
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe Reformatsky analogous reaction for the synthesis of novel β-thioxoesters via using aroyl isothiocyanates under solvent-free ball milling and conventional conditions†
Paula S. Farag,
Magdy M. Hemdan and
Aya I. Hassaballah *
*
Chemistry Department, Faculty of Science, Ain Shams University, Abbassia, 11566, Cairo, Egypt. E-mail: ayaibrahim@sci.asu.edu.eg
First published on 5th April 2022
Abstract
The classical Reformatsky reaction, initially described in 1887, is considered one of the most useful ways of forming C–C bonds. The target of this work includes improving the Reformatsky reaction between aroyl isothiocyanates and α-haloesters using metallic zinc to form β-thioxoesters (3–11). In this procedure, a new metal-mediated carbon–carbon linkage is formed with the formation of an organozinc halide and decomposition due to the presence of dilute acid, affording a good yield of the desired product via conventional techniques and ball milling. The Reformatsky reaction requires no solvent and no inert gases.
Introduction
The Reformatsky reaction1,2 was discovered in 1887 and is still extensively used in the synthesis of heterocyclic compounds because it allows the direct insertion of Zn(0) into α-halogenated esters, and the successive addition of the resulting zinc enolate to ketones, aldehydes, or imines.3,4 The classical form of the Reformatsky reaction is demonstrated, as shown in Scheme 1A,5 through the nucleophilic addition of a zinc enolate intermediate to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of aldehydes or ketones leading to the formation of a β-hydroxy ester. The essential tool in recent organic synthesis is the formation of C–C bonds. For several reactions this consists of the metal-mediated synthesis of complex organic compounds, where nucleophilic addition to electrophiles has been established. However, the high basicity and/or nucleophilicity of some organometallic reagents limits their utility in late-stage modification, where sensitive functional groups may already be present in the chemical structure. On the other hand, organozinc6,7 species are a type of “mild” organometallic compound with great functional group compatibility. Conversely,8 the creation of organozinc species frequently necessitates first gaining access to more reactive organometallics, which are subsequently transmetalized with Zn(II) salts to yield the necessary organozinc reactant. Activated Zn(0) can also be utilized to make oxidative additions to carbon–halogen bonds9 (Scheme 1A). Since solvents must often be distilled and dried before use, inert gases are frequently required and, in the case of organozinc reagents, the form of the bulk metal can play an important role, and chemical additives are typically required to generate the activated zinc species, so the formation and manipulation of organometallic compounds are not particularly clean or green.
O of aldehydes or ketones leading to the formation of a β-hydroxy ester. The essential tool in recent organic synthesis is the formation of C–C bonds. For several reactions this consists of the metal-mediated synthesis of complex organic compounds, where nucleophilic addition to electrophiles has been established. However, the high basicity and/or nucleophilicity of some organometallic reagents limits their utility in late-stage modification, where sensitive functional groups may already be present in the chemical structure. On the other hand, organozinc6,7 species are a type of “mild” organometallic compound with great functional group compatibility. Conversely,8 the creation of organozinc species frequently necessitates first gaining access to more reactive organometallics, which are subsequently transmetalized with Zn(II) salts to yield the necessary organozinc reactant. Activated Zn(0) can also be utilized to make oxidative additions to carbon–halogen bonds9 (Scheme 1A). Since solvents must often be distilled and dried before use, inert gases are frequently required and, in the case of organozinc reagents, the form of the bulk metal can play an important role, and chemical additives are typically required to generate the activated zinc species, so the formation and manipulation of organometallic compounds are not particularly clean or green.
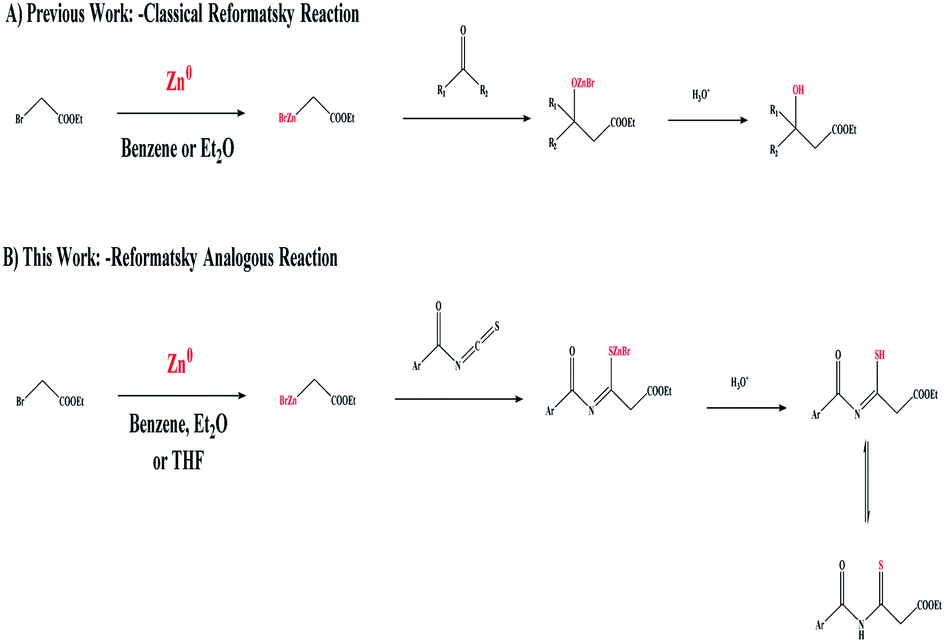 | ||
| Scheme 1 (A) The Reformatsky-reaction-based formation of a β-hydroxy ester. (B) The Reformatsky-analogous-reaction-based formation of a β-thioxoester. | ||
Herein, the formation of our target C–C linkage was obtained through the addition of zinc-enolate to the thiocarbonyl group (CS) of isothiocyanate as the starting organic reagents in the Reformatsky reaction, generating the corresponding organozinc species from the reaction of Zn(0) with α-halogenated esters to give a β-thioxoester (Scheme 1B).
Results and discussion
The research began with the treatment of the model substrates, benzoyl isothiocyanate (1)10–14 (1 mmol) and ethyl 2-bromoacetate (2) (1.5 mmol) with 1 equivalent of zinc (dust <10 mm) heated at 60 °C in benzene (15 ml) for 1–3 h under an N2 atmosphere, as shown in Table 1. The reaction mixture was cooled to 0 °C in ice and then the reaction was quenched with 10% HCl. IR and 1H-NMR spectroscopy of the pure compound confirmed that ethyl 3-benzamido-3-thioxopropanoate (3) was produced in 60% yield (Table 1, entry 1). When the amount of zinc was increased to 1.4 equivalents, the yield of the targeted thioxoester product 3 was greater at 63% (Table 1, entry 2). Adding more zinc with ethyl 2-bromoacetate (2) gave a substantial improvement in yield (Table 1, entries 3–7). With the adjusted reagent ratio in hand, a reaction time study evaluated four different reaction durations of 1, 1.5, 2, and 2.5 h. (Table 1, entries 2 and 5–7), revealing that the reaction requires 2.5 h for complete conversion. The desired product was not produced in a controlled experiment in which zinc was not added to the reaction (Table 1, entry 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Zn [equiv.] | 2 [equiv.] | Solvent | Time [h] | Yield [%] |
| 1 | 1 | 1.5 | Benzene | 2 | 60 |
| 2 | 1.4 | 1.5 | Benzene | 1 | 63 |
| 3 | 1.8 | 1.5 | Benzene | 2 | 68 |
| 4 | 2 | 1.5 | Benzene | 2 | 68 |
| 5 | 2.4 | 2.0 | Benzene | 1.5 | 70 |
| 6 | 2.8 | 2.0 | Benzene | 2 | 74 |
| 7 | 3 | 2.0 | Benzene | 2.5 | 95 |
| 8 | 0 | 2.0 | Benzene | 2.5 | 0 |
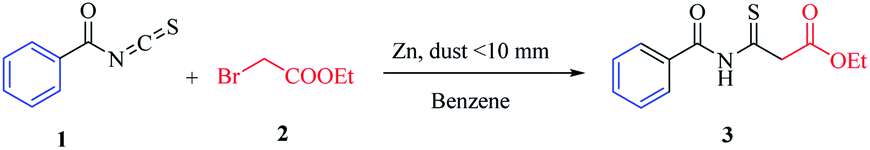
Then we applied the optimized conditions to another 6 different solvents, as shown in Table 1, entry 7, using a fixed equivalent of zinc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl 2-bromoacetate (3:2) for each sample. Luckily, we found that in all cases, regardless of the solvent used, the Reformatsky analogous reaction was successful (Table 2).
:ethyl 2-bromoacetate (3:2) for each sample. Luckily, we found that in all cases, regardless of the solvent used, the Reformatsky analogous reaction was successful (Table 2).
| Entry | Solvent | Time [h] | Yield [%] |
|---|---|---|---|
| 1 | Benzene | 2.5 | 95 |
| 2 | Diethyl ether | 2.5 | 73 |
| 3 | Toluene | 2.5 | 86 |
| 4 | Chloroform | 2.5 | 90 |
| 5 | Dioxane | 2.5 | 83 |
| 6 | Acetonitrile | 2.5 | 89 |
| 7 | Tetrahydrofuran | 2.5 | 80 |
After establishing the optimized conditions, the reactivity with ethyl 2-bromoacetate and zinc (dust <10 mm) under heating was studied (Scheme 2). We found that the Reformatsky analogous reaction showed good functional group tolerance.
 | ||
| Scheme 2 The scope of isothiocyanate electrophiles in the Reformatsky analogous reaction. Reaction conditions: isothiocyanate derivative (1 mmol), ethyl 2-bromoacetate (2 mmol), and zinc (dust, <10 mm, 3 equivalents). Yield refers to the separated products. | ||
The highly hindered substrate 2-chlorobenzoyl isothiocyanate provides the corresponding thioxoester 4 in good yield (72%). Aromatic isothiocyanates were also effective substrates, leading to high yields of isolated products [5–7, 69–81%] (Scheme 2). Under these conditions, the isothiocyanate heterocyclic derivatives were also capable electrophiles, providing isolated products 8 and 9 with 86% and 68% yields, respectively. Aliphatic isothiocyanates such as caproyl and lauroyl isothiocyanates 10 and 11 gave a poor yield of 42% (Scheme 2).
When a solution-based reaction was compared to one performed under ball-milling conditions with comparable chemicals, the convenience of the method was proved (Scheme 3). The solution-based reaction in dry solvent and under a nitrogen atmosphere (N2) at 60 °C with zinc dust yielded 42–95% of the desired products. However, the reaction mixture under ball-milling conditions progressed smoothly without any solvent, inert gas, or additive, giving yields of 50–79% of the desired product (Table 3).
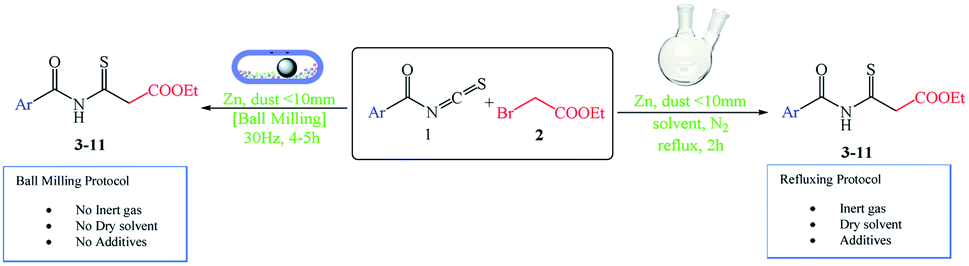 | ||
| Scheme 3 The Reformatsky analogous reaction with zinc dust under different conditions. | ||
|
|||
|---|---|---|---|
| Entry | Ar. | Time [h] | Yield [%] |
| 1 |  |
4.5 | 56 |
| 2 | 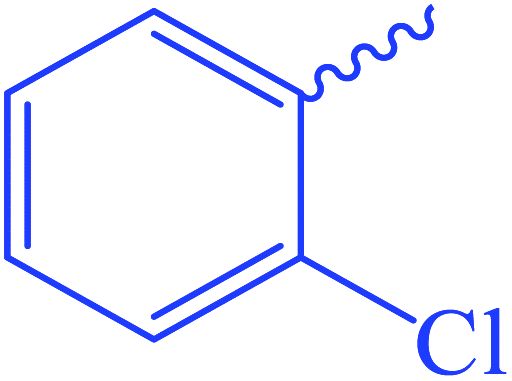 |
4 | 70 |
| 3 | 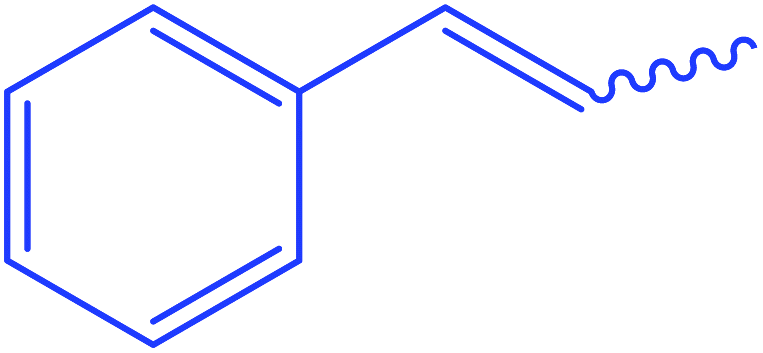 |
5 | 53 |
| 4 | 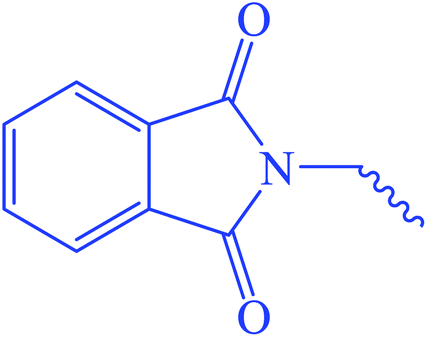 |
4.5 | 76 |
| 5 | 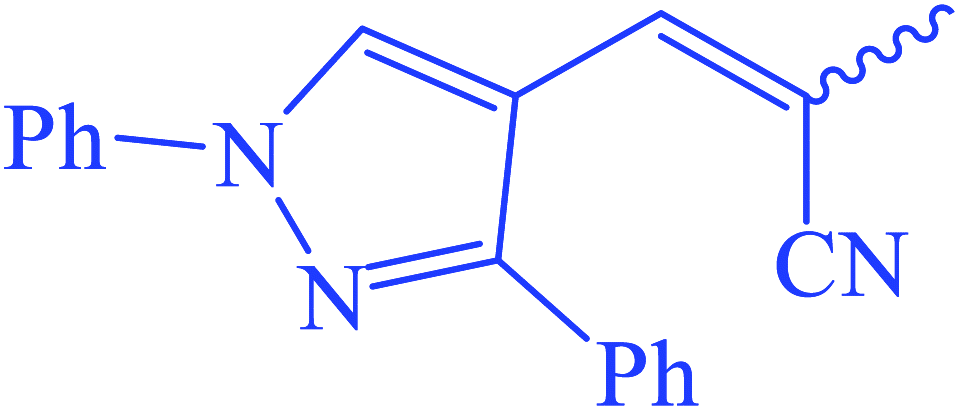 |
4.5 | 55 |
| 6 | 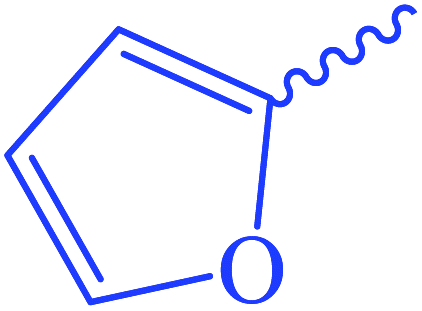 |
4 | 79 |
| 7 |  |
4 | 50 |
| 8 |  |
5 | 48 |
| 9 |  |
5 | 51 |
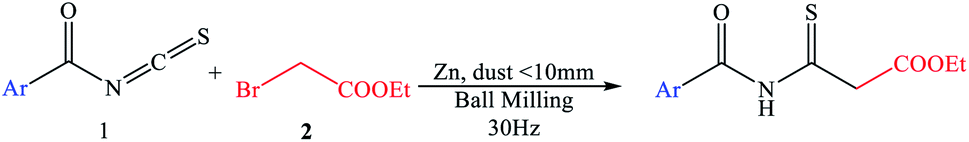
We first compared our mechanochemical technique to the production of such chemicals with a conventional strategy using green metrics, such as atom economy (AE).15,16 The efficiency with which the atoms in the starting components of a reaction are incorporated into the intended product is referred to as the AE17 (i.e., how efficiently a reaction makes use of the reactant atoms).18–20 However, because we employed two different reaction conditions to achieve identical target compounds, the AE values for the mechanochemical and traditional techniques were the same. The theoretical maximum efficiency of reactant usage is given by AE. As a result, we developed yield economy (YE) as a parameter to compare the conversion efficiency of these two different synthetic approaches for the same reaction.17,21,22 The YE is determined using the following equation: [YE = yield (percent)/reaction time (min)]. It basically evaluates how much yield (percent) of the target product is obtained over a given reaction time. A greater YE indicates a higher conversion rate, a considerably more efficient chemical process, and a more cost-effective reaction. In this study, YE was employed to provide a definitive assessment of yields attained under mechanochemical and conventional settings (Table 4).
| Prod. no. | Time (min) | Yield (%) | Yield (YE) economy (% per min) | AE | |||
|---|---|---|---|---|---|---|---|
| B.M. | Con. | B.M. | Con. | B.M. | Con. | ||
| a B.M.: ball milling, Con.: conventional method, AE: atom economy. | |||||||
| 3 | 270 | 150 | 56 | 95 | 20.7 | 63.3 | 76.0 |
| 4 | 240 | 150 | 70 | 72 | 29.1 | 48 | 78.2 |
| 5 | 300 | 150 | 53 | 73 | 17.6 | 48.6 | 77.8 |
| 6 | 270 | 150 | 76 | 69 | 28.1 | 46 | 80.8 |
| 7 | 270 | 150 | 55 | 81 | 20.3 | 54 | 84.8 |
| 8 | 240 | 150 | 79 | 86 | 32.9 | 57.3 | 75.3 |
| 9 | 240 | 150 | 50 | 68 | 20.8 | 45.3 | 76.4 |
| 10 | 300 | 150 | 48 | 42 | 16 | 28 | 75.9 |
| 11 | 300 | 150 | 51 | 42 | 17 | 28 | 80.6 |
Conclusions
The development of an operationally simple one-pot mechanochemical Reformatsky analogous reaction has been completed using isothiocyanate derivatives (CS) instead of aldehydes and/or ketones (CO). This approach eliminates the need for dry solvents, inert gases, and chemical additives via exploiting the organozinc created in situ during milling, resulting in a process with better green metrics.
Experimental
All melting points were determined using uncorrected Griffin and George melting-point equipment (Griffin & George Ltd, Wembley, Middlesex, UK). The quality of the produced compounds was controlled using aluminum sheet silica gel F254 (Merck) thin layer chromatography (TLC). The KBr wafer approach was used to record IR spectra using a Pye Unicam SP1200 spectrophotometer (Pye Unicam Ltd, Cambridge, UK). Ball-milling reactions were made in a MM400 mixer-mill (Retsch GmbH, Haan, Germany), using four 5 mm diameter stainless steel balls in a 5 ml stainless steel jar and were milled vigorously at room temperature with frequency of 1800 rounds per minute (30 Hz). 1H-NMR spectra were obtained on a Bruker Avance III utilizing a Varian Gemini 400 MHz and an internal standard of tetramethylsilane (chemical shifts in scale), whereas 13C-NMR spectra were obtained at 100 MHz in deuterated dimethyl sulfoxide, where TMS was used as an internal standard (DMSO-d6). Elemental studies were performed using a PerkinElmer 2400 CHN elemental analyzer at the Microanalytical Unit, Faculty of Science, Ain Shams University, and satisfactory analytical data (±0.4) were obtained for all substances.General procedures
Ethyl 3-benzamido-3-thioxopropanoate (3). Pale-yellow crystals; m.p 140–142 °C; IR (KBr) (νmax, cm−1): 3168 (NH), 3021 (CH aromatic), 1713 (C
Oester), 1681 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.12 (t, 3H, CH3), 1.72 (q, J = 7.4 Hz, 2H, CH2), 4.89 (s, 2H, CH2), 7.39 (t, J = 8.80 Hz, 1Haromatic), 7.61 (t, J = 4.84 Hz, 2Haromatic), 7.98 (d, J = 1.52 Hz, 2Haromatic), 13.66 (br s, 1H, CON![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) CS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 10, 21, 68, 126, 128, 10, 131, 166, 169, 179. Anal. calcd for C12H13NO3S (251.30) %: C, 57.35; H, 5.21; N, 5.57; found: C, 57.15; H, 5.25; N, 5.60.
CS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 10, 21, 68, 126, 128, 10, 131, 166, 169, 179. Anal. calcd for C12H13NO3S (251.30) %: C, 57.35; H, 5.21; N, 5.57; found: C, 57.15; H, 5.25; N, 5.60.
Ethyl 3-(2-chlorobenzamido)-3-thioxopropanoate (4). Yellow crystals; m.p 152–154 °C; IR (KBr) (νmax, cm−1): 3196 (NH), 30
060, 3003 (CH aromatic), 1713 (COester), 1617 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.59 (t, J = 8.88 Hz, 3H, CH3), 1.82 (q, J = 7.4 Hz, 2H, CH2), 3.778 (s, 2H, CH2), 6.75 (t, J = 13.12 Hz, 1Haromatic), 6.96 (d, J = 8.16 Hz, 1Haromatic), 7.07 (d, J = 9.6 Hz, 1Haromatic), 7.21 (t, J = 13.36 Hz, 1Haromatic), 10.16 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 20, 22, 70, 127, 128, 129, 137, 146, 155, 159, 171. Anal. Calcd for C12H12ClNO3S (285.74) %: C, 50.44; H, 4.23; N, 4.90; found: C, 50.48; H, 4.28; N, 4.95.
Ethyl 3-cinnamamido-3-thioxopropanoate (5). White crystals; m.p 222–224 °C; IR (KBr) (νmax, cm−1): 3265 (NH), 3130 (CH aromatic), 1705 (C
Oester), 1688 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 0.67 (t, J = 15.12 Hz, 3H, CH3), 2.14 (q, J = 5.96 Hz, 2H, CH2), 5.13 (s, 2H, CH2), 6.73 (t, J = 14.48 Hz, 1Haromatic), 6.96 (d, J = 8.72 Hz, 2Haromatic), 7.07 (d, J = 7.72 Hz, 1Holiefinic), 7.21 (t, J = 15.68 Hz, 2Haromatic), 7.60 (d, J = 8.72 Hz, 1Holiefinic), 13.75 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 37, 42, 70, 126.03, 126.71, 128.11, 128.46, 128.55, 129.80, 129.89, 167, 170.62, 170.77. Anal. calcd for C14H15NO3S (277.34) %: C, 60.63; H, 5.45; N, 5.05; found: C, 60.60; H, 5.41; N, 5.08.
Ethyl 3-(2-(1,3-dioxoisoindolin-2-yl)acetamido)-3-thioxopropanoate (6). Yellow crystals; m.p 212–214 °C; IR (KBr) (νmax, cm−1): 3204 (NH), 3063 (CH aromatic), 2921, 2851 (CH aliphatic), 1732 (C
Oester), 1671 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.50 (m, 3H, CH3), 1.70 (q, J = 12.72 Hz, 2H, CH2), 4.89 (s, 2H, CSC2CO), 4.92 (s, 2H, NC2CO), 7.38 (m, 2Haromatic), 7.63 (m, 2Haromatic), 13.69 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 15, 22, 65, 66, 123.33, 123.69, 126, 128, 158, 159, 166, 168, 169. Anal. calcd for C15H14N2O5S (334.35) %: C, 53.89; H, 4.22; N, 8.38; found: C, 53.86; H, 4.25; N, 8.35.
Ethyl(E,Z)-3-(2-cyano-3-(1,3-diphenyl-1H-pyrazol-4-yl)acrylamido)-3-thioxopropanoate (7). Yellow crystals; m.p 278–280 °C; IR (KBr) (νmax, cm−1): 3238 (NH), 3040 (CH aromatic), 2215 (C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1712 (COester), 1667 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.26 (t, J = 6.08 Hz, 3H, CH3), 4.56 (q, J = 15.20 Hz, 2H, CH2), 5.42 (s, 2H, CSC2CO), 7.55 (m, 3Haromatic), 7.67 (m, 3Haromatic), 7.80 (m, 3Haromatic), 8.02 (m, 2Holiefinic and aromatic), 13.75 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 20, 23, 69, 117, 118, 124.32, 124.78, 129, 131, 132, 135, 141, 153, 167, 170. Anal. calcd for C24H20N4O3S (444.51) %: C, 64.85; H, 4.54; N, 12.60; found: C, 64.82; H, 4.50; N, 12.58.
N), 1712 (COester), 1667 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.26 (t, J = 6.08 Hz, 3H, CH3), 4.56 (q, J = 15.20 Hz, 2H, CH2), 5.42 (s, 2H, CSC2CO), 7.55 (m, 3Haromatic), 7.67 (m, 3Haromatic), 7.80 (m, 3Haromatic), 8.02 (m, 2Holiefinic and aromatic), 13.75 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 20, 23, 69, 117, 118, 124.32, 124.78, 129, 131, 132, 135, 141, 153, 167, 170. Anal. calcd for C24H20N4O3S (444.51) %: C, 64.85; H, 4.54; N, 12.60; found: C, 64.82; H, 4.50; N, 12.58.
Ethyl 3-(furan-2-carboxamido)-3-thioxopropanoate (8). Orange crystals; m.p 148–150 °C; IR (KBr) (νmax, cm−1): 3242 (NH), 3051 (CH aromatic), 1710 (C
Oester), 1669 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 2.19 (m, 3H, CH3), 2.59 (m, 2H, CH2), 4.88 (s, 2H, CSC2CO), 7.61 (dd, J = 7.4 and 1.48 Hz, 1Haromatic), 8.01 (t, J = 7.84 Hz, 1Haromatic), 8.63 (d, J = 8.36 Hz, 1Haromatic), 12.34 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 15, 22, 68, 123.33, 123.69, 126, 128, 158, 159, 168, 169. Anal. calcd for C10H11NO4S (241.26) %: C, 49.78; H, 4.60; N, 5.81; found: C, 49.80; H, 4.64; N, 5.83.
Ethyl 3-(thiophene-2-carboxamido)-3-thioxopropanoate (9). Yellow crystals; m.p 212–214 °C; IR (KBr) (νmax, cm−1): 3103 (NH), 3053 (CH aromatic), 1730 (C
Oester), 1698 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.90 (m, 5H, CH2 and CH3), 4.88 (s, 2H, CSC2CO), 6.69 (dd, J = 6.56 and 7.32 Hz, 1Haromatic), 7.13 (t, J = 15.72 Hz, 1Haromatic), 7.73 (dd, J = 2.36 and 2.52 Hz, 1Haromatic), 12.34 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 19, 21, 68, 112, 118, 129.11, 129.50, 149, 169. Anal. calcd for C10H11NO3S2 (257.32) %: C, 46.68; H, 4.31; N, 5.44; found: C, 46.70; H, 4.28; N, 5.40.
Ethyl 3-hexanamido-3-thioxopropanoate (10). White crystals; m.p 88–90 °C; IR (KBr) (νmax, cm−1): 3290 (NH), 2922, 2849 (CH aliphatic), 1711 (C
Oester), 1635 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 0.88 (t, J = 10.96 Hz, 3H, OCH2C3), 1.26 (m, 3Haliphatic), 1.38 (m, 4Haliphatic), 1.64 (m, 4Haliphatic), 4.75 (s, 2H, CSC2CO), 5.16 (q, J = 15.20 Hz, 2H, OC2CH3), 13.69 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 28, 29.29, 29.36, 29.41, 29.60, 29.61, 30, 31, 68, 152, 166, 169. Anal. calcd for C11H19NO3S (245.34) %: C, 53.85; H, 7.81; N, 5.71; found: C, 53.88; H, 7.85; N, 5.73.
Ethyl 3-dodecanamido-3-thioxopropanoate (11). White crystals; m.p 78–80 °C; IR (KBr) (νmax, cm−1): 3171 (NH), 2917, 2849 (CH aliphatic), 1700 (br C
Oester), 1679 (COamide). 1H-NMR (400 MHz, DMSO-d6) δppm: 1.05 (m, 6Haliphatic), 1.56 (m, 20Haliphatic), 3.62 (m, 2H, OC2CH3), 5.01 (s, 2H, CSC2CO), 12.89 (br s, 1H, CONCS, exchangeable with D2O). 13C-NMR (100 MHz, DMSO-d6) δppm: 22, 26, 28, 29.29, 29.36, 29.41, 29.60, 29.61, 30, 31, 65, 161, 169.13, 169.70. Anal. calcd for C17H31NO3S (329.50) %: C, 61.97; H, 9.48; N, 4.25; found: C, 61.95; H, 9.44; N, 4.18.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors would like to extend their sincere appreciation to the Magdy lab in the Chemistry Department, Faculty of Science of Ain Shams University.References
- S. Choppin, L. Ferreiro-Medeiros, M. Barbarotto and F. Colobert, Recent advances in the diastereoselective Reformatsky-type reaction, Chem. Soc. Rev., 2013, 42, 937–949 RSC.
- A. Fürstner, Recent Advancements in the Reformatsky Reaction, Synthesis, 1989, 8, 571–590 CrossRef.
- Y. Segade, M. A. Montaos, J. Rodríguez and C. Jiménez, A Short Stereoselective Synthesis of Prepiscibactin Using a SmI2-Mediated Reformatsky Reaction and Zn2+ Induced Asymmetric Thiazolidine Formation, Org. Lett., 2014, 16, 5820–5823 CrossRef CAS PubMed.
- M. W. Rathke, The Reformatsky Reaction, Org. React., 1975, 22, 423–460 CAS.
- Q. Ca, R. T. Stark, I. A. Fallis and D. L. Browne, A Ball-Milling-Enabled Reformatsky Reaction, ChemSusChem, 2019, 12, 2554–2557 CrossRef PubMed.
- A. Conan, S. Sibille and J. Perichon, Metal exchange between an electrogenerated organonickel species and zinc halide: application to an electrochemical, nickel-catalyzed Reformatsky reaction, J. Org. Chem., 1991, 56, 2018–2024 CrossRef CAS.
- T. Yanagi, R. J. Somerville, K. Nogi, R. Martin and H. Yorimitsu, Ni-Catalyzed Carboxylation of C(sp2)–S Bonds with CO2: Evidence for the Multifaceted Role of Zn, ACS Catal., 2020, 10, 2117–2123 CrossRef CAS.
- E. A. Nikiforovaa, D. V. Baibarodskikha, N. F. Kirillova, M. V. Dmitrieva and D. P. Zverev, Reformatsky Reaction of 1-Aryl-3-(2-hydroxyphenyl)prop-2-en-1-ones with Methyl 1-Bromocyclohexanecarboxylate, Russ. J. Org. Chem., 2020, 56, 2074–2078 CrossRef.
- L. Huck, M. Berton, A. de la Hoz, A. Díaz-Ortiz and J. Alcázar, Reformatsky and Blaise reactions in flow as a tool for drug discovery. One pot diversity oriented synthesis of valuable intermediates and heterocycles, Green Chem., 2017, 19, 1420–1424 RSC.
- P. S. Farag, M. M. Hemdan and A. A. El-Sayed, Nano nickel [1,2,4]-triazole-3-thiones complex: Design, sonochemical synthesis, and antimicrobial evaluation, J. Heterocycl. Chem., 2020, 57, 3428–3441 CAS.
- I. F. Nassar, S. R. Att-Allah and M. M. Hemdan, Utility of thiophene-2-carbonyl isothiocyanate as a precursor for the synthesis of 1,2,4-triazole, 1,3,4-oxadiazole and 1,3,4-thiadiazole derivatives with evaluation of their antitumor and antimicrobial activities, Phosphorus, Sulfur Silicon Relat. Elem., 2018, 193, 630–636 CrossRef CAS.
- M. M. Hemdan and A. A. El-Sayed, Synthesis of Some New Heterocycles Derived from Novel 2-(1,3-Dioxisoindolin-2-yl)Benzoyl Isothiocyanate, J. Heterocycl. Chem., 2016, 487, 487–492 CrossRef.
- M. H. Hekal, P. S. Farag, M. M. Hemdan and W. M. El-Sayed, New N-(1,3,4-thiadiazol-2-yl)furan-2-carboxamide derivatives as potential inhibitors of the VEGFR-2, Bioorg. Chem., 2021, 115, 105176 CrossRef CAS PubMed.
- S. R. Atta-Allah, A. M. AboulMagd and P. S. Farag, Design, microwave assisted synthesis, and molecular modeling study of some new 1,3,4-thiadiazole derivatives as potent anticancer agents and potential VEGFR-2 inhibitors, Bioorg. Chem., 2021, 112, 104923 CrossRef CAS PubMed.
- B. M. Trost, in Handbook of Green Chemistry, ed. Li, C. J., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, vol. 7, pp. 1–88 Search PubMed.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Towards a holistic approach to metrics for the 21st century pharmaceutical industry, Green Chem., 2015, 17, 3111–3121 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Mechanochemistry: opportunities for new and cleaner synthesis, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- A. A. Moores and R. H. Crabtree, Handbook of Green Chemistry, Wiley-VCH Verlag GmbH& Co. KGaA, Weinheim, 2009, vol. 1 Search PubMed.
- N. Kumagai, Development of Atom-Economical Catalytic Asymmetric Reactions under Proton Transfer Conditions: Construction of Tetrasubstituted Stereogenic Centers and Their Application to Therapeutics, Chem. Pharm. Bull., 2011, 59, 1–22 CrossRef CAS PubMed.
- R. A. Sheldon, Fundamentals of green chemistry: efficiency in reaction design, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- A. F. M. Fahmy, S. A. Rizk, M. M. Hemdan, A. A. El-Sayed and A. I. Hassaballah, Efficient Green Synthesis and Computational Chemical Study of Some Interesting Heterocyclic Derivatives as Insecticidal Agents, J. Heterocycl. Chem., 2018, 55, 2545–2555 CrossRef CAS.
- A. F. M. Fahmy, A. A. El-Sayed, M. M. Hemdan, A. I. Hassaballah and A. F. Mabied, Synthesis of N-Containing Heterocycles via Mechanochemical Grinding and Conventional Techniques, Asian J. Chem., 2017, 29, 2679–2686 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/d2ra01154a |
| This journal is © The Royal Society of Chemistry 2022 |