
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzo[de]isoquinoline-1,3-dione condensed asymmetric azaacenes as strong acceptors†
Pengxin Zhou *,
Lanlan Deng,
Zengtao Han,
Xiaolong Zhao,
Zhe Zhang and
Shuhui Huo*
*,
Lanlan Deng,
Zengtao Han,
Xiaolong Zhao,
Zhe Zhang and
Shuhui Huo*
College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, China. E-mail: zhoupx@nwnu.edu.cn; huosh@nwnu.edu.cn
First published on 4th May 2022
Abstract
We have designed and synthesized three novel benzo[de]isoquinoline-1,3-dione (BQD) condensed asymmetric azaacenes, BQD-TZ, BQD-AP and BQD-PA, with different end groups of 1,2,5-thiadiazole, acenaphthylene and phenanthrene. The triisopropylsilylethynyl groups were grafted to increase the solubility and crystallinity of the three molecules. These molecules have high electron affinity values of 3.87, 3.69 and 3.74 eV for BQD-TZ, BQD-AP and BQD-PA, respectively as confirmed by cyclic voltammetry measurements. Single-crystal X-ray diffraction revealed that these molecules have strong π–π stacking with distances of 3.31–3.41 Å and different stacking arrangements.
Introduction
Conjugated molecules are important organic materials because of their outstanding electronic properties and potential applications in organic electronic devices, such as organic field-effect transistors (OFETs) and organic photovoltaics (OPVs).1–7 Currently, a large number of p-type semiconductors have been reported with excellent device performance but the n-type semiconductors are still lagging in comparison.8–14 Therefore, the development of new conjugated molecules with high electron-affinities (EAs) is an important topic in the field of functional π-materials.N-Heteroacenes are among the most important electron-accepting materials.15,16 Compared to the acenes, the N-heteroacenes include unsaturated N atoms in the conjugated backbone; this can lower both the EA and ionic potential (IP), which is beneficial for stabilizing and achieving electron transport for the resulting molecules.17–23 For instance, 6,13-bis(triisopropylsilylethynyl)pentacene (TIPS-PEN) is a typical n-type semiconductor. Miao et al. introduced four unsaturated N atoms to develop an n-type material of 6,13-bis((triisopropylsilyl)ethynyl)-5,7,12,14-tetraazapentacene (TIPS-TAP), which exhibited electron mobility exceeding 10 cm2 V−1 s−1.24 N-heteroacenes usually have large conjugated backbones, which are not beneficial to the structural characteristics and device fabrication via solution processing.25–29 To overcome this issue, triisopropylsilyl (TIPS)-ethynyl groups were introduced by the Stille coupling reaction and the nucleophilic addition reaction.30–33 The studies showed that TIPS-ethynyl units play a vital role not only in improving the solubility and stability of acenes and heteroacenes but also in enhancing their capability for π-stacked arrays.34–37 Larger conjugated N-heteroacenes have been reported due to the employment of TIPS-ethyl groups.28,38–41 However, most of the work focuses on the study of symmetric N-heteroacenes, and the asymmetric N-heteroacenes are rare.
Herein, we report novel benzo[de]isoquinoline-1,3-dione condensed asymmetric azaacenes, which have different end-fused groups. The studies below detail their synthesis, optical and electrochemical properties, and molecular packing, and reveal that the end-fused group plays an important role in tuning the electronic properties and molecular stacking behaviors based on asymmetric azaacenes.
Experimental
Instruments and measurements
1H NMR and 13C NMR spectra of new compounds were recorded on a Varian Mercury-400 Plus or a Varian Mercury-300 Plus and Agilent Technologies DD2 (600 MHz) spectrometer in CDCl3. Chemical shifts (δ) for NMR were quoted in ppm relative to the solvent peak (7.26 ppm for 1H and 77.00 ppm for 13C in CDCl3). High-resolution mass spectra (HRMS) were obtained with a Bruker Daltonics APEXII 47 e FT-ICR, Agilent QTOF 700 or Agilent 1200 spectrometer and Shimadzu MALDI-7090. Thermogravimetric analysis (TGA) was carried out on a Netzsch 5500 thermogravimetry analyser under a nitrogen atmosphere at a flow rate of 100 mL min−1−1, and the heating rate was 10.0 °C min−1−1 from ambient temperature to 800 °C. The UV-vis absorption spectra were recorded on a UV-Vis-NIR spectrophotometer (UV-3600 Plus, Shimadzu, Japan). Fluorescence emission spectra were obtained on a steady-state fluorescence spectrometer (Edinburgh Instruments). Cyclic voltammetry (CV) experiments were performed at room temperature with an electrochemical analyser (CHI 660C, Chenhua Inc., China). Density functional theory (DFT) calculations were performed using the Gaussian 09 program, with the B3LYP hybrid functional and basis set 6-311G (d,p) for the ground-state geometry optimization. Single-crystal data collections were performed with a Bruker Smart Apex CCD X-ray diffractometer (Bruker, Germany) with highly oriented graphite crystal monochromated Mo Kα radiation (λ = 0.071073 nm) using ω and φ scan modes. Unit cell dimensions were obtained with least-squares refinements, and semi-empirical absorption corrections were applied using the SADABS program. The structures were solved by the direct method and refined by the full-matrix least-squares technique based on F2 with the SHELXTL-97 program. All of the atoms were obtained from difference Fourier maps, and all nonhydrogen atoms were refined with atomic anisotropic thermal parameters.Synthetic procedures
All reagents and chemicals were used as received from commercial sources without further purification. Compounds 6, 7 and 9 were synthesized according to the literature.30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 2:1) to obtain the product (5.7 g, 89%). 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 5.5 Hz, 2H), 7.61 (d, J = 5.0 Hz, 2H), 3.61 (s, 4H). 13C NMR (151 MHz, CDCl3) δ 160.8, 155.7, 138.0, 134.9, 129.1, 121.5, 115.4, 31.9.:1) to give the product. Yield (3.06 g, 75%). 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 7.2 Hz, 2H), 7.55 (d, J = 7.6 Hz, 2H), 4.17 (t, J = 7.6 Hz 2H), 3.56 (s, 4H), 1.76–1.67 (m, 2H), 1.47–1.39 (m, 2H), 1.39–1.30 (m, 4H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 164.4, 153.6, 137.7, 132.6, 126.4, 120.8, 119.3, 77.2, 77.0, 76.8, 40.3, 31.61 (d, J = 9.6 Hz), 28.2, 26.86, 22.6, 14.0.:2) as the eluent. Yield (2.54 g, 78%). 1H NMR (400 MHz, CDCl3) δ 8.81 (d, J = 7.2 Hz, 2H), 8.35 (d, J = 7.6 Hz, 2H), 4.22 (t, J = 7.6 Hz 2H), 1.79–1.71 (m, 2H), 1.47–1.41 (m, 2H), 1.38–1.33 (m, 4H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 186.2, 162.5, 143.7, 132.0, 126.8, 126.1, 122.9, 41.1, 31.5, 28.0, 26.7, 22.5, 14.0. HRMS (ESI): m/z [M + Na]+ calc. for C20H17NO4Na: 358.10420; found: 358.10476.:1) to give BQD-TZ (244 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 8.76 (d, J = 7.2 Hz, 2H), 8.46 (d, J = 6.4 Hz, 2H), 4.21 (t, J = 7.6 Hz, 2H), 1.83–1.69 (m, 2H), 1.34 (d, J = 4.5 Hz, 42H), 1.25 (s, 6H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 163.1, 155.4, 154.6, 141.9, 137.6, 136.2, 132.7, 125.4, 124.7, 122.7, 115.9, 110.5, 100.6, 31.5, 29.7, 28.2, 26.8, 22.6, 18.9, 14.0, 11.6. HRMS (ESI): m/z [M + H]+ calc. for C48H60N5O2SSi2: 826.39848; found: 826.39844.:1 to 2:1) to give the corresponding product 8. This crude product was also directly added to acetic acid (10 mL) solutions of the corresponding diketones (148 mg, 0.442 mmol). The mixtures were heated to 110 °C for 7 h under argon. After cooling to room temperature, the mixture was poured into 100 mL of 5% aqueous NaOH and extracted with dichloromethane (3 × 50 mL). The combined organic phases were dried with MgSO4 and filtered. The solvent was concentrated and purified by column chromatography, eluting with petroleum ether/dichloromethane (from 2:1 to 1:1) to give the desired product BQD-AP (166 mg, 59%). 1H NMR (300 MHz, CDCl3) δ 8.74 (d, J = 7.5 Hz, 2H), 8.45 (t, J = 6.8 Hz, 4H), 8.15 (d, J = 8.1 Hz, 2H), 7.91 (t, J = 7.5 Hz, 2H), 4.11 (t, J = 7.5 Hz, 2H), 1.77–1.67 (m, 2H), 1.45 (s, 48H), 1.34 (s, 6H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.2, 155.8, 155.0, 142.6, 141.4, 138.5, 136.8, 136.7, 136.5, 132.7, 131.5, 130.1, 129.0, 125.3, 124.5, 123.9, 122.8, 122.6, 109.0, 101.2, 40.7, 31.5, 28.2, 26.8, 22.6, 19.0, 14.0, 11.8. HRMS (MALDI-TOF) m/z: [M + H]+ calcd for C60H66N5O2Si2, 945.380; found, 945.310.:1 to 2:1) to give the corresponding product of 10. This crude product was also directly added to an acetic acid (10 mL) solution of the corresponding diketone (142 mg, 0.426 mmol). The mixture was heated to 110 °C for 7 h under argon. After cooling to room temperature, the mixture was poured into 100 mL of 5% aqueous NaOH and extracted with dichloromethane (3 × 50 mL). The combined organic phases were dried with MgSO4 and filtered. The solvent was concentrated and purified by column chromatography, eluting with petroleum ether/dichloromethane (from 2:1 to 1:1) to give the final product BQD-PA (190 mg, 68%).1H NMR (300 MHz, CDCl3) δ 9.63 (d, J = 7.2 Hz, 2H), 8.75 (d, J = 7.2 Hz, 2H), 8.54 (d, J = 7.8 Hz, 2H), 8.47 (d, J = 7.2 Hz, 2H), 7.86 (t, J = 7.9 Hz, 2H), 7.75 (t, J = 6.6 Hz, 2H), 4.16 (t, J = 7.4 Hz 2H), 1.79–1.70 (m, 2H), 1.48 (s, 42H), 1.37 (s, 6H), 0.92 (t, J = 6.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.1, 155.2, 144.4, 142.2, 142.0, 137.0, 136.7, 132.8, 132.7, 131.5, 130.1, 128.1, 127.5, 125.3, 124.5, 123.4, 123.0, 122.7, 109.7, 101.6, 40.8, 31.5, 28.2, 26.8, 22.6, 19.0, 14.0, 11.9. HRMS (MALDI-TOF) m/z: [M + H]+ calc. for C62H68N5O2Si2, 971.420; found, 971.315.
:1 to 2:1) to obtain the product (5.7 g, 89%). 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 5.5 Hz, 2H), 7.61 (d, J = 5.0 Hz, 2H), 3.61 (s, 4H). 13C NMR (151 MHz, CDCl3) δ 160.8, 155.7, 138.0, 134.9, 129.1, 121.5, 115.4, 31.9.:1) to give the product. Yield (3.06 g, 75%). 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 7.2 Hz, 2H), 7.55 (d, J = 7.6 Hz, 2H), 4.17 (t, J = 7.6 Hz 2H), 3.56 (s, 4H), 1.76–1.67 (m, 2H), 1.47–1.39 (m, 2H), 1.39–1.30 (m, 4H), 0.88 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 164.4, 153.6, 137.7, 132.6, 126.4, 120.8, 119.3, 77.2, 77.0, 76.8, 40.3, 31.61 (d, J = 9.6 Hz), 28.2, 26.86, 22.6, 14.0.:2) as the eluent. Yield (2.54 g, 78%). 1H NMR (400 MHz, CDCl3) δ 8.81 (d, J = 7.2 Hz, 2H), 8.35 (d, J = 7.6 Hz, 2H), 4.22 (t, J = 7.6 Hz 2H), 1.79–1.71 (m, 2H), 1.47–1.41 (m, 2H), 1.38–1.33 (m, 4H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 186.2, 162.5, 143.7, 132.0, 126.8, 126.1, 122.9, 41.1, 31.5, 28.0, 26.7, 22.5, 14.0. HRMS (ESI): m/z [M + Na]+ calc. for C20H17NO4Na: 358.10420; found: 358.10476.:1) to give BQD-TZ (244 mg, 78%). 1H NMR (400 MHz, CDCl3) δ 8.76 (d, J = 7.2 Hz, 2H), 8.46 (d, J = 6.4 Hz, 2H), 4.21 (t, J = 7.6 Hz, 2H), 1.83–1.69 (m, 2H), 1.34 (d, J = 4.5 Hz, 42H), 1.25 (s, 6H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 163.1, 155.4, 154.6, 141.9, 137.6, 136.2, 132.7, 125.4, 124.7, 122.7, 115.9, 110.5, 100.6, 31.5, 29.7, 28.2, 26.8, 22.6, 18.9, 14.0, 11.6. HRMS (ESI): m/z [M + H]+ calc. for C48H60N5O2SSi2: 826.39848; found: 826.39844.:1 to 2:1) to give the corresponding product 8. This crude product was also directly added to acetic acid (10 mL) solutions of the corresponding diketones (148 mg, 0.442 mmol). The mixtures were heated to 110 °C for 7 h under argon. After cooling to room temperature, the mixture was poured into 100 mL of 5% aqueous NaOH and extracted with dichloromethane (3 × 50 mL). The combined organic phases were dried with MgSO4 and filtered. The solvent was concentrated and purified by column chromatography, eluting with petroleum ether/dichloromethane (from 2:1 to 1:1) to give the desired product BQD-AP (166 mg, 59%). 1H NMR (300 MHz, CDCl3) δ 8.74 (d, J = 7.5 Hz, 2H), 8.45 (t, J = 6.8 Hz, 4H), 8.15 (d, J = 8.1 Hz, 2H), 7.91 (t, J = 7.5 Hz, 2H), 4.11 (t, J = 7.5 Hz, 2H), 1.77–1.67 (m, 2H), 1.45 (s, 48H), 1.34 (s, 6H), 0.90 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.2, 155.8, 155.0, 142.6, 141.4, 138.5, 136.8, 136.7, 136.5, 132.7, 131.5, 130.1, 129.0, 125.3, 124.5, 123.9, 122.8, 122.6, 109.0, 101.2, 40.7, 31.5, 28.2, 26.8, 22.6, 19.0, 14.0, 11.8. HRMS (MALDI-TOF) m/z: [M + H]+ calcd for C60H66N5O2Si2, 945.380; found, 945.310.:1 to 2:1) to give the corresponding product of 10. This crude product was also directly added to an acetic acid (10 mL) solution of the corresponding diketone (142 mg, 0.426 mmol). The mixture was heated to 110 °C for 7 h under argon. After cooling to room temperature, the mixture was poured into 100 mL of 5% aqueous NaOH and extracted with dichloromethane (3 × 50 mL). The combined organic phases were dried with MgSO4 and filtered. The solvent was concentrated and purified by column chromatography, eluting with petroleum ether/dichloromethane (from 2:1 to 1:1) to give the final product BQD-PA (190 mg, 68%).1H NMR (300 MHz, CDCl3) δ 9.63 (d, J = 7.2 Hz, 2H), 8.75 (d, J = 7.2 Hz, 2H), 8.54 (d, J = 7.8 Hz, 2H), 8.47 (d, J = 7.2 Hz, 2H), 7.86 (t, J = 7.9 Hz, 2H), 7.75 (t, J = 6.6 Hz, 2H), 4.16 (t, J = 7.4 Hz 2H), 1.79–1.70 (m, 2H), 1.48 (s, 42H), 1.37 (s, 6H), 0.92 (t, J = 6.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 163.1, 155.2, 144.4, 142.2, 142.0, 137.0, 136.7, 132.8, 132.7, 131.5, 130.1, 128.1, 127.5, 125.3, 124.5, 123.4, 123.0, 122.7, 109.7, 101.6, 40.8, 31.5, 28.2, 26.8, 22.6, 19.0, 14.0, 11.9. HRMS (MALDI-TOF) m/z: [M + H]+ calc. for C62H68N5O2Si2, 971.420; found, 971.315.Synthesis and electronic properties
The synthetic routes of three asymmetric azaacenes are illustrated in Scheme 1. Compound 2 was synthesized from the commercial starting material 1,2-dihydroacenaphthylene by Friedel–Crafts acylation. Compound 2 was then subjected to acidolysis and dehydration to form anhydride 3, which was then transformed into compound 4 by a condensation reaction. Compound 4 was oxidized to give the key intermediate of diketone 5. The condensation reaction was conducted between compound 4 and compound 5 to give the desired asymmetric azaacene of BQD-TZ. Compounds 7 and 9 were reduced to the corresponding diamine compounds by LiAlH4 in a dry THF solution. Finally, the BQD-AP and BQD-PA were also synthesized by a similar reaction to that of BQD-TZ. These compounds have good solubility in common organic solvents such as dichloromethane and chloroform and exhibit excellent thermal stability, with 5% weight loss upon heating up to 368 °C for BQD-TZ, 407 °C for BQD-AP, and 395 °C for BQD-PA (Fig. S1†).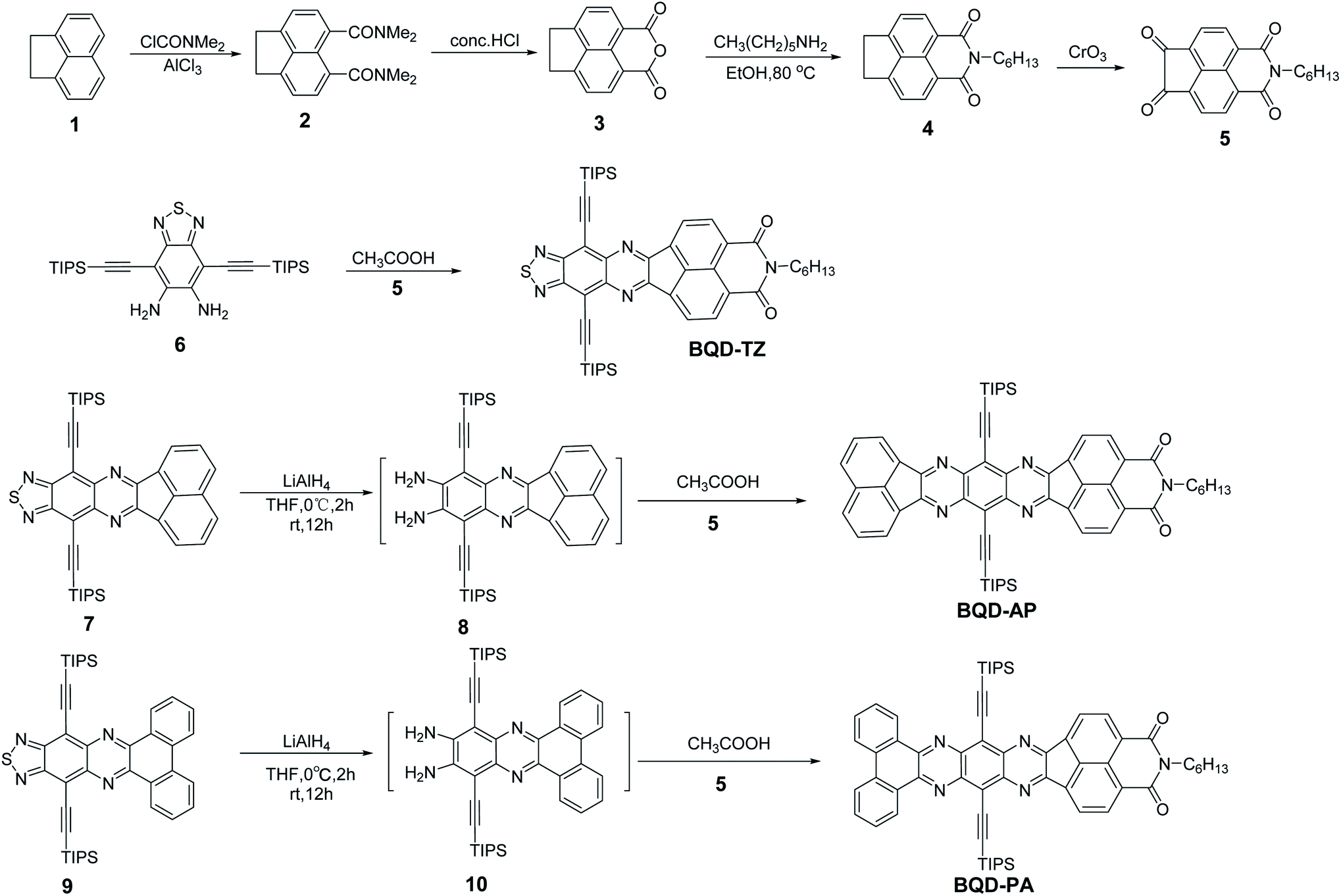 | ||
| Scheme 1 Synthetic routes for BQD-TZ, BQD-AP and BQD-PA. | ||
The absorption and emission spectra of three molecules were recorded in dilute dichloromethane solution (c = 10−5 M, Fig. 1a). BQD-TZ exhibited a similar absorption profile to the previously reported compounds 7 and 9.30 When the end groups were changed from 1,2,5-thiadiazole to acenaphthylene and phenanthrene, the absorption region at 350 nm to 400 nm showed significant differences where the absorption wavelength was gradually red-shifted as π-conjugation increased. This was because this region is contributed by π–π* transitions of the conjugated aromatic segments.42 The maximum absorption wavelengths (λmax) of these molecules are 548, 519, and 554 nm for BQD-TZ, BQD-AP and BQD-PA, respectively. This is attributed to the fused aromatic structures, considering that the three molecules differ only in the electron-donating nature of the aromatic-ring unit. Compared to BQD-TZ and BQD-PA, although the λmax of the BQD-AP has a blue-shift, its emission spectrum shows the largest wavelength, indicating that BQD-AP has the largest Stokes shift as shown in Fig. 1b and Table 1.
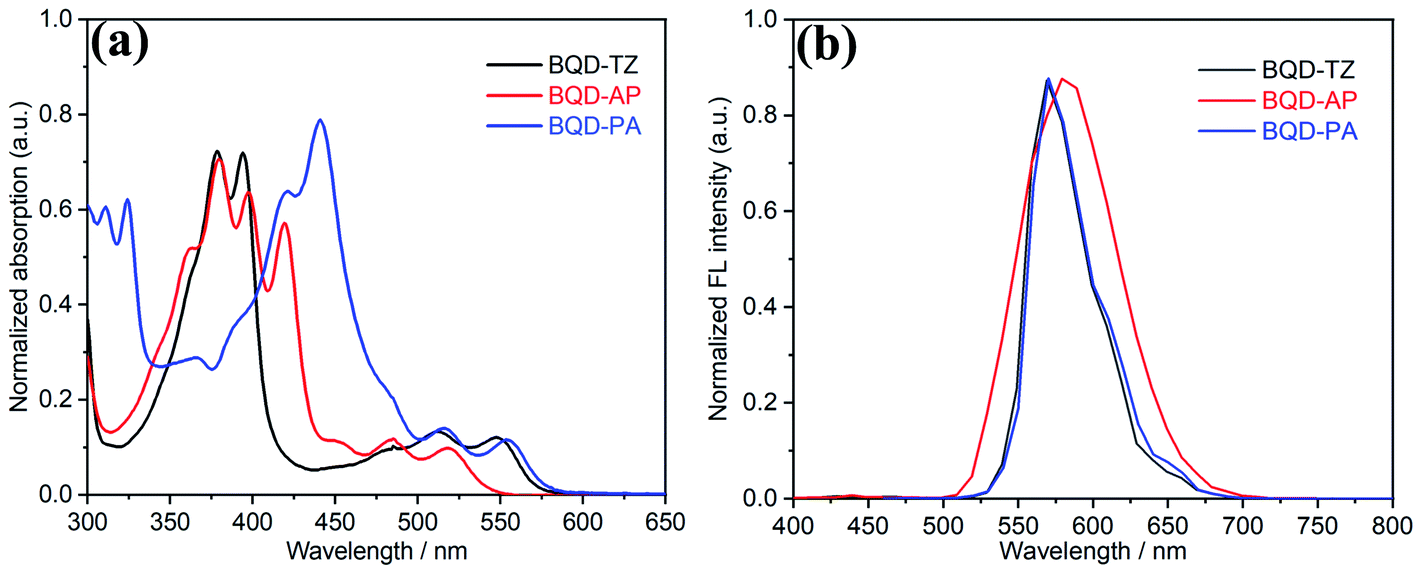 | ||
| Fig. 1 UV-Vis absorption (a) and fluorescence (b) spectra of BQD-TZ, BQD-AP and BQD-PA in dichloromethane. | ||
| Compound | Band Ia (nm) | Band IIa (nm) | λabs/nm (logε) a |
λema,b/nm | Stokes shift/nm | Eopta/eV | EAc/eV | LUMOd/eV | Egd/eV |
|---|---|---|---|---|---|---|---|---|---|
| a Dissolved in dichloromethane (c = 10−5 M).b Excitation wavelengths for the emission measurements are 548, 519, and 554 nm for BQD-TZ, BQD-AP and BQD-PA.c EA was measured by CV.d Calculated energy levels using B3LYP/6-31G, (d, p) ΔEg = LUMO–HOMO. | |||||||||
| BQD-TZ | 350–400 | 500–570 | 548 (7.8 × 104) | 569 | 21 | 2.12 | 3.87 | −3.43 | 2.35 |
| BQD-AP | 350–475 | 480–530 | 519 (6.4 × 104) | 579 | 60 | 2.21 | 3.69 | −3.06 | 2.55 |
| BQD-PA | 425–475 | 500–570 | 554 (8.1 × 104) | 569 | 15 | 2.10 | 3.74 | −3.19 | 2.44 |
The electron affinities (EA) of these molecules were studied by cyclic voltammetry (CV).43 The three compounds exhibited reversible reduction peaks in solution (Fig. 2). Electron affinity (EA) values were estimated from the onsets of the first reduction peak, while the potentials were determined using ferrocene (Fc) as the standard, by empirical formulas, EA = (Eonsetred − EFc/Fc+1/2 + 4.8) eV. The 1,2,5-thiadiazole containing BQD-TZ had the highest EA value of 3.87 eV, while BQD-PA, with phenanthrene, had a slightly higher EA value of 3.74 eV. The major reason is that 1,2,5-thiadiazole is a stronger electron-accepting group as compared to acenaphthylene. When the acenaphthylene was changed to phenanthrene, the conjugated skeleton was enlarged, the electron density was better distributed on the entire conjugated skeleton, and therefore, the BQD-AP provided the lowest EA value of 3.69 eV. It should be noted that the EA values of these molecules are higher than those of most N-heteroarenes due to the introduction of the strong electron-accepting unit of benzo[de]isoquinoline-1,3-dione.32,44–46 These results suggest that by attaching different electron-donating groups, the electron-deficient nature of the azaacene can easily be tuned. In order to combine these results with theoretical predictions, density functional theory (DFT, B3LYP, 6-31G (d,p)) calculations were carried out. The DFT results demonstrated that theoretical predictions exhibited the same tendency as the experimental data by analyzing LUMO levels (−3.43 eV for BQD-TZ, −3.00 eV for BQD-AP, −3.19 eV for BQD-PA, Fig. S2†). It is beneficial for us to predict the electronic properties of such new azaacene derivatives in line with the experimental findings. As shown in Fig. S2,† for the HOMO levels, the three compounds have similar electron density distributions along the conjugated backbone, while for the LUMO levels, the electron density of BQD-AP is mainly distributed on the BQD part, and the electron densities of BQD-TZ and BQD-PA are poor on the BQD part. This might be the main reason why even though BQD-PA has a similar chemical structure to BQD-AP, its absorption and emission spectra are much more similar to those of BQD-TZ than those of BQD-AP.
 | ||
| Fig. 2 The reduction curves of BQD-TZ, BQD-AP and BQD-PA in dichloromethane. | ||
Crystal structure analysis
Single-crystal analysis of molecules is a vital requirement to understand their electronic properties. Single-crystals of three molecules were obtained by slow evaporation from chloroform at room temperature. All of them were examined from X-ray analysis. Their single-crystals are presented in Fig. 3, and the corresponding data are collected in Table S1 (see ESI†). BQD-TZ and BQD-PA crystallize in the triclinic crystal system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) containing two molecules per unit cell. BQD-AP crystallizes in the monoclinic crystal system with space group I2/m containing four BQD-AP units per unit cell. The three molecules have a nearly planar conjugated backbone and an axisymmetric structure. It can be seen from the unit cell stacking that there is a strong π–π interaction between the molecules of these three substances. BQD-TZ has the shortest π-stacking distances of 3.31 Å, suggesting it has the strongest π–π interaction. Introducing the phenanthrene unit slightly increases the π–π distances to 3.38 Å. The largest distances of 3.36 and 3.41 Å for BQD-AP indicate that it possesses the weakest π–π interactions among these molecules. Therefore, the three molecules form a stable, ordered spatial packing. Additionally, the three molecules exhibit very interesting molecular stacking behaviour due to the different end groups. BQD-TA shows the head-to-tail π–π stacking, BQD-PA displays tail-to-centre π–π stacking, and BQD-AP presents the mixture of head-to-tail and tail-to-centre π–π stacking. To understand this issue, the molecular electrostatic potentials (ESPs) of the three molecules were calculated by DFT with the basis set of B3LYP, 6-31G (d,p). For BQD-TZ, the naphthalene unit and the center unit show strong positive and negative ESP, respectively, which make them form strong intermolecular interactions. This is in agreement with its molecular stacking in the single-crystal structure where the naphthalene unit forms strong π–π interactions with the center part. When the end group was changed from 1,2,5-thiadiazole to phenanthrene, BQD-PA showed stronger positive ESP in phenanthrene than that of naphthalene; therefore, the center unit is prone to forming π–π with phenanthrene as compared to naphthalene. This is why we only find the tail-to-center π–π stacking in BQD-PA. However, when the end group was changed from 1,2,5-thiadiazole to acenaphthylene, the BQD-AP also showed the strongest positive and negative ESP in acenaphthylene and center units. The acenaphthylene has a similar molecular size but opposite ESP as compared to the BQD unit, which causes acenaphthylene to form intermolecular interactions with both the center and BQD units. Considering that the center unit has a stronger negative ESP than that of the BQD unit, acenaphthylene forms a stronger intermolecular interaction with the center unit than the BQD unit. This might be the reason the π–π tail-to-center distance (3.36 Å) is slightly smaller than that of π–π head-to-tail distance (3.41 Å) in BQD-AP. Overall, the tuning of the end group in asymmetric N-heteroacenes is a useful strategy for controlling molecular self-organization behaviours.
containing two molecules per unit cell. BQD-AP crystallizes in the monoclinic crystal system with space group I2/m containing four BQD-AP units per unit cell. The three molecules have a nearly planar conjugated backbone and an axisymmetric structure. It can be seen from the unit cell stacking that there is a strong π–π interaction between the molecules of these three substances. BQD-TZ has the shortest π-stacking distances of 3.31 Å, suggesting it has the strongest π–π interaction. Introducing the phenanthrene unit slightly increases the π–π distances to 3.38 Å. The largest distances of 3.36 and 3.41 Å for BQD-AP indicate that it possesses the weakest π–π interactions among these molecules. Therefore, the three molecules form a stable, ordered spatial packing. Additionally, the three molecules exhibit very interesting molecular stacking behaviour due to the different end groups. BQD-TA shows the head-to-tail π–π stacking, BQD-PA displays tail-to-centre π–π stacking, and BQD-AP presents the mixture of head-to-tail and tail-to-centre π–π stacking. To understand this issue, the molecular electrostatic potentials (ESPs) of the three molecules were calculated by DFT with the basis set of B3LYP, 6-31G (d,p). For BQD-TZ, the naphthalene unit and the center unit show strong positive and negative ESP, respectively, which make them form strong intermolecular interactions. This is in agreement with its molecular stacking in the single-crystal structure where the naphthalene unit forms strong π–π interactions with the center part. When the end group was changed from 1,2,5-thiadiazole to phenanthrene, BQD-PA showed stronger positive ESP in phenanthrene than that of naphthalene; therefore, the center unit is prone to forming π–π with phenanthrene as compared to naphthalene. This is why we only find the tail-to-center π–π stacking in BQD-PA. However, when the end group was changed from 1,2,5-thiadiazole to acenaphthylene, the BQD-AP also showed the strongest positive and negative ESP in acenaphthylene and center units. The acenaphthylene has a similar molecular size but opposite ESP as compared to the BQD unit, which causes acenaphthylene to form intermolecular interactions with both the center and BQD units. Considering that the center unit has a stronger negative ESP than that of the BQD unit, acenaphthylene forms a stronger intermolecular interaction with the center unit than the BQD unit. This might be the reason the π–π tail-to-center distance (3.36 Å) is slightly smaller than that of π–π head-to-tail distance (3.41 Å) in BQD-AP. Overall, the tuning of the end group in asymmetric N-heteroacenes is a useful strategy for controlling molecular self-organization behaviours.
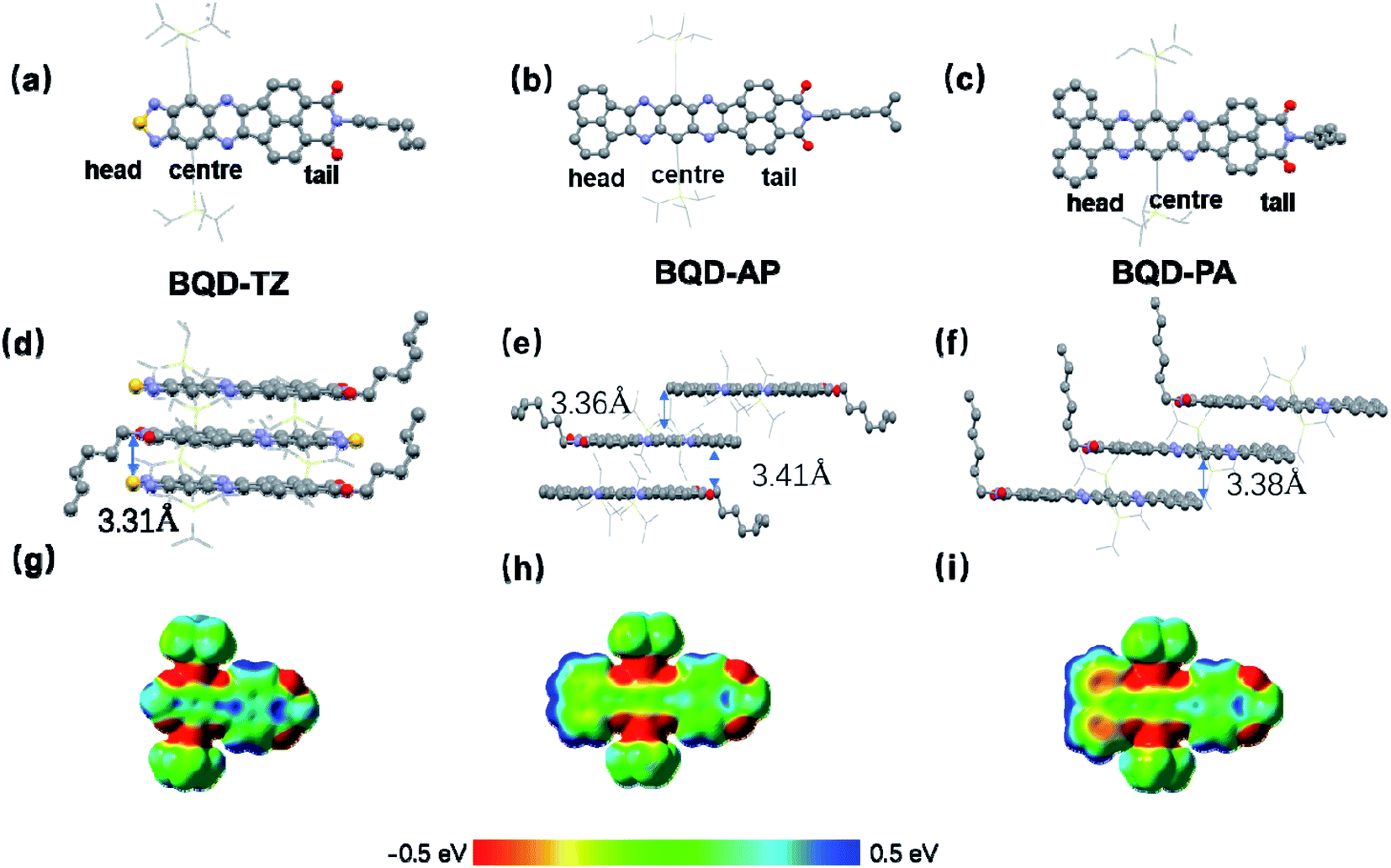 | ||
| Fig. 3 Molecular single-crystal structure of BQD-TZ (a), BQD-AP (b) and BQD-PA (c). The packing of BQD-TZ (d), BQD-AP (e) and BQD-PA (f); view along the b-axis. The packing of BQD-TZ (g), BQD-AP (h) and BQD-PA (f); viewed along the a-axis. The conjugated cores are shown with ball-stick models and the silylethynyl substituents are shown with wireframe models. Hydrogen atoms were removed for clarity. Carbon, nitrogen, oxygen, sulfur and silicon atoms are shown in grey, purple, red, orange, and yellow, respectively. | ||
Conclusions
In conclusion, we have successfully synthesized three novel benzo[de]isoquinoline-1,3-dione (BQD) condensed asymmetric azaacenes, BQD-TZ, BQD-AP and BQD-PA, which have good solubility and high stability. The X-ray single-crystal structure shows that the three molecules have a nearly planar conjugated backbone and strong π–π stacking with distances from 3.31–3.41 Å, which facilitate desirable solid-state molecular packing. Changing the fused aromatic unit in the benzo[de]isoquinoline-1,3-dione condensed asymmetric azaacenes affected the molecule's optoelectronic properties. Cyclic voltammetry results showed that these molecules possess high electron affinity values that are positive for BQD-TZ, BQD-AP and BQD-PA, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No. 21462036, 22066021), West Light Foundation of the Chinese Academy of Sciences (2021) and Industrial Support Plan Project of Colleges in Gansu Province (2021CYZC-17).References
- R. Dheepika, S. Sonalin, P. M. Imran and S. Nagarajan, J. Mater. Chem. C, 2018, 6, 6916–6919 RSC.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Mullen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS PubMed.
- S. Zhang, Y. Guo, Y. Zhang, R. Liu, Q. Li, X. Zhan, Y. Liu and W. Hu, Chem. Commun., 2010, 46, 2841–2843 RSC.
- S. Yang, D. Liu, X. Xu and Q. Miao, Chem. Commun., 2015, 51, 4275–4278 RSC.
- A. Naibi Lakshminarayana, A. Ong and C. Chi, J. Mater. Chem. C, 2018, 6, 3551–3563 RSC.
- J. Niklas and O. G. Poluektov, Adv. Energy Mater., 2017, 7, 1602226 CrossRef.
- B. L. Hu, K. Zhang, C. An, W. Pisula and M. Baumgarten, Org. Lett., 2017, 19, 6300–6303 CrossRef CAS PubMed.
- M. Müller, L. Ahrens, V. Brosius, J. Freudenberg and U. H. F. Bunz, J. Mater. Chem. C, 2019, 7, 14011–14034 RSC.
- E. C. Rudiger, S. Koser, F. Rominger, J. Freudenberg and U. H. F. Bunz, Chemistry, 2018, 24, 9919–9927 CrossRef.
- Q. Ye and C. Chi, Chem. Mater., 2014, 26, 4046–4056 CrossRef CAS.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, K. Appavoo, M. L. Steigerwald, L. M. Campos and M. Y. Sfeir, J. Am. Chem. Soc., 2016, 138, 7289–7297 CrossRef CAS PubMed.
- E. C. Rudiger, M. Muller, S. Koser, F. Rominger, J. Freudenberg and U. H. F. Bunz, Chemistry, 2018, 24, 1036–1040 CrossRef PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS PubMed.
- V. Lami, D. Leibold, P. Fassl, Y. J. Hofstetter, D. Becker-Koch, P. Biegger, F. Paulus, P. E. Hopkinson, M. Adams, U. H. F. Bunz, S. Huettner, I. Howard, A. A. Bakulin and Y. Vaynzof, Sol. RRL, 2017, 1, 1700053 CrossRef.
- S. Zhou, C. An, T. Stelzig, S. R. Puniredd, X. Guo, W. Pisula and M. Baumgarten, New J. Chem., 2015, 39, 6765–6770 RSC.
- P. Biegger, O. Tverskoy, F. Rominger and U. H. Bunz, Chemistry, 2016, 22, 16315–16322 CrossRef CAS PubMed.
- P. Biegger, M. Schaffroth, O. Tverskoy, F. Rominger and U. H. Bunz, Chemistry, 2016, 22, 15896–15901 CrossRef CAS PubMed.
- M. Muller, H. Reiss, O. Tverskoy, F. Rominger, J. Freudenberg and U. H. F. Bunz, Chemistry, 2018, 24, 12801–12805 CrossRef PubMed.
- S. Yang, B. Shan, X. Xu and Q. Miao, Chemistry, 2016, 22, 6637–6642 CrossRef CAS PubMed.
- M. Ganschow, S. Koser, S. Hahn, F. Rominger, J. Freudenberg and U. H. Bunz, Chemistry, 2017, 23, 4415–4421 CrossRef CAS PubMed.
- J. I. Martinez, J. P. Mora-Fuentes, M. Carini, A. Saeki, M. Melle-Franco and A. Mateo-Alonso, Org. Lett., 2020, 22, 4737–4741 CrossRef CAS PubMed.
- H. Reiss, L. Ji, J. Han, S. Koser, O. Tverskoy, J. Freudenberg, F. Hinkel, M. Moos, A. Friedrich, I. Krummenacher, C. Lambert, H. Braunschweig, A. Dreuw, T. B. Marder and U. H. F. Bunz, Angew. Chem., Int. Ed., 2018, 57, 9543–9547 CrossRef CAS PubMed.
- X. Xu, Y. Yao, B. Shan, X. Gu, D. Liu, J. Liu, J. Xu, N. Zhao, W. Hu and Q. Miao, Adv. Mater., 2016, 28, 5276–5283 CrossRef CAS PubMed.
- D. Cortizo-Lacalle, C. Gozalvez, M. Melle-Franco and A. Mateo-Alonso, Nanoscale, 2018, 10, 11297–11301 RSC.
- P.-Y. Gu, Z. Wang, G. Liu, H. Yao, Z. Wang, Y. Li, J. Zhu, S. Li and Q. Zhang, Chem. Mater., 2017, 29, 4172–4175 CrossRef CAS.
- B. Kohl, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2015, 54, 6051–6056 CrossRef CAS PubMed.
- B.-L. Hu, K. Zhang, C. An, D. Schollmeyer, W. Pisula and M. Baumgarten, Angew. Chem., Int. Ed., 2018, 57, 12375–12379 CrossRef CAS PubMed.
- Z. Wang, P. Gu, G. Liu, H. Yao, Y. Wu, Y. Li, G. Rakesh, J. Zhu, H. Fu and Q. Zhang, Chem. Commun., 2017, 53, 7772–7775 RSC.
- C. An, S. Zhou and M. Baumgarten, Cryst. Growth Des., 2015, 15, 1934–1938 CrossRef CAS.
- C. An, X. Guo and M. Baumgarten, Cryst. Growth Des., 2015, 15, 5240–5245 CrossRef CAS.
- P. Biegger, M. Schaffroth, K. Brödner, O. Tverskoy, F. Rominger and U. H. F. Bunz, Chem. Commun., 2015, 51, 14844–14847 RSC.
- S. Miao, A. L. Appleton, N. Berger, S. Barlow, S. R. Marder, K. I. Hardcastle and U. H. F. Bunz, Chem. - Eur J., 2009, 15, 4990–4993 CrossRef CAS PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed.
- B. D. Lindner, J. U. Engelhart, O. Tverskoy, A. L. Appleton, F. Rominger, A. Peters, H.-J. Himmel and U. H. F. Bunz, Angew. Chem., Int. Ed., 2011, 50, 8588–8591 CrossRef CAS PubMed.
- M. L. Tang, A. D. Reichardt, N. Miyaki, R. M. Stoltenberg and Z. Bao, J. Am. Chem. Soc., 2008, 130, 6064–6065 CrossRef CAS PubMed.
- M. L. Tang, A. D. Reichardt, P. Wei and Z. Bao, J. Am. Chem. Soc., 2009, 131, 5264–5273 CrossRef CAS.
- B.-L. Hu, C. An, M. Wagner, G. Ivanova, A. Ivanova and M. Baumgarten, J. Am. Chem. Soc., 2019, 141, 5130–5134 CrossRef CAS PubMed.
- C. Wang, J. Zhang, G. Long, N. Aratani, H. Yamada, Y. Zhao and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 6292–6296 CrossRef CAS PubMed.
- Q. Zhang, W. Chen, X. Li, G. Long, W. Gao, Y. Li, R. Ganguly, M. Zhang, N. Aratani, H. Yamada and M. Liu, Angew. Chem., 2018, 130, 13743–13747 CrossRef.
- R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, J. Am. Chem. Soc., 2021, 143, 6593–6600 CrossRef CAS PubMed.
- G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, D. Ma and Z. Y. Wang, J. Phys. Chem. C, 2009, 113, 1589–1595 CrossRef CAS.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- S. Miao, A. L. Appleton, N. Berger, S. Barlow, S. R. Marder, K. I. Hardcastle and U. H. F. Bunz, Chem. - Eur J., 2009, 15, 4990–4993 CrossRef CAS PubMed.
- Z. X. Liang, Q. Tang, R. X. Mao, D. Q. Liu, J. B. Xu and Q. Miao, Adv. Mater., 2011, 23, 5514–5518 CrossRef CAS PubMed.
- Z. L. Wang, J. W. Miao, G. K. Long, P. Y. Gu, J. B. Li, N. Aratani, H. Yamada, B. Liu and Q. C. Zhang, Chem.–Asian J., 2016, 11, 482–485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2150585, 2150586, 2150828. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2ra01074g |
| This journal is © The Royal Society of Chemistry 2022 |