
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of ZrO2–WO3 solid acid catalysts in a Friedel–Crafts reaction through post-hydrothermal treatment†
Sha Li *,
Ruopeng Yu,
Bonan Xu,
Zhikun Wang,
Chunzheng Wu* and
Jianzhong Guo*
*,
Ruopeng Yu,
Bonan Xu,
Zhikun Wang,
Chunzheng Wu* and
Jianzhong Guo*
Key Laboratory of Chemical Utilization of Forestry Biomass in Zhejiang Province, College of Chemistry and Materials Engineering, Zhejiang A & F University, Hangzhou 311300, China. E-mail: shali@zafu.edu.cn; wucz@zafu.edu.cn; guojianzhong@zafu.edu.cn
First published on 4th May 2022
Abstract
ZrO2–WO3 mixed oxide plays an essential role in the chemical and petroleum industries. So far, very little work has paid attention to the activation of the low activity of ZrO2–WO3 catalysts. In this work, poorly reactive ZrO2–WO3 was prepared as a model catalyst by a sol–gel method and it was accompanied by post-hydrothermal treatment with various solutions. The catalytic results in the Friedel–Crafts reaction of anisole and benzyl alcohol showed that the post-hydrothermal treatment with ethylenediamine or ammonium hydroxide solutions dramatically improved the activity of ZrO2–WO3, while the hydrothermal treatments with water or ammonia chloride solution resulted in poorer activity and selectivity. The former treatments were found to induce a huge transformation of the ZrO2 crystal from monoclinic to tetragonal as well as a significant increase in acidic WOx clusters that anchored onto ZrO2. The generation of the WOx clusters was responsible for the activation of ZrO2–WO3.
Introduction
Solid acid catalysts are of great importance in the chemical and petroleum industries not only because of their advantages in the separation of products, but also due to their good stability against corrosion. Among the abundant solid acid catalysts, WO3–ZrO2, that was first reported by Hino and Arata,1–3 has attracted much attention in last decades due to its high activity in various acid reactions and high mechanical and thermal stability that allows it to work in harsh conditions (like oxidative/reductive and H2O environments). Basically, the acidity and activity of WO3–ZrO2 originate from sub-nanometer WOx clusters, which could be largely impacted by the phase of the ZrO2,4,5 the content of tungsten,6–11 calcination temperature12,13 or more generally the preparation methods applied.14–16The hydrothermal method is one of the most popular ways for the synthesis of ZrO2–WO3. It usually results in a stronger zirconium–tungsten interaction compared to precipitation–calcination and sol–gel methods.17 This strong interaction is proposed to cause a better dispersion of WO3 and generates more acid sites.18,19 On the other hand, hydrothermal conditions affect the size, morphology, composition, and structure of the products. For instance, hydrothermal treatment at improved temperatures was reported by Zhou et al. to affect the structural deformation of ZrO2 (i.e., causing more tetragonal ZrO2), which resulted in stronger surface acidity.20
Noteworthy, most of the reported hydrothermal method was applied to the synthesis process of catalysts and very little attention has been paid to the post-activation of a poor catalyst. In the present work, we found for the first time that a suitable post-hydrothermal treatment could activate a poor ZrO2–WO3 catalyst in the Friedel–Crafts reaction. Contrary trends were observed when using different solutions: the post-hydrothermal treatments with water or aqueous ammonium chloride reduced the acidity and activity, whereas, the treatments with ammonia solution or ethylenediamine solution largely improved the performance. The mechanism behind these changes was carefully addressed.
Experimental
Catalyst preparation
ZrO2–WO3 catalyst samples were synthesized by sol–gel process.21 Firstly, 9 mmol Zr(OBu)4 (TBOZ) and 1 mmol WCl6 were dissolved in 30 mL ethanol containing 4.6 mL acetic acid and 2 mL hydrochloric acid at 25 °C. The sol was stirred for 2 h and evaporated at 30 °C to get the gel. After that, it was aged for 24 h at 65 °C to get the as-synthesized ZrO2–WO3 (A-ZrO2–WO3). Finally, the sample was calcined in air at 550 °C for 5 h. The resulting catalyst was denoted as ZrO2–WO3.Post-synthesis treatment of ZrO2–WO3 by hydrothermal process (ZrO2–WO3-S, “S” indicates the solute, AH: ammonium hydroxide, EDA: ethylenediamine, W: water, AC: ammonium chloride). The suspension of 3.0 g A-ZrO2–WO3 and 30 mL solution was transferred into a stainless Teflon-lined 100 mL capacity autoclave. The autoclave was subjected to hydrothermal treatment at 200 °C for 24 h. The products after the hydrothermal treatment was filtered and thoroughly washed with deionized water till the pH value of the filtered liquid was 7, and the powder was calcined in air at 550 °C for 5 h. The pH value for ammonium hydroxide, ethylenediamine, water and ammonium chloride aqueous solution is 12, 12, 7 and 5 respectively.
Characterization methods
X-ray diffraction (XRD) patterns were recorded on an Ultima IV diffractometer with Cu Kα radiation at 3 KW, a step size of 0.02°, scanning speed is 10° min−1. Raman spectroscopy was obtained on HORIBA Scientific LabRAM HR Evolution with a visible (514.532 nm) laser excitation. The visible excitation was generated by an Ar excitation light source. The laser power was adjusted to 10 mW. Nitrogen adsorption–desorption analysis was carried out at 77 K on Nova 4200 e. All samples were pretreated at 300 °C under vacuum condition for 3 h and then they went through N2 adsorption and desorption. X-ray photoelectron spectroscopy (XPS) was carried out on a Thermo Scientific K-Alpha spectrometer equipped with Al Kα X-ray source (1486.6 eV). The base pressure during the analysis was 5 × 10−7 mBar. The C 1s line at 284.80 eV was used as an internal standard for the correction of binding energies (BE). Transmission electron microscopy analysis (HRTEM) of the samples was conducted on a JEOL 2010 transmission electron microscope operating at 200 kV. The samples were dispersed in high-purity ethanol and supported on carbon-coated copper grids for TEM. High angle annular dark field (HAADF) STEM was acquired with an aberration-corrected Titan ChemiSTEM equipped with a probe corrector (CEOS). This microscope was operated at 200 kV with an optimum attainable resolution of 0.078 nm.The acid properties of the samples were determined by temperature-programmed desorption of ammonia (NH3-TPD). NH3-TPD was carried out on the Bel Cata II with a thermal conductivity detector. The 125 mg samples were pretreated at 573 K in dry air for 1 h and then cooled in flowing helium at 50 °C for 1 h. The ammonia adsorption was carried out for 1 h, using a NH3/He (10% v/v) mixture gas. Weakly bound ammonia was removed by blowing-off with helium at a temperature of 50 °C for 1 h. NH3-TPD was conducted in the temperature range of 50–550 °C with heating rate 10 °C min−1.
Temperature-programmed reduction (TPR) was performed using an AutoChem1 II 2920 instrument (Thermo Fisher). The 0.050 g fresh sample was treated in a stream of He (50 mL min−1), ramping the temperature at 10 °C min−1 from room temperature to 300 °C and maintaining 300 °C for 1 h, and then cooled to 50 °C. The TPR measurement was carried out using H2/Ar (10% v/v) as reducing gas mixture, flowing at 50 mL min−1. The heating rate was 10 °C min−1 from 50 °C to 800 °C.
Acid-catalyzed reaction
The Friedel–Crafts reaction of anisole and benzyl alcohol (BA for short) was carried out in a three-necked flask reactor at 122 °C. The reaction system was 0.1 g of catalyst, 50 mmol of anisole, and 5 mmol of BA. The concentration of each substrate was analyzed by a gas chromatograph (GC-2014, Shimadzu) equipped with a capillary column with n-decane as an internal standard. The BA conversion and selectivity were calculated by the following equation by internal standard method.21where Ai.Res is residual BA peak area, Ai.Ini is initial BA peak area.


Results and discussion
A ZrO2–WO3 sample was firstly prepared by a sol–gel method and then treated in a hydrothermal condition with an aqueous solution of ammonia (the obtained sample was named ZrO2–WO3-AH). The XRD patterns in Fig. 1a show that the prepared ZrO2–WO3 contains both tetragonal ZrO2 (t-ZrO2, patterns at 30.1° and 35.1°) and monoclinic ZrO2 (m-ZrO2, patterns at 28.2° and 31.4°), and the ratio of t-ZrO2/m-ZrO2 is 3.26. After the post-hydrothermal treatment, the intensity of m-ZrO2 was almost diminished while that of t-ZrO2 was enhanced, showing a final t-ZrO2/m-ZrO2 ratio of 6.85. No evident signal from tungsten-based materials (such as WO3) could be observed despite that a considerable amount of W precursor (i.e., atomic ratio W/Zr = 1/9) was added in the synthesis. This indicated a poor crystallinity or a high dispersion of W species in the product.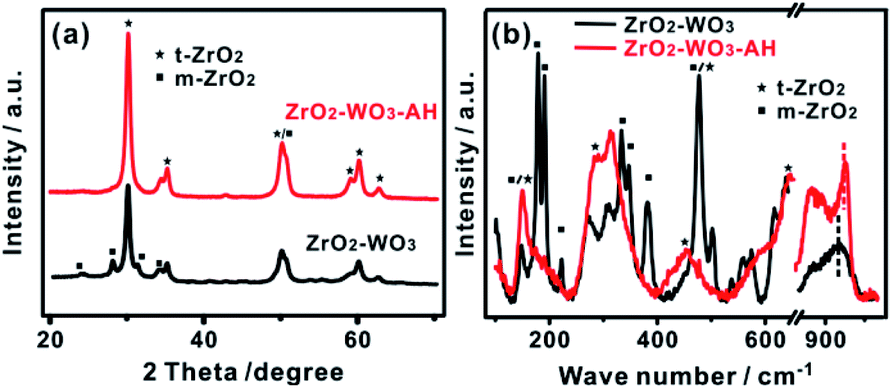 | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of ZrO2–WO3 and ZrO2–WO3-AH. | ||
The catalysts were further characterized by Raman spectroscopy, which is more sensitive than XRD in characterizing the short-range order of nano-materials. As the results in Fig. 1b shown, t-ZrO2 and m-ZrO2 phases shared strong overlapping bands at 478 and 637–647 cm−1, ZrO2–WO3 presented obvious m-ZrO2 phase (characteristic peaks are at 179, 191, 220, 383 and 638 cm−1), while ZrO2–WO3-AH contained the features of crystalline t-ZrO2 phase with bands at 148, 285, 315 and 460 cm−1, consistent with the XRD results.22 Moreover, no crystalline WO3 bands at 800, 708 and 265 cm−1 were observed in both samples, but only a broad band at 910–950 cm−1 that assigned to the W–O–Zr bond vibrations23 and a peak at 950–1020 cm−1 that related to the stretching mode of terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds formed.24 This confirmed the presence of W species that had a chemical interaction with ZrO2. The Raman band shifts from 956 cm−1 in ZrO2–WO3 to 976 cm−1 in ZrO2–WO3-AH reflected the transformation of surface mono-tungstate to poly-tungstate species.25–27 XRD and Raman results give valid evidence for the structural change of ZrO2–WO3 under hydrothermal treatment.
O bonds formed.24 This confirmed the presence of W species that had a chemical interaction with ZrO2. The Raman band shifts from 956 cm−1 in ZrO2–WO3 to 976 cm−1 in ZrO2–WO3-AH reflected the transformation of surface mono-tungstate to poly-tungstate species.25–27 XRD and Raman results give valid evidence for the structural change of ZrO2–WO3 under hydrothermal treatment.
The morphology of both samples was characterized by electron microscopy. The representative HRTEM image of ZrO2–WO3 in Fig. 2a and ZrO2–WO3-AH in Fig. 2d show uniform lattice fringes: the lattice spacing of 0.294 and 0.303 nm were corresponded to the (110) planes of the t-ZrO2, while the lattice spacing of 0.368 and 0.375 nm were corresponded to the (111) planes of m-ZrO2. No tungsten oxide could be seen in this HRTEM mode. However, by changing to a STEM-HAADF mode, one can clearly see a spontaneous dispersion of bright spots on the surface of both samples (Fig. 2b and e).These spots with brighter contrast were proposed to be W species as W atoms are heavier than Zr atoms. It was important to note that the clear edge of the spots in ZrO2–WO3 became fuzzy after the hydrothermal treatment, possibly forming WOx–Zr clusters at the ZrO2–WO3 interface. The elemental mapping in Fig. 2c and f shows a homogenous dispersion of Zr on the sample, while the W signal was relatively stronger in the regions with bright spots. Consistent with the HAADF results, the W dispersed more evenly on ZrO2 after the hydrothermal treatment. The XPS analysis of both samples indicated a 6+ state of W, suggesting that the W-rich clusters were dominated by WO3.26 (Fig. S1†).
 | ||
| Fig. 2 (a and d) TEM, (b and e) HAADF and EDS (c and f) of ZrO2–WO3 and ZrO2–WO3-AH. White/black circles indicate WOx clusters with diameters of about 0.8 nm. | ||
The catalytic performance of the two catalysts were evaluated in the classic F–C reaction of anisole and benzyl alcohol (BA for short) to produce ortho-benzylanisole and para-benzylanisole, which are key industrial compounds as pharmaceutical intermediates or fine chemicals.28 Basically, the activity of this reaction was determined by the strength or amount of Brønsted acid sites on the catalyst.29 The catalytic results showed that ortho-benzylanisole, para-benzylanisole and dibenzylether (a byproduct) were all produced. The activity that calculated based on the conversion of BA after 0.5 h (Fig. 3a) indicated that ZrO2–WO3-AH (32.4%) was 8.76 times higher than that of the ZrO2–WO3 (3.7%). The activity of ZrO2–WO3-AH is comparable to the ZrO2–WO3 with hexadecyl trimethyl ammonium bromide as surfactant (39.2%) synthesized in our previous paper,30 lower than sulfated zirconia.31 But, ZrO2–WO3 is much more stable than the sulfated ones and less deactivate during catalytic reaction. Some other solid catalysts already known to catalyze Friedel–Crafts arylation reaction of anisole with benzyl alcohol are shown in Table S1.† ZrO2–WO3-AH exhibited lower price or higher activity. The five-cycle stability tests of ZrO2–WO3-AH sample in Fig. S2† showed almost no loss of both the activity and selectivity, indicating good stability and recyclability of the catalyst.
 | ||
| Fig. 3 (a) Friedel–Crafts reactions of anisole and benzyl alcohol and (b) NH3-TPD of ZrO2–WO3 and ZrO2–WO3-AH. | ||
Considering the strong correlation between the activity and the Brønsted acid sites on the catalysts, we firstly tried to analyze the acidity of the catalysts before and after the hydrothermal treatment by using NH3-TPD, for which, higher desorption temperature of NH3 indicated stronger acidity. As the results shown in Fig. 3b, the NH3 desorption profile could be roughly divided into two regions: <320 °C and >320 °C, which could be defined as “weak” and “strong” acid sites, respectively. The post-hydrothermal treatment with ammonia resulted in stronger desorption of NH3 but with peaks at the same temperatures. This possibly indicated an increased amount of acid sites, rather than stronger acidity of each site. The concentration of weak and strong acid sites increased roughly by 40.6% and 34.0%, respectively, calculated based on the areas of the peaks.
To find the correlation between the catalytic activity and the hydrothermal solutions used, the ZrO2–WO3 was post-treated with a series of other solutions, including pure water (sample named ZrO2–WO3-W), ammonium chloride (sample named ZrO2–WO3-AC), and ethylenediamine (sample named ZrO2–WO3-EDA). Strong base sodium hydroxide was not considered as hydrothermal medium because it would lead to the leaching of WO3. The catalytic performance of these materials is displayed in Fig. 4a and Table 1. Basically, ZrO2–WO3-EDA and ZrO2–WO3-AH showed superior conversions of BA and similar selectivity to ZrO2–WO3, while both the conversion of BA and selectivity over ZrO2–WO3-W and ZrO2–WO3-AC were lower. The same trend was observed in yield of all catalysts as BA conversion. The yield values follow the order ZrO2–WO3-EDA > ZrO2–WO3-AH ≫ ZrO2–WO3 > ZrO2–WO3-W ∼ ZrO2–WO3-AC. Such a big impact of hydrothermal solutions in tailoring the catalytic performance of ZrO2–WO3 has never been reported to the best of our knowledge.
 | ||
| Fig. 4 (a) Friedel–Crafts reactions of anisole and benzyl alcohol (30 min), (b) XRD patterns for ZrO2–WO3 and ZrO2–WO3 post-treated with different solutions. | ||
| Catalyst | BA% | S% | Y%a | It/mb | t-ZrO2 sizec (nm) | Surface area (m2 g−1) |
|---|---|---|---|---|---|---|
| a Y% = conversion of BA × selectivity = BA% × S%.b Calculated by the equation It/m = It/(Im1 + Im2) where It, Im1, and Im2 represents the relative diffraction intensity of (101) plane of tetragonal zirconia, (111) plane and (111) plane of monoclinic zirconia.c Analyzed from XRD patterns. | ||||||
| ZrO2–WO3 | 3.7 | 78.7 | 2.9 | 3.26 | 13.1 | 35.5 |
| ZrO2–WO3-AH | 32.4 | 77.7 | 25.2 | 6.85 | 12.2 | 54.0 |
| ZrO2–WO3-EDA | 43.9 | 78.2 | 34.3 | 5.88 | 10.9 | 65.2 |
| ZrO2–WO3-W | 1.97 | 62.7 | 1.2 | ∼0 | — | 74.9 |
| ZrO2–WO3-AC | 1.86 | 64.6 | 1.2 | ∼0 | — | 70.0 |
We firstly tried to understand the effects of surface area and valence state of W. The surface areas that calculated based on N2 adsorption–desorption curves (Fig. S3a†) showed a significant increase after all the post-hydrothermal treatments no matter what solution was used (Table 1). However, ZrO2–WO3-W and ZrO2–WO3-AC with the largest surface areas performed the worst activity, claiming that the increase of surface area did not directly contribute to the reaction. In addition, the pore size was not likely proportional to the catalytic activity difference of all catalysts either (Fig. S3b†). On the other hand, the presence of ammonia species was reported to favour the formation of W5+ ions during annealing at high temperatures and these W5+ ions were responsible for the acidic properties as well as the activity towards the F–C reaction. However, our XPS analysis on ZrO2–WO3 and ZrO2–WO3-AH clearly showed that only W6+ was present in both samples (Fig. S1†), also ruling out this possibility.
We then focused on the structural changes after post-hydrothermal treatments. The XRD patterns and Raman spectra of all the samples are displayed in Fig. 4b and 5. Both results showed that ZrO2–WO3-AH and ZrO2–WO3-EDA were dominated by t-ZrO2, whereas ZrO2–WO3-W and ZrO2–WO3-AC contained almost only m-ZrO2. This converse structures and their good correlation with the catalytic performance highly suggested that t-ZrO2 was beneficial to the F–C reaction. Comparing to ZrO2–WO3, the frequency shift of the Raman bands from 956 to 967 cm−1 with the hydrothermal under EDA indicated that the W interaction species are polymeric (Fig. 5), similar to ZrO2–WO3-AH; whereas no obvious difference at this peak is detected in ZrO2–WO3-AC and ZrO2–WO3-W. To further verify the effect of hydrothermal solution on the crystal structure and the activity, we performed the same post-hydrothermal treatments on a ZrO2–WO3 that was calcined at 800 °C (sample named as ZrO2–WO3-800), a temperature that was high enough to caused crystalline WO3 (XRD in Fig. S4†). As the results shown in Table S2 and Fig. S4,† the changes of the ZrO2 structure and the catalytic activity were basically the same with ZrO2–WO3 when treating with the same solutions. It is interesting to note that the crystalline WO3 in ZrO2–WO3-800 was retained after the post-hydrothermal treatments with water or NH4Cl, yet it was not detected at all in ZrO2–WO3-EDA-800 and ZrO2–WO3-AH-800. This suggested a re-dispersion of WO3 accompanied with the transformation of ZrO2 crystalline phase in the EDA or ammonia solution.
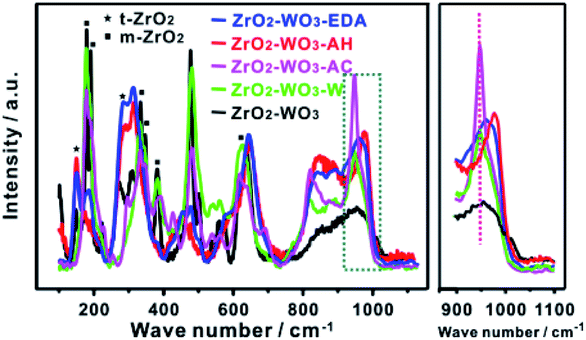 | ||
| Fig. 5 Raman spectra of ZrO2–WO3 and ZrO2–WO3-S. | ||
We postulated that the formation of WOx clusters and t-ZrO2 was highly related to the pH value of the aqueous solution, as both the ammonia and EDA are alkaline solutions, whereas the water and NH4Cl are neutral or acidic medium. Ethanediamine (EDA) can create hydroxyl ions due to thermal degradation during hydrothermal process. Ammonium hydroxide is a typical weak base and produces hydroxide ions. At high pH, the high concentration of hydroxide ion (OH−) contributed to the formation of WOx clusters. WOx was thought to be synthesized through reactions (1) and (2).32 These OH− ions reacted with formed HCl and generated water (H2O) which induced the generation of WOx. What's more, OH− ions would promote linking of WOx units, and led to more polymerized WOx clusters.33 The effect of OH− was further confirmed by the particle size change of t-ZrO2 crystals results from XRD analyses. In XRD patterns, the tetragonal ZrO2 percentage increases and the crystallite size decreases with post-treated by ammonium hydroxide or ethylenediamine (Table 1). It is suggested WOx surface species inhibit ZrO2 crystallite sintering, stabilize t-ZrO2 crystallites and lead to smaller size of t-ZrO2 than in pure ZrO2–WO3.34,35
| WCl6 + aCH3CH2OH → (CH3CH2O)a–WCl6−a + aHCl | (1) |
| (CH3CH2O)a–WCl6−a + H2O → WOx + aCH3CH2OH + 6-aHCl | (2) |
The formation of more WOx clusters induced by OH− ions can be verified by H2-TPR. As observed, a peak centered on 406 °C in ZrO2–WO3 and ZrO2–WO3-AH could be attributed to first step of WO3 reduction (Fig. 6). While, two peaks of ZrO2–WO3-AH sample, about 640 °C and 720 °C, ascribed to reduction of WO3 species (amorphous and non-stoichiometric WO3)36,37 shift to lower reduction temperature, compared with the peak above 800 °C of ZrO2–WO3. The two reduction step suggested a growth of WOx with W–O–W linkages; because linked WOx are reduced at lower temperatures than isolated WOx species.38 TPR results confirmed that larger and more interconnected WOx clusters of ZrO2–WO3-AH formed after ammonium hydroxide hydrothermal treatment.
 | ||
| Fig. 6 H2-TPR for ZrO2–WO3 and ZrO2–WO3-AH. | ||
Conclusions
In conclusion, post-hydrothermal treatments with different solutions were performed on a poor ZrO2–WO3 solid acid catalyst. It was found that the treatment with ammonium hydroxide or ethylenediamine significantly affected the structure and acidity of ZrO2–WO3, resulting in higher activity in F–C reaction. The treatment with water or ammonium chloride, in opposite, led to deactivation. High pH values were proposed to play the essential role in the regulation of crystal structure and acidic sites, and improved the uniformity of WOx species and prevented the formation of m-ZrO2 during calcination. Our work demonstrated that even a low activity of ZrO2–WO3 catalyst can be activated by performing a suitable hydrothermal treatment. The formation of these acidic sites controlled by adjusting a hydrothermal condition rationally is of great importance in the design of solid acid catalyst.Author contributions
Sha Li designed the experiment, analysed the data, wrote the manuscript and provided the funding acquisition. Ruopeng Yu, Bonan Xu, Zhikun Wang, conducted the experiments. Chunzheng Wu, Jianzhong Guo designed the experiment and reviewed the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22002141, 21677132), the Research and Development Fund of Zhejiang A & F University (2014FR061), and Fund of Zhejiang Provincial Key Laboratory of Chemical Utilization of Forestry Biomass (2020CUFB002).References
- M. Hino and K. Arata, Chem. Lett., 1979, 8, 477–480 CrossRef.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1988, 18, 1259–1260 RSC.
- M. Hino and K. Arata, J. Chem. Soc., Chem. Commun., 1988, 17, 1168–1169 RSC.
- W. J. Ji, J. Q. Hu and Y. Chen, Catal. Lett., 1998, 53, 15–21 CrossRef CAS.
- N. Soultanidis, W. Zhou, A. C. Psarras, A. J. Gonzalez, E. F. Iliopoulou, C. J. Kiely, I. E. Wachs and M. S. Wong, J. Am. Chem. Soc., 2010, 132, 13462–13471 CrossRef CAS PubMed.
- J. G. Santiesteban, J. C. Vartuli, S. Han, R. D. Bastian and C. D. Chang, J. Catal., 1997, 168, 431–441 CrossRef CAS.
- R. Kourieh, S. Bennici, M. Marzo, A. Gervasini and A. Auroux, Catal. Commun., 2012, 19, 119–126 CrossRef CAS.
- E. I. Ross-Medgaarden, W. V. Knowles, T. Kim, M. S. Wong, W. Zhou, C. J. Kiely and I. E. Wachs, J. Catal., 2008, 256, 108–125 CrossRef CAS.
- R. Sakthivel, H. Prescott and E. Kemnitz, J. Mol. Catal. A: Chem., 2004, 223, 137–142 CrossRef CAS.
- T. Y. Kim, D. S. Park, Y. Choi, J. Baek, J. R. Park and J. Yi, J. Mater. Chem., 2012, 22, 10021–10028 RSC.
- M. Scheithauer, R. K. Grasselli and H. Knozinger, Langmuir, 1998, 14, 3019–3029 CrossRef CAS.
- M. A. Cortes-Jacome, C. Angeles-Chavez, E. Lopez-Salinas, J. Navarrete, P. Toribio and J. A. Toledo, Appl. Catal., A, 2007, 318, 178–189 CrossRef CAS.
- W. D. Sun, Z. B. Zhao, C. Guo, X. K. Ye and Y. Wu, Ind. Eng. Chem. Res., 2000, 39, 3717–3725 CrossRef CAS.
- P. Afanasiev, C. Geantet, M. Breysse, G. Coudurier and J. C. Vedrine, J. Chem. Soc., Faraday Trans., 1994, 90, 193–202 RSC.
- M. Hartmanova, I. Travenec, A. A. Urusovskaya, K. Putyera, D. Tunega and I. I. Korobkov, Solid State Ionics, 1995, 76, 207–214 CrossRef CAS.
- G. K. Chuah, S. Jaenicke and B. K. Pong, J. Catal., 1998, 175, 80–92 CrossRef CAS.
- H. Armendariz, M. A. Cortes, I. Hernandez, J. Navarrete and A. Vazquez, J. Mater. Chem., 2003, 13, 143–149 RSC.
- H. Wang, Y. Wu, L. He and Z. Liu, Energy Fuels, 2012, 26, 6518–6527 CrossRef CAS.
- M. A. Cortes-Jacome, J. A. Toledo, C. Angeles-Chavez, M. Aguilar and J. A. Wang, J. Phys. Chem. B, 2005, 109, 22730–22739 CrossRef CAS PubMed.
- Y. Song, C. Kang, Y. Feng, F. Liu, X. Zhou, R. Dong and L. Xu, Chin. J. Catal., 2008, 29, 1196–1198 CrossRef CAS.
- S. Li, H. Zhou, C. Jin, N. Feng, F. Liu, F. Deng, J. Wang, W. Huang, L. Xiao and J. Fan, J. Phys. Chem. C, 2014, 118, 6283–6290 CrossRef CAS.
- M. J. Li, Z. H. Feng, G. Xiong, P. L. Ying, Q. Xin and C. Li, J. Phys. Chem. B, 2001, 105, 8107–8111 CrossRef CAS.
- W. Zhou, E. I. Ross-Medgaarden, W. V. Knowles, M. S. Wong, I. E. Wachs and C. J. Kiely, Nat. Chem., 2009, 1, 722–728 CrossRef CAS PubMed.
- I. E. Wachs, Catal. Today, 1996, 27, 437–455 CrossRef CAS.
- R. A. Boyse and E. I. Ko, J. Catal., 1997, 171, 191–207 CrossRef CAS.
- J. R. Sohn and M. Y. Park, Langmuir, 1998, 14, 6140–6145 CrossRef CAS.
- K. Song, H. Zhang, Y. Zhang, Y. Tang and K. Tang, J. Catal., 2013, 299, 119–128 CrossRef CAS.
- B. Yuan, W. Zhao, F. Yu and C. Xie, Catal. Commun., 2014, 57, 89–93 CrossRef CAS.
- C. Tagusagawa, A. Takagaki, A. Iguchi, K. Takanabe, J. N. Kondo, K. Ebitani, S. Hayashi, T. Tatsumi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 1128–1132 CrossRef CAS PubMed.
- S. Li, C. Jin, N. Feng, F. Deng, L. Xiao and J. Fan, Catal. Commun., 2019, 123, 54–58 CrossRef CAS.
- Z. Miao, J. Zhou, J. Zhao, D. Liu, X. Bi, L. Chou and S. Zhuo, Appl. Surf. Sci., 2017, 411, 419–430 CrossRef CAS.
- Y. Wang, X. Wang, Y. Xu, T. Chen and J. Liu, Small, 2017, 13, 1603689 CrossRef PubMed.
- G. Y. Guo, Y. L. Chen and W. J. Ying, Mater. Chem. Phys., 2004, 84, 308–314 CrossRef CAS.
- F. Di Gregorio and V. Keller, J. Catal., 2004, 225, 45–55 CrossRef CAS.
- L. M. Hernández-Pichardo, A. J. Montoya, P. del Angel, A. Vargas and J. Navarrete, Appl. Catal., A, 2008, 345, 233–240 CrossRef.
- A. M. Garrido Pedrosa, M. J. B. Souza, S. H. Lima, D. M. A. Melo, A. G. Souza and A. S. Araújo, J. Therm. Anal. Calorim., 2007, 87, 703–707 CrossRef CAS.
- D. G. Barton, S. L. Soled, G. D. Meitzner, G. A. Fuentes and E. Iglesia, J. Catal., 1999, 181, 57–72 CrossRef CAS.
- A. Martinez, G. Prieto, M. A. Arribas, P. Concepcion and J. F. Sanchez-Royo, J. Catal., 2007, 248, 288–302 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XPS, repeated experiments, N2 adsorption–desorption curve, XRD and Tables S1 and S2. See https://doi.org/10.1039/d2ra00519k |
| This journal is © The Royal Society of Chemistry 2022 |