
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective monoallylation of anilines to N-allyl anilines using reusable zirconium dioxide supported tungsten oxide solid catalyst†
Yoshihiro Kon *a,
Shota Tsurumia,
Shunsuke Yamadab,
Toshiyuki Yokoib and
Tadahiro Fujitania
*a,
Shota Tsurumia,
Shunsuke Yamadab,
Toshiyuki Yokoib and
Tadahiro Fujitania
aInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: y-kon@aist.go.jp
bNanospace Catalysis Unit, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
First published on 19th April 2022
Abstract
The monoallylation of aniline to give N-allyl aniline is a fundamental transformation process that results in various kinds of valuable building block allyl compounds, which can be used in the production of pharmaceuticals and electronic materials. For decades, sustainable syntheses have been gaining much attention, and the employment of allyl alcohol as an allyl source can follow the sustainability due to the formation of only water as a coproduct through dehydrative monoallylation. Although the use of homogeneous metal complex catalysts is a straightforward choice for the acceleration of dehydrative monoallylation, the use of soluble catalysts tends to contaminate products. We herein present a 10 wt% WO3/ZrO2 catalyzed monoallylation process of aniline to give N-allyl anilines in good yields with excellent selectivity, which enables the continuous selective flow syntheses of N-allyl aniline with 97–99% selectivity. The performed detailed study about the catalytic mechanism suggests that the dispersed WO3 with the preservation of the W(VI) oxidation state of 10 wt% WO3/ZrO2 with appropriate acidity and basicity is crucial for the monoallylation. The inhibition of the over allylation of the N-allyl anilines is explained by the unwilling contact of the N-allyl aniline with the active sites of WO3/ZrO2 due to the steric hindrance.
Introduction
Among various kinds of N-allyl compounds,1 N-allyl anilines are known as important building blocks that can add another substituent to N–H and/or allyl moieties to give various value-added fine chemicals, which are applicable to pharmaceuticals and electronic materials.1–3 While various allylic electrophiles, such as allyl chloride, allyl acetate, and allyl silane, are frequently used in allylation,4 they contaminate the waste coproducts derived from the chloride, acetate, and silyl substituents of allyl sources. The employment of allyl alcohol (1) is a promising pathway to produce N-allyl anilines because the resulting coproduct is only water3,5,6 and the alcohol 1 can be supplied from biomass glycerol by catalytic processes.7The transformation method of 1 with aniline (2) to N-allyl aniline (3) has significantly been studied due to its versatility and broad application.2,3 There are many examples of the monoallylation of 2 with 1 to produce 3 in excellent yields by Pd,8 Ir,9 Pt10 and Ni11 complex catalysts through the formation of π-allyl metal intermediates.6,8–11 However, these complex catalysts are complicated to treat and costly. Moreover, it is hard to apply metal complex catalysts to practical syntheses because the complex catalysts are solvated and contamination with products. From these perspectives, the expectations of using solid catalysts have been increasing due to the reusability of such catalysts and their applicability to flow reactors.12,13
Kaneda and Motokura demonstrated the monoallylation of 1 using Brønsted acidic montmorillonite catalysts, in which the monoallylation of 1 with 2 was performed to give 3 in around 30% yield.14 However, there are no subsequent reports to increase the yield of 3 with excellent selectivity. Recently, we reported a titanium dioxide supported with molybdenum oxide (MoO3/TiO2) for the allylation of 2.15 However, the MoO3/TiO2-catalyzed allylation of 2 resulted in 3 with only 4% yield and N,N-diallyl aniline (4) was formed in 85% yield via the formation of an allyl-oxo molybdenum intermediate, which was caused by the reduction of Mo(VI) to Mo(V).15,16
To avoid diallylation, we inspired to use tungsten oxide (WO3) as a reactive site with the support of metal oxides. WO3/ZrO2 is known as a solid superacid,17 and there are many useful WO3/ZrO2-catalyzed reactions, such as alkane isomerization, Friedel–Crafts acylation, alkene trimerization, and biomass conversion.17,18 However, to the best of our knowledge, there are no examples of the WO3/ZrO2-catalyzed monoallylation of anilines utilizing a stable W(VI) oxidation state of WO3. We herein report the development of zirconium dioxide supported with a 10 wt% of WO3 (10 wt% WO3/ZrO2), which can catalyze the selective dehydrative monoalkylation of anilines and can be applied to continuous flow reactions.
Experimental
Catalyst preparation
The preparation of metal oxide catalysts, such as silicon dioxide supported WO3 (WO3/SiO2), aluminum oxide supported WO3 (WO3/Al2O3), WO3/TiO2, magnesium oxide supported WO3 (WO3/MgO), and WO3/ZrO2, was performed according to a modified impregnation method, where citric acid was used as a pH controlling agent to prevent the formation of tungsten oxide clusters on the surfaces of the supports. For the preparation of 10 wt% WO3/ZrO2, 1.80 g of ZrO2 were added to a diluted aqueous solution (1 mL) of (NH4)10W12O41(H2O)5 (235.3 mg, 75.1 mmol) with citric acid. The solution was subjected to ultrasonic irradiation for 10 min to ensure a mixture with good homogeneity. After 1 h, the impregnated catalyst was dried at 100 °C for 24 h and then calcined under static air from 25 °C to 500 °C with an increasing rate of 10 °C min−1. Then, after reaching 500 °C, it was calcined at 500 °C for 3 h.Monoallylation of aniline (2) with allyl alcohol (1) using a WO3/ZrO2 catalyst (as standard reaction conditions)
A pressure-resistant glass tube equipped with a magnetic stirring bar was loaded with WO3/ZrO2 (100 mg), n-octane (0.25 mL), 1 (116 mg, 2.0 mmol), and 2 (93.0 mg, 1.0 mmol). The vessel was tightly sealed by a screw cap, and the mixture was stirred (500 rpm) in an oil bath maintained at 140 °C for 24 h. After the reaction, the solution was cooled to room temperature and then diluted with 10 mL of ethyl acetate. Biphenyl (77 mg, 0.50 mmol) was added to the solution as an internal standard for the gas chromatography (GC) analysis. The solution was then placed under ultrasonic irradiation for 10 min to ensure a mixture with good homogeneity. The conversion and yield were determined based on 2 from the analysis of the mixture by GC. The yields of the monoallyl aniline (3) and diallyl aniline (4) were 71% and 7%, respectively. The conversion of 2 was 78%, and the selectivity of 3 [(yield of 3)/(conversion of 2) × 100] was 91%. The isolation of 3 was performed through column chromatography (eluent: n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene = 8:1) to give 3 with a 58% isolation yield. From an inductively coupled plasma analysis (ICP), the leaching of the W species was found to be 0.35 mg. This value was calculated as 0.4 wt% of the tungsten species in WO3/ZrO2 (the calculation was performed by regarding the tungsten species as WO3).
:toluene = 8:1) to give 3 with a 58% isolation yield. From an inductively coupled plasma analysis (ICP), the leaching of the W species was found to be 0.35 mg. This value was calculated as 0.4 wt% of the tungsten species in WO3/ZrO2 (the calculation was performed by regarding the tungsten species as WO3).
Retesting of the spent catalyst
The reaction conditions at cycle number 1 (original catalyst) are as follows. 1 (2.0 mmol), 2 (1.0 mmol), and the spent 10 wt% WO3/ZrO2 catalyst (100 mg) were stirred in a pressure-resistant glass tube at 140 °C for 24 h. Four batch reactions of the same batch reaction conditions were performed all at once, and the yields of 3 in the three batch reactions were comparable (average 69% yields). The used WO3/ZrO2 was recovered by centrifugation, washed with ethyl acetate, dried at 100 °C for 24 h, and then calcined at 500 °C for 3 h before the next reaction.Monoallylation of aniline (2) with allyl alcohol (1) using a continuous flow reactor for 15 h
A mixture of 10 wt% WO3/ZrO2 (1.00 g) and sea sand (1.29 g) (diluent) was packed in a column reactor (length: 100 mm, inner diameter: 5 mm) equipped with a plunger pump, which was used for passing a mixture of 1 (0.2 mol L−1), 2 (0.1 mol L−1), and biphenyl (0.05 mol L−1) to an n-octane solution. The reaction was initiated at a flowrate of 0.1 mL min−1 under atmospheric pressure, and the residence time was estimated to be 2.2 min at the column reactor. The column was heated at 140 °C, and the 1.5–6.0 mL solution was corrected each reaction time to check the product yields using GC. The reaction time was set to 0 h when the reaction solution was stabilized to flow. The yields of 3 were in the range of 8–16%, and the selectivities at each reaction time were over 97% from 0 to 15 h.Hot filtration test
Hot filtration tests were executed for the reaction mixture of 10 wt% WO3/ZrO2 (100 mg), n-octane (0.25 mL), 1 (116 mg, 2.0 mmol), and 2 (93.1 mg, 1.0 mmol). The mixture was stirred in a pressure-resistant glass tube at 140 °C and was then temporally stopped at 4 h. Afterward, the mixture was subjected to quick filtration under reduced pressure, and centrifugation was performed to separate the solid catalysts from the reaction solution. Then, the filtrate was additionally stirred at 140 °C for 4 h without the addition of any new catalysts. The conversion and yield were determined by GC. The yield of 3 was 21%, and the conversion of 2 was 39%. The reaction of 2 at 140 °C for 8 h without hot filtration resulted in 3 with a yield of 48%.Materials and methods
Results and discussion
Screening of the catalysts
We screened the catalytic activities of various metal oxide-supported WO3, H-montmorillonite, heteropolyacid, and solid acid polymer (Table 1). WO3 supported by SiO2, Al2O3, TiO2, MgO, and ZrO2 were prepared using the modified impregnation method, as shown in the experimental section. The crystal structures of WO3/SiO2, WO3/Al2O3, WO3/TiO2, WO3/MgO, and WO3/ZrO2 were checked using XRD analyses to confirm the preservation of their support metal oxide crystal structures (Fig. S1†). In the XRD, there were no characteristic signals of 2θ = 23.2, 23.7, and 24.4°, which are generally assigned to the WO3 monoclinic crystal structures.19 Therefore, the prepared WO3/SiO2, WO3/Al2O3, WO3/TiO2, WO3/MgO, and WO3/ZrO2 showed the dispersed WO3 nature on the surface of the solid catalysts.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (100 mg) | Conv. of 2b (%) | Yieldsb (%) | Selectivityc (%) | |
| 3 | 4 | ||||
| a Reaction conditions: 1 (2.0 mmol), 2 (1.0 mmol), solid catalyst (100 mg), n-octane (0.25 mL), 140 °C, 500 rpm, 4.0 h, unless otherwise stated.b Conversion and yield were on the basis of 2, determined by GC analysis using biphenyl as an internal standard.c Selectivity = (yield of 3)/(conversion of 2) × 100 (%). | |||||
| 1 | 10 wt% WO3/SiO2 | 11 | 4 | 0 | 36 |
| 2 | 10 wt% WO3/Al2O3 | 11 | 2 | 0 | 18 |
| 3 | 10 wt% WO3/TiO2 | 15 | 6 | 0 | 40 |
| 4 | 10 wt% WO3/MgO | 7 | 0 | 0 | 0 |
| 5 | 10 wt% WO3/ZrO2 | 38 | 31 | 0.6 | 82 |
| 6 | WO3 | 6 | 1 | 0 | 17 |
| 7 | ZrO2 | 5 | 0 | 0 | 0 |
| 8 | — | 4 | 0 | 0 | 0 |
| 9 | H-montmorillonite | 23 | 5 | 0 | 22 |
| 10 | H3PW12O40(H2O)n | 18 | 2 | 0 | 13 |
| 11 | Amberlyst 15DRY | 50 | 0 | 0 | 0 |
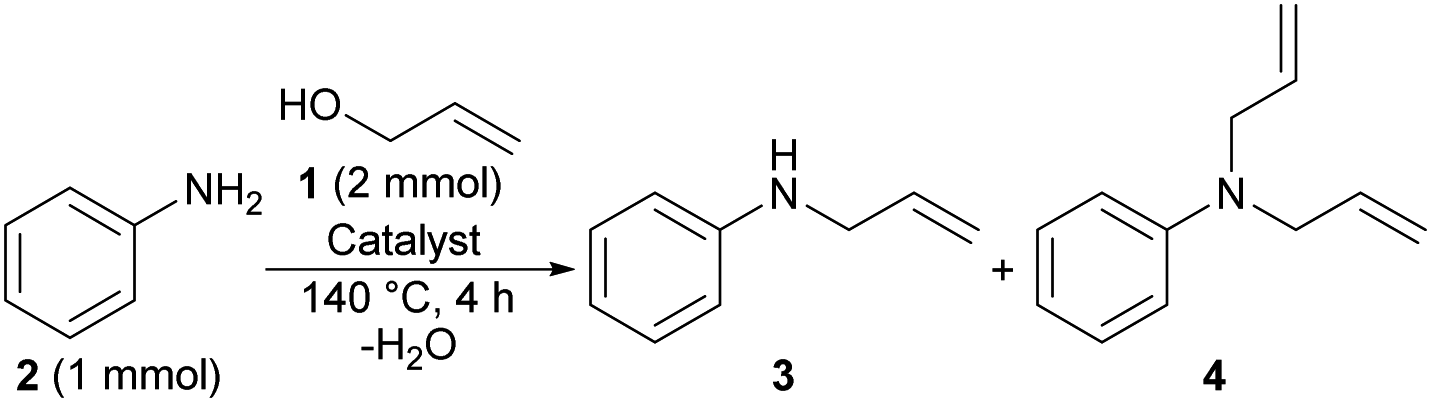
Table 1 indicates the results of the dehydrative monoallylation of 2 with 1 in the presence of various solid catalysts at 140 °C for 4 h. Although solid acids such as WO3/SiO2, WO3/Al2O3, and WO3/TiO2 were estimated to be reactive solid acids,17 they gave 3 in only 2–6% of the yields with low selectivity (Table 1, entries 1–3). In the case of using the WO3/MgO catalyst, in which MgO is known as a basic support, the allylation of 2 did not proceed (Table 1, entry 4). Only the case of using WO3/ZrO2 indicated good reactivity to give 3 in 31% yield with 82% selectivity, and the coproduction of 4 was given in only 0.6% yield (Table 1, entry 5). When the reactions were performed with WO3 or ZrO2 only, they did not proceed (Table 1, entries 6 and 7). Also, the reactions did not proceed without catalysts (Table 1, entry 8). H-montmorillonite, which is known as an effective solid acid catalyst,14 gave 3 in 5% yield, and the heteropolyacid (H3PW12O40(H2O)n) catalyst20 resulted in the production of 3 in 2% yield (Table 1, entries 9 and 10). Amberlyst 15DRY, as an example of solid acid polymers that contain –SO3H groups on the surface,21 showed 50% conversion of 2 without the detection of any products by GC analysis because aniline was adsorbed on its surface (Table 1, entry 11).22 The adsorption of 2 on the catalyst surface was also estimated for other solid catalysts (Table 1, entries 1–3, 9 and 10).
Effect of supports (SiO2, Al2O3, TiO2, and ZrO2)
The screening data with the BET surface area, acid amounts measured by NH3-TPD, acid density calculated from the BET surface area, and acid amounts of 10 wt% WO3/SiO2, 10 wt% WO3/Al2O3, 10 wt% WO3/TiO2, and 10 wt% WO3/ZrO2 are shown in Table 2. As shown in the correlation between the acid densities and yields of 3, 10 wt% WO3/ZrO2 showed a specifically high yield (Fig. S2†). Although 10 wt% WO3/TiO2 showed a higher acid density than that of 10 wt% WO3/ZrO2, it produced 3 in only 6% yield. Comparing the acid strengths of 10 wt% WO3/TiO2 and 10 wt% WO3/ZrO2 by NH3-TPD, a signal derived from a relatively strong acid at 300 to 500 °C was observed in 10 wt% WO3/ZrO2, but hardly observed in 10 wt% WO3/TiO2 (Fig. S3†). The moderate acid density with relatively strong acid of 10 wt% WO3/ZrO2 was effective in proceeding the monoallylation of 2. The dispersed tungsten species in 10 wt% WO3/ZrO2 was checked by HAADF-STEM EDS mappings as well as XRD patterns, as shown in Fig. S1,† where the WO3/ZrO2 catalyst showed well-dispersed tungsten species with a particle size of under 4 nm (Fig. S4†).| Catalystsa | BET (m2 g−1) | Acid amount (mmol g−1) | Acid densityb (mmol m−2) | Yield of 3c,d (%) |
|---|---|---|---|---|
| a 10 wt% WOx was supported.b Acid density = (acid amount)/BET.c Reaction conditions: 1 (2.0 mmol), 2 (1.0 mmol), catalyst (100 mg), 140 °C, 500 rpm, 4.0 h.d Yields were on the basis of 2, determined by GC analysis using biphenyl as an internal standard. | ||||
| WO3/SiO2 | 99 | 0.195 | 1.97 × 10−3 | 4 |
| WO3/Al2O3 | 178 | 0.364 | 2.04 × 10−3 | 2 |
| WO3/TiO2 | 55 | 0.221 | 4.02 × 10−3 | 6 |
| WO3/ZrO2 | 105 | 0.301 | 2.87 × 10−3 | 31 |
Screening the amount of tungsten oxides in WO3/ZrO2
Next, the correlation between the amounts of WO3 in the WO3/ZrO2 catalyst and their reactivities was investigated using the 1, 2.5, 5, 7.5, and 10 wt% WO3/ZrO2 catalysts (Fig. 1).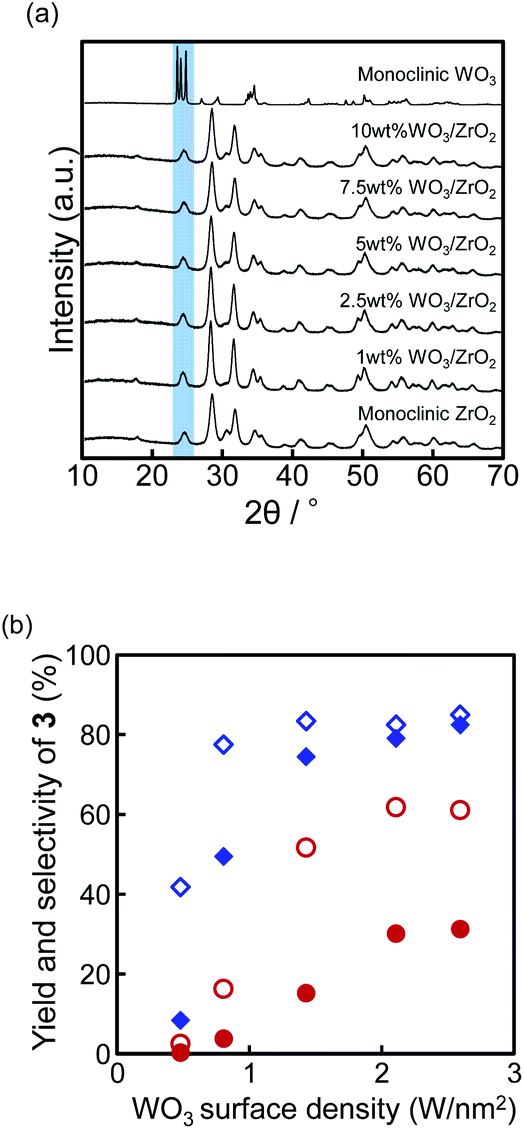 | ||
| Fig. 1 (a) XRD patterns of 1, 2.5, 5, 7.5 and 10 wt% WO3/ZrO2 species. Pale blue highlight shows the WO3 monoclinic characteristic patterns; (b) correlation between the W surface density (W nm−2) and the yields (selectivities) of 3. Yields and selectivities at 4 h were indicated by the solid red circle and solid blue diamond, respectively. Yields and selectivities at 24 h were indicated by the hollow red circle and hollow blue diamond, respectively. | ||
The characteristic sharp signals of the WO3 monoclinic crystal structures were not observed at the XRD spectra of all the employed WO3/ZrO2 catalysts, which shows the good dispersion nature of WO3 on the ZrO2 surface (highlighted in pale blue, Fig. 1a).19 The characteristic patterns assigned to the monoclinic ZrO2 structure in the XRD spectra were shown (Fig. 1a).19 The WO3 surface density is known to be a good indicator to express the surface structures in WO3/ZrO2 catalysts.19,23 We calculated the WO3 surface density of the prepared 1, 2.5, 5, 7.5, and 10 wt% WO3/ZrO2 from the ICP and BET data, and the calculated values were found to be 0.48, 0.81, 1.43, 2.11, and 2.59 W nm−2, respectively (Table S1†).23 The correlation between the WO3 surface density and the yields of 3 is shown in Fig. 1b.
All the catalyst samples were distributed in the WO3 surface density range of 0–3 W nm−2, which indicates that there were no WO3 crystallites and that less W–O–W bonds were formed on the surface of the employed WO3/ZrO2.19a Also, the WO3 surface density range below 4 W nm−2 shows that the surface WO3 were in the levels of monolayer coverage.19b The WO3 species in the range below 4 W nm−2 are reported to be stabilized through multiple W–O–Zr bonds between each WO6 octahedra and the ZrO2 surface.19 Also, the increase in the WO3 surface density corresponded to the increase in the W–OH–Zr active acidic sites.19 Moreover, the yield of 3 was saturated at over 2.11 W nm−2 for 24 h (hollow red circle, Fig. 1b). However, the selectivity of 3 was improved when the WO3 surface density of WO3/ZrO2 increased from 2.11 to 2.59 W nm−2 (hollow blue diamonds, Fig. 1b). This data can be explained by considering the balance of the acidic sites and basic sites of WO3/ZrO2. ZrO2 is known to show the bifunctional characters of relatively stable solid acids and solid bases,24 and the basic sites would adsorb 2 through the hydrogen bonding between H–N and the basic surface of ZrO2. An increase in the WO3 surface density signifies an increase in the W–OH–Zr active acidic sites, which results in a relative decrease in the basic sites of ZrO2. A decrease in the basic sites of ZrO2 was observed by CO2-TPD (Table S1, Fig. S5†). The presence of basic sites in WO3/ZrO2 was effective for the acceleration of the monoallylation process because of the adsorption of 2 to the basic sites on the catalyst surface. However, the presence of excess basic sites reduced selectivity because 2 was strongly adsorbed on the basic sites and could not be recovered from WO3/ZrO2. From these aspects, the optimized WO3 surface density on 10 wt% WO3/ZrO2 showing 2.59 W nm−2 supplied appropriate distribution of the acid and basic active sites on the 10 wt% WO3/ZrO2, which were crucial for proceeding the highly selective monoallylation of 2.
The measurement of the W 4f7/2 and 4f5/2 profiles of 10 wt% WO3/ZrO2 was performed by the XPS (Fig. 2). By referencing to C 1s (284.6 eV), the peaks at the 35.0 eV and 37.0 eV binding energies (BEs) were ascribed to W 4f7/2 and 4f5/2, which were corresponding to W(VI).25 After the reaction, the peaks slightly changed (35.1 eV and 37.3 eV for W 4f7/2 and 4f5/2, respectively) with the same shape of the spectra, and the lower BEs assigned to W(V) and W(IV) were not observed at all. From the XPS analysis, we confirmed that the oxidation state of tungsten(VI) did not change during the reaction.
 | ||
| Fig. 2 XPS image of WOx/ZrO2 before and after the monoallylation. | ||
Optimization of the reaction conditions using the WO3/ZrO2 catalyst
The developed 10 wt% WO3/ZrO2-catalyzed monoallylation was optimized by adjusting the solvents, reaction temperature, and reaction time (Tables S2, S3† and 3). The monoallylation of 2 was inhibited using polar solvents, such as ketone, amide, and ether, to give 3 in 0–12% yields (Table S2†). However, the alkanes and aromatic solvents showed good reactivity for the monoallylation of 2 to give 3 in 25–32% yields (Table S2†). The reactivity decreased under solvent-free conditions to give 3 in 13% yield (Table S2†). The yields of 3 increased from 14% to 67% with increasing the reaction temperature from 130 to 160 °C, respectively (Table S3†). We selected the reaction temperature of 140 °C for the monoallylation process because the reaction over 150 °C caused the formation of the undesired product 4.
|
||||
|---|---|---|---|---|
| Reaction time (h) | Conversion of 2b (%) | Yield of 3b (%) | Yield of 4b (%) | Selectivityc (%) |
| a The base reaction conditions are as follows: 1 (2.0 mmol), 2 (1.0 mmol), 10 wt% WO3/ZrO2 (100 mg), n-octane (0.25 mL), 140 °C, 500 rpm, 4.0 h.b Determined by GC analysis based on 2, the yields are the average of the results of three experiments.c Selectivity = (yield of 3)/(conversion of 2) × 100 (%). | ||||
| 2 | 22 | 14 | 0 | 66 |
| 4 | 32 | 25 | 0 | 79 |
| 8 | 57 | 48 | 2 | 85 |
| 24 | 78 | 71 | 7 | 91 |
| 48 | 87 | 70 | 10 | 80 |
The time course reaction profile of the allylation using 10 wt% WO3/ZrO2 showed steady progress of forming 3 until 8 h. Then, the reaction proceeded slowly to reach 71% yield for 24 h (Table 3). The selectivity of 3 was maintained over 80% at 8 h and 24 h, and the slight decrease in the selectivity of 3 was observed at 48 h because of the increase of 4. The allylation of the mixture of 1 (1.5 mmol), 2 (0.5 mmol), and 3 (0.5 mmol) was performed as a competitive reaction of 2 and 3 (Scheme S1†). After 4 h, products with a molar ratio of 2:3:4 = 0.33:0.66:0.02 were observed, and the ratio of the products reached 2:3:4 = 0.18:0.71:0.05 (mmol) at 24 h (Scheme S1†). The results indicated that 2 was more reactive than 3, and were in good agreement with the production of 3 in 71% yield with 91% selectivity, as shown in Table 3 and Fig. 1. A catalytic reaction was assumed to have proceeded on the surface of the solid catalyst since the reaction was terminated when the catalyst was removed by hot filtration during the reaction (Table S4†).
Reuse of WO3/ZrO2 and continuous synthesis using WO3/ZrO2
The developed 10 wt% WO3/ZrO2 was easily separated from the reaction solution, and it could be reused at least three times before seeing a significant loss in yields of 3 (Table S5 and Fig. S6†). Continuous flow monoallylation of 2 with 1 was demonstrated using a flow reactor equipped with a glass column (length: 100 mm, inner diameter: 5 mm) and packed with 10 wt% WO3/ZrO2, which was diluted with sea sand (Fig. 3).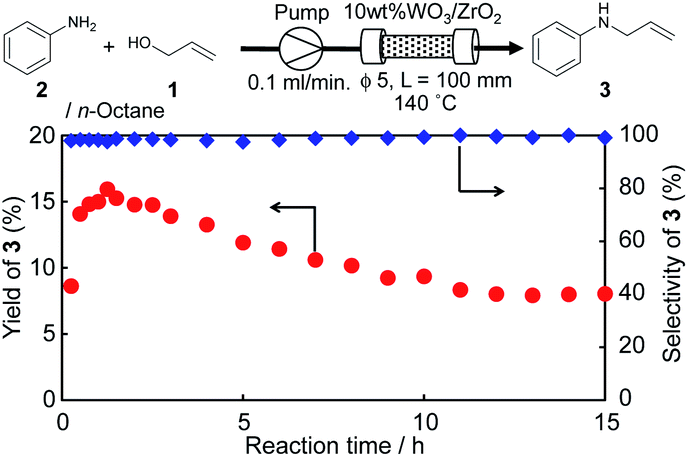 | ||
| Fig. 3 Monoallylation of 2 with 1 in the presence of WO3/ZrO2 using a flow reactor. | ||
A mixture of 1 and 2 in an n-octane solution was sent to the catalyst column at a flow rate of 0.1 mL min−1 under atmospheric pressure. Continuous flow synthesis of 3 was performed at 140 °C for 15 h, and 3 was continuously produced in around 10–15% yields with 97–99% selectivity without detection of 4 (Fig. 3 and Table S6†). The estimated contact time in the catalyst column was 2.2 min, and the calculated TOF per W was around 20 (h−1).19,26
Scope and limitation of the substrates
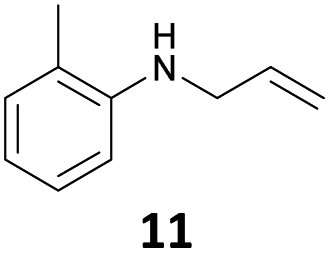
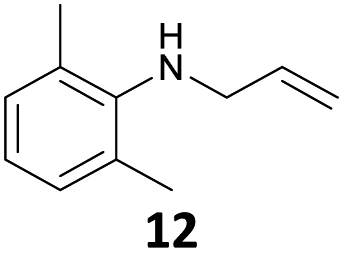
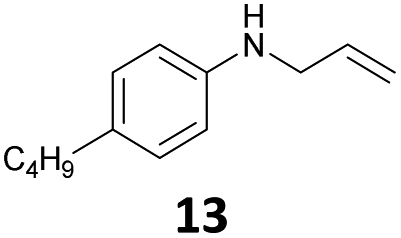
The developed 10 wt% WO3/ZrO2 catalyzed the monoallylation of various anilines with 1 (Table 4). Besides 2, the 4-fluoro-, 4-chloro-, and 4-bromo-substituted anilines reacted with 1 in the presence of the 10 wt% WO3/ZrO2 catalyst to give the corresponding N-allyl anilines 3, 5, 6, and 7 in 58, 33, 43, and 45% isolated yields, respectively (Table 4). The electron-deficient substrates, such as the 4-nitro-substituted aniline, decreased the reactivity to give 8 in 25% yield. The monoallylation of the 4-amino-substituted aniline gave 9 in 30% yield without the production of N,N′-diallyl aniline. Also, the allylation of the 4-methyl-, 2-methyl-, and 2,6-dimethyl anilines gave the corresponding N-allyl anilines 10, 11 and 12 in 29, 54, and 17% yields, respectively. The electronic effects at the 4-position of the anilines were less correlated with the isolated yields of the N-allyl anilines (Table 3, 3 and 5–10). On the other hand, the sterically bulky 2,6-dimethyl aniline showed lower reactivity than that of the 2-methyl aniline (Table 3, 11 and 12). The elongation of the alkyl chain at the 4-position from the methyl to n-butyl in the aniline did not affect the reactivity to give 13 in 29% yield. Various N-allyl anilines were selectively synthesized at the molar ratio of 1:aniline = 2:1, and the selectivity (mono:di) of N-allyl anilines were calculated to be over 94% (Table 4).
|
||
|---|---|---|
| Products | Yieldsb (%) of N-allyl anilines | Selectivity (mono:di)c (%) |
| a Reaction conditions: 1 (2.0 mmol), aniline (1.0 mmol), WO3/ZrO2 (100 mg), n-octane (0.25 mL), 140 °C, 24 h.b Isolated yield.c Selectivity (mono:di) = ((isolated yield of N-allyl aniline)/[(isolated yield of N-allyl aniline) + (isolated yield of N,N-diallyl aniline)]) × 100 (%).d Small amounts of the other compounds were contaminated. |
||
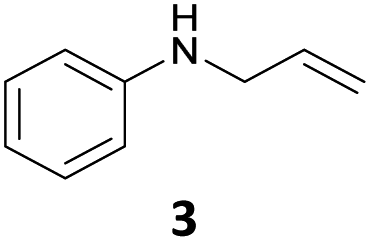 |
58 | 98 |
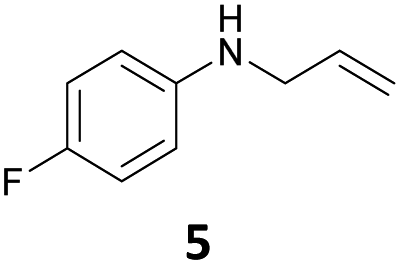 |
33 | 97 |
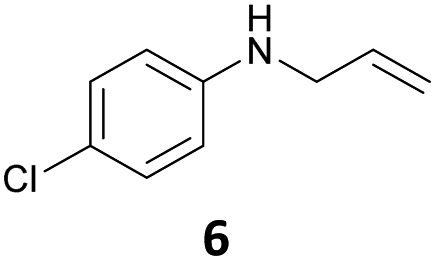 |
43 | 96 |
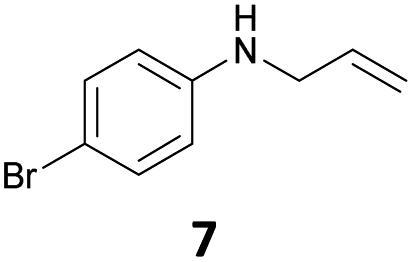 |
45 | 98 |
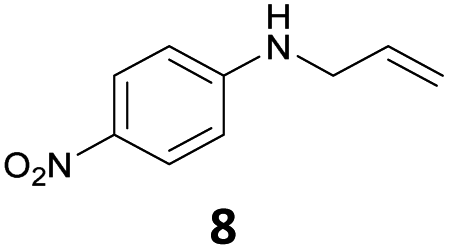 |
25 | 96 |
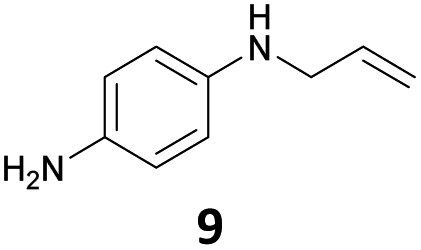 |
30 | —d |
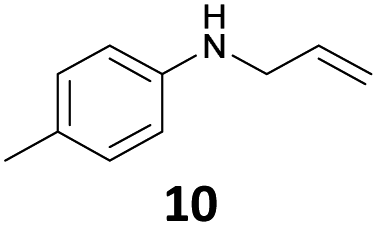 |
29 | 97 |
 |
54 | 95 |
 |
17 | 94 |
 |
29 | —d |
 |
63 | —d |
 |
29 | —d |
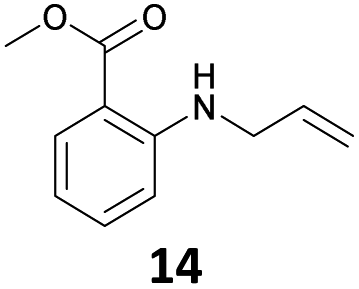
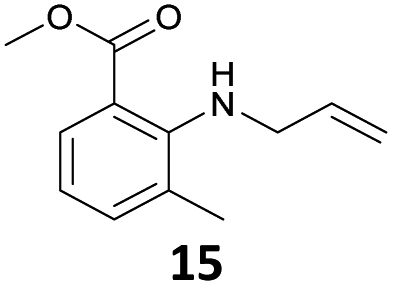
The steric effect is clearly shown in the comparison of the syntheses of 14 and 15. The monoallylation of methyl anthranilate gave 14 in 63% yield, which indicated that the reactivity did not reduce because monoallylation occurred at the sterically less hindered N–H bond. In addition, the stabilization by the intramolecular hydrogen bonding between the carbonyl oxygen and hydrogen at H–N accelerated the monoallylation of the aniline with the stabilization of the given 14. On the contrary, the reaction of the methyl 2-amino-3-methylbenzoate with 1 showed the low yield of 15 in 29% yield. This was because the reaction site of the aniline was sterically hindered by the methyl substituent at the 3-position of the methyl benzoate.
The reaction was not affected by atmospheric conditions. The average yields of 3 in the two experiments were 64% (under Ar) and 61% (under air), respectively (Table S7†). The developed 10 wt% WO3/ZrO2 catalyst did not show any reactivity for the N-allylation of the aliphatic amines, such as n-propyl amine, n-dodecyl amine, benzylamine, and cyclohexylamine, because they would be strongly adsorbed on the 10 wt% WO3/ZrO2 catalyst to deactivate the W–OH–Zr active acidic sites (Table S8†). Also, the developed 10 wt% WO3/ZrO2 catalyst did not apply to the selective monoallylation using other allylic alcohols, for example, the reaction using trans-2-buten-1-ol instead of 1 resulted in the formation of complex mixtures.
Reaction mechanism
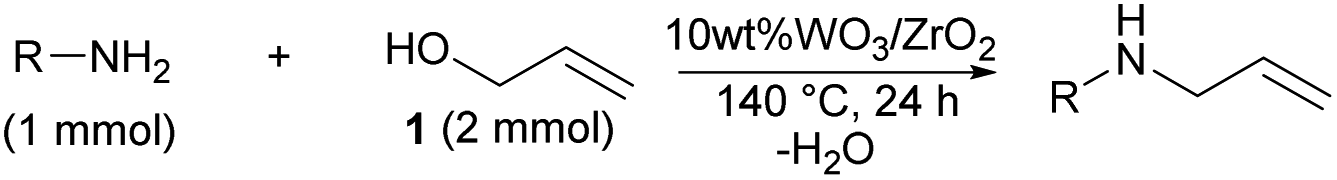
The reaction mechanism was considered based on the probe experiments and the screening of the anilines. Benzyl alcohol reacted with 2 to give benzyl aniline (16) in 62% yield (Scheme 1(a)). The developed catalytic reaction was not only allowed to 1 but also allowed to the electron-deficient carbon compounds such as benzyl alcohol by the nucleophilic attack of the N-atom of 2. The allylation of 3 with 1 at 140 °C for 4 h did not give 4 in good yield, but the recovery of 3 was observed in 95% yield (Scheme 1(b)). The steric repulsion between the allyl moiety of 3 and the surface of 10 wt% WO3/ZrO2 hindered over allylation.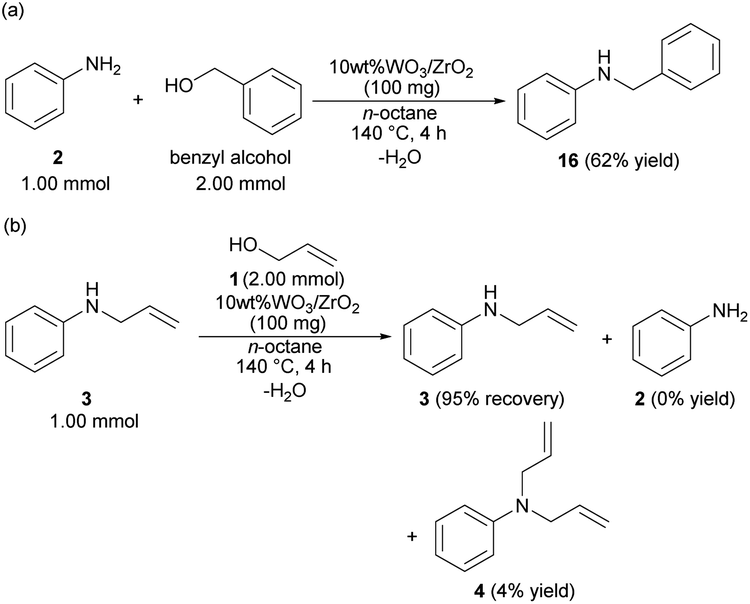 | ||
| Scheme 1 Probe experiments using 10 wt% WO3/ZrO2; (a) reaction of 2 with benzyl alcohol; (b) reaction of 3 with 1. | ||
The employed 10 wt% WO3/ZrO2 catalyst was estimated to form W–OH–Zr structures on its surface (Fig. 1).19a,b Besides, the support ZrO2 is known to work as a bifunctional solid acid and solid base.24 From these aspects with the probe reactions and the screening of the anilines, the allylation process seemed to proceed through the nucleophilic attack of the N-atom of 2 to the allyl moiety of 1.
Also, the activation of the N-atom of 2 by the basic site of ZrO2 and the activation of the allyl moiety of 1 by the Brønsted acidic site of W–OH–Zr would be effective in accelerating the nucleophilic attack of 2 (Fig. 4a). Therefore, 3 did not proceed the allylation with 1, as 3 could not access the surface of ZrO2 due to the steric repulsion between its allyl moiety and the surface of 10 wt% WO3/ZrO2 (Fig. 4b). In the case of MoO3/TiO2, the allyl-oxo molybdenum species had high nucleophilicity to receive the attack from the N-atom of 2 without any activation of aniline.15 However, 10 wt% WO3/ZrO2 did not form an allyl-oxo tungstate intermediate. And a direct contact between aniline and ZrO2 was needed for proceeding the allylation process. Therefore, the hindered random surface structure of 10 wt% WO3/ZrO2 directly affected the selective synthesis of 3 (Fig. 4).
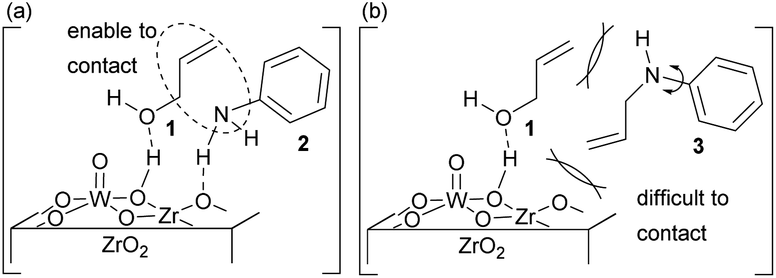 | ||
| Fig. 4 Active sites of 10 wt% WO3/ZrO2 during the allylation of (a) 2 with 1 and (b) 3 with 1. | ||
Conclusions
In this study, selective dehydrative monoallylation of anilines was performed by using optimized amounts of WO3 supported with a ZrO2 catalyst (10 wt% WO3/ZrO2). Due to the preservation of the optimized acid density of the WO3 on ZrO2 and the support of the basic sites on ZrO2 during the monoallylation process, 10 wt% WO3/ZrO2 could catalyze the allylation of 2 to give a corresponding monoallyl aniline 3 up to 78% yield with 93% selectivity. The developed 10 wt% WO3/ZrO2 catalyst could proceed the monoallylation process in a fixed-bed flow reactor for 15 hours to continuously produce 3 with over 97% selectivity. From the performed XPS analyses before and after the reaction, the preservation of WO3 with the W(VI) oxidation state was confirmed during the allylation process. Considering the probe reactions and substrate scope, the steric repulsion between the allyl substituent of the monoallyl aniline and the random structure of the 10 wt% WO3/ZrO2 surface prevented the over allylation of the monoallyl aniline.Author contributions
Conceptualization, investigation, and writing, Y. K.; investigation, S. T. and S. Y. formal analysis, T. Y.; supervision and formal analysis, T. F.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is partly based on results obtained from a project, JPNP20005, subsidized by the New Energy and Industrial Technology Development Organization (NEDO), Japan. We express our thanks to Mr Takuya Nakashima and Mr Makoto Kitai for useful discussion.Notes and references
-
(a) M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689 CrossRef CAS PubMed
; (b) L. Krähling, J. Krey, G. Jakobson, J. Grolig and L. Miksche, “Allyl Compounds” Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed
- Recent examples of transformations from monoallyl anilines, see:
(a) H. Egami, S. Kawamura, A. Miyazaki and M. Sodeoka, Angew. Chem., Int. Ed., 2013, 52, 7841 CrossRef CAS PubMed
- J. Muzurt, Eur. J. Org. Chem., 2007, 3077 CrossRef
-
(a) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed
- Y. Tamaru, Eur. J. Org. Chem., 2005, 2647 CrossRef CAS
-
(a) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467 RSC
- For recent reviews for the formation of allyl alcohol from glycerol, see:
(a) S. Raju, M.-E. Moret and R. J. M. K. Gebbink, ACS Catal., 2015, 5, 281 CrossRef CAS
-
(a) F. Ozawa, H. Okamoto, S. Kawagishi, S. Yamamoto, T. Minami and M. Yoshifuji, J. Am. Chem. Soc., 2002, 124, 10968 CrossRef CAS PubMed
- S. T. Madrahimov, D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7228 CrossRef CAS PubMed
-
(a) T. Ohshima, Y. Miyamoto, J. Ipposhi, Y. Nakahara, M. Utsunomiya and K. Mashima, J. Am. Chem. Soc., 2009, 131, 14317 CrossRef CAS PubMed
-
(a) Y. Kita, H. Sakaguchi, Y. Hoshimoto, D. Nakauchi, Y. Nakahara, J.-F. Carpentier, S. Ogoshi and K. Mashima, Chem.–Eur. J., 2015, 21, 14571 CrossRef CAS PubMed
- The reuses of Pd complex catalyst and Pt homogeneous catalyst have been carried out8g,10d A. S. Alshammari, K. Natte, N. V. Kalevaru, A. Bagabas and R. V. Jagadeesh, J. Catal., 2020, 382, 141 CrossRef CAS
-
(a) K. Masuda, T. Ichitsuka, N. Koumura, K. Sato and S. Kobayashi, Tetrahedron, 2018, 74, 1705 CrossRef CAS
- K. Motokura, N. Nakagiri, T. Mizugaki, K. Ebitani and K. Kaneda, J. Org. Chem., 2007, 72, 6006 CrossRef CAS PubMed
- Y. Kon, T. Nakashima, T. Fujitani, T. Murayama and W. Ueda, Synlett, 2019, 30, 287 CrossRef CAS
-
(a) S. Matsubara, T. Okazoe, K. Oshima, K. Takai and H. Nozaki, Bull. Chem. Soc. Jpn., 1985, 58, 844 CrossRef CAS
- K. Arata, “Preparation of Superacidic Metal Oxides and Their Catalytic Action” Metal Oxide Catalysis, Wiley-VCH, Weinheim, 2009, vol. 2 Search PubMed
- For examples;
(a) J. Chai, S. Zhu, Y. Cen, J. Guo, J. Wang and W. Fan, RSC Adv., 2017, 7, 8567 RSC
-
(a) D. G. Barton, M. Shtein, R. D. Wilson, S. L. Soled and E. Iglesia, J. Phys. Chem. B, 1999, 103, 630 CrossRef CAS
-
(a) Y. Izumi, K. Matsuo and K. Urabe, J. Mol. Catal., 1983, 18, 299 CrossRef CAS
- Reviews for cationic ion exchange resins, see: A. Chakrabarti and M. M. Sharma, React. Polym., 1993, 20, 1 CrossRef CAS
- The 50% conversion of 2 (0.5 mmol) in Table 1, entry 11 was in good accordance with the amounts of active –SO3H groups (> 0.47 mmol) in amberlyst 15DRY (100 mg) calculated from the ion exchange capacity (>4.7 mol kg−1)..
- W surface densities (W nm−2) were calculated by the following equation considering the paper from C. Thomas, J. Phys. Chem. C, 2011, 115, 2253 and ref. 19d: W surface density (W nm−2) = [(wt% W/100)/MW × 6.023 × 1023]/[S.A. × 1018/(1 − (wt% W/100)) × (MWO3/MW)]..
- K. AlGhamdi, J. S. J. Hargreaves and S. D. Jackson, “Base Catalysis with Metal Oxides” Metal Oxide Catalysis, Wiley-VCH, Weinheim, 2009, vol. 2 Search PubMed
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder, Handbook of X-ray photoelectron spectroscopy, ed. G. E. Mullemberg, PerkinElmer, Eden Prairie, MN, 1978 Search PubMed
- TOF (h−1) was calculated as the following equation: [3 obtained from the allylation of 2 with 1 by passing through a catalyst column for 1 h (mmol)]/[WO3 in a catalyst column (mmol)]..
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00198e |
| This journal is © The Royal Society of Chemistry 2022 |