young's modulus decreased from 3.48 to 0.40 GPa. The terpolymers with higher mol% of PA exhibited higher ε and lower E values, presumably due to their greater abundance of non-heterocyclic PA components that are expected to be more flexible than the heterocyclic PBO components.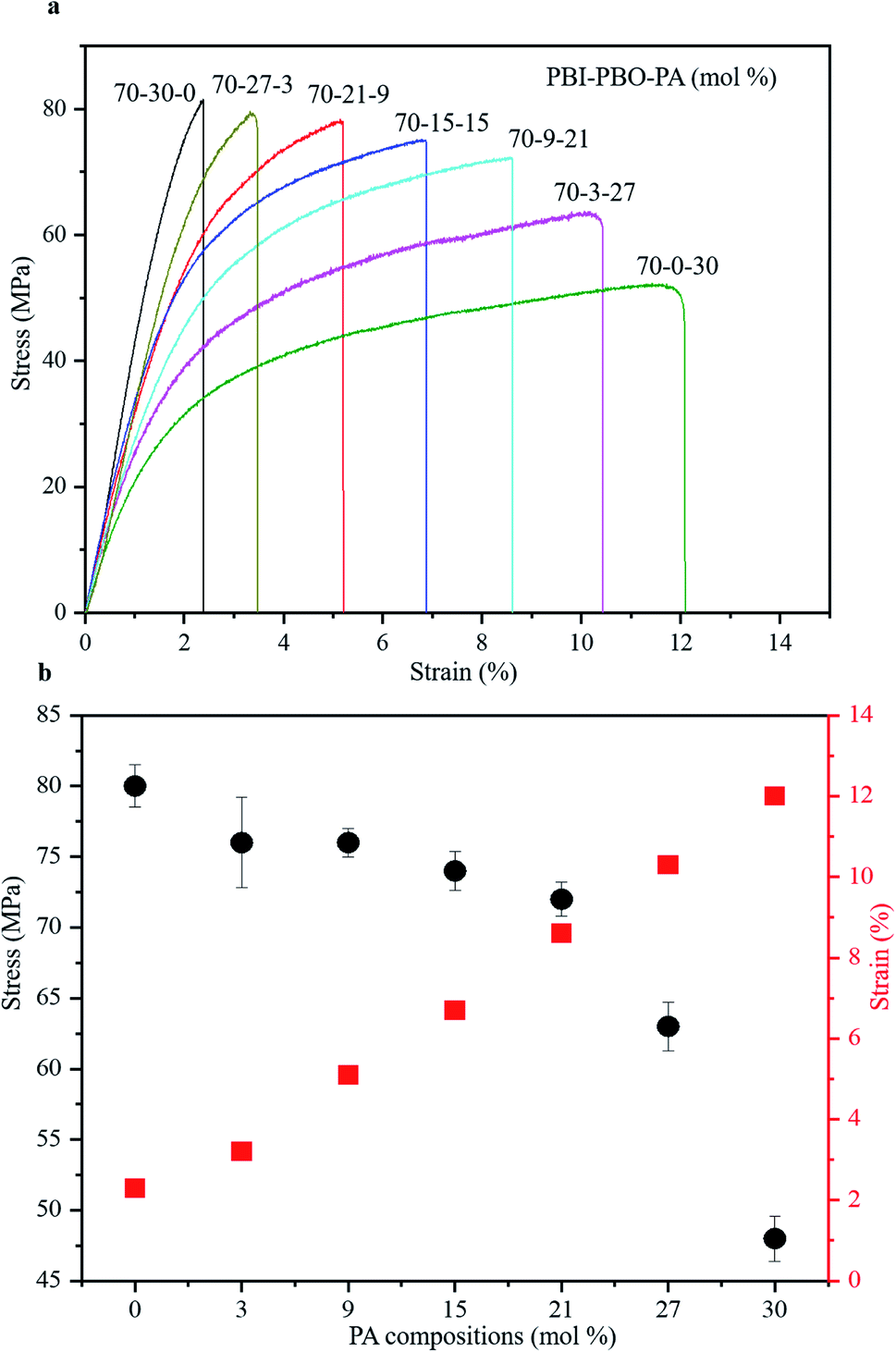 | ||
| Fig. 5 Mechanical properties of terpolymers. (a) Stress–strain curves of terpolymers. (b) Tensile stress and strain values in different PA compositions. | ||
3.4. Dielectric properties
Dielectric measurements were performed using the films obtained from P(BI-b-BO-A) synthesized by stepwise polymerization. The thicknesses of the terpolymer films were maintained between 7.0 to 10.0 μm for the dielectric measurements. The values of the different parameters obtained from these measurements, for the terpolymer films of different compositions, have been listed in Table 3. Fig. S2a† shows the volume resistivity for dried P(BI-b-BO-A) films as a function of PA composition. The volume resistivity for terpolymer with a PA mole composition (rPA) of 3% was equal to that for P(BI-b-BO) (rPA = 0) at approximately 6.22 × 1011 Ω cm and it significantly increased to 1.24 × 1014 Ω cm at rPA = 9%, suggesting that the terpolymer changed to highly insulative. However, it suddenly decreased to 2.67 × 1010 Ω cm at rPA = 15%, showing that the terpolymer transforms to a low insulator where the resistivity is more than an order of magnitude lower than that of P(BI-b-BO) (rPA = 0). At a region above 15%, the volume resistivity was independent of the PA composition. This drastic change in volume resistivity in the vicinity of rPA = 10% is considered to be a structural transition with nano- or submicron- scale although there was no remarkable change in the appearance or transparency of the films. The structural transition was confirmed through X-ray diffraction in Fig. S3 and S4.† In electrically heterogeneous materials, even if morphologically uniform, the bulk electrical resistivity is strongly dominated by the local resistivity. Therefore, it can be considered that the low resistivity at rPA > 15% was caused by the occurrence of a continuous phase with low resistivity having a wide distribution of resistance in a highly insulative matrix.22,23 On the contrary, it can be considered that the significant increase in the resistivity at rPA < 9% is due to an electrically homogeneous and high resistivity phase rapidly develops in the whole bulk of terpolymer by incorporating PA and reducing PBO. Thus, in order to understand the complex behavior of the volume resistivity, it is necessary to consider the electrical homogeneity of the resulting terpolymers.Fig. S2b† shows the relative dielectric constant at 1 MHz for dried P(BI-b-BO-A) films as a function of PA ratio. The dielectric constant seems to have increased monotonously with the PA ratio although there was some variation. PBO units have a rigid aromatic backbone, resulting in a low mobility of charge in response to electric field, and the lower polarizability of the oxazoles contributed to the lower dielectric constant values for the terpolymers with higher mol% of PBO (and lower mol% of PA). It is shown below that the dielectric constant has to be considered for electrical uniformity in the same way as the resistivity. The observed dielectric constant kobs for a dielectric substance consisting of several components with different dielectric property can be explained as the following equation when a parallel connection of capacitances is hypothesized,
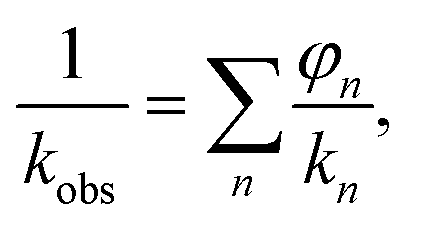 | (1) |
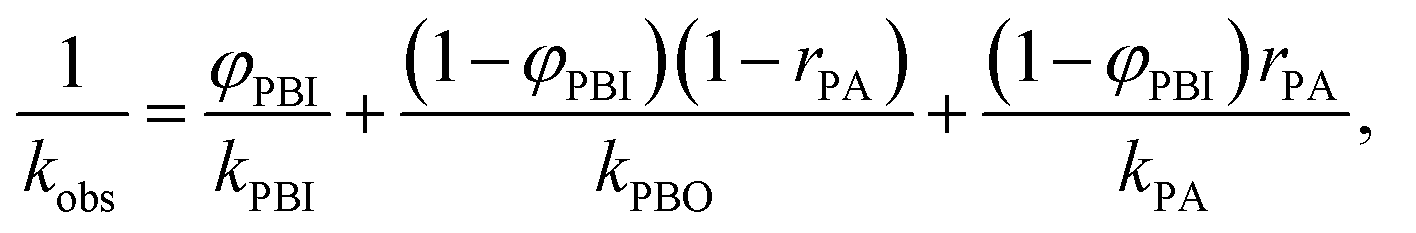 | (2) |
Fig. 6 shows a plot of the dielectric constant versus 1% thermal degradation temperature (Td1) of the obtained terpolymers alongside other commercially available polymer materials. Generally, polymer materials with low dielectric constants show relatively low thermal resistance, while those with higher dielectric constants exhibit higher thermal resistance. For example, in the same condition as terpolymer was measured, polypropylene (PP), polystyrene (PS), and poly(butyl methacrylate) (PBMA) which have low dielectric constant values (2.2, 2.6, and 2.7 respectively), also have low Td1 values (310, 375, and 350 °C respectively).24–27 In contrast, polymers like poly(phenylene sulfide) (PPS), polyimide (Kapton™), and poly(ether ether ketone) (PEEK), which exhibit high thermal resistance with high Td1 values (495, 500, and 525 °C respectively), also have relatively high dielectric constant values (3.8, 3.3, and 3.4, respectively). Compared to the aforementioned polymers, Zylon exhibits a relatively low dielectric constant and a high thermal resistance, presumably due to the effects of its benzoxazole structure.28–38 The lowest dielectric constant among the terpolymers synthesized in the present work was found to be 2.4, which is extremely low compared to that of different commercially available polymer materials. Considering its outstanding thermal resistance, we can conclude that this terpolymer exhibits the best performance among all the polymers that have been compared. Overall, the P(BI-b-BO-A) films can be considered reliable as thermoresistant insulators that can withstand ultrahigh operating voltages.
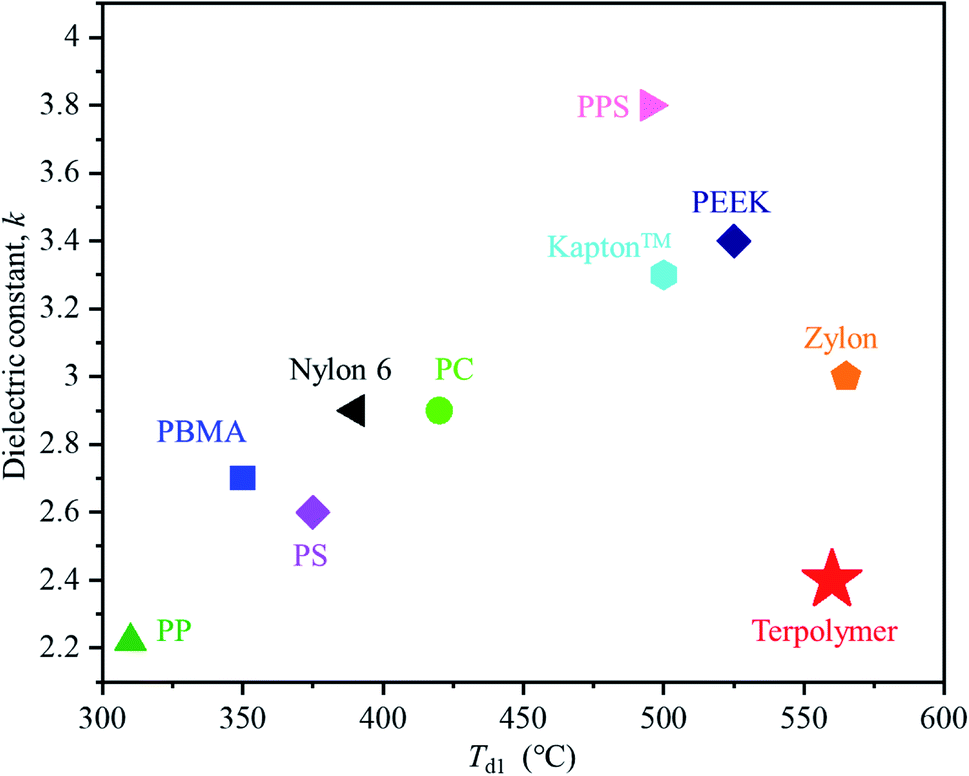 | ||
| Fig. 6 Plot of dielectric constant versus 1% thermal degradation temperature (Td1) for the terpolymer and other common commercial polymer materials:polypropylene (PP),24,25 poly(butyl methacrylate) (PBMA),26,27 polystyrene (PS),28,29 Nylon 6,30,31 polycarbonate (PC),32 Kapton™,33,34 polyphenylene sulfide (PPS)35, polyether ether ketone (PEEK)36,37 and Zylon.38 | ||
3.5. Coating
A coating experiment was conducted to investigate the applicability of the obtained P(BI-b-BO-A) as an insulator. For this, a solution of P(BI-b-BO-A) (mol% composition of 70–15–15) in TFA (70 mg/3 mL) was prepared, and a spiral coil of copper wire was soaked in it for 2 min. On removing from the solution and drying in air, a thin layer of the polymer was coated onto the surface of the copper wire (Fig. 7a). To investigate its dielectric performance, the coil of copper wire coated with P(BI-b-BO-A) was equipped with a magnet to assemble a mini motor model (Fig. 7b). The coil rotated successfully after being powered on, indicating that the coating layer functioned as an insulator. In addition, the thermal resistance of the P(BI-b-BO-A)-coated copper wire was compared with commercial copper wire (polyurethane enameled copper wire) by simultaneously applying a direct current (16 A, 10 V). Since the electric circuit is parallel and the length of the copper wires is same, the electric current and the resultant Joule heat should be the same for both samples. When the current was gradually increased, the commercial copper wire started giving off smoke while the P(BI-b-BO-A)-coated one remained stable (Fig. 7c). Both coils were observed using SEM to check the condition of their surfaces after the experiment. The coating layer of the commercial copper wire presented a significant amount of damage (Fig. 7d), whereas no significant damage was observed in the P(BI-b-BO-A) coating layer (Fig. 7e). Thus, we can confirm that P(BI-b-BO-A) works as a thermostable insulating coating material. | ||
| Fig. 7 a) Process of coating copper wire with terpolymer P(BI-b-BO-A). (b) Running experiment of motor, where the coil was assembled by a P(BI-b-BO-A) coated copper wire. (c) Voltage applied across the copper wires in direct current mode (upper: commercially available copper wire, lower: terpolymer coated copper wire). (d) SEM image of commercially available copper wire after applying voltage. (e) SEM image of terpolymer coated copper wire after applying voltage. | ||
4. Conclusions
A series of bio-based terpolymers P(BI-b-BO-A) having different molar compositions of PBI, PBO and PA were successfully synthesized through a stepwise polymerization procedure, which involved the pre-polymerization of DABA followed by an additional terpolymerization with AHBA and PABA. The terpolymers thus prepared exhibited outstanding thermal, mechanical, and dielectric properties. For 30 mol% of PA, the Td10 reached ∼760 °C, which is the highest reported for any petroleum- or bio-based plastic developed so far, and is comparable to the melting temperature of light metals. DFT calculations of the hydrogen bonding enthalpy between imidazole rings revealed how the inter-chain interactions within the polymer were strengthened by incorporating PA, which could be the reason for such ultrahigh thermoresistance of the prepared terpolymers. The highest tensile strength and strain at break for the terpolymers were found to reach up to 80 MPa and 12%, respectively, which were regulated by the molar composition of the individual monomers. The strain at break increased regularly with increase in mol% of PA. The lowest dielectric constant was found to be 2.4, which is much lower than that of most commercially available thermoresistant insulators, and comparable to that of inorganic materials such as silica. Among all the compositions, the terpolymer in composition of 70–15–15 (mol%) exhibited a Td10 of 710 °C and a dielectric constant of 2.6, the appropriate amount of oxazole groups of PBO decreased the polarizability and the H-bond brought by PA increased the thermal stability, thus synergistically balanced the thermal stability and low-k performance and was considered to the most extraordinary one in all the compositions. Therefore, P(BI-b-BO-A) can be useful as an excellent insulating material for coating purposes, as was also confirmed from our coating experiment. Overall, we can conclude from all our results that the bio-based P(BI-b-BO-A) terpolymers have great potential to be utilized as coating materials for high power coil of high-speed electric vehicles, in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JAIST Research Grants (Houga) 2020. This work was also supported by the Cabinet Office, Government of Japan, Cross-ministerial Strategic Innovation Promotion Program (SIP), “Technologies for Smart Bio-industry and Agriculture” (funding agency: Bio-oriented Technology Research Advancement Institution, NARO, Grant No. 18087978), and partially supported by a JSPS KAKENHI Grant-in-aid for Scientific Research A (22H00332). The computations in this work were performed using the facilities provided by the Research Center for Advanced Computing Infrastructure at JAIST.References
- K. K. Sadasivuni, P. Saha, J. Adhikari, K. Deshmukh, M. B. Ahamed and J. Cabibihan, Polym. Compos., 2020, 41, 32–59 CrossRef CAS.
- X.-Y. Zhao and H.-J. Liu, Polym. Int., 2010, 59, 597–606 CAS.
- M. G. A. Vieira, M. A. Da Silva, L. O. Dos Santos and M. M. Beppu, Eur. Polym. J., 2011, 47, 254–263 CrossRef CAS.
- J. Rajesh Banu, S. Kavitha, R. Yukesh Kannah, T. Poornima Devi, M. Gunasekaran, S.-H. Kim and G. Kumar, Bioresour. Technol., 2019, 290, 121790 CrossRef CAS PubMed.
- S. Park, H. Jeon, H. Kim, S. Shin, S. Choy, D. S. Hwang, J. M. Koo, J. Jegal, S. Y. Hwang, J. Park and D. X. Oh, Nat. Commun., 2019, 10, 2601 CrossRef PubMed.
- O. Valerio, M. Misra and A. K. Mohanty, RSC Adv., 2017, 7, 38594–38603 RSC.
- T. P. Dawin, Z. Ahmadi and F. A. Taromi, Prog. Org. Coat., 2018, 119, 23–30 CrossRef CAS.
- Q. Li, F.-Z. Yao, Y. Liu, G. Zhang, H. Wang and Q. Wang, Annu. Rev. Mater. Res., 2018, 48, 219–243 CrossRef CAS.
- A. Nag, M. A. Ali, M. Watanabe, M. Singh, K. Amornwachirabodee, S. Kato, T. Mitsumata, K. Takada and T. Kaneko, Polym. Degrad. Stab., 2019, 162, 29–35 CrossRef CAS.
- K. L. Gordon, J. H. Kang, C. Park, P. T. Lillehei and J. S. Harrison, J. Appl. Polym. Sci., 2012, 125, 2977–2985 CrossRef CAS.
- Z.-J. Liu, C.-G. Yin, V. Cecen, J.-C. Fan, P.-H. Shi, Q.-J. Xu and Y.-L. Min, Polymer, 2019, 179, 121613 CrossRef CAS.
- D. Li, D. Shi, Y. Xia, L. Qiao, X. Li and H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 8742–8750 CrossRef CAS PubMed.
- A. Nag, M. A. Ali, H. Kawaguchi, S. Saito, Y. Kawasaki, S. Miyazaki, H. Kawamoto, D. T. N. Adi, K. Yoshihara, S. Masuo, Y. Katsuyama, A. Kondo, C. Ogino, N. Takaya, T. Kaneko and Y. Ohnishi, Adv. Sustainable Syst., 2020, 2000193 Search PubMed.
- J. D. Moon, A. T. Bridge, C. D'Ambra, B. D. Freeman and D. R. Paul, J. Membr. Sci., 2019, 582, 182–193 CrossRef CAS.
- S. W. Chuang, S. L. C. Hsu and Y. H. Liu, J. Membr. Sci., 2007, 305, 353–363 CrossRef CAS.
- F. Yao, W. Xie, M. Yang, H. Zhang, H. Gu, A. Du, N. Naik, D. P. Young, J. Lin and Z. Guo, Mater. Today Phys., 2021, 21, 100502 CrossRef CAS.
- A. Nag, M. A. Ali, A. Singh, R. Vedarajan, N. Matsumi and T. Kaneko, J. Mater. Chem. A, 2019, 7, 4459–4468 RSC.
- A. A. Arbuzov, V. E. Muradyan, B. P. Tarasov, E. A. Sokolov and S. D. Babenko, Russ. J. Phys. Chem. A, 2016, 90, 907–910 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024–11031 CrossRef CAS.
- X. Xu, Q. Fu, H. Gu, Y. Guo, H. Zhou, J. Zhang, D. Pan, S. Wu, M. Dong and Z. Guo, Polymer, 2020, 188, 122129 CrossRef CAS.
- P. Wang, L. Yang, S. Gao, X. Chen, T. Cao, C. Wang, H. Liu, X. Hu, X. Wu and S. Feng, Adv. Compos. Hybrid Mater., 2021, 4, 639–646 CrossRef CAS.
- X. Yuan, Y. Matsuyama and T. C. M. Chung, Macromolecules, 2010, 43, 4011–4015 CrossRef CAS.
- E. Esmizadeh, C. Tzoganakis and T. H. Mekonnen, Polymers, 2020, 12, 1627 CrossRef CAS PubMed.
- S. Strella and R. Zand, J. Polym. Sci., 1957, 25, 105–114 CrossRef CAS.
- Z. Czech, K. Agnieszka, P. Ragańska and A. Antosik, J. Therm. Anal. Calorim., 2015, 119, 1157–1161 CrossRef CAS.
- S. Yu, P. Hing and X. Hu, J. Appl. Phys., 2000, 88, 398–404 CrossRef CAS.
- T. ARII, J. Mass Spectrom. Soc. Jpn., 2003, 51, 235–241 CrossRef CAS.
- H. Gu, X. Xu, J. Cai, S. Wei, H. Wei, H. Liu, D. P. Young, Q. Shao, S. Wu, T. Ding and Z. Guo, Chem. Commun., 2019, 55, 10068–10071 RSC.
- Matweb, Overview of Materials for Nylon 6/66, https://www.matweb.com/search/datasheet_print.aspx?matguid=26386631ec1b49eeba62c80a49730dc4, accessed: March 2021.
- Overview of materials for Polycarbonate, High Heat, https://www.matweb.com/search/DataSheet.aspx?MatGUID=bfcedbb55b164a68a93acfa854fa2fe0, accessed: March 2021.
- A. C. Lua and J. Su, Polym. Degrad. Stab., 2006, 91, 144–153 CrossRef CAS.
- T. Kuroki, Y. Tanaka, T. Hokudoh and K. Yabuki, J. Appl. Polym. Sci., 1997, 65, 1031–1036 CrossRef CAS.
- W. Tanthapanichakoon, M. Furuuchi, K. Nitta, M. Hata, S. Endoh and Y. Otani, Polym. Degrad. Stab., 2006, 91, 1637–1644 CrossRef CAS.
- H. Sun, Y. Lv, C. Zhang, X. Zuo, M. Li, X. Yue and Z. Jiang, RSC Adv., 2018, 8, 7753–7760 RSC.
- P. Patel, T. R. Hull, R. W. McCabe, D. Flath, J. Grasmeder and M. Percy, Polym. Degrad. Stab., 2010, 95, 709–718 CrossRef CAS.
- ToyoboZylon® https://www.matweb.com/search/DataSheet.aspx?MatGUID=140045b4f06c4aea89f8900f4c7bb4fc, accessed: March 2021.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01488b |
| This journal is © The Royal Society of Chemistry 2022 |