
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal-catalyzed remote C–H functionalization of thioethers
Xiao-Qing Feng
a,
He-Cheng Wang
a,
Zhi Li
b,
Long Tang
a,
Xiaoqiang Sun
b and
Ke Yang
 *b
*b
aSchool of Pharmacy & School of Medicine, Changzhou University, Changzhou, Jiangsu 213164, China
bJiangsu Key Laboratory of Advanced Catalytic Materials & Technology, School of Petrochemical Engineering, Changzhou University, Changzhou, Jiangsu 213164, China. E-mail: keyang@cczu.edu.cn
First published on 7th April 2022
Abstract
In the last decade, transition-metal-catalyzed direct C–H bond functionalization has been recognized as one of most efficient approaches for the derivatization of thioethers. Within this category, both mono- and bidentate-directing group strategies achieved the remote C(sp2)–H and C(sp3)–H functionalization of thioethers, respectively. This review systematically introduces the major advances and their mechanisms in the field of transition-metal-catalyzed remote C–H functionalization of thioethers from 2010 to 2021.
 Xiao-Qing Feng | Xiao-Qing Feng got her master's degree at Jiangnan University. Next, she received her Ph.D. degree from Fudan University in 2010. Then, she joined Changzhou University. Her research focuses on pharmaceutical chemistry and organic synthesis. |
 He-Cheng Wang | He-Cheng Wang graduated and got his Bachelor's degree from Taizhou Institute of Science and Technology, Nanjing University of Science and Technology. Since 2020, he began his research as a master's degree candidate at Changzhou University. Currently, his research interests involve pharmaceutical chemistry and organic synthesis. |
 Zhi Li | Zhi Li completed his B.E. degree in Applied Chemistry in Changshu Institute of Technology. Since 2020, he has begun his master's degree studies in the School of Petrochemical Engineering at Changzhou University and has joined the Yang group. Currently, his research interests involve novel transition metal-catalyzed C–H bond functionalization. |
 Long Tang | Long Tang received his master's degree in Organic Chemistry from Changzhou University. Since 2019, he began to study for a doctorate at the Chinese Academy of Forestry. At present, his research interest involves biomass organic synthetic chemistry. |
 Xiaoqiang Sun | Xiaoqiang Sun obtained his Ph.D. degree from Nanjing University in 1988. Then he became a postdoctoral fellow at the University of Sheffield and University of Birmingham under the supervision of Prof. J. Fraser Stoddart. After that, he became a Professor at Changzhou University in 1997. His current research interests involve pharmaceutical design and synthesis, and supramolecular chemistry. |
 Ke Yang | Ke Yang obtained his Ph.D. degree from Nanjing University in 2014. After that, he became a postdoctoral fellow at Nanjing University with Professor Guigen Li. In 2015, he joined in the group of Professor Haibo Ge at the Department of Chemistry and Chemical Biology, Indiana University – Purdue University Indianapolis (IUPUI) in USA. In 2017, he moved to Changzhou University and started his independent academic career. His research interests involve novel transition metal catalysis, green organic synthesis and pharmaceutical chemistry. |
1. Introduction
In the last decade, transition-metal-catalyzed C–H bond functionalization stands out as one of the most efficient and economical approaches for the C–C bond or C–X bond constructions.1 Different kinds of mono- and bidentate-directing groups have been employed to control selectivity and enhance the efficiency of C–H bond functionalization.2,3 In this field, remote C–H bond functionalization is performed by using transition-metal catalysts and thioether directing groups. In this process, the transition-metals can coordinate with the internal sulfur atoms to achieve this issue. More importantly, as a traceless directing group, the thioether group is easily removed or transformed to other useful functional groups, which dramatically improves the practicability of this method.4Meanwhile, thioethers are also important ubiquitous skeletons in a variety of pharmaceutical drugs, natural products, and organic materials.5 Furthermore, thioethers can also be used as transition-metal ligands and organo-catalysts in synthetic chemistry.6 However, previous reviews on thioethers mainly focused on the C–S bond activation and transformations.7 In comparison, only a few related reviews highlighted the remote C–H functionalization of thioethers. In 2019, the Sun group introduced different S-containing directing groups, including thioether, thioamide, sulfoxide, thioketone and alkoxythiocarbonyl, in transition metal-catalyzed C–H functionalization reactions.8a Very recently, Zhao group also provided a similar review.8b However, the above two reviews only briefly describe the recent advances on thioethers, and there are no detailed mechanism discussions. In addition, the direct α-C(sp3)–H functionalization of thioethers also represents another important strategy to prepare novel thioethers.9
In this review, we will systematically introduce and discuss the significant progress made in the field of transition-metal-catalyzed remote C–H functionalization of thioethers by using thioethers themselves as directing groups from 2010 to 2021. Meanwhile, the detailed mechanisms for these reactions are also disclosed. Furthermore, thioether-directed asymmetric C–H bond functionalization is firstly introduced.
2. Remote C(sp2)–H functionalization
Transition metal catalyzed/mediated selective functionalization of either C(sp2)–H or C(sp3)–H bonds by employing thioethers as directing groups via coordination of the internal sulfur atom to transition metal species has been well developed. This strategy enables direct remote C–H bond functionalization of thioethers to efficiently construct a series of novel thioether compounds. In 2012, the Zhang group reported a thioether-directed Pd(II)-catalyzed C(sp2)–H olefination reaction of unactivated arenes (Scheme 1a).10 This is the first example of thioether utilization as a sole guiding group. Different acrylates were selectively incorporated into thioether compounds in moderate to good yields. In this work, an acetate-bridged dinuclear cyclopalladation intermediate 1A was isolated and had its structure confirmed by single crystal X-ray diffraction, providing strong evidence on directed C–H bond activation. Furthermore, mechanistic studies suggest that the C(sp2)–H cleavage process may be a turnover-limiting step (Scheme 1b). This study proposes a plausible catalytic cycle involving Pd(II)/Pd(0) catalysis for this transformation (Scheme 1c). The thioether-directed C–H bond cleavage of 1a with the Pd(OAc)2 catalyst provides the dinuclear cyclopalladation intermediate 1A. Next, alkene insertion takes place to produce the carbopalladation intermediate 1B. Then, β-hydride elimination of the intermediate 1B and subsequent reductive elimination afford the desired product 3a and regenerate the Pd(0) catalyst. Finally, the Pd(0) catalyst is re-oxidized to Pd(II) by AgOTFA.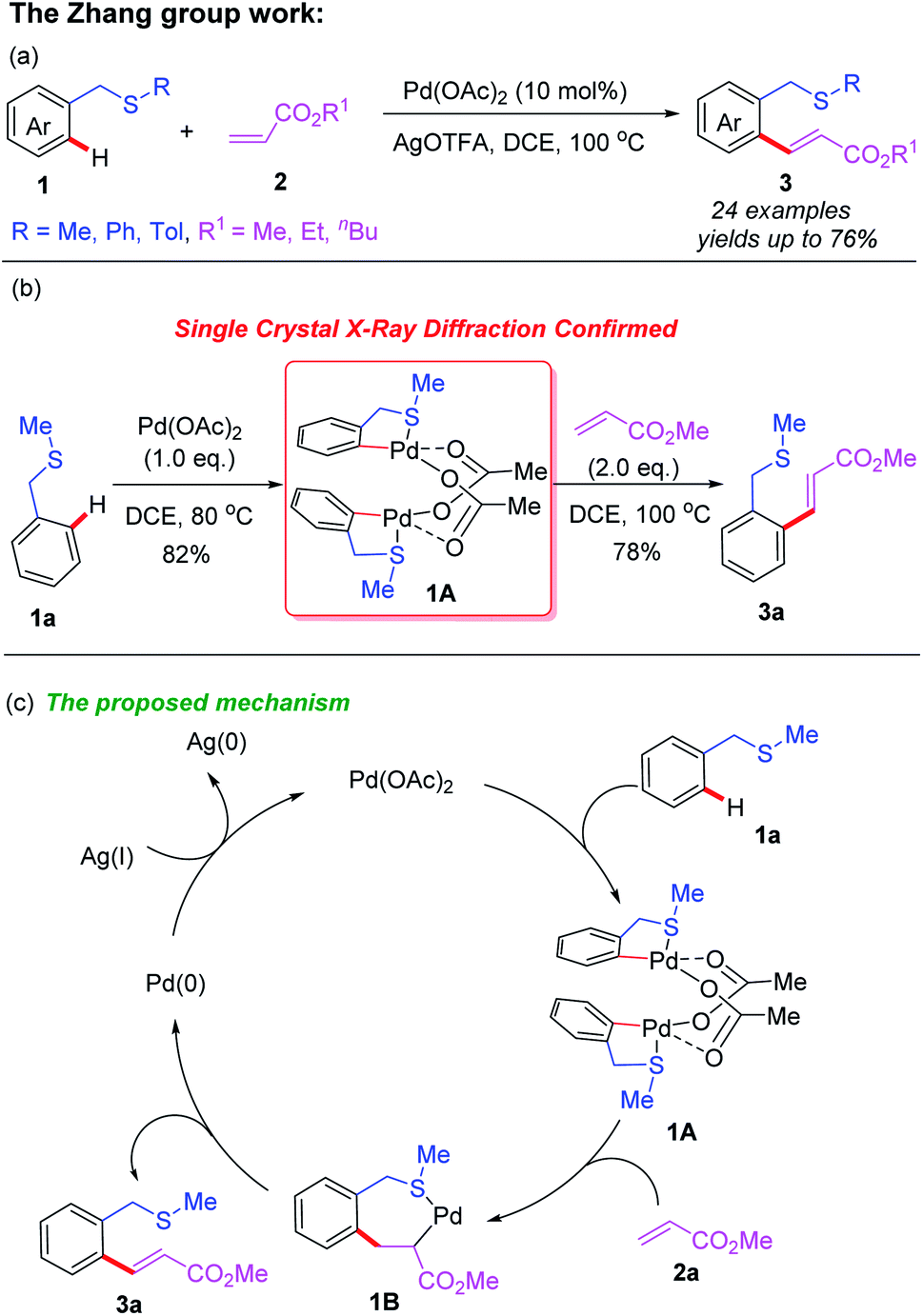 | ||
| Scheme 1 (a) Thioether-directed Pd(II)-catalyzed C(sp2)–H olefination of unactivated arenes. (b) Single crystal X-ray diffraction experiment. (c) The proposed mechanism. | ||
On the basis of the aforementioned research, a thioether-directed C(sp2)–H arylation reaction of unactivated arenes catalyzed by a palladium(II) catalyst was also reported by the same group (Scheme 2a).11 Various potassium aryltrifluoroborates were proven to be suitable coupling partners, leading to desired biaryl thioether derivatives. Notably, p-benzoquinone (BQ) was shown to be an important additive for the reductive elimination step in this reaction. Subsequently, they demonstrated an efficient Pd(II)-catalyzed acetoxylation of aromatic C(sp2)–H bonds by employing thioether as a sole directing group (Scheme 2b).12 Both benzyl and phenethyl sulfides reacted effectively with PhI(OAc)2 and Ac2O to provide the desired acetoxylated products in moderate to good yields. Furthermore, a plausible catalytic cycle involving Pd(II)/Pd(IV) catalysis is also proposed in Scheme 3. First, the coordination of substrate 6 to Pd(OAc)2 followed by a thioether-directed C–H bond activation produces the Pd(II) species 2A. Next, the oxidative addition of the intermediate 2A with PhI(OAc)2 and Ac2O generates the Pd(IV) complex 2B. Finally, the reductive elimination of intermediate 2B affords the desired product 7 and releases the active Pd(II) species.
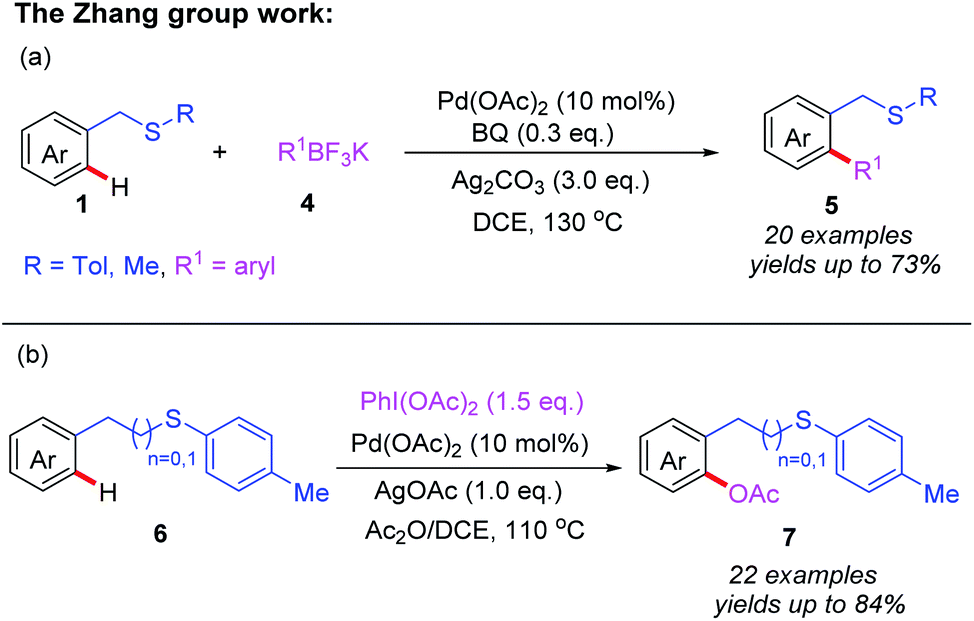 | ||
| Scheme 2 (a) Thioether-directed Pd(II)-catalyzed C(sp2)–H arylation of unactivated arenes. (b) Thioether-directed Pd(II)-catalyzed C(sp2)–H acetoxylation of unactivated arenes. | ||
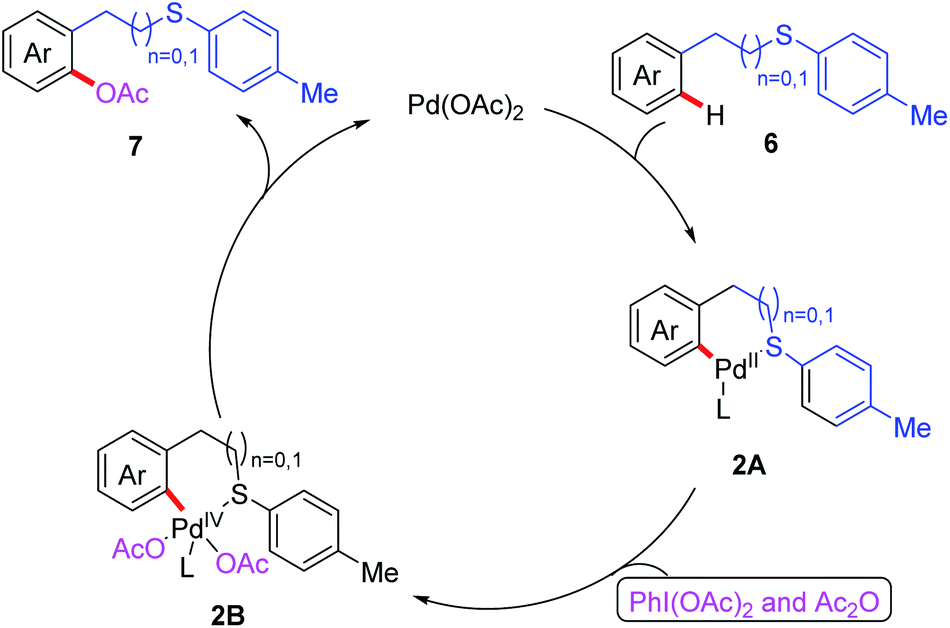 | ||
| Scheme 3 Proposed mechanism of thioether-directed Pd(II)-catalyzed C(sp2)–H acetoxylation reaction. | ||
Moreover, the Kuang group also reported a similar protocol to achieve Pd(II)-catalyzed C(sp2)–H acylation directed by a thioether group (Scheme 4a).13 In this reaction, thioethers bearing an electron-donating (Me, i-Pr, or MeO) or electron-withdrawing (F or Cl) group were well-tolerated and provided the corresponding acylated products with good yields. However, only trace products were detected with thioethers containing a strong electron-withdrawing (NO2 or CN) group. In addition, a plausible catalytic cycle is also shown in Scheme 4b. First, the thioether-directed coordination of substrate 1 to Pd(OAc)2 with a subsequent cyclopalladation process afford the Pd(II) complex 3A. Meanwhile, a silver-mediated decarboxylation of α-oxocarboxylic acid 8 produces acyl silver species 3B. After that, transmetalation of the Pd(II) complex 3A with the acyl silver species 3B gives the Pd(II) complex 3C. Reductive elimination of 3C generates the desired product 9, and the Pd(0) species. Finally, re-oxidation of the Pd(0) species by Ag2CO3 regenerates the Pd(II) catalyst. In addition, the Shi group also reported a thioether-directed and oxidant-controlled selective mono- or di-alkenylation of C(sp2)–H bonds of biaryl compounds (Scheme 4b).14 When AgOTFA was employed as an oxidant, mono-alkenylated products were obtained in moderate to good yields, whereas di-alkenylated products were generated with AgNO3 as an oxidant instead.
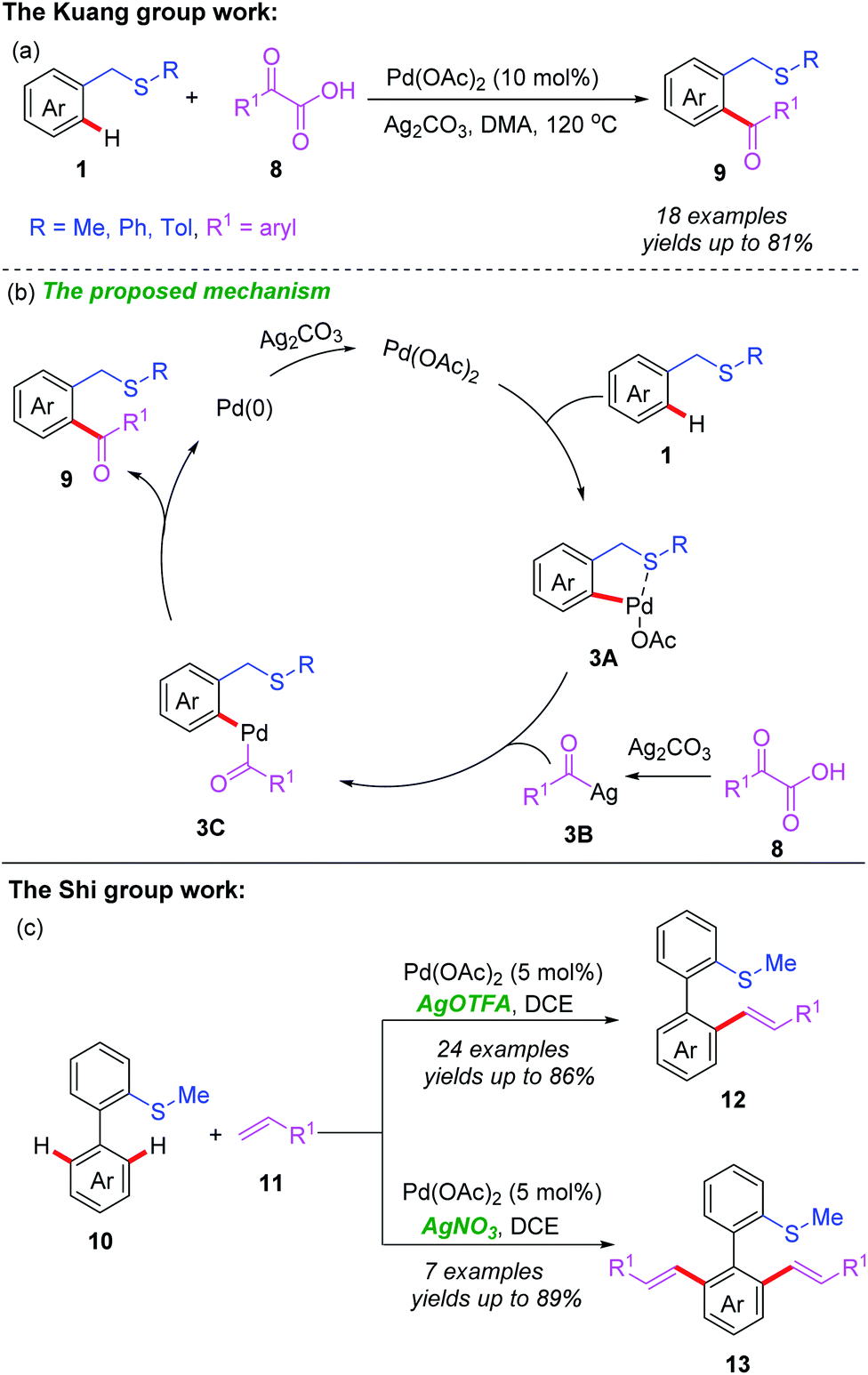 | ||
| Scheme 4 (a) Pd(II)-catalyzed C(sp2)–H acylation reactions directed by a thioether group. (b) The proposed mechanism of acylation. (c) Pd(II)-catalyzed C(sp2)–H alkenylation reactions directed by a thioether group. | ||
In addition to palladium catalysts, other transition metal catalysts, such as Rh, Ru and Ir, could also be utilized in the C(sp2)–H functionalization reactions.15 In 2013, the Shi group reported the first example of Rh(III)-catalyzed thioether-directed C(sp2)–H alkenylation reactions (Scheme 5).16 The substrate scope indicated that both mono- and di-substituted alkenylated products could be obtained in good yields by tuning the solvents. Later, a catalytic cycle is proposed (Scheme 6): the initial pre-coordination of the Rh(III) catalyst to the substrate 1 generates the rhodium intermediate 4A. Next, in the presence of the OAc anion, a thioether-directed C–H bond activation step provides the 5-membered intermediate 4C through a CMD pathway. The subsequent alkene insertion followed by a β-hydride elimination process affords the final product 14 and releases the rhodium intermediate 4A.
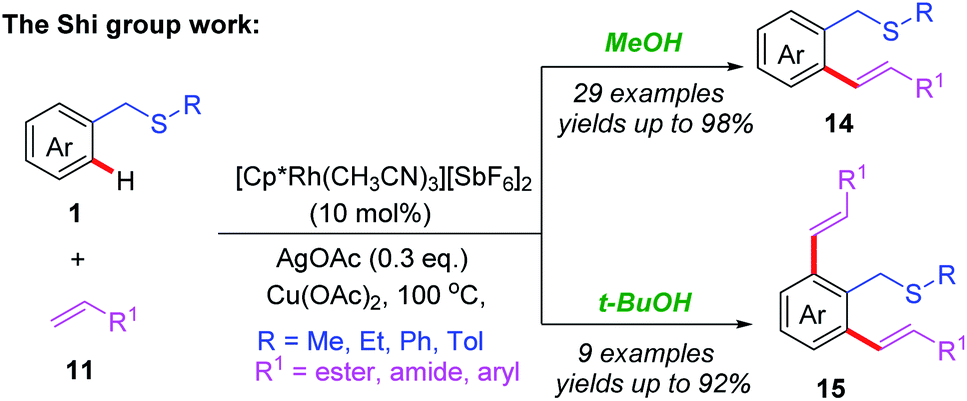 | ||
| Scheme 5 Rh(III)-catalyzed and thioether-directed C(sp2)–H alkenylation reactions. | ||
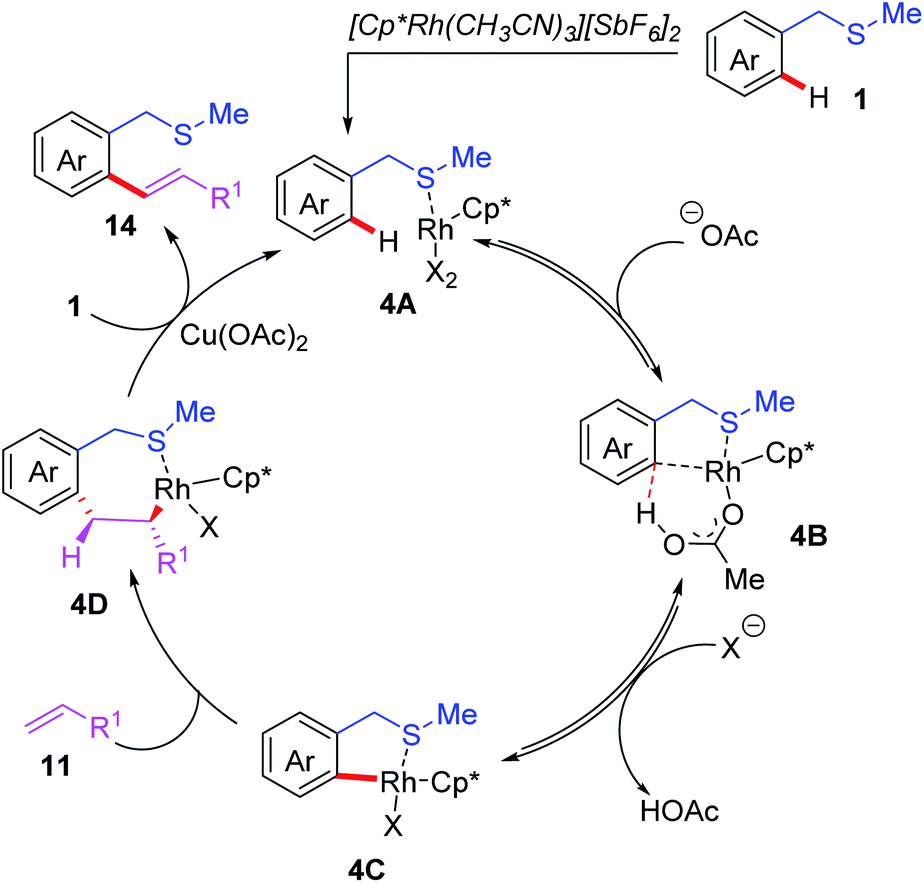 | ||
| Scheme 6 Proposed mechanism of Rh(III)-catalyzed and thioether-directed C(sp2)–H alkenylation reactions. | ||
In 2018, the Miura group reported the first example of rhodium(I)-catalyzed alkenylation of 1-(methylthio)naphthalene with alkynes and alkenes through a thioether-directed C–H bond functionalization process (Scheme 7a).17 This reaction was selectively carried out in the peri-position of the naphthalene ring. When alkynes were used as coupling counterparts, a mixture of E/Z-isomers were produced, while alkene couples afforded the E-configuration products. Later, they reported a Rh(I)-catalyzed direct C–H arylation reaction of naphthalene derivatives by employing a thioether directing group (Scheme 7b).18 The reaction exhibited an excellent peri-selectivity and broad functional group compatibility. Furthermore, the arylated product was very susceptible to the sulfur-containing poly aromatic compound. Additionally, the same group demonstrated a thioether-directed selective C–H alkenylation of indoles under rhodium catalysis (Scheme 7c).19 Using different thioether directing groups including MeS, CyS, and PhS, more than 36 examples of C4-alkenylated indoles were isolated in excellent yields (up to 97%). The authors speculated that the formation of 5-membered metallacycle intermediates was the key for this transformation. Moreover, a plausible catalytic cycle involving Rh(III)/Rh(I) catalysis is depicted in Scheme 8. First of all, the initial coordination of substrate 16 and Cp*Rh(III) catalyst gives the intermediate 5A. Next, the C–H bond activation assisted by thioether group provides the intermediate 5B. Subsequently, the alkene insertion with a β-hydride elimination generates the desired product 20 with Cp*Rh(I) catalyst.
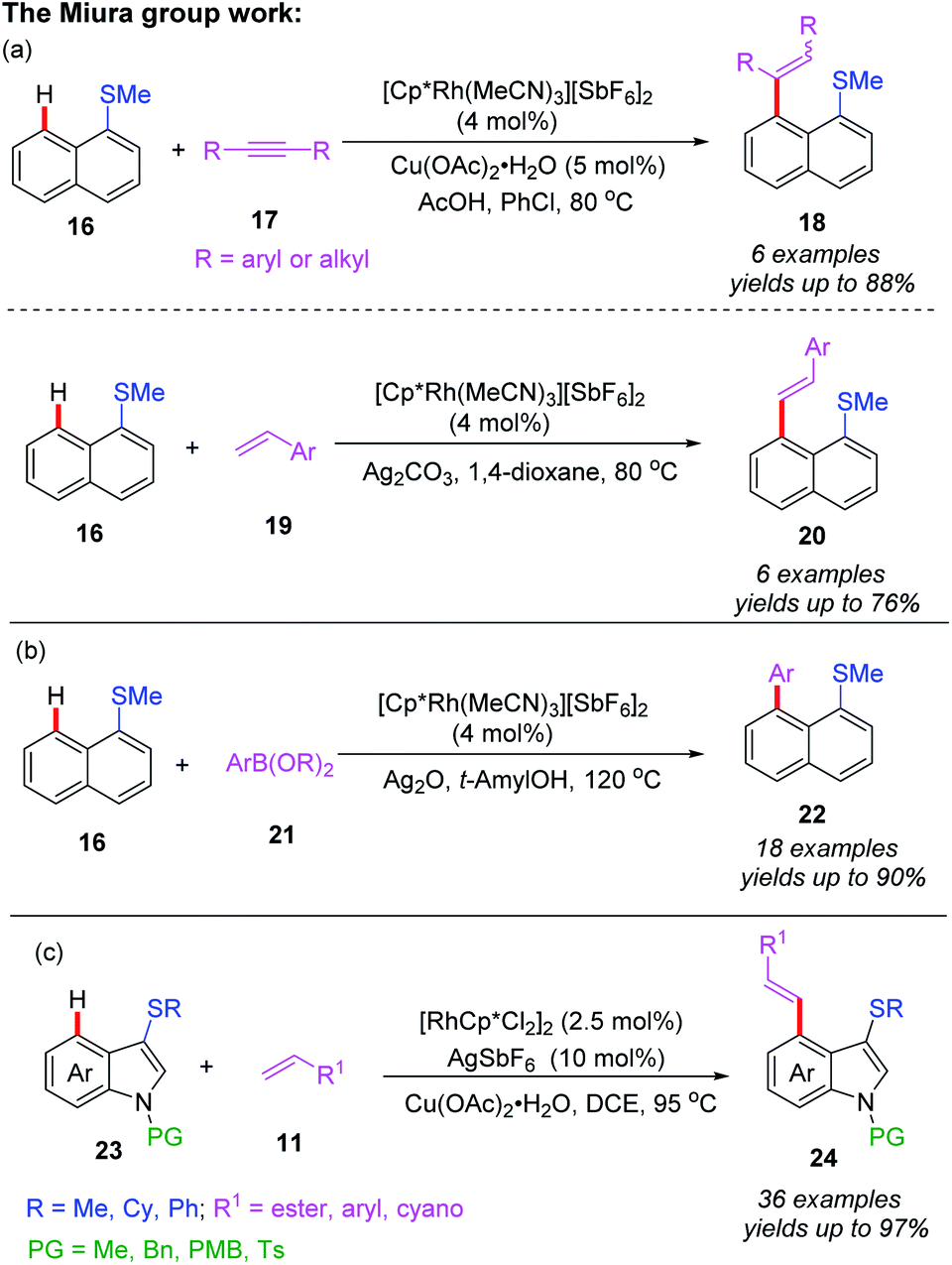 | ||
| Scheme 7 (a) Rh(III)-catalyzed and thioether-directed C–H alkenylation of 1-(methylthio)naphthalene. (b) Rh(III)-catalyzed and thioether-directed C–H arylation of 1-(methylthio)naphthalene. (c) Rh(III)-catalyzed and thioether-directed C–H alkenylation of indole derivatives. | ||
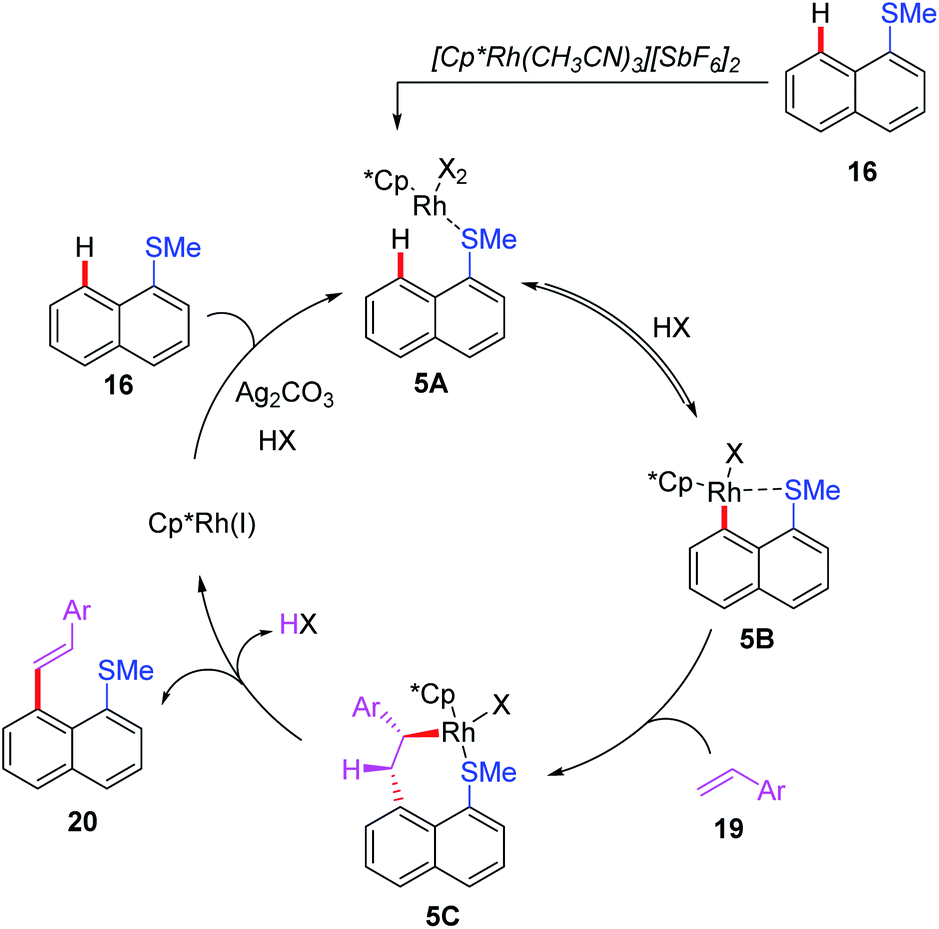 | ||
| Scheme 8 Proposed mechanism of Rh(III)-catalyzed and thioether-directed C–H alkenylation of 1-(methylthio)naphthalene. | ||
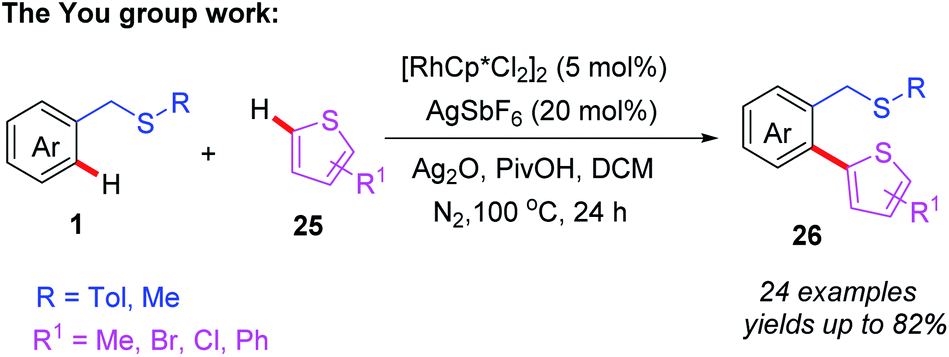
Recently, the You group reported an efficient Rh(III)-catalyzed thioether-directed C–H/C–H cross coupling of benzylthioethers with thiophenes to construct various ortho-functionalized 2-aryl thiophenes (Scheme 9).20 The substrate scope studies indicated that both ortho- and meta-substituted substrates produce mono-heteroarylated benzylsulfides products, while para-substituted substrates mainly gave rise to the di-heteroarylated products. Moreover, a plausible catalytic cycle involving Rh(III)/Rh(I) catalysis is depicted (Scheme 10). First, coordination of the Rh(III) catalyst to the thioether 1 followed by thioether-directed C–H bond activation provides the rhodium intermediate 6A. Then, the intermediate 6A reacts with the thiophene to generate the rhodium intermediate 4B. Next, reductive elimination of intermediate 6B affords the desired product 26 and generates the Rh(I) catalyst. In the presence of a silver salt, the Rh(I) catalyst is then oxidized to Rh(III) for the following catalytic cycle.
 | ||
| Scheme 9 Rh(III)-catalyzed and thioether-directed C–H/C–H cross coupling of benzylthioethers with thiophenes. | ||
 | ||
| Scheme 10 The proposed mechanism of Rh(III)-catalyzed and thioether-directed C–H/C–H cross coupling of benzylthioethers with thiophenes. | ||
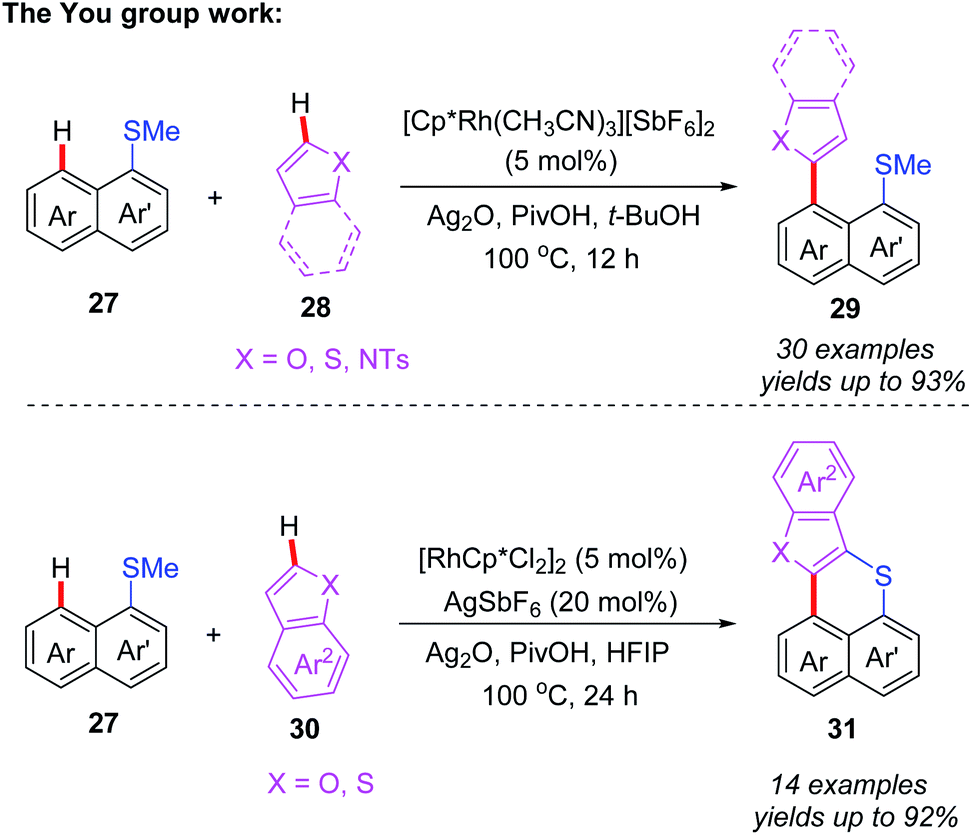
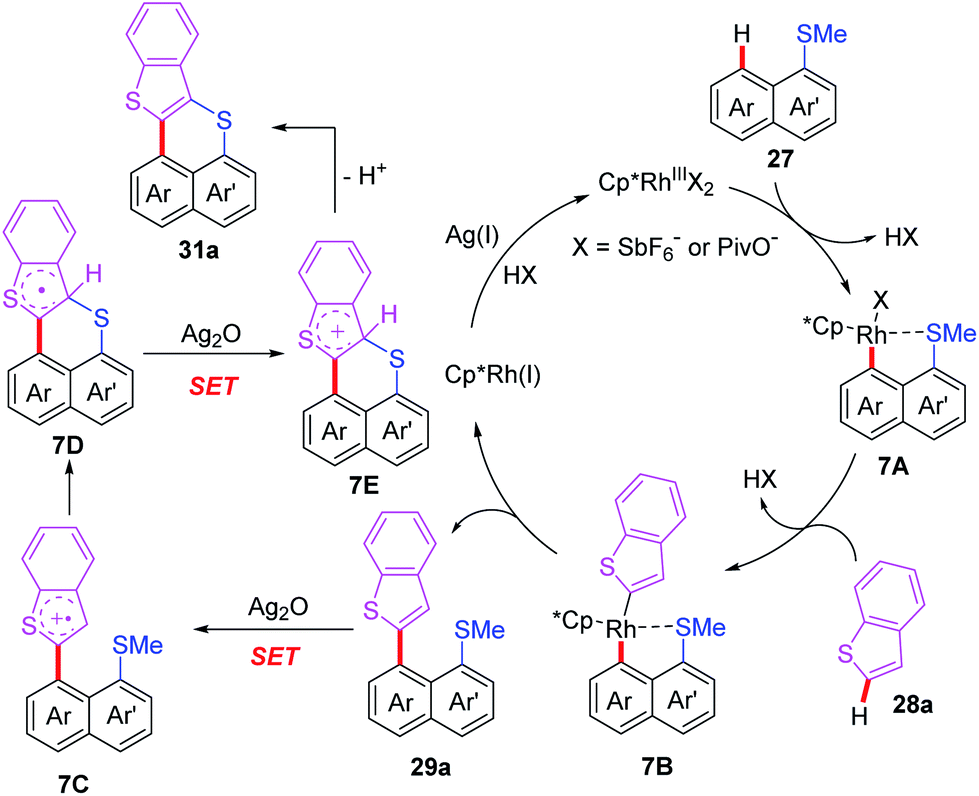
After that, the same group also reported a Rh-catalyzed peri-selective heteroarylation of 1-(methylthio)naphthalenes and aromatic heterocyclic compounds (Scheme 11).21 Furthermore, when HFIP was instead of t-BuOH, benzo[de]thioacenes were formed through a subsequent Ag-mediated intramolecular cyclization process. Different from a traditional acid-mediated sulfoxide cyclization reaction, the detailed mechanism studies indicate that this process involves a SET cyclization of cationic radical and methanethiol group. Furthermore, a plausible mechanism is also proposed (Scheme 12). The rhodium intermediate 7A is formed through the coordination of Cp*Rh(III) catalyst and substrate 27 with a subsequent thioether-directed peri-C–H bond activation. Next, the reaction of benzothiophene with intermediate 7A affords the rhodium intermediate 7B, which can be further converted to the coupled product 29a and Cp*Rh(I) species through a reductive elimination process. The Cp*Rh(I) species can be re-oxidized to Cp*Rh(III) catalyst for the next catalytic cycle. It's worth noting that, when HFIP is reaction solvent, the coupled product 29a can be transformed to cation-radical intermediate 7C through a Ag2O-mediated SET process. The subsequent electrophilic cyclization and demethylation provide intermediate 7D. Next, the second SET oxidation and followed by a deprotonation generate the final product 31a.
 | ||
| Scheme 11 Rh-catalyzed peri-selective heteroarylation/Ag-mediated SET intramolecular cyclization sequence of 1-(methylthio)naphthalenes and heteroatom-embedded analogues. | ||
 | ||
| Scheme 12 Proposed mechanism of Rh-catalyzed peri-selective heteroarylation/Ag-mediated SET intramolecular cyclization reactions. | ||
Normally, the system of iridium/bipyridine-catalyzed C–H borylation often achieves meta- and para-(C–H) functionalization reactions.22 However, the Kanai and Kuninobu groups developed an iridium/bipyridine-catalyzed ortho-C–H borylation of methylthiomethyl protected phenol and aniline derivatives (Scheme 13).23 In this work, a methylthiomethyl group was utilized as a fixed directing group, and bipyridine-type ligand L1 as a chiral ligand. The desired borylated products were obtained in good yields with good functional group tolerance. Furthermore, the authors propose two possible reaction mechanisms. The first pathway undergoes outer spherical Lewis acid–base interaction between the boron-based ligand and the thioether group (8A). The second proceeds through the coordination of the thioether auxiliary, as a directing group, to the iridium catalyst (8B).
 | ||
| Scheme 13 Iridium/bipyridine-catalyzed ortho-C–H borylation of methylthiomethyl protected phenol and aniline derivatives. | ||
Recently, the Miura group also reported an Ir-catalyzed, thioether-directed, C4-selective acyl-methylation of indoles employing α-carbonyl sulfoxonium ylides as carbene precursors (Scheme 14a).24 Both electron-withdrawing and electron-donating groups on the aromatic rings of indoles and α-carbonyl sulfoxonium ylides were well-tolerated in this reaction. A catalytic cycle is proposed in Scheme 14b. The initial cyclometalation of indole substrate 23 with Ir catalyst generates the intermediate 9A together with HX. Next, the coordination of sulfoxonium ylide affords the species 9B. A reactive carbene species 9C is then formed undergoes α-elimination of DMSO. The subsequent migratory insertion followed by a protonolysis process gives the desired product 35 and regenerates the active catalyst.
 | ||
| Scheme 14 (a) Ir-catalyzed and thioether-directed C4-selective acylmethylation of indoles. (b) The proposed mechanism. | ||
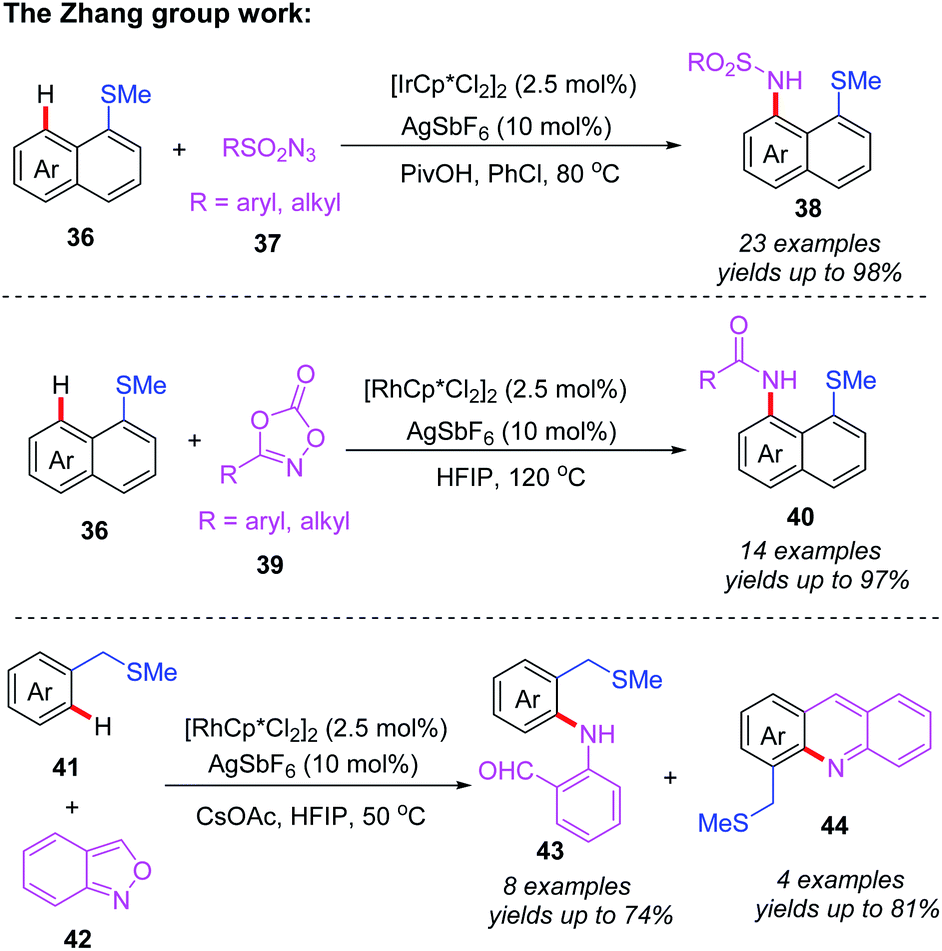
Very recently, the Zhang group reported thioether-directed, Cp*Ir(III)- and Cp*Rh(III)-catalyzed C(sp2)–H amination of unactivated arenes by using different nitrogen sources (Scheme 15).25 Three types of nitrene precursor reagents inducing dioxazolone, anthranil and azide compounds, were converted to the desired amidated products in moderate to good yields. It should be noted that it is first time to use Cp*Ir(III)- and Cp*Rh(III) catalysts for thioether-directed C–H amination reactions. The further mechanistic experiments show that the C–H bond cleavage process may not be involved in a rate-determining step. In addition, a plausible catalytic mechanism is outlined in Scheme 16. The initial ligand exchange and thioether-directed C–H bond activation produce the cyclometallated intermediate 10A. Then, the resulting intermediate 10A reacts with nitrene precursors, affording the intermediate 10B. After that, α-migratory insertion followed by a protonation process provides the final product. Besides, compound 43 can be transformed to product 44 through a Friedel–Crafts cyclization and subsequent dehydration.
 | ||
| Scheme 15 Thioether-directed, Cp*Ir(III)- and Cp*Rh(III)-catalyzed C(sp2)–H amination of unactivated arenes by using different nitrogen sources. | ||
 | ||
| Scheme 16 (a) Proposed mechanism of thioether-directed Cp*Ir(III)- and Cp*Rh(III)-catalyzed C(sp2)–H amination reactions. (b) Friedel–Crafts cyclization and subsequent dehydration. | ||
In 2015, Ru-catalyzed, thioether-directed regioselective C–H hydroarylation of alkynes with benzylthioethers was developed by Urriolabeitia and Villuendas (Scheme 17a).26 A variety of alkenylated benzylthioethers were prepared from thioether substrates and electron-rich alkynes in good yields under microwave irradiation and short reaction times. A plausible catalytic cycle is depicted in Scheme 17b. First, the pre-coordination of KPF6 to the Ru catalyst provides the Ru species 11A. Meanwhile, the coordination of the substrate 1 to Cu(OAc)2 forms intermediate 11B which prevented the poisoning of the Ru species. Next, the reaction of intermediate 11B and Ru species 11A followed by a selective C–H bond cleavage produces the Ru intermediate 11D, which reacts with the internal alkyne through π-bonding and migratory syn-insertion, leading to the formation of Ru intermediate 11E. Finally, the desired product 46 and Ru species 11A were yielded through the reductive elimination of 11E.
 | ||
| Scheme 17 (a) Ru-catalyzed and thioether-directed regioselective C–H hydroarylation of alkynes with benzylthioethers. (b) The proposed mechanism. | ||
The above reports indicate that thioether can act as a directing group for C–H bond functionalization through the coordination of the internal sulfur atom to transition metal centers, however, this is not the only possible catalysis pathway. In 2019, Engle and co-workers developed a Pd-catalyzed directed oxidative Heck reaction assisted by a weakly coordinating benzothiazole thioether (BTS) directing group (Scheme 18a).27 Employing this directing group, the Pd(II)-catalyzed C(sp2)–H olefination of arenes has also been reported (Scheme 18b). The control experiments and DFT calculations suggested that the Pd catalyst was coordinated to the nitrogen atom of the benzothiazole thioether directing group. A general catalytic cycle has been proposed (Scheme 18c). It is believed that the initial transmetalation of Pd(II) with arylboronic acid provides the aryl-Pd species, which is further converted into intermediate 12A via a substrate binding step. Next, the subsequent migratory insertion process generates the palladacycle intermediate 12B, upon which, the β-H elimination step affords the desired product 49 and the Pd–H species. Finally, Pd–H is transformed Pd(II) via reductive elimination and reoxidation.
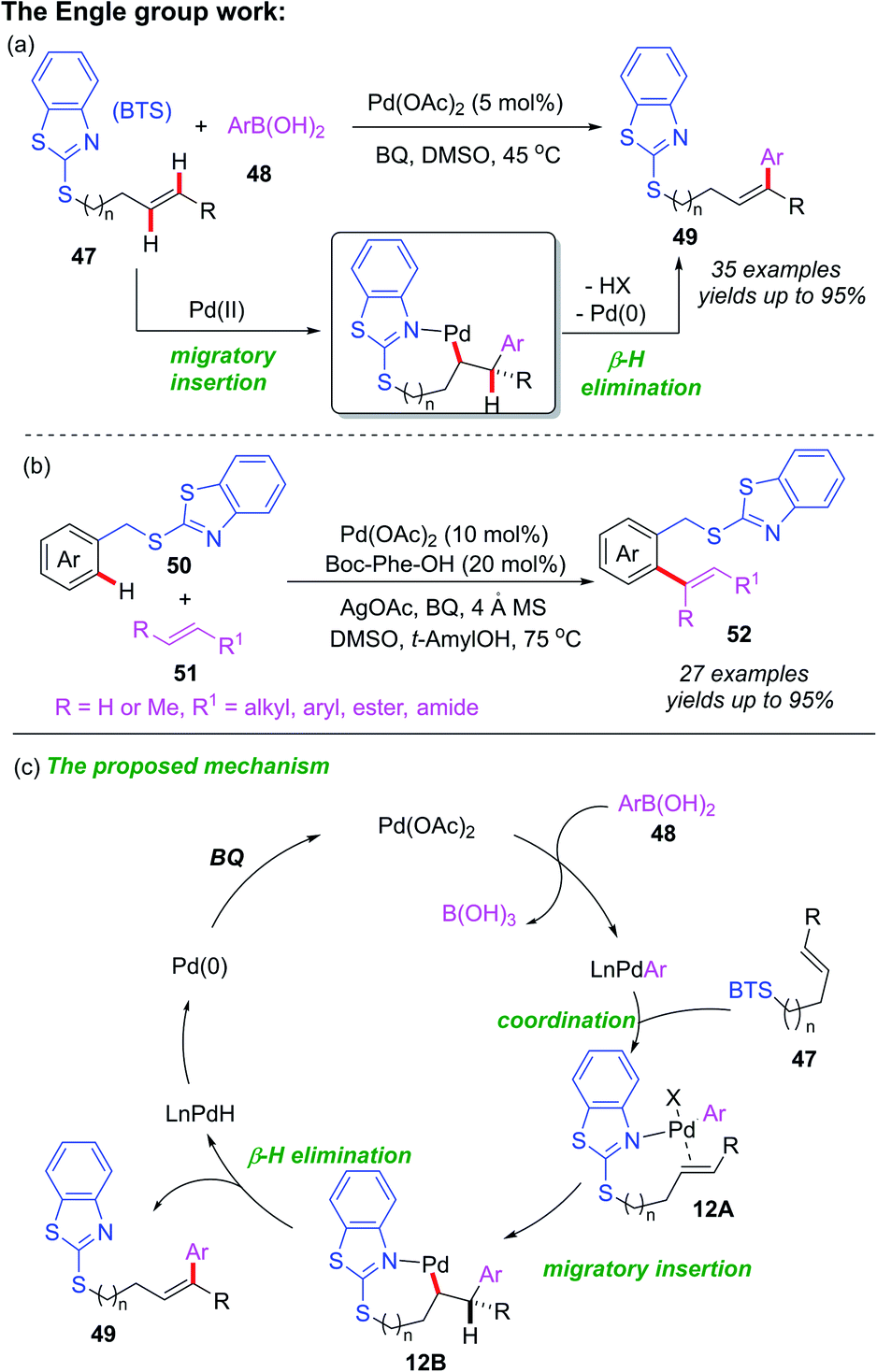 | ||
| Scheme 18 (a) Palladium-catalyzed directed oxidative Heck reactions assisted by a BTS directing group. (b) Palladium-catalyzed directed olefination reactions assisted by a BTS directing group. (c) The proposed mechanism of Heck reaction. | ||
In the past ten years, directing group assisted palladium-catalyzed asymmetric C–H bond functionalization has attracted considerable attention.28 Very recently, Shi and co-workers developed a palladium-catalyzed and thioether-directed atroposelective C–H olefination by employing a novel chiral spiro phosphoric acid ligand SPA (Scheme 19a).29 More than 69 examples of axially chiral styrenes with a conjugated 1,3-diene scaffolds were obtained in good yields (up to 99% yields) with excellent enantioselectivities (up to 99% ee) and complete Z-selectivity control. Subsequently, they also used the same chiral spiro phosphoric acid ligand SPA to prepare axially chiral biaryls through thioether-directed Pd-catalyzed atroposelective C–H olefination (Scheme 19b).30 A variety of axially chiral biaryls containing thioether motifs were isolated in excellent yields (up to 99% yields) and enantioselectivities (up to 99% ee). In this process, the authors used DFT calculations to optimize reaction conditions and propose possible mechanism (Scheme 19c). The selective C–H bond activation of biaryl 55 with chiral spiro phosphoric acid ligand SPA and Pd(OAc)2 catalyst generates the Pd(II) intermediate 13A. The subsequent R-configuration alkene insertion forms the intermediate 13B. Next, the β-H elimination and a rapid reductive elimination produce the final product 56.
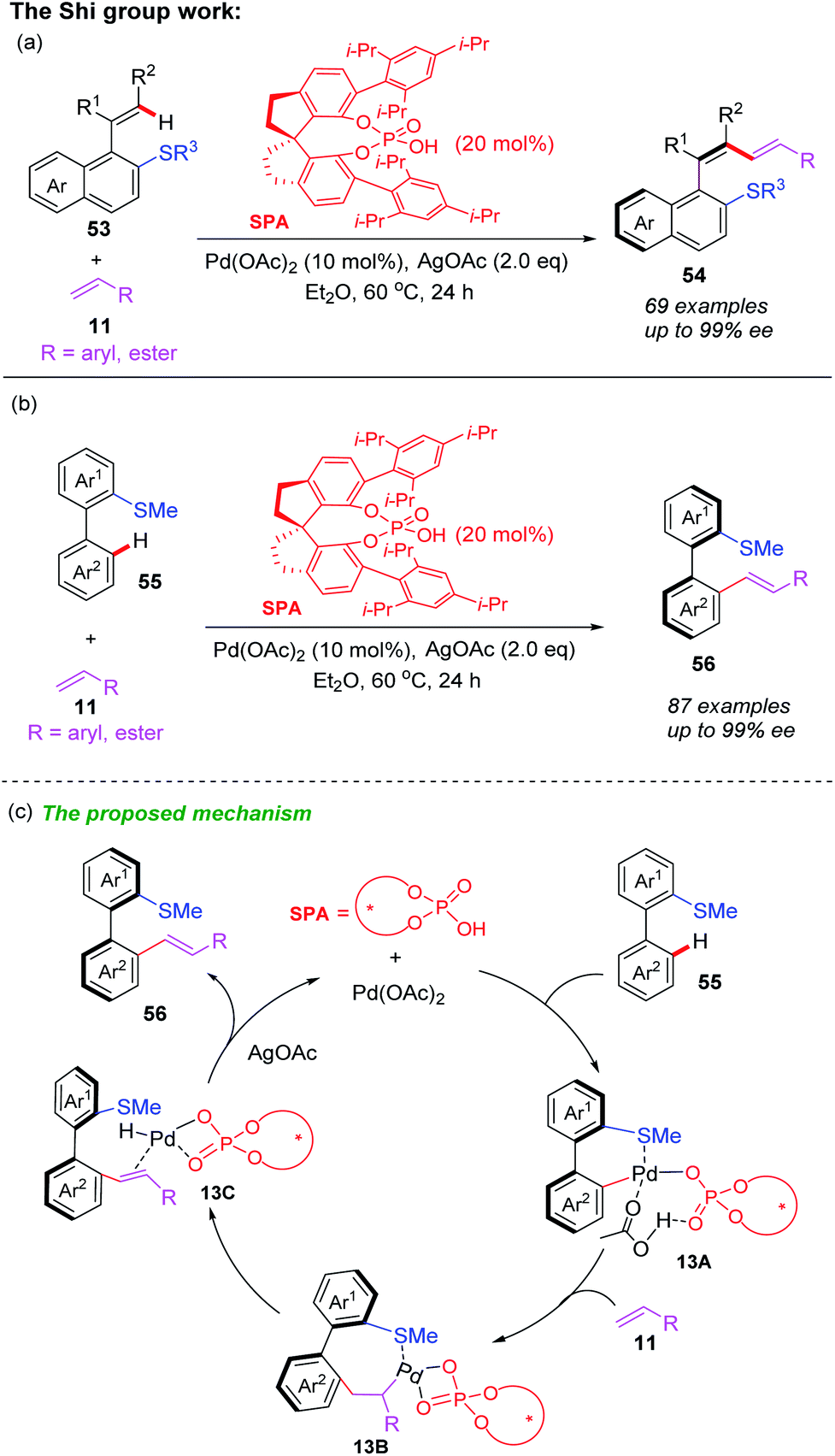 | ||
| Scheme 19 (a) Palladium-catalyzed and thioether-directed atroposelective C–H olefination by employing a SPA ligand for preparing chiral styrenes with a conjugated 1,3-diene scaffolds. (b) Palladium-catalyzed and thioether-directed atroposelective C–H olefination by employing a SPA ligand for preparing chiral biaryls. (c) The proposed mechansim for preparing chiral biaryls. | ||
3. Remote C(sp3)–H functionalization
Compared to C(sp2)–H bond functionalization, C(sp3)–H bond functionalization is more challenging because the formation of alkyl-metal bonds is more difficult than aryl-metal bonds. Usually, bidentate directing groups show higher reactivity than their monodentate counterparts.31 Therefore, the catalytic functionalization of C(sp3)–H bonds of thioethers could be achieved by using a S,N- or S,O-bidentate directing group.In 2010, Daugulis and Shabashov developed a method for palladium-catalyzed bidentate-directed β-arylation of C(sp3)–H bonds (Scheme 20a).32 Using 2-methylthioaniline as a bidentate directing group and aryl iodide as a coupling partner, a variety of mono-arylated products were isolated in moderate to good yields through a primary sp3 C–H bond activation process. A generalized mechanism is proposed (Scheme 20b). It is believed that this reaction is initiated by the coordination of the substrate 57 to Pd(OAc)2, providing the palladium intermediate 14A. Next, the C–H bond cyclometalation of intermediate 14A produces the cyclopalladium intermediate 14B. The subsequent oxidative addition of aryl iodide to intermediate 14B affords intermediate 14C, which is further converted to the desired product 59 through reductive elimination followed by a ligand exchange process.
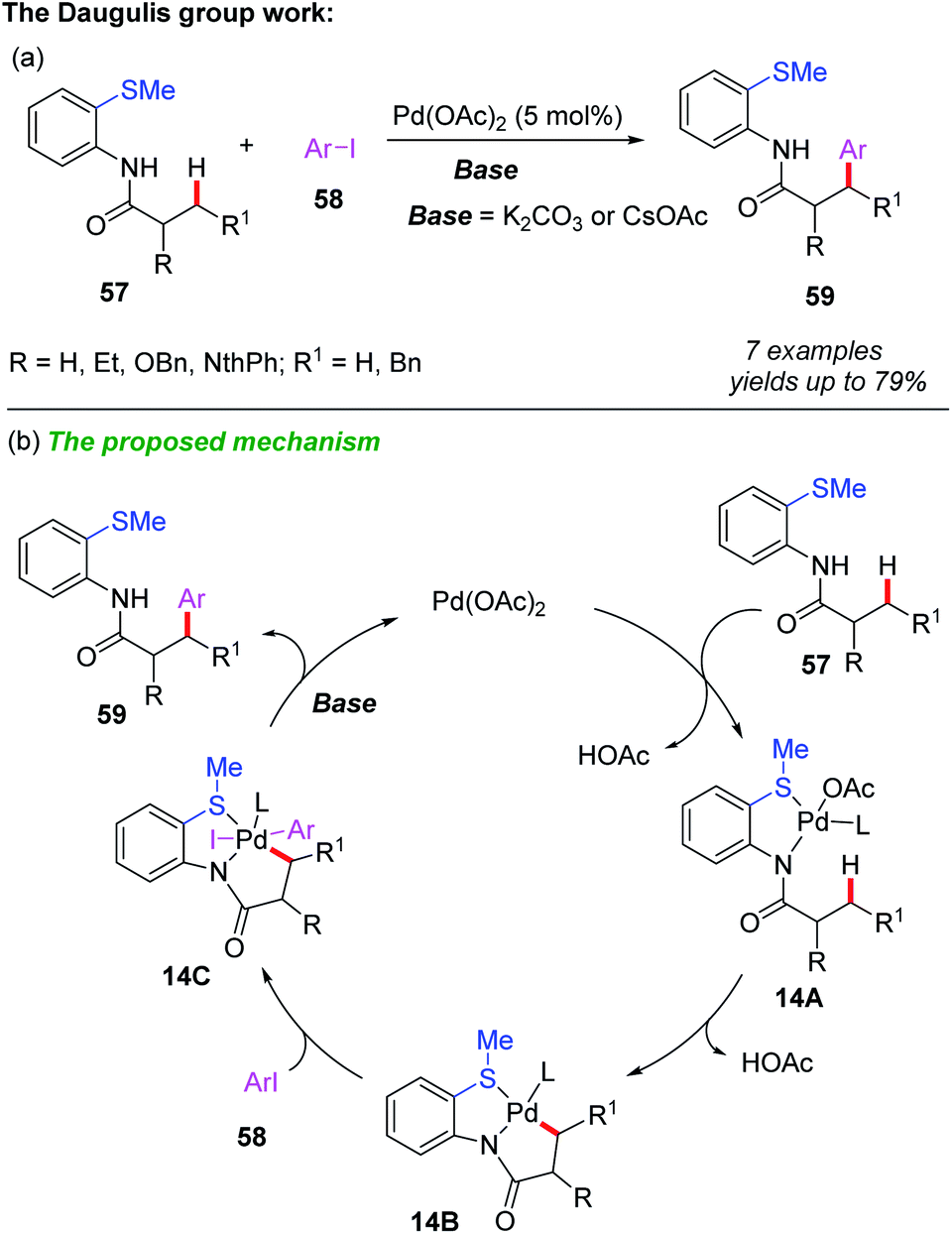 | ||
| Scheme 20 (a) Palladium-catalyzed and 2-methylthioaniline-directed arylation of C(sp3)–H bonds. (b) The proposed mechanism. | ||
Recently, Dong and co-workers reported a Pd-catalyzed γ-C(sp3)–H arylation of thioethers using a S,O-bidentate directing group (Scheme 21a and b).33 Both benzylic and alkyl C–H bonds could be activated to react with various aryl iodides, providing the desired arylated thioether derivatives in moderate to good yields. In this catalytic system, the C–H metalation step could be accelerated by using 3-nitrobenzoic acid and 5-(trifluoromethyl)-pyridin-2-ol as ligands.
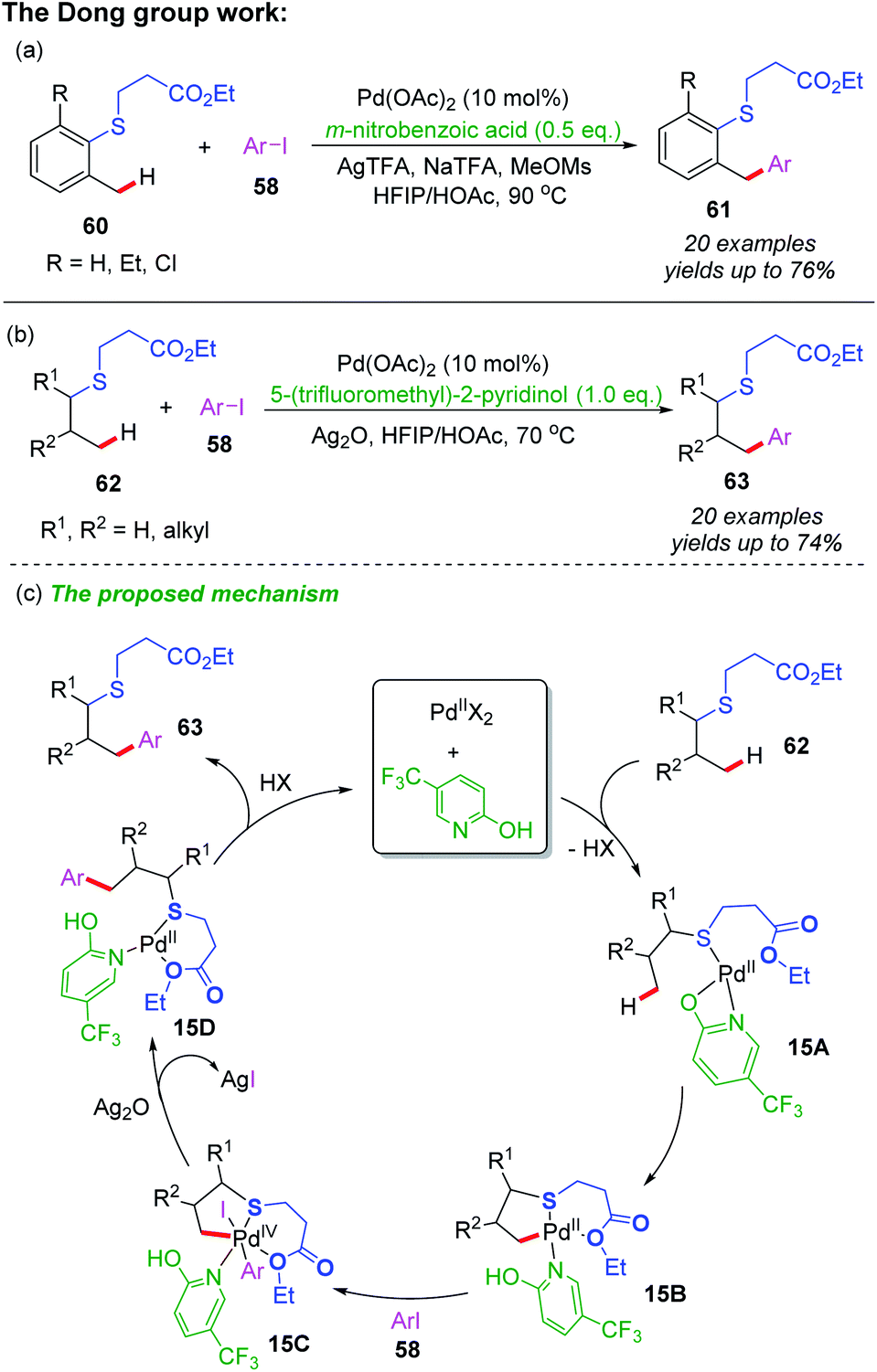 | ||
| Scheme 21 (a) Pd-catalyzed benzylic C–H arylation of thioethers using a S,O-bidentate directing group. (b) Pd-catalyzed alkyl C–H arylation of thioethers using a S,O-bidentate directing group. (c) The proposed mechanism of alkyl C–H arylation. | ||
In additional, a proposed mechanism is also shown in Scheme 21c. The initiated coordination of thioether substrate 62, 5-(trifluoromethyl)-pyridin-2-ol ligand and Pd(II) catalyst affords the cyclic palladium intermediate 15A. Subsequently, 5-(trifluoromethyl)-pyridin-2-ol ligand assisted β-C(sp3)–H bond activation of the intermediate 15A provides a [5,6]-bicyclic palladium intermediate 15B. Then, oxidative addition of the intermediate 15B with an aryl iodide followed by a reductive elimination process produces intermediate 15D. Finally, the desired arylated product 63 is formed through a ligand dissociation process, and Pd(II) catalyst and 5-(trifluoromethyl)-pyridin-2-ol ligand recovered.
4. Conclusions and outlook
In this review, we provide significant recent advances on the directing group assisted C–H functionalization of thioethers enabled by different transition metals. This review mainly introduces the transition-metal-catalyzed remote C(sp2)–H functionalization of thioethers through the coordination of the internal sulfur or nitrogen atom with transition metals. Additionally, remote C(sp3)–H bond functionalization of thioethers via an S,N- or S,O-bidentate directing group strategy has also been included. While significant progress has been made in this area, there is still much room for improvement and utilization, including: (1) C–H functionalization of thioethers using photo-chemistry or electro-chemistry approaches; (2) thioether-directed transition metal-catalyzed asymmetric C(sp2)–H and C(sp3)–H bond functionalization. Finally, we hope this review will provide readers with some insights and inspire them to discover more innovative strategies in the transition metal-catalyzed remote C–H functionalization of thioethers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from Changzhou University.Notes and references
- (a) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, 6543 CrossRef PubMed; (b) J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723 CrossRef CAS PubMed; (c) E. Nobile, T. Castanheiro and T. Besset, Angew. Chem., Int. Ed., 2021, 60, 12170 CrossRef CAS PubMed; (d) F. Liu, Z. Zhang, H. Diao and Z.-J. Shi, ChemCatChem, 2021, 13, 1475 CrossRef CAS; (e) Q. Zhang and B.-F. Shi, Chem. Sci., 2021, 12, 841 RSC; (f) A. J. Rago and G. Dong, Green Synthesis and Catalysis, 2021, 2, 216 CrossRef; (g) B. Li, A. I. M. Ali and H. Ge, Chem, 2020, 6, 2591 CrossRef CAS; (h) J. Das, S. Guin and D. Maiti, Chem. Sci., 2020, 11, 10887 RSC; (i) K. Yang, M. Song, Z. Ma, Y. Li, Z. Li and X. Sun, Org. Chem. Front., 2019, 6, 3996 RSC; (j) K. Yang, B. Niu, Z. Ma, H. Wang, B. Lawrence and H. Ge, J. Org. Chem., 2019, 84, 14045 CrossRef CAS PubMed; (k) K. Yang, D. Li, L. Zhang, Q. Chen and T. Tang, RSC Adv., 2018, 8, 13671 RSC.
- (a) S. Sasmal, U. Dutta, G. K. Lahiri and D. Maiti, Chem. Lett., 2020, 49, 1406 CrossRef CAS; (b) K. Ramakrishna, J. P. Biswas, S. Jana, T. K. Achar, S. Porey and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13808 CrossRef CAS PubMed; (c) S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrashekar, S. S. Anjana, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633 CrossRef CAS PubMed; (d) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (e) W.-B. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435 RSC; (f) A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147 RSC; (g) Z.-K. Chen, B.-J. Wang, J.-T. Zhang, W.-L. Yu, Z.-X. Liu and Y.-H. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- (a) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955 CrossRef CAS PubMed; (b) Q. Zhao, T. Polsson, X. Pannecouke and T. Besset, Synthesis, 2017, 49, 4808 CrossRef CAS; (c) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (d) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456 RSC; (e) S. St John-Campbell, J. Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582 RSC.
- (a) N. Barbero and R. Martin, Org. Lett., 2012, 14, 796 CrossRef CAS PubMed; (b) J. F. Hooper, A. B. Chaplin, C. González-Rodríguez, A. L. Thompson, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 2906 CrossRef CAS PubMed; (c) A. Senthilmurugan and I. S. Aidhen, Eur. J. Org. Chem., 2010, 555 CrossRef CAS.
- (a) P. Li, Y. Yang, X. Wang and X. Wu, J. Heterocycl. Chem., 2021, 58, 1225 CrossRef CAS; (b) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed; (c) K. L. Dunbar, D. H. Scharf, A. Litomska and C. Hertweck, Chem. Rev., 2017, 117, 5521 CrossRef CAS PubMed; (d) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200 CrossRef CAS PubMed.
- (a) R. Gómez Arrayás and C. C. Carretero, Chem. Commun., 2011, 47, 2207 RSC; (b) M. Mellah, A. Voituriez and E. Schultz, Chem. Rev., 2007, 107, 5133 CrossRef CAS PubMed; (c) H. Pellissier, Tetrahedron, 2007, 63, 1297 CrossRef CAS.
- (a) J. Lou, Q. Wang, P. Wu, H. Wang, Y.-G. Zhou and Z.-K. Yu, Chem. Soc. Rev., 2020, 49, 4307 RSC; (b) S. Otsuka, K. Nogi and H. Yorimitsu, Top. Curr. Chem., 2018, 376, 199 Search PubMed; (c) L. Zhang, J. Dong, X. Xu and Q. Liu, Chem. Rev., 2016, 116, 22 Search PubMed; (d) F. Pan and Z.-J. Shi, ACS Catal., 2014, 4, 280 CrossRef CAS; (e) S. G. Modha, V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042 RSC; (f) L. D. Wang, W. He and Z.-K. Yu, Chem. Soc. Rev., 2013, 42, 599 RSC.
- (a) K.-X. Tang, C.-M. Wang, T.-H. Gao, L. Chen, L. Fan and L.-P. Sun, Adv. Synth. Catal., 2019, 361, 26 CrossRef CAS; (b) X. Tang and Y. Zhao, Mini-Rev. Org. Chem., 2021, 18, 902 CrossRef CAS.
- (a) L. Tang, Q. Hu, K. Yang, M. Elsaid, C. Liu and H. Ge, Green Synthesis and Catalysis, 2022 DOI:10.1016/j.gresc.2022.02.002; (b) A. M. F. Phillips and A. J. L. Pombeiro, ChemCatChem, 2018, 10, 3354 CrossRef.
- M. Yu, Y. Xie, C. Xie and Y. Zhang, Org. Lett., 2012, 14, 2164 CrossRef CAS PubMed.
- J. Yao, M. Yu and Y. Zhang, Adv. Synth. Catal., 2012, 354, 3205 CrossRef CAS.
- B. Wang, C. Lin, Y. Liu, Z. Fan, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 973 RSC.
- B. Xu, W. Liu and C. Kuang, Eur. J. Org. Chem., 2014, 2576 CrossRef CAS.
- X.-S. Zhang, Y.-F. Zhang, K. Chen and Z.-J. Shi, Org. Chem. Front., 2014, 1, 1096 RSC.
- (a) X.-T. Cao, S.-N. Wei, H.-T. Sun, M. Li, Z.-L. Zheng and G. Wang, RSC Adv., 2021, 11, 22000 RSC; (b) H. Xu, Y. Zhu, X. Chai, J. Yang and L. Dong, Green Synthesis and Catalysis, 2020, 1, 167 CrossRef; (c) H. Shen, D. Cheng, Y. Li, T. Liu, X. Yi, L. Liu, F. Ling and W. Zhong, Green Synthesis and Catalysis, 2020, 1, 175 CrossRef; (d) Y. Rao, G. Shan and X. Yang, Sci. China Chem., 2014, 57, 930 CrossRef CAS.
- X.-S. Zhang, Q.-L. Zhu, Y.-F. Zhang, Y.-B. Li and Z.-J. Shi, Chem.–Eur. J., 2013, 19, 11898 CrossRef CAS PubMed.
- M. Shigeno, Y. Nishii, T. Satoh and M. Miura, Asian J. Org. Chem., 2018, 7, 1334 CrossRef CAS.
- S. Moon, Y. Nishii and M. Miura, Org. Lett., 2019, 21, 233 CrossRef CAS PubMed.
- C. N. Kona, Y. Nishii and M. Miura, Org. Lett., 2018, 20, 4898 CrossRef CAS PubMed.
- S. Yang, R. Cheng, T. Zhao, A. Luo, J. Lan and J. You, Org. Lett., 2019, 21, 5086 CrossRef CAS PubMed.
- S. Yang, R. Cheng, M. Zhang, Z. Bin and J. You, ACS Catal., 2019, 9, 6188 CrossRef CAS.
- (a) M. E. Hoque, R. Bisht, C. Haldar and B. J. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745 CrossRef CAS PubMed; (b) L. Yang, K. Semba and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 4853 CrossRef CAS PubMed; (c) L. Zhu, X. Qi, Y. Li, M. Duan, L. Zou, R. Bai and Y. Lan, Organometallics, 2017, 36, 2107 CrossRef CAS; (d) B. E. Haines, Y. Saito, Y. Segawa, K. Itami and D. G. Musaev, ACS Catal., 2016, 6, 7536 CrossRef CAS; (e) Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193 CrossRef CAS PubMed; (f) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712 CrossRef CAS PubMed.
- H. L. Li, M. Kanai and Y. Kuninobu, Org. Lett., 2017, 19, 5944 CrossRef CAS PubMed.
- C. N. Kona, Y. Nishii and M. Miura, Org. Lett., 2020, 22, 4806 CrossRef CAS PubMed.
- H. Xie, M. Zhong, X.-T. Wang, J.-Q. Wu, Y.-Q. Cai, J. Liu, B. Shu, T. Che and S.-S. Zhang, Org. Chem. Front., 2021, 8, 635 RSC.
- P. Villuendas and E. P. Urriolabeitia, Org. Lett., 2015, 17, 3178 CrossRef CAS PubMed.
- A. M. Romine, K. S. Yang, M. K. Karunananda, J. S. Chen and K. M. Engle, ACS Catal., 2019, 9, 7626 CrossRef CAS PubMed.
- (a) K. Yang, M. Song, H. Liu and H. Ge, Chem. Sci., 2020, 11, 12616 RSC; (b) Y. Dong, R. Liu and W. Wang, Green Synthesis and Catalysis, 2020, 1, 83 CrossRef; (c) S. Li, Q. Chen, Z.-M. Zhang and J. Zhang, Green Synthesis and Catalysis, 2021, 2, 374 CrossRef; (d) A. A. Zhang, C. Chen, Y. Gao, M. Mo, R.-Z. Shen, Y.-H. Zhang, N. Ishida, M. Murakami and L. Liu, Green Synthesis and Catalysis, 2021, 2, 311 CrossRef.
- L. Jin, P. Zhang, Y. Li, X. Yu and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 12335 CrossRef CAS PubMed.
- G. Liao, T. Zhang, L. Jin, B.-J. Wang, C.-K. Xu, Y. Lan, Y. Zhao and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202115221 CrossRef CAS PubMed.
- (a) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS PubMed; (b) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed.
- L. Jin, J. Wang and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 12352 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |