DOI:
10.1039/D2RA01127A
(Paper)
RSC Adv., 2022,
12, 13412-13427
Enriching NLO efficacy via designing non-fullerene molecules with the modification of acceptor moieties into ICIF2F: an emerging theoretical approach†
Received
20th February 2022
, Accepted 19th April 2022
First published on 4th May 2022
Abstract
Non-fullerene (NF)-based compounds have attracted much attention as compared to fullerene-based materials because of their promising optoelectronic properties, lower synthetic cost and greater stability. Usually, the end-capped groups have a promising impact in magnifying the nonlinear optical (NLO) characteristics in the non-fullerene molecules. Based on this, a series of new NLO active non-fullerene molecules (NFAD2–NFAD6) have been established. The non-fullerene molecules (NFAD2–NFAD6) were designed by end-capped modification in acceptor moieties of the reference (NFAR1), while donor and π-bridge moieties were kept the same in the entire series. Quantum chemistry-based calculations at the M06/6-311G(d,p) level were done to determine the NLO characteristics and for other supportive analyses. The acceptor and donor moieties were utilized at the opposite terminals of NFAD2–NFAD6, which proved to be an effective approach in tuning the FMO band gap. Overall the results of natural bond orbital (NBO), density of state (DOS) and transition density matrices (TDMs) analyses supported the NLO properties of the designed compounds. Among all the studied compounds, NFAD4 was proven to be the most suitable candidate due to its promising NLO properties, well supported by a lower bandgap of 1.519 eV and a maximum absorption wavelength of 999.550 nm. Therefore, NFAD4 was reported with greater amplitude of dipole polarizability (10.429 e.s.u), average polarizability (2.953 × 10−22 e.s.u), first hyperpolarizability (13.16 × 10−27 e.s.u.) and second hyperpolarizability (2.150 × 10−31 e.s.u.) than other derivatives and NFAR1. Subsequently, the present study depicted the significance of utilizing different non-fullerene (NF)-based acceptor moieties to achieve the promising NLO material. This computational study may lead towards new plausible pathways for researchers to design potent NLO substances for impending hi-tech applications.
Introduction
In the past few decades, nonlinear optical (NLO) materials exhibited remarkable advancements in the area of fiber optics, data transformation, photonic laser, electro-optics, and data storage in the field of wireless communication.1 Nowadays, it is challenging and the most important subject for researchers to design high-performance NLO materials using organic and inorganic systems.2 For most of the period, inorganic materials have been used in many commercial NLO applications, as they allow anisotropic ion-exchange and provide waveguide structures.3 However, organic compounds are easy to synthesize and have lower dielectric coefficients, which have tremendously shifted away the attention of researchers from inorganics towards organic systems during the last fifteen years.4,5 The organic chromophores are considered as vital NLO materials these days owing to their thermal stability, intrinsic rapid reaction rates, appropriate refinement, high molecular susceptibility, excellent absorption co-efficient, high response time, tunable band and ease of molecular designing compared to inorganic molecules.6–8 To increase the charge transference ability, organic compounds are coupled with fullerene-based acceptor units.9,10 However, fullerene-based compounds are described accompanying a few defects: low absorption in the visible region and poor photo-stability.11,12 Now, the focus of researchers is shifted from fullerene towards non-fullerene (NF) chromophores due to their ability to modify chemical structures and electron affinities over a wide range, tunable energy levels and facile synthesis.13–16 As they have shown an exceptional stability, hence their electronic and optical characteristics can easily be tuned for the sake of promising results17–19 via structural modifications.
The acceptor–donor-acceptor (A-D-A)-type chromophores have arisen as outstanding electron acceptors in the field of organic OPV materials. Additionally, owing to a highly π-conjugated system, robust ICT and greater electronic charge movement, the A-D-A kind molecules are considered to express efficient NLO characteristics.20 The ICT behavior of molecules has been governed not only by the inherent properties of their component moieties but also by their molecular packing.21 Therefore, the structure–property relationship or molecular architecture should be another significant factor to enriched NLO performance.22,23 Literature study reveals that the organic frameworks possess remarkable NLO results generally through the push–pull model (D–π–A) configuration that increases the charge transfer process. For this, the NLO chromophores with pull–push schemes have drained the attention of researchers.24,25 Numerous structures incorporating donor–acceptor, donor–π-spacer–acceptor, donor–π-linker–acceptor–π-spacer–donor, acceptor–π-spacer–donor–π-linker–acceptor, donor–π–π–acceptor, donor–acceptor–π–acceptor and donor–donor–π–acceptor are reported in the literature.26–28 The nature of substituents and degree of π conjugation greatly influence the NLO characteristics of the compounds. The intramolecular charge transfer (ICT) via π-linkers exists between donor and acceptor moieties of a molecule, accomplishing marvelous NLO responses.23,24 In fact, the π-linkers in the A–π–D–π–D approach act as connectors between acceptor and donor groups, shift the absorption spectra towards the longer wavelength, enhance the charge density and decrease the HOMO–LUMO bandgap.29,30 Moreover, the extended π-conjugation with intense absorption parameters of fullerene-free chromophores have been enhanced via attachment of electron-deficient terminal groups (cyano, nitro and halogens groups) over the skeleton of the non-fullerene acceptors (NFAs).31,32 Furthermore, the nature and conjugated length of donor and acceptor parts are also important to design interesting NLO materials.33
The synthesized compound (ICIF2F) is reported in the literature34 and according to our best insights, its NLO study has not yet been published. NFAR1 is a non-fullerene (A1–D–π–A2)-based compound that comprises (2-(2-methylene-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile) as a first acceptor, (2,4,4,7,9,9-hexamethyl-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene) as a donor, (methyl 3-fluoro-6-methylthieno[3,4-b]thiophene-2-carboxylate) as a π-linker and (2-(5,6-difluoro-2-methylene-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile) as a second acceptor. As the literature review exploits that NF chromophores with the push–pull configuration possess outstanding NLO properties, in this study, we designed a series of five A–π–D–π–D-type NF-based chromophores in order to obtain significant NLO materials. The D and π-spacers are kept unchanged throughout the design, while A groups are tailored. Various parameters for all designed compounds (NFAD2–NFAD6) and the reference molecule (NFAR1) were calculated and compared to evaluate the performance of the newly engineered molecules as efficient NLO materials. These NLO-based outcomes might provide a clue for the engineered of D–π–A architecture-based novel fullerene-free organic entities and also support the experimental communities to synthesize promising NLO materials for up-to-date anticipations.
Computational procedure
DFT/TDDFT calculations were carried out using the Gaussian 0935 program package in conjugation with the M06/6-311G(d,p) basis set to investigate all the quantum chemical computations of designed NFAR1 and NFAD2–NFAD6 compounds. The Gauss View 6.1 program36 was used to make input files. The DOS analysis (to calculate the electronic charge density of the studied molecules) was interpreted using the PyMOlyze 2.0 program.37 The absorption properties of the aforesaid chromophore in the excited state were calculated in dichloromethane using the polarizable continuum (CPCM) model.38 The FMO analysis was performed to compute the lowest unoccupied molecular orbital (LUMO), highest occupied molecular orbital (HOMO) and the band gap energies. Various software programs like Avogadro39 and Chemcraft40 were employed to interpret the results from output files. Dipole moment (μ),41 average polarizability 〈α〉,42 first hyperpolarizability (βtotal),42 and second hyperpolarizability (γtotal)42 values were calculated using eqn (1)–(4):| | |
μ = (μx2 + μy2 + μz2)1/2
| (1) |
| | |
〈α〉 = 1/3(αxx + αyy + αzz)
| (2) |
| | |
βtotal = [(βx2 + βy2 + βz2)]1/2
| (3) |
where (βx = βxxx + βxyy + βxzz, βy = βyyy + βxxy + βyzz, βz = βzzz + βxxz + βyyz) tensors were obtained using eqn (3):| |
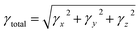 | (4) |
where. 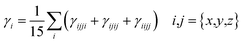 .
.
Results and discussion
The synthesized compound (ICIF2F) consists of A1–D–π–A2 configuration as reported in the literature.34 In the present study, different acceptors were utilized on the chemical framework of designed compounds to understand their influence for the NLO activity. In ICIF2F, an alkyl chain (R = –C8H17) was substituted on the donor unit and an EH = –2-ethylhexyl bulky group was incorporated into the π-linker, which were exchanged with methyl groups (–CH3) to reduce the steric hindrance of long alkyl chains and to avoid the computational cost. After this minor substitution modification, the name of the compound is changed from “ICIF2F” to “NFAR1” for distinguishing purpose (Fig. 1).
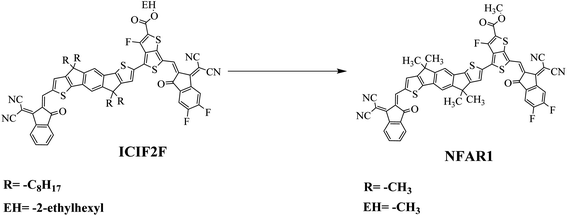 |
| | Fig. 1 Structural modifications in the parent compound (ICIF2F). | |
In the current investigation, NFAR1 was considered as a reference compound after modifications in the parent compound (ICIF2F).34 The end-capped acceptor “A1” group of the NFAR1 molecule was replaced with N,N-dimethylaniline and acceptor “A2” was modified with different fluoro (–F), cyano (–CN) and nitro (–NO2) groups to find the electronic and optical properties of the tailored chromophores (Fig. 2). Further, from the reference (NFAR1) compound, five derivatives (NFAD2–NFAD6) were designed with donor–π–acceptor configuration, as shown in Fig. 3(a and b). The recent work is based on the theoretical fabrication of novel materials, which show efficient NLO characteristics. Their results are organized in different subsections for better understanding as HOMO/LUMO orbital energy gaps, NLO properties, TDMs, DOS, NBO, and UV-visible spectra.
 |
| | Fig. 2 Sketch map of designed compounds (NFAD2–NFAD6). | |
 |
| | Fig. 3 (a) Structure of NFAR1 (A1–D–π–A2) and designed compounds; NFAD2–NFAD6 (D–π–A). (b) Push–pull mechanism of designed compounds. | |
Electronic structures
FMO analysis enables us to determine significant quantum chemistry parameters: electronic properties, chemical stability, electron transfer properties, and reactivity of investigated compounds.43 The obtained energy difference (Egap = ELUMO − EHOMO) of designed compounds is directly associated with their chemical and kinetic stability.44 Normally, HOMO entails the electron-donating tendency, while LUMO focuses on the electron accepting nature. FMOs also play an important role in shaping the UV-visible spectrum and the reaction mechanism study of compounds.45 Molecules having a higher bandgap are considered chemically hard, more stable, and less reactive, whereas molecules having smaller bandgaps are considered unstable, highly polarizable, and soft compounds; consequently, such compounds exhibit impressive NLO response.23–26 DFT computations were executed to determine EHOMO, ELUMO, and Egap of NFAD2–NFAD6, and their obtained values are displayed in Table 1.
Table 1 Computed energies as EHOMO, ELUMO and bandgaps of said chromophoresa
| Compounds |
EHOMO |
ELUMO |
Egap |
| Units in eV. |
| NFAR1 |
−5.877 |
−3.563 |
2.314 |
| NFAD2 |
−5.325 |
−3.411 |
1.914 |
| NFAD3 |
−5.333 |
−3.454 |
1.879 |
| NFAD4 |
−5.384 |
−3.777 |
1.607 |
| NFAD5 |
−5.390 |
−3.772 |
1.618 |
| NFAD6 |
−5.362 |
−3.614 |
1.748 |
Table 1 describes the calculated HOMO/LUMO energy values of the reference molecule (NFAR1) as −5.877/−3.563 eV, which are found in concordance with the experimental values, i.e. −5.45/−4.02 eV of the parent molecule.34 These results indicated that the DFT methodology, i.e., the M06 level with the 6-311G(d,p) basis set, is an appropriate functional to investigate quantum chemical calculations of NFAD2–NFAD6. Among all the designed compounds, the reference compound has the largest bandgap value of 2.314 eV. This energy gap value reduces to 1.914 eV for NFAD2 due to the presence of N,N-dimethylaniline on (4,4,9,9-tetramethyl-4,5,9,10-tetrahydro-s-indaceno[1,2-b:5,6-b′]dithiophene) as a donor moiety, which boosts the electron-donating capability of the D group towards the A group (2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile) by creating a strong push–pull mechanism. The Egap value in NFAD3 is further lowered, which might be due to the substitution of one more fluoro group on the acceptor part as (2-(4,5,6-trifluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile). As it is well known that the fluoro group is highly electronegative among all halogens, it improves the electron-withdrawing ability of the acceptor moieties, which may cause a reduction in the energy gap value of NFAD3, i.e. 1.879 eV. The HOMO–LUMO band gap in NFAD4 is observed to be even smaller, i.e. 1.607 eV, than that in NFAD3 owing to its chemical structure that is designed by incorporating the three cyano (–CN) groups on the acceptor part of NFAD3. The –CN group is more electron withdrawing and has a larger negative inductive effect than that of the –F group, which strengthens the charge transfer process and further reduces the energy gap value. This is the smallest value of Egap amongst all the aforesaid compounds. Similarly, the geometry of NFAD5 is tailored by exchanging the three cyano (–CN) units with nitro (–NO2) moieties in the acceptor region of NFAD4 (Fig. 4). The bandgap value 1.618 eV of NFAD5 was found to be slightly larger than that of NFAD4. This variation in bandgap is due to –NO2, which deactivates the aromatic ring through the resonance and negative inductive effect (–I). Although the negative inductive effect of the –NO2 group is greater than that of the –CN group, it is more stable. This is the main reason behind the variation in the bandgap of NFAD4 and NFAD5. Furthermore, the bandgap value of NFAD6 was found to be 1.748 eV owing to the presence of three –CF3 groups on the acceptor part as (2-(3-oxo-4,5,6-tris(trifluoromethyl)-2,3-dihydro-1H-inden-1-ylidene)malononitrile). The energy gap (Egap) of NFAD6 was larger than that of NFAD5 because it has the –CF3 group on the acceptor part. Although three powerful fluoro atoms attached to the –CF3 group enhance its withdrawing capacity, –NO2 has both –I and –R effects. The group that has both –I and –R effects might be more electron-withdrawing than the group that has only the –I effect. Hence, the LUMO–HOMO energy band gap is successfully narrowed down in all the designed compounds.
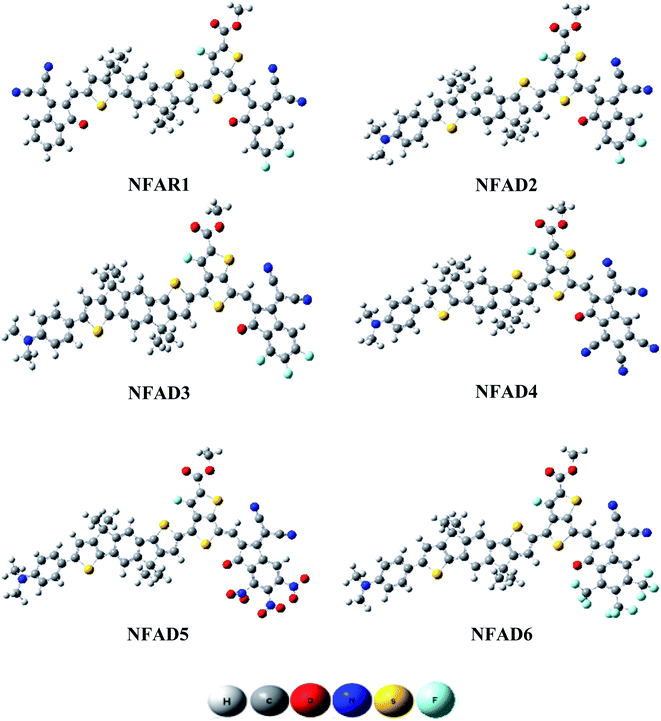 |
| | Fig. 4 Optimized geometries of NFAR1 and NFAD2–NFAD6. | |
The descending order of the energy band gap was studied as follows: NFAR1 > NFAD2 > NFAD3 > NFAD6 > NFAD5 > NFAD4. This order illustrates that the incorporation of different electronegative substituents on the acceptor part of the designed compounds would be an important aspect to obtain significant NLO responses.24
The electronic charge distribution in NFAR1 and NFAD2–NFAD6 on their respective HOMO/LUMO levels is depicted in Fig. 5. The significant CT proved that the above-mentioned chromophores might be impressive NLO materials.24,25 In NFAR1, the electronic cloud for HOMO is localized mainly over (4,4,9,9-tetramethyl-4,5,9,10-tetrahydro-s-indaceno[1,2-b:5,6-b′]dithiophene), while for LUMO, it is over the acceptor part (2-(5,6-difluoro-3-oxo-2,3-dihydro-1H-inden-1-ylidene)malononitrile) and partly over the π-linker. In the tailored compounds, NFAD2–NFAD6 charge density for HOMO is mainly over (N,N-dimethyl-4-(4,4,9,9-tetramethyl-4,5,9,10-tetrahydro-s-indaceno[1,2-b:5,6-b′]dithiophen-2-yl)aniline). The LUMOs are presented mainly on the electron-withdrawing moieties and π-linker. The ICT from the donor to acceptor part is indicated by the separation between HOMO and LUMO when the molecules are excited. This assistance of charge confirms that all designed compounds could be promising NLO active materials.
 |
| | Fig. 5 HOMOs and LUMOs of NFAR1 and designed molecules (NFAD2–NFAD6). | |
Density of state
The density of state (DOS) exploration was achieved to support the results obtained by FMO analysis of studied compounds (NFAR1 and NFAD2–NFAD6) at the M06/6-311G(d,p) level. By changing the terminal acceptor parts, the electronic charge density distribution is shifted in different patterns around HOMO and LUMO, as can be visualized in Fig. 6. The DOS exposes the distribution of electrons from HOMO to LUMO of the molecule.14 From Fig. 6, it is cleared that the electronic cloud of HOMO in the reference compound is only focused on the donor moiety, whereas the acceptor moiety has major LUMO charge density and minimal LUMO concentration is presented on the π-core. In the entitled molecules (NFAD2–NFAD6), a similar charge distribution phenomenon of HOMO and LUMO density is noted, i.e., major concentration in the acceptor region and minor concentration over the π-core. The photovoltaic and electronic characteristics of the NFAD2–NFAD6 compounds are efficiently improved due to powerful shifting of the electron densities, as confirmed by the scattering configurations of HOMO and LUMO.
 |
| | Fig. 6 Density of states (DOS) of NFAR1 and NFAD2–NFAD6. | |
For the reference compound (NFAR1), the highest charge density of HOMO is found to be −5.877, and it is located on the donor part, while the highest charge density for LUMO is found to be −3.563 eV by the acceptor group and smaller contributions are also seen by the π-spacer. In NFAR1, acceptor 1 and acceptor 2 contributions collectively are found to be 24.5% to HOMO and 52.3% contributions to LUMO, while the donor contributes 54.4% to HOMO and 23.8% to LUMO. These contributions are changed effectively in tailored compounds due to the use of different acceptor groups.
Herein, the donor contributes 90.7%, 90.6%, 90.3%, 90.9%, and 90.8%, to HOMO and 21.1%, 21.1%, 17.3%, 10.9%, and 20.6% to LUMO in NFAD2–NFAD6, respectively. Acceptors contribute 4.3%, 4.4%, 5.2%, 4.8% and 4.7% to HOMO and 50.3%, 51.3%, 63.6%, 76.9% and 54.2% to LUMO in NFAD2–NFAD6, respectively. Similarly, π-spacers contribute 5.0%, 4.9%, 4.5%, 4.3%, and 4.5% to HOMO and 28.5%, 27.7%, 19.1%, 12.2% and 25.1% to LUMO in NFAD2–NFAD6, respectively.
In NFAD4, the concentrated HOMO charge cloud is examined by both donors and the LUMO distributions seemed to be on acceptor moiety. Approximately the same charge distribution between HOMO and LUMO is inspected for compounds NFAD5 and NFAD6. All plotted charge distributions reveal that the compound NFAD4 transfers the charge most efficiently among all the tailored compounds. Therefore, effective charge transfers occur via the π-bridge in all the fabricated molecules, suggested that these NFA-based molecules could be efficient NLO compounds.
Global reactivity depicters
The energy values of derivatives were computed by the DFT method to examine global reactivity variables such as electronegativity, ionization potential, hardness, electron affinity, softness, electrophilicity and chemical potential. These parameters have been defined by Koopmans' equations.46 The ionization potential (IP), electronegativity (X), electron affinity (EA),47 global hardness (η),48 global softness (σ), chemical potential (μ) and electrophilicity index (ω)49 were calculated using eqn (5–11), respectively:
The results obtained from these equations are presented in Table 2.
Table 2 Global reactivity descriptors of reference and designed compoundsa
| Compounds |
IP |
EA |
X |
η |
μ |
ω |
σ |
| Units in eV. |
| NFAR1 |
5.877 |
3.563 |
4.720 |
1.157 |
−4.720 |
9.628 |
0.432 |
| NFAD2 |
5.325 |
3.411 |
4.368 |
0.957 |
−4.368 |
9.968 |
0.522 |
| NFAD3 |
5.333 |
3.454 |
4.394 |
0.940 |
−4.394 |
10.273 |
0.532 |
| NFAD4 |
5.384 |
3.777 |
4.581 |
0.804 |
−4.581 |
13.056 |
0.622 |
| NFAD5 |
5.390 |
3.772 |
4.581 |
0.809 |
−4.581 |
12.970 |
0.618 |
| NFAD6 |
5.362 |
3.614 |
4.488 |
0.874 |
−4.488 |
11.523 |
0.572 |
The HOMO and LUMO energies of the above-mentioned chromophores are expressed by IP and EA, respectively. Indeed, the electron donating and accepting property of aforesaid molecules could be determined by IP and EA values.27 Further, in our investigated compounds (NFAD2–NFAD6), the IP values (5.325–5.390 eV) are smaller than those of their parent chromophore (5.877 eV), which indicated easier release of electron and lower energy required to make them polarized than NFAR1. Similarly, greater EA values of derivatives excluding NFAD2 and NFAD3 than those of the reference chromophore exploited their greater tendency to accept electron because of the presence of robust acceptor moieties. Likewise, the η values of NFAD2–NFAD6 are found to be much smaller (0.957–0.804 eV) with larger σ (0.522–0.622 eV) than those of NFAR1 (η = 1.157 and σ = 0.432 eV) which expressed the greater chemical reactivity with a higher rate of polarizability, which, in turn, enhanced the NLO properties of said chromophores.50 The influence of the A segments in assembling the μ values is more negative, which sequentially makes the chromophore more reactive, highly polarizable and least stable, as manifested in the aforementioned chromophores. Overall, this discussion described the greater charge mobility capability of chromophores amongst their LUMO and HOMO orbitals, resulting in enhanced polarizability and appropriated NLO responses.
UV-vis analysis
The UV-Vis absorption spectra for NFAR1 and NFAD2–NFAD6 in a dichloromethane (DCM) solvent were accomplished at TD-DFT/M06/6-311G(d,p) level. The transition energy E (eV), oscillator strength (fos), maximum absorption wavelength λmax (nm) and molecular transitions of investigated compounds (NFAR1) and (NFAD2–NFAD6) are presented in Table S1,† and the values with oscillator strengths (fos) are put on view in Table 3.
Table 3 Excitation energy (E), oscillator strengths (fos), wavelengths (λmax), and MO contributions of NFAR1 and NFAD2–NFAD6a
| Compounds |
λ (nm) |
E (eV) |
fos |
MO contributions |
| H = HOMO, H−1 = HOMO−1, L = LUMO, L−1 = LUMO−1. |
| NFAR1 |
704.336 |
1.760 |
2.645 |
H → L (95%), H−1 → L+1 (3%) |
| NFAD2 |
797.069 |
1.556 |
1.588 |
H → L (94%), H−1 → L (4%) |
| NFAD3 |
810.301 |
1.530 |
1.578 |
H → L (95%), H−1 → L (4%) |
| NFAD4 |
946.155 |
1.310 |
1.325 |
H → L (96%), H−1 → L (3%) |
| NFAD5 |
936.861 |
1.323 |
0.872 |
H → L (95%), H−1 → L (4%) |
| NFAD6 |
868.662 |
1.427 |
1.533 |
H → L (96%), H−1 → L (3%) |
The calculated λmax of all studied compounds (NFAD2–NFAD6) is found to be greater than that of the reference compound (NFAR1), as observed in the absorption spectra in Fig. 7. The obtained maximum absorption wavelength value is reported to be 946.155 nm in NFAD4, and the absorption wavelength could be influenced by the substituents. Using electron withdrawing/electronegative groups in the acceptor region in NFAD2–NFAD6, λmax is successfully increased. The λmax value of designed compounds decreases in the following order: NFAD4 > NFAD5 > NFAD6 > NFAD3 > NFAD2 > NFAR1. This increasing order of λmax is attained to be similar to the decreasing band gap order of all the investigated molecules. The NFAR1–NFAD6 chromophores consisting of least energy gaps demand lower energy for electronic transitions. Actually, bathochromic absorption spectra are found in NFAR1–NFAD6 due to smaller exciton energy values. Furthermore, the excitation transition of NFAR1 and NFAD2–NFAD6 is found to be in the range of 1.310–1.760 eV. The lower excitation energy that defined the higher charge transference ability is seen in NFAD4 owing to the presence of a strong electron-withdrawing terminal moiety. The ascending order of excitation energies is NFAD4 < NFAD5 < NFAD6 < NFAD3 < NFAD2 < NFAR1, which is similar to the oscillator strength. The exciton energy is directly related to the charge transfer and power conversion efficacy, so the above discussion exploits that transition energy, absorption spectra and oscillator strength of NFAD2–NFAD6 are much better than those of NFAR1. Enhancement in λmax and lower excitation energy values revealed that the aforesaid chromophores validating larger CT facility, consequently, easy excitation might occur among HOMO and LUMO. Prior HOMO/LUMO analysis of entitled molecules supports absorption data with respect to minimal excitation energy and broader λmax spectra, which aid them to improve their photo electric properties. The results proposed that such designed compounds possessing D–π–A configuration have marvelous NLO properties, which are greatly influenced by the electronegative groups on the molecules. To sum things up, NFAD4 has ability for the minimum transition energy, lowermost band gap and maximum λmax value, which might be contemplated like a suitable material for utilizing its photo-electronic characteristics in the NLO field.
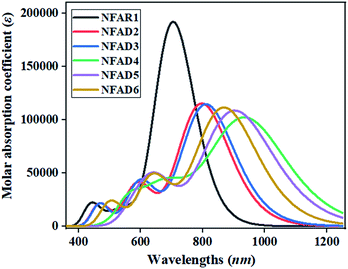 |
| | Fig. 7 UV-visible absorption spectra of NFAR1 and NFAD2–NFAD6. | |
NBO analysis
Natural bond orbital analysis (NBO) explains the nature of inter- and intramolecular interactions, charge distribution, nature of bonding and interactions in the entitled compounds.51 One of the most important aspects of NBO analysis is that it explores the delocalization of electronic charges, and the charge density transfer from the donor to the acceptor region of the system with the D–π–A architecture.52,53 The hyperconjugation and electron density transfer (EDT) from filled to vacant orbitals resulted in the intermolecular attractions.54 We have performed NBO analysis to determine the charge density of the studied compounds (NFAD2–NFAD6) and the results are tabulated in Table 4.
Table 4 Computed NBO charges for donor, π-spacer and acceptor of reference and designed compounds
| Compounds |
Donors |
π-spacers |
Acceptors |
| NFAD2 |
0.234 |
0.056 |
−0.291 |
| NFAD3 |
0.246 |
0.064 |
−0.311 |
| NFAD4 |
0.342 |
0.105 |
−0.447 |
| NFAD5 |
0.202 |
0.110 |
−0.444 |
| NFAD6 |
0.290 |
0.096 |
−0.386 |
In the reference compound (NFAR1), first acceptor, donor, π-linker, and second acceptor are reported with −0.200, 0.363, 0.081 and −0.243, respectively. The donor always has a positive value, which represents its ability to donate electrons as compared to the acceptor moiety. Contrarily, NBO charges with negative values reveal the effectiveness of the acceptor moiety to accept electrons. A little bit of variation was found in the case of π-conjugated linkers with either positive or negative NBO charge values, but in our study, the π-linker is found with positive values, which reveal that they only facilitate the electron transfer from donors towards the acceptor portion in the molecule. All the investigated compounds show positive NBO values of donors and π-linkers and negative values for acceptor units. For this, donors and π-linkers act as electron-pushing units, whereas acceptor moieties act as electron-pulling units.
Natural bond orbital computations were carried out using the DFT-optimized geometric structures by the NBO 6.0 method55 for investigating the interactions among bonds and a satisfactory explanation for charge transfer between the filled and vacant orbitals. It also elucidated a precise picture for exploring the intermolecular delocalization from donors to acceptors in D–π–A structures. The stabilization energy of molecules in the NBO analysis can be determined using second-order perturbation theory using the following equation:56
| |
 | (12) |
where i represents the donor, j represents the acceptor,
E(2) means the stabilization energy, and
qi,
εj,
εi and
Fi,j represent the ith donor orbital occupancy, diagonal, off-diagonal NBO Fock matrix elements respectively. Therefore, NBO analysis was performed using M06/6-31G(d,p) on optimized structures of
NFAD2–NFAD6, and the results are presented in Tables S2–S7
† and the major values are enlisted in
Table 5.
Table 5 Natural bond orbital analysis for reference and designed compounds with their representative values
| Compounds |
Donor (i) |
Type |
Acceptor (j) |
Type |
E(2)a |
E(j)E(i)b |
F(i,j)c |
| E(2) means the energy of hyper conjugative interaction (stabilization energy in kcal mol−1). Energy difference between donor and acceptor i and j NBO orbitals. F(i;j) is the Fock matrix element between i and j NBO orbitals. |
| NFAR1 |
C14–C15 |
π |
C39–C47 |
π* |
31.09 |
0.31 |
0.088 |
| C91–N92 |
π |
C93–N94 |
π* |
0.69 |
0.47 |
0.016 |
| C74–H75 |
σ |
C63–S65 |
σ* |
10.37 |
0.72 |
0.077 |
| C14–S36 |
σ |
C14–C39 |
σ* |
0.50 |
1.21 |
0.022 |
| O69 |
LP(2) |
C67–O68 |
π* |
49.01 |
0.37 |
0.121 |
| O68 |
LP(2) |
C67–O69 |
σ* |
34.23 |
0.66 |
0.136 |
| NFAD2 |
C48–C50 |
π |
C61–C69 |
π* |
30.51 |
0.32 |
0.089 |
| C78–N79 |
π |
C80–N81 |
π* |
0.71 |
0.47 |
0.016 |
| C61–H62 |
σ |
C50–S52 |
σ* |
10.61 |
0.72 |
0.078 |
| C14–S36 |
σ |
C9–C13 |
σ* |
0.52 |
1.11 |
0.022 |
| N86 |
LP(1) |
C41–C42 |
π* |
48.7 |
0.29 |
0.109 |
| O55 |
LP(2) |
C54–O56 |
σ* |
34.33 |
0.66 |
0.136 |
| NFAD3 |
C48–C50 |
π |
C61–C69 |
π* |
31.03 |
0.32 |
0.089 |
| C77–N78 |
π |
C79–N80 |
π* |
0.72 |
0.47 |
0.017 |
| C61–H62 |
σ |
C50–S52 |
σ* |
10.69 |
0.72 |
0.078 |
| C14–S36 |
σ |
C9–C13 |
σ* |
0.52 |
1.11 |
0.022 |
| N85 |
LP(1) |
C41–C42 |
π* |
48.92 |
0.29 |
0.11 |
| O55 |
LP(2) |
C54–O56 |
σ* |
34.33 |
0.66 |
0.136 |
| NFAD4 |
C48–C49 |
π |
C1–C51 |
π* |
28.81 |
0.27 |
0.083 |
| C54–O55 |
π |
C47–C79 |
π* |
3.52 |
0.41 |
0.037 |
| C61–H62 |
σ |
C50–S52 |
σ* |
11.02 |
0.72 |
0.079 |
| C14–S36 |
σ |
C9–C13 |
σ* |
0.52 |
1.11 |
0.022 |
| O56 |
LP(2) |
C54–O55 |
π* |
48.91 |
0.37 |
0.121 |
| O55 |
LP(2) |
C54–O56 |
σ* |
34.26 |
0.66 |
0.135 |
| NFAD5 |
C48–C50 |
π |
C61–C69 |
π* |
33.72 |
0.31 |
0.092 |
| N98–O99 |
π |
C66–C67 |
π* |
3.22 |
0.48 |
0.039 |
| C61–H62 |
σ |
C50–S52 |
σ* |
11.02 |
0.71 |
0.079 |
| N92–O93 |
σ |
N92–O94 |
σ* |
0.50 |
1.59 |
0.026 |
| N83 |
LP(1) |
C41–C42 |
π* |
49.79 |
0.29 |
0.110 |
| O56 |
LP(2) |
C54–O56 |
σ* |
34.25 |
0.66 |
0.136 |
| NFAD6 |
C61–C69 |
π |
C73–C74 |
π* |
30.71 |
0.29 |
0.084 |
| C74–C77 |
π |
C77–N78 |
π* |
8.22 |
1.61 |
0.103 |
| C47–S53 |
σ |
C79–F80 |
σ* |
6.61 |
0.95 |
0.071 |
| C77–N78 |
σ |
C73–C74 |
σ* |
0.50 |
1.65 |
0.026 |
| N83 |
LP(1) |
C41–C42 |
π* |
49.41 |
0.29 |
0.110 |
| O55 |
LP(2) |
C54–O56 |
σ* |
34.25 |
0.66 |
0.136 |
From Table 5, it is clear that the maximum value of π → π* transition for the reference compound (NFAR1) occurs at 31.09 kcal mol−1 of π(C14–C15) → π*(C39–C47), while the minimum value of stabilization energy of π → π*is observed as 0.69 kcal mol−1 of π(C91–N92) → π*(C93–N94). Furthermore, σ(C74–H75) → σ*(C63–S65) exhibits the highest value of σ → σ* transition at a stabilization energy of 10.37 kcal mol−1. The lowest value of the same transition is observed by σ(C14–S36) → σ*(C14–C39). The highest value of LP2 → π* transition is observed at 49.01 kcal mol−1 energy of stabilization by LP2(O69) → π* (C67–O68). The lowest value of the same transition is observed at 0.51 kcal mol−1 energy of stabilization by LP1(S65) → σ*(C63–C74).
For the tailored compound NFAD2, the highest value of π → π* transitions are observed at 30.51 kcal mol−1 energy of stabilization by π(C48–C50) → π*(C61–C69). The lowest value of π → π* transitions is observed by π(C78–N79) → π*(C80–C81), which exhibited the lowest stabilization energy of 0.71 kcal mol−1. Moreover, the maximum energy value of 10.61 kcal mol−1 for σ → σ* transition is observed by σ(C61–H62) → σ*(C50–S52), while the lowest σ → σ* energy transition value is 0.52 kcal mol−1 exhibited by σ(C14–S36) → σ*(C9–C13). The highest value of LP → π* transition is observed at 48.70 kcal mol−1 energy of stabilization by LP1(N86) → π* (C41–C42). The lowest value of the same transition is observed at 0.52 kcal mol−1 energy of stabilization by LP1(S52) → σ*(C50–C61).
The maximum value of π → π* transition for tailored compound NFAD3 occurs at 31.03 kcal mol−1 of π(C48–C50) → π*(C61–C69), whereas the lowest value of stabilization energy for π → π* transition is perceived as 0.72 kcal mol−1 by π(C77–N78) → π*(C79–N80). Furthermore, σ(C61–H62) → σ*(C50–S52) reveals the highest value of σ → σ* transition at a stabilization energy of 10.61 kcal mol−1. The lowest value of the same transition is observed by σ(C14–S36) → σ*(C9–C13). The highest value of LP1 → π* transition is observed at 48.70 kcal mol−1 energy of stabilization by LP1(N86) → π* (C41–C42). The lowest value of the same transition is observed at 0.53 kcal mol−1 energy of stabilization by LP1(S52) → σ*(C50–C61).
For compound NFAD4, the maximum value of π → π* transition energy is trailed at 28.81 kcal mol−1 due to π(C48–C49) → π*(C1–C51). However, the lowest value of stabilization energy for π → π* specious is 3.52 kcal mol−1 due to π(C54–O55) → π*(C47–C79). Furthermore, σ(C61–H62) → σ*(C50–S52) reveals the highest value of σ → σ* transition at a stabilization energy of 11.02 kcal mol−1. The lowest value of σ → σ* transition is observed by σ(C14–S36) → σ*(C9–C13) at 0.52 kcal mol−1. The highest value of LP1 → π* transition is observed at 100.26 kcal mol−1 energy of stabilization by LP1(C69) → π* (C50–C61). The lowest value of the same transition is observed at 0.54 kcal mol−1 energy of stabilization by LP1(S52) → σ*(C50–C61).
For compound NFAD5, the maximum value of π → π* transition energy is observed at 33.72 kcal mol−1 due to π(C48–C50) → π*(C61–C69). However, the lowest value of stabilization energy for π → π* transition is obtained at 3.22 kcal mol−1 by π(N98–O99) → π*(C66–C67). Moreover, σ(C61–H62) → σ*(C50–S52) exhibited the highest value of σ → σ* transition at a stabilization energy of 11.02 kcal mol−1. The lowest value of σ → σ* transition is detected by σ(N92–O93) → σ*(N92–O94) at 0.50 kcal mol−1. The highest value of LP → π* transition is observed at 49.79 kcal mol−1 energy of stabilization by LP1(N83) → π* (C41–C42). The lowest value of the same transition is observed at 0.50 kcal mol−1 energy of stabilization by LP1(O56) → σ*(C47–C54).
For the tailored compound NFAD6, the highest value of π → π* transitions are observed at 30.71 kcal mol−1 energy of stabilization by π(C61–C69) → π*(C73–C74). The lowest value of π → π* transitions is observed by π(C54–O55) → π*(C47–C79), which exhibits the lowest stabilization energy of 3.52 kcal mol−1. Moreover, the maximum energy value of 10.96 kcal mol−1 for σ → σ* transitions is observed by σ(C61–H62) → σ*(C50–S52), while the lowest σ → σ* transition energy value is 0.50 kcal mol−1 exhibited by σ(C77–N78) → σ*(C73–C74). The highest value of LP1 → π* transition is observed at 49.41 kcal mol−1 energy of stabilization by LP1(N83) → π* (C41–C42). The lowest value of the same transition is observed at 0.51 kcal mol−1 energy of stabilization by LP1(F101) → σ*(C66–C94). NBO investigations of the aforementioned chromophores showed that the prolonged hyper-conjugation and large intramolecular charge mobility rate play a good role in stabilizing the above-mentioned chromophores. Moreover, it enhances the CT properties that are significant for the NLO response.
Non-linear optics
Usually, organic compounds gained potential applications in the field of NLO materials,57 photonic materials,58 photonic devices,59 optical devices,60 and electrochemical sensors, and in ultrafast optical signal processing.61 The NLO character is related to the computed values explaining the structural properties, electronic behavior, linear polarizability (〈α〉), first hyperpolarizability (βtotal), and second hyperpolarizability γtotal, which describe the molecular structure and bandgap. Hence, to evaluate how donor and acceptor moieties influenced the linear and nonlinear responses of NFAR1 and NFAD2–NFAD6 as their 〈α〉, βtotal and γtotal were calculated, and their simulated values are expressed in Tables S8–S10† and major tensors are presented in Table 6.
Table 6 Computed dipole polarizability (μtotal), average polarizability 〈α〉, first hyperpolarizability (βtotal), and second hyperpolarizability γtotal of the studied compoundsa
| Compounds |
μtotal × 10−22 |
〈α〉 × 10−22 |
βtotal × 10−27 |
γtotal × 10−31 |
| Units in e.s.u. |
| NFAR1 |
2.304 |
2.559 |
0.553 |
0.417 |
| NFAD2 |
7.588 |
2.373 |
6.699 |
0.927 |
| NFAD3 |
7.368 |
2.413 |
7.192 |
1.007 |
| NFAD4 |
10.429 |
2.953 |
13.16 |
2.150 |
| NFAD5 |
10.269 |
2.864 |
12.15 |
1.924 |
| NFAD6 |
8.899 |
2.665 |
9.770 |
1.463 |
The dipole moment values of NFAR1 along with it derivatives (NFAD2–NFAD6) in DCM solvent are studied to be 2.304 × 10−22, 7.588 × 10−22, 7.368 × 10−22, 10.429 × 10−22, 10.269 × 10−22 and 8.899 × 10−22 e.s.u., respectively.
Among the reference compound and NFAD2–NFAD6, NFAD4 revealed the highest value of μtotal and this may be due to the strong electronegative nature of terminal acceptor moieties. All the engineered compounds expressed large μtotal as compared to the reference compound. The calculated dipole moments are arranged in the descending order as follows: NFAD4 > NFAD5 > NFAD6 > NFAD2 > NFAD3 > NFAR1. Hence, it is clear from the results that the greater the values of μtotal, the more would be the ICT, and the greater the electron transportation rate, and the afore-said chromophores are enriched in this aspect. The linear polarizability values (2.373–2.95310−22 e.s.u.) of derivatives are almost the same as studied for the reference chromophore (2.55910−22 e.s.u.)
The first hyperpolarizability is related to the ICT that occurs from the donor moiety to the acceptor moiety through the π-spacer. The stronger the ICT, the greater would be the NLO response, as all the designed compounds (NFAD2–NFAD6) exhibited the stronger ICT. The βtotal value of NFAD2 is examined to be 6.699 × 10−27 e.s.u, which is the lowest, while NFAD4 is found to have the highest value (13.16 × 10−27 e.s.u.) of βtotal among all the studied molecules. The incorporation of strong electron-withdrawing substituents in the acceptor region in designed compounds (NFAD2–NFAD6) has a significant influence on the βtotal values, which proves that NFAs are very effective in tuning the NLO responses. The βtotal values of NFAD2–NFAD6 are observed to be 6.699 × 10−27, 7.192 × 10−27, 13.16 × 10−27, 12.15 × 110−27, and 9.770 × 10−27 e.s.u., respectively. The βtotal values were found to be in the decreasing order as follows: NFAD4 > NFAD5 > NFAD6 > NFAD3 > NFAD2 > NFAR1. This order indicates that NFAD4 is the most suitable designed structure, which shows the maximum NLO value.
Urea is utilized as the standard compound (βtotal = 0.3728 × 10−30 e.s.u.) for the exploration of hyperpolarizability properties.62 The hyperpolarizability values of NFAR1 and NFAD2–NFAD6 are greater than those of urea. For example, the calculated βtotal value of NFAD6 is 2.272![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 093 × 10−28 times greater than that of the urea molecule.61 Similarly, the value of NFAD2 is 1.557907 × 10−28, NFAD3 is 1.672558 × 10−28, NFAD4 is 3.060465 × 10−28 and NAFD5 is 2.825581 × 10−28 times greater than the value of urea. The second hyperpolarizability of compounds NFAD2–NFAD6 is thought to be based on the two-photon absorption (TPA) phenomena in NLO materials. The maximum value of the second hyperpolarizability is observed in NFAD4 as 2.150 × 10−31 e.s.u. The overall decreasing order of all the studied compounds is as follows: NFAD2 > NFAR1 > NFAD4 > NFAD5 > NFAD6 > NFAD3. For comparative analysis, we also compared the NLO behavior of NFAR1 and NFAD2–NFAD6 with that of a reported fullerene-free FH chromophore [(βtotal = 0.169 × 10−27 e.s.u. or 19556.9 a.u), (γtotal = 3.123 × 10−32 e.s.u. or 6.2 × 107 a.u.)] used as a standard chromophore.63 The first hyperpolarizability response of NFAR1 and NFAD2–NFAD6 chromophores is 3.27, 39.64, 42.56, 77.86, 71.90 and 57.81 times greater than that of FH, respectively. Similarly, the second hyperpolarizability behavior is 1.26, 2.96, 3.22, 6.88, 6.16 and 4.68 times larger than that of the FH molecule, respectively. The comparative analysis with the FH molecule proves all investigated compounds to be potential NLO materials. Further, it may be comprehended from the above discussion that various types of acceptors with π-conjugation played a pivotal role and yielded remarkable NLO amplitudes.
093 × 10−28 times greater than that of the urea molecule.61 Similarly, the value of NFAD2 is 1.557907 × 10−28, NFAD3 is 1.672558 × 10−28, NFAD4 is 3.060465 × 10−28 and NAFD5 is 2.825581 × 10−28 times greater than the value of urea. The second hyperpolarizability of compounds NFAD2–NFAD6 is thought to be based on the two-photon absorption (TPA) phenomena in NLO materials. The maximum value of the second hyperpolarizability is observed in NFAD4 as 2.150 × 10−31 e.s.u. The overall decreasing order of all the studied compounds is as follows: NFAD2 > NFAR1 > NFAD4 > NFAD5 > NFAD6 > NFAD3. For comparative analysis, we also compared the NLO behavior of NFAR1 and NFAD2–NFAD6 with that of a reported fullerene-free FH chromophore [(βtotal = 0.169 × 10−27 e.s.u. or 19556.9 a.u), (γtotal = 3.123 × 10−32 e.s.u. or 6.2 × 107 a.u.)] used as a standard chromophore.63 The first hyperpolarizability response of NFAR1 and NFAD2–NFAD6 chromophores is 3.27, 39.64, 42.56, 77.86, 71.90 and 57.81 times greater than that of FH, respectively. Similarly, the second hyperpolarizability behavior is 1.26, 2.96, 3.22, 6.88, 6.16 and 4.68 times larger than that of the FH molecule, respectively. The comparative analysis with the FH molecule proves all investigated compounds to be potential NLO materials. Further, it may be comprehended from the above discussion that various types of acceptors with π-conjugation played a pivotal role and yielded remarkable NLO amplitudes.
Transition density matrices (TDMs) and binding energy (Eb) analysis
The transition density matrix (TDM) is an important tool used to find the electronic charge transfer in designed compounds (NFAD2–NFAD6) and the reference compound (NFAR1).64 The TDM of investigated compounds was computed by the M06/6-311G(d, p) method. TDM helps to evaluate: (a) the relationship between donor and acceptor moieties in the excited state, (b) electronic charge excitation phenomena, and (c) localization and delocalization of electro–hole pairs.14 In the current report, the effect of the hydrogen atom is omitted due to its little contribution to electronic transitions. The TDM diagram of all the investigated compounds shows the nature of transition in these compounds. The TDM results of all the designed compounds are displayed in Fig. 8. To understand the transference of the charge density, we divided each designed compound into three portions i.e., D, π-linker, and A. TDM maps demonstrated an effective diagonal charge transfer (CT) coherence in all the above-mentioned chromophores. In NFAR1, NFAD2 and NFAD3, the higher electronic cloud can be seen on donor and acceptor parts of the heat maps as expressed by green and red spots. However, a little charge can be observed over the π-linker, which exploited the electron coherence successfully transferred from D to the π-linker, which excellently facilitated the shifting of electron density towards A without trapping. In NFAD4–NFAD6, the excellent charge cloud is noted at the donor and π-linker, while a little charge is examined on A. This unique behaviour may be due to the presence of highly electron-deficient groups (–CN,–NO2, and –CF3) on acceptors. The findings of TDM heat maps imply a facile and greater exciton dissociation in the excited state, which is very important for NLO material development. The binding energy is the difference between electrical and optical bandgap energies and is an important tool for the estimation of optoelectronic properties of the studied compounds. Eqn (13) is used for the theoretical estimation of the binding energy of reference and investigated compounds.14,62where Eb is the binding energy, EH–L is the bandgap and Eopt is the first excitation energy.65,66 The computed results of binding energy are presented in Table 7.
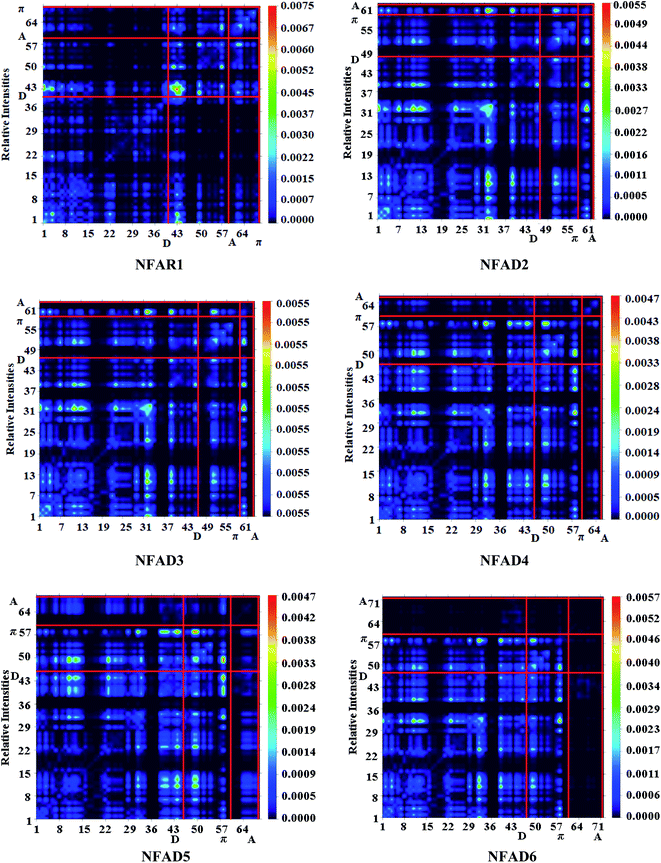 |
| | Fig. 8 Transition density matrix (TDM) graphs of reference (NFAR1) and designed compounds (NFAD2–NFAD6). | |
Table 7 HOMO–LUMO = energy gap = EH–L, Eopt, and (Eb) of investigated compoundsa
| Compounds |
EH–L |
Eopt |
Eb |
| Units in eV. |
| NFAR1 |
2.314 |
1.760 |
0.554 |
| NFAD2 |
1.914 |
1.556 |
0.358 |
| NFAD3 |
1.879 |
1.530 |
0.349 |
| NFAD4 |
1.607 |
1.310 |
0.297 |
| NFAD5 |
1.618 |
1.323 |
0.295 |
| NFAD6 |
1.748 |
1.427 |
0.321 |
The above-mentioned data revealed that all the designed chromophores exhibited lower values of binding energy (0.358–0.295 eV) than those of their reference chromophore (0.554 eV). These lower binding energies might be due to the change in the configuration, which created a strong push–pull configuration. Similarly, excitation energy values of NFAD2–NFAD6 are lower than those of NFAR1 with the same pattern in the HOMO–LUMO band gap. This smaller binding energy with lower excitation and band gap energy values supports the higher excitation dissociation and significantly larger charge mobility rate with better optoelectronic properties.14 The values of binding energy for the reference and the designed compounds decrease in the following order: NFAR1 > NFAD2 > NFAD3 > NFAD6 > NFAD4 > NFAD5. Low binding energy values have a direct relation with polarizability and molecules with 1.9 eV Eb are considered as perfect photonic materials with significant NLO response.67 Interestingly, all our derivatives expressed a binding energy lower than 1.9 eV with high charge separation in excited states illustrating them to be significant NLO materials.
Conclusion
In this research, first-, second- and third-order NLO behaviors of NFA-based chromophores have been examined. The five non-fullerene acceptor molecules NFAD2–NFAD6 are designed based on the D–π–A topology by structural modeling of the acceptor in the reference (NFAR1) molecule. Quantum chemical approach has been performed to elucidate the electronic and optical characteristics of NFAR1 and NFAD2–NFAD6 compounds. The investigated compounds showed low transition energy with the absorption maxima wavelength in the visible region, smaller energy bandgap values than those of the reference compound, which justified the efficiency of acceptor molecules. Exceedingly, smaller binding energy (0.358–0.295 eV) with larger μtotal (7.368–10.429 e.s.u.) for studied chromophores are examined, which predict larger excitation dissociation and greater polarity. Among the aforesaid chromophores, maximum red-shifted absorption spectra, greater electron mobility, and the smallest energy band gap are observed for NFAD4 owing to the presence of strong electronegative cyano (–CN) units substituted in the end-capped region. Interestingly, remarkable NLO findings such as βtotal and γtotal values are noted to be 13.16 × 10−26 e.s.u. and 2.150 × 10−31 e.s.u., respectively for NFAD4, which are significantly greater in all derivatives. Moreover, a comparative study with urea demonstrated that the entitled chromophores showed excellent NLO responses. Overall, the results indicated that the replacement of fullerene acceptors with non-fullerene small acceptor molecules is an efficient approach for improving the optoelectronic properties of chemically designed compounds. All the investigated compounds should be targeted for synthesis investigation. Thus, this work will trigger researchers to synthesize these systems for high-performance NLO responses.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant no. (D-579-130-1443). The authors, therefore, gratefully acknowledge DSR technical and financial support.
References
- F. Zafar, et al., End-capped engineering of truxene core based acceptor materials for high performance organic solar cells: theoretical understanding and prediction, Opt. Quant. Electron., 2021, 53(2), 1–24 CrossRef.
- P. A. Franken, A. E. Hill, C. e. Peters and G. Weinreich, Generation of optical harmonics, Phys. Rev. Lett., 1961, 7, 118 CrossRef.
- J. T. Lin and C. Chen, Choosing a nonlinear crystal, Lasers Optron., 1987, 6, 59–63 Search PubMed.
- Y. Wang, W. Tam, S. H. Stevenson, R. A. Clement and J. Calabrese, New organic non-linear optical materials of stilbene and diphenylacetylene derivatives, Chem. Phys. Lett., 1988, 148, 136–141 CrossRef CAS.
- W. Tam, Y. Wang, J. C. Calabrese & R. A. Clement, Organometallics And Organics For Nonlinear Optics: New Material For Second Harmonic Generation, in Nonlinear Optical Properties of Organic Materials, SPIE, 1988, vol. 971, pp. 107–112 Search PubMed.
- D. J. Williams, Organic polymeric and non-polymeric materials with large optical nonlinearities, Angew Chem. Int. Ed. Engl., 1984, 23, 690–703 CrossRef.
- D. S. Chemla, Nonlinear Optical Properties of Organic Molecules and Crystals V1, Elsevier, 2012, vol. 1 Search PubMed.
- W. E. Moerner and S. M. Silence, Polymeric photorefractive materials, Chem. Rev., 1994, 94, 127–155 CrossRef CAS.
- Y. Zhang, et al., Preparation of non-fullerene acceptors with a multi-asymmetric configuration in a one-pot reaction for organic solar cells, J. Mater. Chem. C, 2020, 8(48), 17229–17236 RSC.
- Y. He and Y. Li, Fullerene derivative acceptors for high performance polymer solar cells, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- A. Wadsworth, et al., Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- D. M. Guldi, Fullerenes: three dimensional electron acceptor materials, Chem. Commun., 2000, 321–327 RSC.
- M. N. Arshad, I. Shafiq, M. Khalid and A. M. Asiri, Exploration of the Intriguing Photovoltaic Behavior for Fused Indacenodithiophene-Based A–D–A Conjugated Systems: A DFT Model Study, ACS Omega, 2022, 7(14), 11606–11617 CrossRef CAS PubMed.
- M. Khalid, I. Shafiq, M. Zhu, M. U. Khan, Z. Shafiq, J. Iqbal and M. Imran, Efficient tuning of small acceptor chromophores with A1-π-A2-π-A1 configuration for high efficacy of organic solar cells via end group manipulation, J. Saudi Chem. Soc., 2021, 25(8), 101305 CrossRef CAS.
- A. Wadsworth, et al., Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells, Chem. Soc. Rev., 2019, 48, 1596–1625 RSC.
- Y. Lin and X. Zhan, Non-fullerene acceptors for organic photovoltaics: an emerging horizon, Mater. Horiz., 2014, 1, 470–488 RSC.
- A. Mahmood and J. L. Wang, A time and resource efficient machine learning assisted design of non-fullerene small molecule acceptors for P3HT-based organic solar cells and green solvent selection, J. Mater. Chem. A, 2021, 9(28), 15684–15695 RSC.
- A. Mahmood, A. Irfan and J. L. Wang, Machine learning and molecular dynamics simulation-assisted evolutionary design and discovery pipeline to screen efficient small molecule acceptors for PTB7-Th-based organic solar cells with over 15% efficiency, J. Mater. Chem. A, 2022, 10(8), 4170–4180 RSC.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Next-generation organic photovoltaics based on non-fullerene acceptors, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- Y. Huang, W. Zhou, X. Li, L. Jiang and Y. Song, Highly broadband NLO response of acceptor–donor–acceptor materials with a planar conformation, Mater. Adv., 2021, 2(6), 2097–2103 RSC.
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed.
- T. G. Allen, S. Benis, N. Munera, J. Zhang, S. Dai, T. Li and S. R. Marder, Highly Conjugated, Fused-Ring, Quadrupolar Organic Chromophores with Large Two-Photon Absorption Cross-Sections in the Near-Infrared, J. Phys. Chem. A, 2020, 124(22), 4367–4378 CrossRef CAS PubMed.
- M. U. Khan, M. Khalid, I. Shafiq, R. A. Khera, Z. Shafiq, R. Jawaria and C. Lu, Theoretical investigation of nonlinear optical behavior for rod and T-Shaped phenothiazine based D-π-A organic compounds and their derivatives, J. Saudi Chem. Soc., 2021, 25(10), 101339 CrossRef CAS.
- M. Khalid, M. U. Khan, I. Shafiq, R. Hussain, K. Mahmood, A. Hussain and Y. Li, NLO potential exploration for D–π–A heterocyclic organic compounds by incorporation of various π-linkers and acceptor units, Arab. J. Chem., 2021, 14(8), 103295 CrossRef CAS.
- M. Khalid, M. U. Khan, I. Shafiq, R. Hussain, A. Ali, M. Imran and M. S. Akram, Structural modulation of π-conjugated linkers in D–π–A dyes based on triphenylamine dicyanovinylene framework to explore the NLO properties, R. Soc. Open Sci., 2021, 8(8), 210570 CrossRef CAS PubMed.
- M. U. Khan, M. Ibrahim, M. Khalid, M. S. Qureshi, T. Gulzar, K. M. Zia and M. R. S. A. Janjua, First theoretical probe for efficient enhancement of nonlinear optical properties of quinacridone based compounds through various modifications, Chem. Phys. Lett., 2019, 715, 222–230 CrossRef CAS.
- M. Haroon, R. Mahmood and M. R. S. A. Janjua, An Interesting Behavior and Nonlinear Optical (NLO) Response of Hexamolybdate Metal Cluster: Theoretical Insight into Electro-Optic Modulation of Hybrid Composites, J. Cluster Sci., 2017, 28, 2693–2708 CrossRef CAS.
- I. Khan, M. Khalid, M. Adeel, S. I. Niaz, I. Shafiq, S. Muhammad and A. A. C. Braga, Palladium-catalyzed synthesis of 5-(arylated) pyrimidines, their characterization, electronic communication, and non-linear optical evaluations, J. Mol. Struct., 2021, 1237, 130408 CrossRef CAS.
- D. M. Guldi, Fullerenes: three dimensional electron acceptor materials, Chem. Commun., 2000, 321–327 RSC.
- Y. He and Y. Li, Fullerene derivative acceptors for high performance polymer solar cells, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- G. Y. Ge, J. T. Li, J. R. Wang, M. Xiong, X. Dong, Z. J. Li and J. L. Wang, Unveiling the Interplay among End Group, Molecular Packing, Doping Level, and Charge Transport in N-Doped Small-Molecule Organic Semiconductors, Adv. Funct. Mater., 2022, 32(7), 2108289 CrossRef CAS.
- L. Wang, Q. An, L. Yan, H. R. Bai, M. Jiang, A. Mahmood and J. L. Wang, Non-fullerene acceptors with hetero-dihalogenated terminals induce significant difference in single crystallography and enable binary organic solar cells with 17.5% efficiency, Energy Environ. Sci., 2022, 15(1), 320–333 RSC.
- M. Khalid, A. Ali, R. Jawaria, M. A. Asghar, S. Asim, M. U. Khan and M. S. Akram, First principles study of electronic and nonlinear optical properties of A–D–π–A and D–A–D–π–A configured compounds containing novel quinoline–carbazole derivatives, RSC Adv., 2020, 10(37), 22273–22283 RSC.
- Y. Zhang, Y. Wang, Z. Xie, T. Shan, L. Zhu, F. Liu and H. Zhong, Preparation of non-fullerene acceptors with a multi-asymmetric configuration in a one-pot reaction for organic solar cells, J. Mater. Chem. C, 2020, 8(48), 17229–17236 RSC.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji, et al., Gaussian 09, Revision A.02, 2013 Search PubMed.
- R. D. Dennington; T. A. Keith and J. M. MillamGaussView 5.0, Gaussian. Inc., Wallingford, 2008 Search PubMed.
- N. M. O’boyle, A. L. Tenderholt and K. M. Langner, Cclib: A library for package-independent computational chemistry algorithms, J. Comput. Chem., 2008, 29(5), 839–845 CrossRef PubMed.
- V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102(11), 1995–2001 CrossRef CAS.
- M. D. Hanwell, et al., Avogadro: An advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4(1), 17 CAS.
- G. A. Zhurko & D. A. Zhurko. ChemCraf, version 1.6. URL: http://www.chemcrafprog.com, 2009.
- J. C. Kromann, C. Steinmann and J. H. Jensen, Improving Solvation Energy Predictions Using the SMD Solvation Method and Semiempirical Electronic Structure Methods, J. Chem. Phys., 2018, 149(10), 104102 CrossRef PubMed.
- F. Ullah, K. Ayub and T. Mahmood, Remarkable Second and Third Order Nonlinear Optical Properties of Organometallic C 6 Li 6–M 3 O Electrodes, New J. Chem., 2020, 44(23), 9822–9829 RSC.
- M. Khalid, H. M. Lodhi, M. U. Khan and M. Imran, Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: A DFT approach, RSC Adv., 2021, 11, 14237–14250 RSC.
- P. Ferdowsi, et al., Molecular Design of Efficient Organic D–A—-A Dye Featuring Triphenylamine as Donor Fragment for Application in Dye-Sensitized Solar Cells, ChemSusChem, 2018, 11, 494–502 CrossRef CAS PubMed.
- S. S. Amiri, et al., Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1, 3, 4-oxadiazol-2-yl) thio) benzene-1, 2-diol, J. Mol. Struct., 2016, 1119, 18–24 CrossRef.
- T. Koopmans, Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms, Phys, 1934, 1(1–6), 104–113 Search PubMed.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, Electronegativity: The Density Functional Viewpoint, J. Chem. Phys., 1978, 68(8), 3801–3807 CrossRef CAS.
- R. G. Parr and R. G. Pearson, Absolute Hardness: Companion Parameter to Absolute Electronegativity, J. Am. Chem. Soc., 1983, 105(26), 7512–7516 CrossRef CAS.
- P. K. Chattaraj and D. R. Roy, Update 1 of: Electrophilicity Index, Chem. Rev., 2007, 107(9), 46–74 CrossRef.
- M. U. Khan, M. Khalid, R. A. Khera, M. N. Akhtar, A. Abbas, M. F. ur Rehman and C. Lu, Influence of Acceptor Tethering on the Performance of Nonlinear Optical Properties for Pyrene-Based Materials with A-π-D-π-D Architecture, Arab. J. Chem., 2022, 103673 CrossRef CAS.
- P. Dhamodharan, K. Sathya and M. Dhandapani, Physico-chemical characterization, density functional theory (DFT) studies and Hirshfeld surface analysis of a new organic optical material: 1H-benzo [d] imidazole-3-ium-2, 4, 6-trinitrobenzene-1, 3 bis (olate), J. Mol. Struct., 2017, 1146, 782–792 CrossRef CAS.
- M. I. Nan, et al., Mono-and di-substituted pyrene-based donor-π-acceptor systems with phenyl and thienyl π-conjugating bridges, Dyes Pigm., 2020, 181, 108527 CrossRef CAS.
- P. Goszczycki, et al., Synthesis, crystal structures, and optical properties of the π-π interacting pyrrolo [2, 3-b] quinoxaline derivatives containing 2-thienyl substituent, J. Mol. Struct., 2017, 1146, 337–346 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, Natural bond orbital methods, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2(1), 1–42 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: Natural bond orbital analysis program, J. Comput. Chem., 2013, 34(16), 1429–1437 CrossRef CAS PubMed.
- M. P. Costa, et al., Interaction of polyelectrolyte complex between sodium alginate and chitosan dimers with a single glyphosate molecule: A DFT and NBO study, Carbohydr. Polym., 2018, 198, 51–60 CrossRef CAS PubMed.
- I. Sheikhshoaie and W. M. Fabian, Quantum chemical study on the electronic structure and second-order nonlinear optical properties of salen-type Schiff bases, Dyes Pigm., 2006, 70(2), 91–98 CrossRef CAS.
- A. Iwan and D. Sek, Polymers with triphenylamine units: Photonic and electroactive materials, Prog. Polym. Sci., 2011, 36(10), 1277–1325 CrossRef CAS.
- M. Morks, Magnesium phosphate treatment for steel, Mater. Lett., 2004, 58(26), 3316–3319 CrossRef CAS.
- C. Adant, M. Dupuis and J. Bredas, Ab initio study of the nonlinear optical properties of urea: Electron correlation and dispersion effects, Int. J. Quantum Chem., 1995, 56(S29), 497–507 CrossRef.
- S. N. Margar and N. Sekar, Nonlinear optical properties of curcumin: solvatochromism-based approach and computational study, Mol. Phys., 2016, 114(12), 1867–1879 CrossRef CAS.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects, Chem. Rev., 1994, 94(1), 195–242 CrossRef CAS.
- M. N. Arshad, M. Khalid, M. Asad, A. M. Asiri, M. M. Alotaibi, A. A. Braga and A. Khan, Donor moieties with D–π–a framing modulated electronic and nonlinear optical properties for non-fullerene-based chromophores, RSC Adv., 2022, 12(7), 4209–4223 RSC.
- M. Ans, et al., Designing Three-dimensional (3D) Non-Fullerene Small Molecule Acceptors with Efficient Photovoltaic Parameters, ChemistrySelect, 2018, 3(45), 12797–12804 CrossRef CAS.
- M. Naeem, et al., Tuning of optoelectronic properties of triphenylamines-based donor materials for organic solar cells, J. Theor. Comput. Chem., 2019, 18(07), 1950036 CrossRef CAS.
- B. G. Kim, et al., Organic Dye Design Tools for Efficient Photocurrent Generation in Dye-Sensitized Solar Cells: Exciton Binding Energy and Electron Acceptors, Adv. Funct. Mater., 2012, 22(8), 1606–1612 CrossRef CAS.
- M. Khalid, M. U. Khan, E. T. Razia, Z. Shafiq, M. M. Alam, M. Imran and M. S. Akram, Exploration of efficient electron acceptors for organic solar cells: rational design of indacenodithiophene based non-fullerene compounds, Sci. Rep., 2021, 11(1), 1–15 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Muhammad Nadeem Arshadbc,
Shahzad Murtazaa,
Iqra Shafiqa,
Muhammad Haroond,
Abdullah M. Asiri
*a,
Muhammad Nadeem Arshadbc,
Shahzad Murtazaa,
Iqra Shafiqa,
Muhammad Haroond,
Abdullah M. Asiri
 .
.








