
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved hydrogen evolution performance by engineering bimetallic AuPd loaded on amino and nitrogen functionalized mesoporous hollow carbon spheres†
Lenan Wangab,
Zhankui Zhaoa,
Hongli Wang *a and
Yue Chi*ab
*a and
Yue Chi*ab
aCollege of Material Science and Engineering, Key Laboratory of Advanced Structural Materials, Ministry of Education, Changchun University of Technology, Changchun, 130012, China. E-mail: yuechi@ccut.edu.cn; wanghongli@ccut.edu.cn
bAdvanced Institute of Materials Science, Changchun University of Technology, Changchun, 130012, China
First published on 19th April 2022
Abstract
A highly efficient heterogeneous catalyst was synthesized by delicate engineering of NH2-functionalized and N-doped hollow mesoporous carbon spheres (NH2–N-HMCS), which was used for supporting AuPd alloy nanoparticles with ultrafine size and good dispersion (denoted as AuPd/NH2–N-HMCS). Without using any additives, the prepared AuPd/NH2–N-HMCS catalytic formic acid dehydrogenation possesses superior catalytic activity with an initial turnover frequency value of 7747 mol H2 per mol catalyst per h at 298 K. The excellent performance of AuPd/NH2–N-HMCS derives from the unique hollow mesoporous structure, the small particle sizes and high dispersion of AuPd nanoparticles and the modified Pd electronic structure in the AuPd/NH2–N-HMCS composite, as well as the synergistic effect of the modified support.
Introduction
With the rapid development of nanotechnology and catalysis science, metal nanoparticle catalysts have been widely used in industry and the laboratory.1 Recently, considerable progress has revealed that Pd, Au, and their bimetallic nanomaterials are the most active catalysts for dehydrogenation of formic acid (HCOOH, FA),2–4 which is a safe and convenient H2 carrier with high volumetric H2 density (53 g L−1).2 Pd alloying with Au can tune the surface electronic and geometric effects to optimize the catalysis, thus the bimetallic AuPd usually exhibits enhanced catalytic performance.5,6 Although AuPd catalysis can enable the dehydrogenation of FA to proceed at milder conditions and with lower molar amounts of metal components, the reported catalytic performance is still far from being satisfactory even with assistance of additional additives.3The introduction of catalyst supports with delicately designed nanostructures has constituted a reasonable strategy in improving the performance of metal catalysts. An appropriate support material not only acts as a physical carrier to disperse and stabilize the active metal, but interacts with the supported metal thus boosting the catalytic activity.7 Up to now, the performance of supported metal catalysts can be enhanced by using different support materials, such as titanium dioxide,8 zeolite,9,10 porous carbon11 and graphene oxide.12 Among various supported metal catalysts, carbon-supported metal catalysts have recently attracted particular research interest, owing to the carbon frame employed as a substrate for growing supported metal, which also substantially influence the properties of supported metals through electronic interaction.7 Particularly, hollow mesoporous carbon spheres (HMCS) would be attractive as catalyst supports for H2 evolution from FA dehydrogenation comparing to conventional carbon supports. Such a structure provides a large void space, where reactants can freely diffuse in the cavity through mesoporous channels to participate in reaction.13 The mesoporous shell not only induces the “confinement effect” to limit the growth and aggregation of supported metal, but also confines and enriches the reactants around the supported metal, leading to a locally increased reactant concentration and accelerating catalytic reaction.13,14
Unfortunately, the electronic interaction is usually weak between supported metal and a dopant-free carbon support, resulting in the aggregation of metal nanoparticles and the degradation of catalytic performance.15 Introduction of heteroatom nitrogen brings more active π electrons to the carbon surface, which can not only in situ anchor the active metal nanoparticles, but also achieve the fine modulation of electronic structure of metal particles and maximize the strong metal-to-support interaction and thus enhance the catalytic performance.16–18 However, N-doped hollow mesoporous carbon spheres (N-HMCS) can afford very limited application in FA system, as its nonpolar nature forces its tendency to aggregate and stack in aqueous solution, and unfavorable to adsorb polarity-featured reactants. To get enhanced catalytic performance, a further step beyond the preparation of N-HMCS is to introduce polar groups, such as –NH2, which can endow the N-HMCS with hydrophilicity as well as impede aggregation of supported metal.19 Furthermore, the –NH2 groups can accelerate the cleavage of O–H bond in FA, thus accelerate the process of FA dehydrogenation and greatly improve the H2 evolution activity of catalyst.20,21
In this work, we prepared NH2-functionalized and N-doped hollow mesoporous carbon sphere (NH2–N-HMCS) as a novel support for AuPd nanoparticles, and the resultant AuPd/NH2–N-HMCS was used as catalyst for H2 evolution from FA dehydrogenation. Benefitting from its spherical morphology, large cavity, high surface area, rich mesoporous structure, well-designed surface chemistry properties, metal–support interactions and synergistic catalysis of AuPd alloy nanoparticles, the AuPd/NH2–N-HMCS exhibits excellent catalytic activity (up to 7747 mol H2 per mol catalyst per h) and stability at room temperature without any additives.
Experimental
Materials
Resorcinol (Xilong Science Co., Ltd, ≥99.5%), formaldehyde (Sinopharm Chemical Reagent Co., Ltd, 37.0%), tetrapropyl orthosilicate (TPOS, Shanghai Mackin Biochemical Co., Ltd, 97%), ammonia solution (NH3·H2O, Beijing Chemical Works, 25–28%), hydrofluoric acid (HF, Sinopharm Chemical Reagent Co., Ltd, ≥40%), sulfuric acid (H2SO4, Beijing Chemical Works, 95–98%), ammonium persulfate (APS, Tianjin Xinbote Chemical Co., Ltd, ≥98%), 3-aminopropyl trimethoxysilane (APTMS, Aladdin Chemistry Co., Ltd, 97%), palladium chloride (PdCl2, Sinopharm Chemical Reagent Co., Ltd, ≥59%), sodium chloride (NaCl, Sinopharm Chemical Reagent Co., Ltd, ≥99.5%), tetrachloroauric(III) acid (HAuCl4·4H2O, Sinopharm Chemical Reagent Co., Ltd, Au > 47.8%), sodium borohydride (NaBH4, Sinopharm Chemical Reagent Co., Ltd, ≥96%), formic acid (HCOOH, FA, Aladdin Chemistry Co., Ltd, ≥96%) and ethanol (CH3CH2OH, Tianjin Xinbote Chemical Co., Ltd, ≥99.7%) were used as received without further purification. High purified water from a Millipore Milli-Q water purification system was used in all the experiments.Preparation of catalyst
HMCS were synthesized by a previously reported the one-pot and surfactant-free approach.22,23 Firstly, 3.4 mL of TPOS was added into the mixed solution, which consisted of 70 mL ethanol, 10 mL distilled water and 3 mL ammonia, and stirred at 298 K for 15 minutes. Then, 0.4 g resorcinol and 0.5 mL formaldehyde were added to the solution in turn, and the stirring was continued for 24 h. The product was collected by centrifugation and washed six times alternately with water and ethanol, and dried at 323 K for 24 h. The dried specimen was calcined at 973 K under argon atmosphere for 5 h in a tubular furnace. Finally, the black products were desiliconized by hydrofluoric acid (5%) under strong magnetic stirring at room temperature for 24 h, and the reaction was repeated twice. The final product was washed with water and dried at 323 K for 12 h and denoted as HMCS.Subsequently, 200 mg HMCS powder was evenly dispersed into 5 mL APS and H2SO4 mixed solution, and stirred at 298 K for 18 h. The concentrations of APS and H2SO4 in the solution were 1.7 M and 2 M respectively. The precipitates were centrifuged and washed with distilled water for 6 times. After drying at 373 K for 4 h, the surface oxidized mesoporous carbon hollow spheres were obtained and named as HMCS-O. Then, 0.2 mL APTMS was added into the pre ultrasonic HMCS-O aqueous solution (1.25 mg mL−1, 35 mL), the –NH2 functionalized and N-doped hollow mesoporous carbon spheres (NH2–N-HMCS) were obtained after 10 min of ultrasonic treatment at 298 K.
AuPd nanoparticles were then introduced via impregnation reduction. Typically, for preparation of Au0.3Pd0.7/NH2–N-HMCS, 2.5 mmol NaCl and 1.25 mmol PdCl2 were put into 50.0 mL H2O and stirred magnetically for 10 h to obtain Na2PdCl4 (0.025 M) solution. The HAuCl4·4H2O (0.02 M, 1.5 mL) and Na2PdCl4 (0.025 M, 2.8 mL) solution were added to the NH2–N-HMCS aqueous solution with magnetic stirring at 298 K for 3 h. And then, NaBH4 (40.5 mg) was added into the above mixed solution and stirred for 30 min. Finally, the resultant Au0.3Pd0.7/NH2–N-HMCS product was separated and washed with water for several times.
In order to study the effect of different molar ratios of Au and Pd on dehydrogenation of formic acid, AuxPd1−x/NH2–N-HMCS [x = 0, 0.1, 0.3, 0.5, 0.7, 0.9 and 1.0 calculated from the equation of x = nAu/(nAu + nPd)] were synthesized in the similar method without changing the total molar weight of AuPd particles. For comparison, Au0.3Pd0.7/HMCS (the non-modified support material) and Au0.3Pd0.7 nanoparticles without any support were synthesized.
For comparison, Au0.3Pd0.7/N-HMCS was synthesized. Firstly, 100 mg HMCS was added into 10 mL ultrapure water and sonicated for 10 min. Then, 70 mL NH3·H2O was poured into the mixture. Finally, the mixture was transferred to a Teflon-lined autoclave and heated at 473 K for 12 h to get N-HMCS. The N-HMCS were collected by centrifugation (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 3 min) and washed repeatedly with ultrapure water and ethanol and dried at 333 K for 12 h. In order to introduce AuPd nanoparticles, 40 mg of N-HMCS was added into 35 mL H2O for 10 min of ultrasonic treatment, then the HAuCl4·4H2O (0.02 M, 1.5 mL) and Na2PdCl4 (0.025 M, 2.8 mL) solution were added to the mixed solution with 3 h of magnetic stirring at 298 K. Finally, NaBH4 (40.5 mg) was added as the reducing agent to obtain the Au0.3Pd0.7/N-HMCS product, the product was centrifuged and washed several times with distilled water.
000 rpm, 3 min) and washed repeatedly with ultrapure water and ethanol and dried at 333 K for 12 h. In order to introduce AuPd nanoparticles, 40 mg of N-HMCS was added into 35 mL H2O for 10 min of ultrasonic treatment, then the HAuCl4·4H2O (0.02 M, 1.5 mL) and Na2PdCl4 (0.025 M, 2.8 mL) solution were added to the mixed solution with 3 h of magnetic stirring at 298 K. Finally, NaBH4 (40.5 mg) was added as the reducing agent to obtain the Au0.3Pd0.7/N-HMCS product, the product was centrifuged and washed several times with distilled water.
Characterization
X-ray diffraction (XRD) patterns were collected using a Rigaku D/max 2500 vpc X-ray diffractometer operating at 40 kV and 100 mA with Cu Kα X-ray source. Field-emission scanning electron microscopy (FESEM) images were acquired on Zeiss model Gemini Supra 40 field emission scanning electron microscope. The transmission electron microscope (TEM), energy-dispersive X-ray spectroscopy (EDS) and high-resolution transmission electron microscopy (HRTEM) images were obtained with an FEI-Talos-F200S microscope. The surface chemical properties of the specimens were measured using Fourier transform infrared spectrum (FTIR, Bio-Rad, FTS 6000). The nitrogen adsorption and desorption isotherms were measured at 77 K on a Micromeritics ASAP 2020 plus HD88 instrument (Micromeritics Instrument Corp, Norcross, GA). The Brunauer–Emmett–Teller (BET) specific surface area was calculated using desorption data. The pore size distribution curves were calculated based on the analysis of the desorption branch of the isotherm using the Barrett–Joyner–Halenda (BJH) algorithm. The Au and Pd content in the specimens were measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Thermo Jarrell Ash (TJA) Atom scan Advantage). X-rays photoelectron spectroscopy (XPS) were acquired with an ESCALABMKLL (Vacuum Generators) spectrometer using Al Kα X-rays.Catalytic activities
The as-prepared catalyst was put into a double neck round-bottom flask, one neck was connected to a gas burette, the other neck was connected to a pressure-equalizing funnel which contained FA solution (1 M, 5 mL). The reaction took place when the FA solution was added into the flask under magnetic stirring at 298 K. The gases generated were monitored by the gas burette, and the catalytic tests were carried out under the same conditions.In order to investigate the stability of the catalyst, the cyclic catalytic experiments were carried out without changing the temperature and stirring speed. After the first catalytic reaction of Au0.3Pd0.7/NH2–N-HMCS was thoroughly completed, an equivalent amount of FA aqueous solution (1 M, 5 mL) was injected into the flask to produce H2 under magnetic stirring. The generation of gas was monitored by the gas burette.
TOF calculation
The initial TOF was calculated using the following equation:where Patm is the atmospheric pressure, VH2 is the volume of the generated H2 when the conversion reaches 20%, R is the universal gas constant, T is the room temperature (298 K), ncatalyst is the measured amount of AuPd catalyst using ICP-OES, and t is the reaction time when the conversion reaches 20%.

Results and discussion
Preparation and characterization of catalysts
A multi-step process was used to synthesize AuPd/NH2–N-HMCS. First, the HMCS was prepared through an in situ free assembly method, where the SiO2@RF core–shell structure material was achieved by employing TPOS as the silica source for SiO2 particles and resorcinol and formaldehyde as carbon sources. After pyrolysis to carbonize the organics and etching in HF to remove the SiO2 template, the hollow structured HMCSs were obtained. The HMCS was blended with a mixture of APS and H2SO4 solution, after which the surface of HMCS was equipped with oxy functional groups, forming oxidized HMCS (HMCS-O). Then APTMS was introduced into the surface oxidized HMCS aqueous solution by sonication stirring at 298 K, during this process the oxy functional groups (–COOH and –OH) on the HMCS-O condensed with the silicon hydroxyl groups generated by hydrolysis of APTMS to form surface amine groups, and reacted with amine groups of APTMS to generate amide and C–N bonds, leading to NH2-functionalized and N-doped hollow mesoporous carbon sphere (NH2–N-HMCS). And then a small amount solution of HAuCl4 and Na2PdCl4 (with an Au:Pd molar ratio of 0.3:0.7) was added into the above mixed solution with magnetic stirring. Finally, NaBH4 was dissolved into the above-mentioned system, and the Au0.3Pd0.7/NH2–N-HMCS could be promptly obtained. AuPd/N-HMCS and AuPd/HMCS were also prepared for comparison. Besides, the molar ratio of Au:Pd in AuPd/NH2–N-HMCS has been changed by several values (0:1, 0.1:0.9, 0.3:0.7, 0.5:0.5, 0.7:0.3, 0.9:0.1, 1:0).
As shown in Fig. S1,† the HMCS are uniform hollow spheres with a disordered mesoporous channeled-wall structure, which can serve as a reservoir for metal precursors. After surface modification and metal loading, the Au0.3Pd0.7/NH2–N-HMCS perfectly retain the original structure of the HMCS (Fig. S2†). The corresponding EDX of Au0.3Pd0.7/NH2–N-HMCS displays all elements of C, N, Au, Pd, O and Si (Fig. S3†), and the molar ratio of Au:Pd is quantitatively determined to be 0.2924:0.7076 by the ICP-OES, in good agreement with the designed ratio. The Au0.3Pd0.7/NH2–N-HMCS exhibits porous hollow sphere morphology with an average diameter and wall thickness of 450 nm and 75 nm (Fig. S2†), respectively. Differently, the surface of Au0.3Pd0.7/NH2–N-HMCS with metal nanoparticles loading becomes coarse (Fig. 1a). Fig. 1b clearly shows that ultrafine AuPd nanoparticles are uniformly dispersed on the support. The average size of AuPd nanoparticles is estimated to be 2.2 nm by random count in TEM. While in Au0.3Pd0.7/N-HMCS, not only the AuPd nanoparticles supported on N-HMCS are well dispersed, but a mixture of various large-sized AuPd nanoparticles can be observed. This difference verifies the strong interaction between AuPd nanoparticles and NH2–N-HMCS support, proving that the NH2-functionalized and N-doped hollow mesoporous carbon sphere we reported is more effective for encapsulation of highly dispersed and ultrafine AuPd nanoparticles. As revealed in Fig. 1c, the Au0.3Pd0.7 nanoparticles have shown a typical crystalline character. The lattice spacing of the nanoparticle is measured to be 0.234 nm. This value is between the lattice spacing of (111) crystal planes in cubic Au (0.235 nm) and Pd (0.224 nm), confirming the formation of AuPd alloy. The wide-angle XRD pattern of the Au0.3Pd0.7/NH2–N-HMCS (Fig. 1d) shows a broad diffraction peak that is corresponding to (111) crystal plane and located between the diffraction peak of Au (JCPDS card no. 65-8601) and Pd (JCPDS card no. 65-2867), indicating that the Au0.3Pd0.7 nanoparticles in Au0.3Pd0.7/NH2–N-HMCS are formed in alloy structure, which is well consistent with the HRTEM result.24 Compared with the XRD patterns of Au0.3Pd0.7/N-HMCS and Au0.3Pd0.7/HMCS (Fig. S4†), the peak of Au0.3Pd0.7/NH2–N-HMCS centered around 39° is significantly broader, which further confirms the ultrafine size of Au0.3Pd0.7 nanoparticles in Au0.3Pd0.7/NH2–N-HMCS. Fig. 1e shows the high-angle annular dark-field scanning transmission electron (HAADF-STEM) image and the corresponding Au, Pd, C and N EDX mapping images of Au0.3Pd0.7/NH2–N-HMCS, where many isolated bright dots can be observed, demonstrating the presence of AuPd nanoparticles and their distribution on the NH2–N-HMCS support. In addition, the elements of Au and Pd are co-distributed and homogenously dispersed on the positions of N and C elements, further evincing the uniform dispersion of AuPd nanoparticles in Au0.3Pd0.7/NH2–N-HMCS.
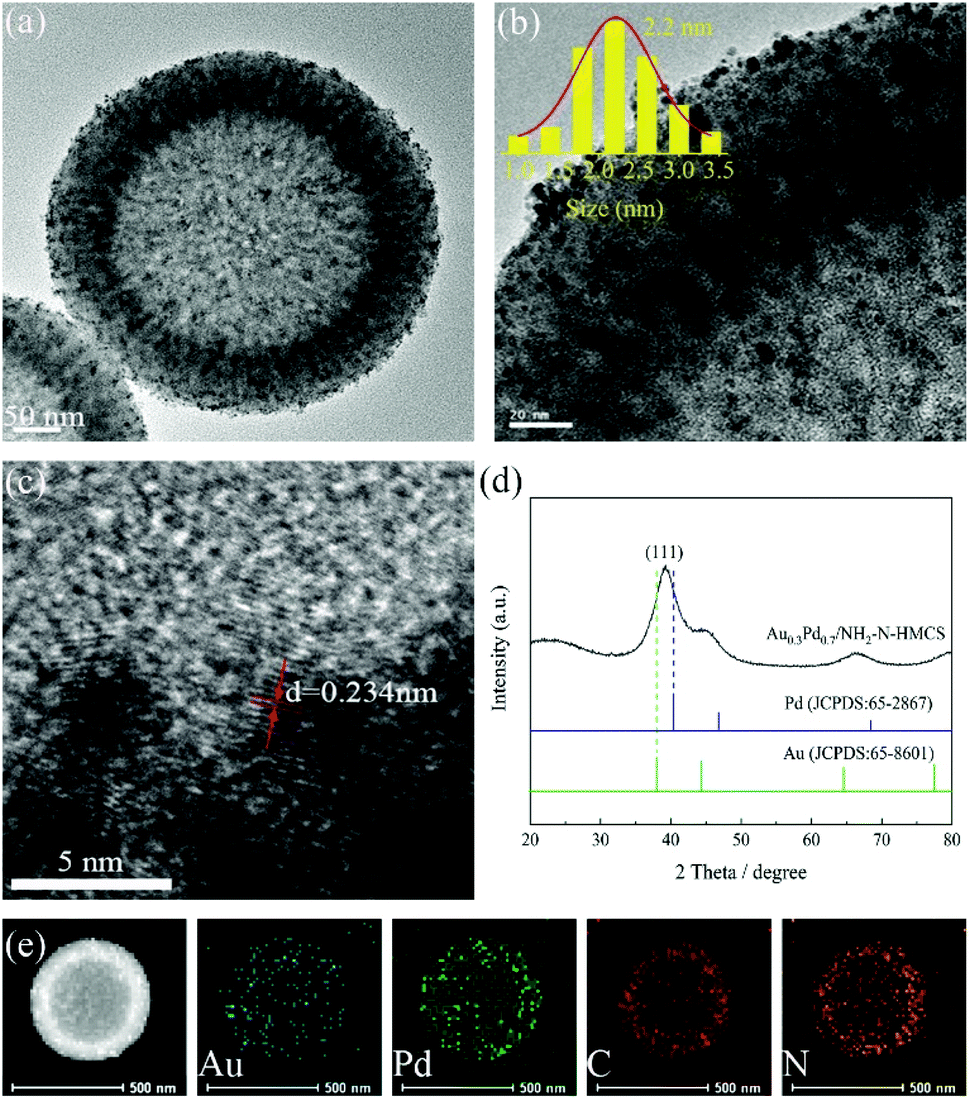 | ||
| Fig. 1 TEM image and the size distribution of Au0.3Pd0.7/NH2–N-HMCS (a and b), HRTEM image of Au0.3Pd0.7/NH2–N-HMCS (c), XRD pattern of Au0.3Pd0.7/NH2–N-HMCS (d), HAADF-STEM and the corresponding elemental mapping images of Au0.3Pd0.7/NH2–N-HMCS (e). | ||
The N2 adsorption–desorption measurements of Au0.3Pd0.7/NH2–N-HMCS exhibit a type IV adsorption isotherm similar to that of HMCS (Fig. 2), indicating the mesoporous structure has remained after surface modification and incorporation of AuPd nanoparticles. However, the surface area, pore size and total pore volume of Au0.3Pd0.7/NH2–N-HMCS are calculated to be 593 m2 g−1, 4.0 nm and 0.58 cm3 g−1 respectively (Table 1), which significantly reduced compared to that of HMCS. According to previous studies,25 loading metal in the pore channels would lead to remarkable perturbation of the pore size, surface area, and pore volume. Such phenomenon was also observed in this study, indicating the successful loading of AuPd nanoparticles in the mesoporous carbon support.
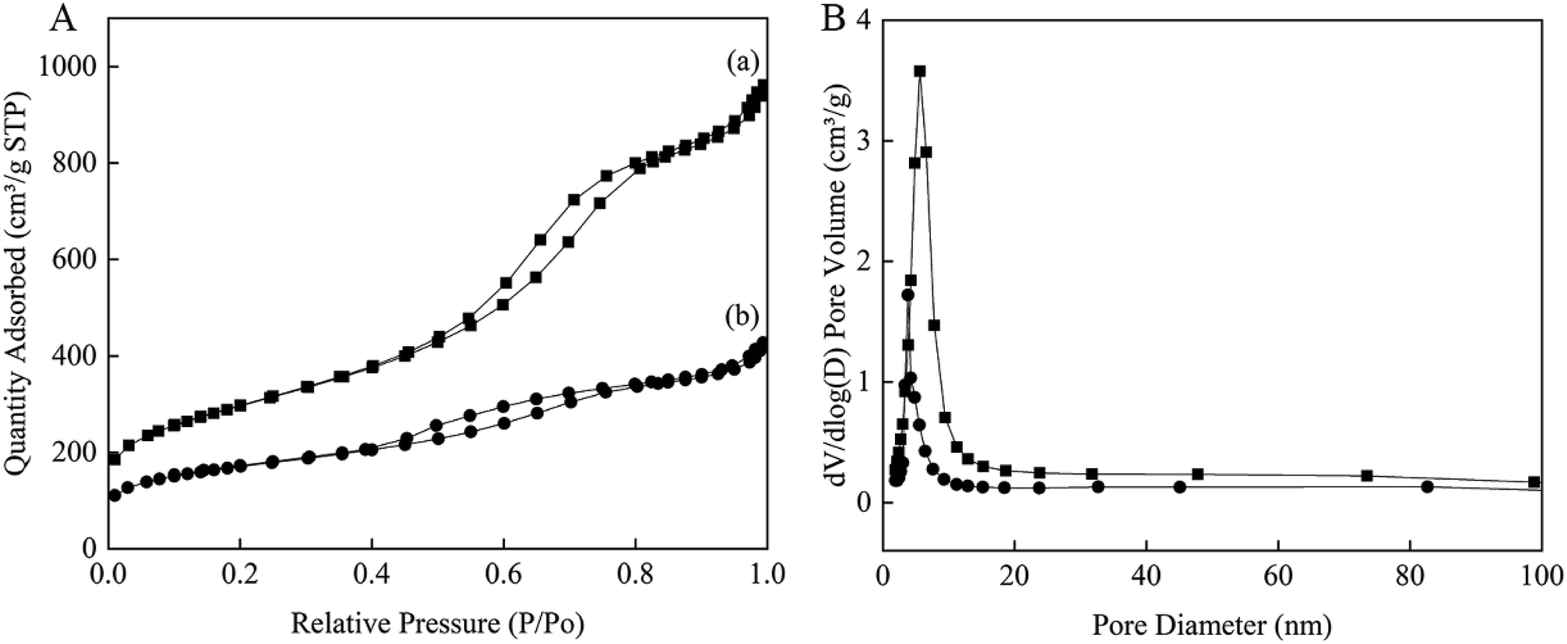 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms (A) and the corresponding pore size distributions (B) of the samples of (a) HMCS and (b) Au0.3Pd0.7/NH2–N-HMCS. | ||
| Sample | Pore size (nm) | BET surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|---|
| HMCS | 5.2 | 1051 | 1.35 |
| Au0.3Pd0.7/NH2–N-HMCS | 4.0 | 593 | 0.58 |
FTIR spectra were carried out to verify the functional groups of Au0.3Pd0.7/NH2–N-HMCS (Fig. 3A). For HMCS, only one absorption at 1606 cm−1 corresponding to the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration can be observed, indicating there is no other element or group modification on the HMCS. For HMCS-O, apart from the peak of CC, it presents another three bands centered around 1719, 1401 and 1262 cm−1 that are not observed for the untreated HMCS.26 A typical band at 1719 cm−1 assigned to –COOH groups is clearly visible, which suggests the carboxylic functional groups were successfully introduced upon surface oxidizing. In addition, the band at 1401 cm−1 can be assigned either to carboxyl–carbonate structure or aromatic C–C bonds whereas the band at 1262 cm−1 can be attributed to C–O–C vibrations in ether groups on the surface of HMCS-O. With APTMS treatment, several new absorption bands at 1203, 1111, 916 and 689 cm−1 were observed in the spectrum of Au0.3Pd0.7/NH2–N-HMCS, related to N–H groups,27 Si–O–C stretching vibration,28 C–H bending vibration and C–N stretching vibration,29,30 confirming the existence of –NH2 groups and N doping in the NH2–N-HMCS. Interestingly, compared with HMCS-O, the band at 1719 cm−1 is barely visible for Au0.3Pd0.7/NH2–N-HMCS. The result further proves that the APTMS is grafted over the carboxylate groups on the surface of HMCS-O, through which the –NH2 groups and N doping has been successfully introduced to the support. Moreover, the band centered at 680 cm−1 corresponding to C–N stretching vibration can be observed in the FTIR spectrum of Au0.3Pd0.7/N-HMCS, proving that N element has been successfully doped in the support of this specimen.
C stretching vibration can be observed, indicating there is no other element or group modification on the HMCS. For HMCS-O, apart from the peak of CC, it presents another three bands centered around 1719, 1401 and 1262 cm−1 that are not observed for the untreated HMCS.26 A typical band at 1719 cm−1 assigned to –COOH groups is clearly visible, which suggests the carboxylic functional groups were successfully introduced upon surface oxidizing. In addition, the band at 1401 cm−1 can be assigned either to carboxyl–carbonate structure or aromatic C–C bonds whereas the band at 1262 cm−1 can be attributed to C–O–C vibrations in ether groups on the surface of HMCS-O. With APTMS treatment, several new absorption bands at 1203, 1111, 916 and 689 cm−1 were observed in the spectrum of Au0.3Pd0.7/NH2–N-HMCS, related to N–H groups,27 Si–O–C stretching vibration,28 C–H bending vibration and C–N stretching vibration,29,30 confirming the existence of –NH2 groups and N doping in the NH2–N-HMCS. Interestingly, compared with HMCS-O, the band at 1719 cm−1 is barely visible for Au0.3Pd0.7/NH2–N-HMCS. The result further proves that the APTMS is grafted over the carboxylate groups on the surface of HMCS-O, through which the –NH2 groups and N doping has been successfully introduced to the support. Moreover, the band centered at 680 cm−1 corresponding to C–N stretching vibration can be observed in the FTIR spectrum of Au0.3Pd0.7/N-HMCS, proving that N element has been successfully doped in the support of this specimen.
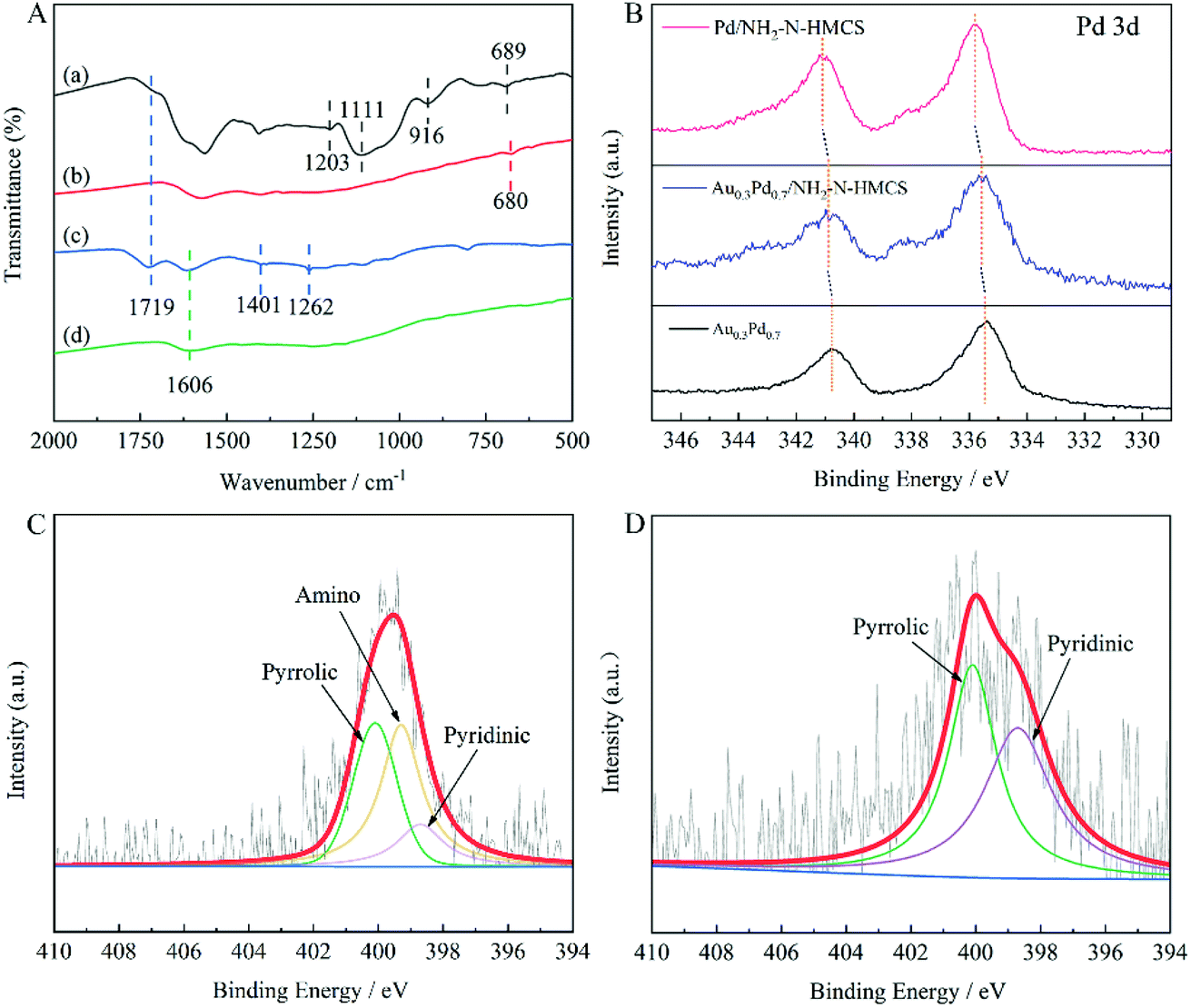 | ||
| Fig. 3 (A) FTIR spectra of Au0.3Pd0.7/NH2–N-HMCS (a), Au0.3Pd0.7/N-HMCS (b), HMCS-O (c) and HMCS (d), (B) the high-resolution XPS spectra of Pd 3d for Pd/NH2–N-HMCS, Au0.3Pd0.7/NH2–N-HMCS and Au0.3Pd0.7, (C) the high-resolution XPS spectrum of N 1s for Au0.3Pd0.7/NH2–N-HMCS and (D) Au0.3Pd0.7/N-HMCS. | ||
To better understand surface composition and bonding environment of the prepared heterostructure, the Au0.3Pd0.7/NH2–N-HMCS was studied by XPS analysis. Fig. S5† shows the fully scanned spectrum of Au0.3Pd0.7/NH2–N-HMCS in the range of 0–1200 eV. The peaks on the curve of Au0.3Pd0.7/NH2–N-HMCS are assigned to C, O, Au, Pd, Si and N elements, which is consistent with the EDX result. The electron structures of Pd and Au in the Au0.3Pd0.7/NH2–N-HMCS catalyst were also analyzed by XPS, where the Pd 3d and Au 4f XPS spectra of Au0.3Pd0.7/NH2–N-HMCS indicate that Pd and Au elements are mostly in metallic state, the minor portion of oxidized state of Pd(II) and Au(I) can be attributed to surface oxidation during sample preparation for XPS (Fig. 3B and S6†). Besides, the XPS results of Pd 3d in Au0.3Pd0.7/NH2–N-HMCS, Pd/NH2–N-HMCS and unsupported Au0.3Pd0.7 were also measured for comparison (Fig. 3B). Compared with the unsupported Au0.3Pd0.7, the Pd 3d peak in Pd/NH2–N-HMCS shifted to a higher binding energy (from 335.5 to 335.8 eV), which can be attributed to the partial transfer of electrons from Pd to the NH2–N-HMCS support caused by the strong interactions between metal and support.
In addition, the peaks of Pd 3d in Au0.3Pd0.7/NH2–N-HMCS shifted toward lower binding energies in comparison with that of Pd/NH2–N-HMCS (from 335.8 to 335.6 eV), indicating the electron structure of Pd can be adjusted by adding Au through electron synergetic effect, which further confirms the formation of alloy structure in AuPd nanoparticles. It is also noteworthy that in comparison with the unsupported Au0.3Pd0.7, both peaks of Pd 3d in Au0.3Pd0.7/NH2–N-HMCS shifted toward higher binding energies, owing to the metal–support interactions and electron synergetic effect. This shift reveals more electron depletion state of Pd in Au0.3Pd0.7/NH2–N-HMCS, which can endow Au0.3Pd0.7/NH2–N-HMCS with excellent catalytic activity for H2 generation from FA as a result of the acceleration for C–H cleavage in the adsorbed formate.31
The high resolution XPS spectra confirm the chemical states of N in Au0.3Pd0.7/NH2–N-HMCS and Au0.3Pd0.7/N-HMCS. As shown in Fig. 3C and D, the N 1s peaks of Au0.3Pd0.7/NH2–N-HMCS and Au0.3Pd0.7/N-HMCS are centered at ≈398.7 and ≈400.1 eV, respectively, indicating different chemical states of N in the sample. The N 1s XPS spectrum of Au0.3Pd0.7/NH2–N-HMCS can be fitted into three components with binding energy of 398.7, 399.3, 400.1 eV corresponding to the pyridinic N, amino N, and pyrrolic N, respectively, while that in Au0.3Pd0.7/N-HMCS can be assigned to pyridinic N (398.7 eV) and pyrrolic N (400.1 eV).32,33 Taking both XPS and FTIR results into account, N in Au0.3Pd0.7/N-HMCS is proven to be the doping atoms, while N in Au0.3Pd0.7/NH2–N-HMCS exists in forms of doping atoms and –NH2 group.
Catalytic activity
The as-prepared Au0.3Pd0.7/NH2–N-HMCS catalyst has excellent catalytic activity for H2 generation from FA (1.0 M, 5 mL) without any additives at 298 K under ambient atmosphere (Fig. 4a). Obviously, the Au0.3Pd0.7/NH2–N-HMCS exhibits the highest catalytic activity for FA decomposition among all the prepared catalysts, with which 224 mL of gas can be released within 67 s, corresponding 100% conversion. To understand the effect of different Au/Pd molar ratio on the dehydrogenation activity towards FA, various AuxPd1−x/NH2–N-HMCS (0 ≤ x ≤ 1.0) with different Au:Pd molar ratios were synthesized and used for dehydrogenation of FA. As shown in Fig. S7,† the Au/NH2–N-HMCS sample shows very low catalytic activity for FA, and the catalytic activities of nanocomposites containing AuPd binary alloy (AuPd/NH2–N-HMCS) are higher than those containing single metal (Au/NH2–N-HMCS and Pd/NH2–N-HMCS).34–37 This phenomenon can be attributed to the electron synergetic effect between Pd and Au, which has been proven by fore-mentioned XPS analysis. In addition, the molar ratio of Au to Pd has also been found to be critical in the catalytic dehydrogenation reaction. With the increase of the Au content, the catalytic activity of AuxPd1−x/NH2–N-HMCS first increases, exhibiting the maximum initial turnover frequency (TOF) value of 7747 mol H2 per mol catalyst per h at x = 0.3. However, the further increase in Au content results in a negative impact on the catalytic dehydrogenation reaction, which is due to the fact that Au does not have high catalytic activity toward FA. The excellent catalytic activity of the Au0.3Pd0.7/NH2–N-HMCS results from the modulation of the electronic structure of Pd by introducing a proper amount of Au and the synergistic catalysis between the carrier and the metal.
 | ||
| Fig. 4 Gas generation from the dehydrogenation of FA (1.0 M, 5.0 mL) versus time (a), initial TOF values of Au0.3Pd0.7/NH2–N-HMCS, Au0.3Pd0.7/N-HMCS, Au0.3Pd0.7 and Au0.3Pd0.7/HMCS (b). | ||
To further understand the mechanism for the enhanced catalytic activity towards FA decomposition by Au0.3Pd0.7/NH2–N-HMCS, the catalytic performance of unsupported Au0.3Pd0.7 nanoparticles, Au0.3Pd0.7/HMCS (without any functionalization), and Au0.3Pd0.7/N-HMCS in H2 generation are given for comparison and reference (Fig. 4a), which clearly shows that the Au0.3Pd0.7 nanoparticles (50 mL, 53 min) and Au0.3Pd0.7/HMCS (20 mL, 10 min) have much lower catalytic activities. For Au0.3Pd0.7/HMCS, the HMCS without any functionalization cannot provide sufficient coordination sites to facilitate the formation of AuPd nanoparticles, thus resulting in the low particle dispersity (Fig. S8†) and finally causes severe degradation of catalytic performance. For Au0.3Pd0.7/N-HMCS, it can release 180 mL gas from FA within 23 min, giving an 80% conversion. The hollow morphology and interconnected mesopores of N-HMCS can facilitate mass diffusion and accessibility, which helps in improving the catalytic performance. In addition, the N-doped N-HMCS is conducive to the formation of highly dispersed Au0.3Pd0.7 nanoparticles (Fig. S9†), thus expose more active sites and enhance its catalytic performance. However, a few of large and agglomerated Au0.3Pd0.7 nanoparticles were also detected in the Au0.3Pd0.7/N-HMCS, which impedes their catalytic performance. All in all, the Au0.3Pd0.7/N-HMCS exhibits much lower catalytic activities compared with Au0.3Pd0.7/NH2–N-HMCS. The significant enhancement on the catalytic performance of Au0.3Pd0.7/NH2–N-HMCS can be ascribed to the introduction of amine groups that improve the wettability of the carbon materials and thus lead to the ultrafine particle sizes and good dispersion. Besides, it has been reported that amine groups in supported Pd-based catalysts can facilitate the FA adsorption and the cleavage of O–H bond in FA molecules, and thereby greatly improve the catalytic activities for H2 evolution from FA dehydrogenation.38,39 On top of that, the Au0.3Pd0.7/NH2–N-HMCS has outstanding catalytic activity for heterogeneously catalyzed FA dehydrogenation ever reported without any additives,3,17,33,40 and moreover, could be comparable to the most active catalysts with additives under elevated temperatures (Table S1†).6,9,11,41
The reusability of the catalysts Au0.3Pd0.7/NH2–N-HMCS was also tested. The recycling stability of the Au0.3Pd0.7/NH2–N-HMCS is evaluated by adding the same amount of the FA solution to the reaction vessel after completion of the previous run. There is a slight decay in catalytic activity after four cycles (Fig. S10†). The TEM image of the sample after the 4th run shows that the nanoparticles still maintain good dispersion with a slight increase in the particle size (from 2.2 to 2.7 nm; Fig. S11†), which maybe the reason for the decrease in catalytic activity. The above analysis indicates the good recycling stability of Au0.3Pd0.7/NH2–N-HMCS towards FA dehydrogenation reaction.
Based on the comparative experimental investigations and results, a possible mechanism for Au0.3Pd0.7/NH2–N-HMCS catalyzed FA dehydrogenation has been proposed (Fig. 5). After adsorption of FA molecules on Au0.3Pd0.7/NH2–N-HMCS, the amine groups can donate electrons and act as a proton scavenger to promote O–H cleavage and produce H+. Subsequently, HCOO− adsorbs on the surface of metal nanoparticles and forms the metal-formate intermediates. Then, the rate determining step is promoted by the Pd with more electron depletion state in Au0.3Pd0.7/NH2–N-HMCS, which can accelerate C–H cleavage in the adsorbed formate to form H− and release CO2. Finally, the generated H− combines with H+ to produce H2, along with the regeneration of active centers.40,42 The excellent catalytic performance of this synthesized Au0.3Pd0.7/NH2–N-HMCS nanocomposites can be ascribed to the following four factors. Firstly, the hollow morphology and interconnected mesopores can not only facilitate mass diffusion and accessibility but also confine and enrich the reactants around the supported metal, which helps in improving the catalytic performance. Secondly, NH2-functionalized and N-doped support could effectively immobilize the metal ion by chemical complexation and favor the formation of highly dispersed and small sized metal nanoparticles. Thirdly, the –NH2 group can act as a proton scavenger and facilitate the cleavage of O–H bond in FA molecules. Fourthly, the more electron depletion state of Pd in Au0.3Pd0.7/NH2–N-HMCS caused by metal–support interactions and electron synergetic effect can accelerate the C–H cleavage in the adsorbed formate and the formation of H−.43–45
 | ||
| Fig. 5 Reaction pathway of the dehydrogenation of FA catalyzed by Au0.3Pd0.7/NH2–N-HMCS. | ||
Conclusions
In summary, a novel nanocomposite Au0.3Pd0.7/NH2–N-HMCS has been designed and synthesized in this work, which shows highly efficient catalytic activity for hydrogen generation from aqueous solutions of formic acid, and affords an excellent TOF value as high as 7747 mol H2 per mol catalyst per h at 298 K without any additives. The novel NH2–N-HMCS substrate with spherical morphology, large surface area, sufficient pore volume, rich mesoporous structure, nitrogen doping and –NH2 functionalized surface endows the in situ formed Au0.3Pd0.7 nanoparticles with ultrafine size (2.2 nm) and high dispersity, and the occurring of metal–support interactions and electron transfer between metals, which are the key factors responsible for the excellent catalytic activity of Au0.3Pd0.7/NH2–N-HMCS. This work sheds light on designing highly efficient and durable heterogeneous catalysts through nanostructure engineering for promising applications towards formic acid as a hydrogen storage power source.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Research Project of the Education Department of Jilin Province (No. JJKH20210728KJ) and the National Natural Science Foundation of China (No. 51608050).Notes and references
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- Z. Li and Q. Xu, Acc. Chem. Res., 2017, 50, 1449–1458 CrossRef CAS PubMed.
- M. Wen, K. Mori, Y. Kuwahara and H. Yamashita, ACS Energy Lett., 2017, 2, 1–7 CrossRef CAS.
- Z. Wang, C. Wang, S. Mao, Y. Gong, Y. Chen and Y. Wang, J. Mater. Chem. A, 2019, 7, 25791–25795 RSC.
- Z. Wang, S. Liang, X. Meng, S. Mao, X. Lian and Y. Wang, Appl. Catal., B, 2021, 291, 120140 CrossRef CAS.
- Q. Wang, L. Chen, Z. Liu, N. Tsumori, M. Kitta and Q. Xu, Adv. Funct. Mater., 2019, 29, 1903341 CrossRef.
- M. Navlani-García, K. Mori, Y. Kuwahara and H. Yamashita, NPG Asia Mater., 2018, 10, 277–292 CrossRef.
- Z. Zhang, S. Cao, Y. Liao and C. Xue, Appl. Catal., B, 2015, 162, 204–209 CrossRef CAS.
- N. Wang, Q. Sun, R. Bai, X. Li, G. Guo and J. Yu, J. Am. Chem. Soc., 2016, 138, 7484–7487 CrossRef CAS PubMed.
- Q. Sun, B. W. J. Chen, N. Wang, Q. He, A. Chang, C. Yang, H. Asakura, T. Tanaka, M. J. Hülsey, C. Wang, J. Yu and N. Yan, Angew. Chem., Int. Ed., 2020, 132, 20358–20366 CrossRef.
- J. Cheng, X. Gu, X. Sheng, P. Liu and H. Su, J. Mater. Chem. A, 2016, 4, 1887–1894 RSC.
- M. Khalid, X. Zarate, M. Saavedra-Torres, E. Schott, A. M. B. Honorato, M. R. Hatshan and H. Varela, Chem. Eng. J., 2021, 421, 129987 CrossRef CAS.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G. H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS PubMed.
- Y. Li and J. Shi, Adv. Mater., 2014, 26, 3176–3205 CrossRef CAS PubMed.
- T. Fu, M. Wang, W. Cai, Y. Cui, F. Gao, L. Peng, W. Chen and W. Ding, ACS Catal., 2014, 4, 2536–2543 CrossRef CAS.
- Q. Wang, N. Tsumori, M. Kitta and Q. Xu, ACS Catal., 2018, 8, 12041–12045 CrossRef CAS.
- Q. Bi, J. Lin, Y. Liu, H. He, F. Huang and Y. Cao, Angew. Chem., Int. Ed., 2016, 55, 11849–11853 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- J. Cheng, X. Gu, P. Liu, T. Wang and H. Su, J. Mater. Chem. A, 2016, 4, 16645–16652 RSC.
- H. Wang, Y. Chi, D. Gao, Z. Wang, C. Wang, L. Wang, M. Wang, D. Cheng, J. Zhang, C. Wu and Z. Zhao, Appl. Catal., B, 2019, 255, 117776 CrossRef CAS.
- K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114–1119 CrossRef CAS.
- H. Zhang, O. Noonan, X. Huang, Y. Yang, C. Xu, L. Zhou and C. Yu, ACS Nano, 2016, 10, 4579–4586 CrossRef CAS PubMed.
- B. Shao, Z. Liu, G. Zeng, Y. Liu, X. Yang, C. Zhou, M. Chen, Y. Liu, Y. Jiang and M. Yan, J. Hazard. Mater., 2019, 362, 318–326 CrossRef CAS PubMed.
- W. Nie, Y. Luo, Q. Yang, G. Feng, Q. Yao and Z. Lu, Inorg. Chem. Front., 2020, 7, 709–717 RSC.
- Y. Zhang, F. Jiang, D. Huang, S. Hou, H. Wang, M. Wang, Y. Chi and Z. Zhao, RSC Adv., 2018, 8, 31348–31357 RSC.
- Y. Chi, L. Zhao, Q. Yuan, X. Yan, Y. Li, N. Li and X. Li, J. Mater. Chem., 2012, 22, 13571–13577 RSC.
- V. Duraisamy and R. Krishnan, ACS Appl. Nano Mater., 2020, 3, 8875–8887 CrossRef CAS.
- M. Singh and S. Chauhan, J. Appl. Polym. Sci., 2007, 104, 3261–3268 CrossRef CAS.
- T. Gallardo-Velázquez, G. Osorio-Revilla, M. Zuñiga-deLoa and Y. Rivera-Espinoza, Food Res. Int., 2009, 42, 313–318 CrossRef.
- Z. Lin, M. Song, Y. Ding, Y. Liu, M. Liu and C. Wong, Phys. Chem. Chem. Phys., 2012, 14, 3381–3387 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- C. Zhang, R. Hao, H. Liao and Y. Hou, Nano Energy, 2013, 2, 88–97 CrossRef CAS.
- J. Yan, S. Li, S. Yi, B. R. Wulan, W. Zheng and Q. Jiang, Adv. Mater., 2018, 30, 1703038 CrossRef PubMed.
- P. Liu, X. Gu, H. Zhang, J. Cheng, J. Song and H. Su, Appl. Catal., B, 2017, 204, 497–504 CrossRef CAS.
- X. Yang, P. Pachfule, Y. Chen, N. Tsumori and Q. Xu, Chem. Commun., 2016, 52, 4171–4174 RSC.
- L. Xu, F. Yao, J. Luo, C. Wan, M. Yea, P. Cui and Y. An, RSC Adv., 2017, 7, 4746–4752 RSC.
- A. Al-Nayili and M. Albdiry, New J. Chem., 2021, 45, 10040–10048 RSC.
- A. Bulut, M. Yurderi, Y. Karatas, Z. Say, H. Kivrak, M. Kaya, M. Gulcan, E. Ozensoy and M. Zahmakiran, ACS Catal., 2015, 5, 6099–6110 CrossRef CAS.
- Y. Karatas, A. Bulut, M. Yurderi, I. E. Ertas, O. Alal, M. Gulcan, M. Celebi, H. Kivrak, M. Kaya and M. Zahmakiran, Appl. Catal., B, 2016, 180, 586–595 CrossRef CAS.
- J. Cheng, X. Gu, P. Liu, H. Zhang, L. Ma and H. Su, Appl. Catal., B, 2017, 218, 460–469 CrossRef CAS.
- Y. Chen, Q. Zhu, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2015, 137, 106–109 CrossRef CAS PubMed.
- D. Gao, Z. Wang, C. Wang, L. Wang, Y. Chi, M. Wang, J. Zhang, C. Wu, Y. Gu and H. Wang, Chem. Eng. J., 2019, 361, 953–959 CrossRef CAS.
- L. Zhang, J. Zhu, X. Li, S. Mu, F. Verpoort, J. Xue, Z. Kou and J. Wang, Interdiscip. Mater., 2022, 1, 51–87 CrossRef.
- T. Wang, P. Wang, W. Zang, X. Li, D. Chen, Z. Kou, S. Mu and J. Wang, Adv. Funct. Mater., 2022, 32, 2107382 CrossRef CAS.
- T. Wang, P. Wang, Y. Pang, Y. Wu, J. Yang, H. Chen, X. Gao, S. Mu and Z. Kou, Nano Res., 2022 DOI:10.1007/s12274-022-4072-5.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra01191c |
| This journal is © The Royal Society of Chemistry 2022 |