
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemplated synthesis of microparticles with carbonaceous skeletal structures using polymer cubosomes as templates†
Jeongeun Song,
Subin Choi,
Jongwoo Lim * and
Kyoung Taek Kim*
* and
Kyoung Taek Kim*
Department of Chemistry, Seoul National University, Seoul 08826, Korea. E-mail: ktkim72@snu.ac.kr; jwlim@snu.ac.kr
First published on 16th March 2022
Abstract
Polymer cubosomes (PCs) are bicontinuous mesoporous colloidal particles that feature high surface areas and an extremely ordered crystalline pore network. PCs have attracted tremendous attention because of their potential applications in many fields. Herein, we obtained new microparticles with carbonaceous reticulated networks via templated synthesis using PCs as templates. The water-channel networks of the PCs were translated into a carbonaceous skeletal cubic structure. Carbon precursors were polymerized inside the water-channel networks of the PCs under acidic conditions without collapsing the internal crystalline mesophases. The carbonaceous interconnected networks created by the templated synthesis exhibited cubic crystalline skeletal networks similar to those of the PCs. These cubic-ordered mesoporous carbon (cOMC) microparticles exhibited several properties in electrochemical experiments. In addition, the nanoscopic structures and surfaces of these microparticles sustain electrochemically perturbing environments, and thus retain more than 90% capacitance after 1000 charge–discharge cycles.
Introduction
Ordered mesoporous carbon (OMC) materials with three-dimensionally (3D) connected pore networks have attracted increasing interest in fields such as adsorbents,1–3 separation,4,5 drug delivery,6,7 and energy storage devices8–10 due to their chemical and physical properties, such as electrical conductivity, superior chemical and mechanical stability, and high surface area, diffusivity, and biocompatibility. The diffusivity of OMCs can be significantly affected by the lattice symmetry and size of internal reticulated pore networks. The lattice symmetry of the internal porous networks of the OMCs can be inherited via templated synthesis by using ordered mesoporous structures, such as mesoporous silica,11–13 anodic alumina,14,15 and zeolites,16,17 as hard templates. The carbonization of the carbonaceous precursors contained in these hard templates yielded OMCs as a negative-tone replica of the template. However, the desired OMCs can only be obtained after the removal of the hard templates under harsh chemical conditions. In contrast, ordered structures of the carbon precursors can be obtained in situ by the incorporation of the precursors in the coronal domain of the self-assembled structures of amphiphiles or block copolymers.18–21 This soft-templating strategy allows the direct creation of OMCs by the calcination of the self-assembled structures without requiring a selective etching of the templates.PCs are colloidal microparticles comprising the triply periodic minimal surfaces (TPMSs) of block copolymer bilayers, which internalize two non-interpenetrating water channel networks arranged in a cubic symmetry.22,23 The pore size and symmetry of the cubic pore networks of the PCs can be controlled by adjusting the structural parameters of their block copolymer building blocks, such as a molecular weight and block ratio (a weight fraction of the hydrophilic polymer block).24–27 These structural characteristics make PCs promising materials for nanoreactors,22,28 storage vehicles for large biomolecules,29 and photonic crystals.30 PCs can potentially serve as unique templates for the synthesis of the microparticles with carbonaceous skeletal structures because they internalize the crystalline pore networks. In addition, PCs can accommodate a variety of chemistries required for the polymerization and cross-linking of precursor molecules for carbonization within the template. In contrast to conventional hard templates approaches, PCs can be readily removed under mild chemical conditions without requiring strong acids or high temperatures. Previously, PCs have served as ideal templates for creating periodic nanostructures of inorganic oxides,28,30 transition metals,31 and metal-phenolic networks29 without requiring template removal under harsh chemical conditions.
In this study, we report the synthesis of the cubic-ordered mesoporous carbon (cOMC) microparticles composed of single cubic networks of nanoskeletal carbons using PCs as easily removable hard templates. A mixture of water and furfuryl alcohol, a hydrophilic monomer (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 v/v), was used as the carbonaceous precursor.32–35 This carbonaceous mixture could be polymerized and cross-linked in the presence of an acidic vapor at ambient temperature.36,37 The cross-linked carbonaceous materials contained in the PC templates were fully cured and carbonized under high temperatures (>900 °C), during which the PC templates were removed. Two types of cOMC microparticles with different pore and lattice sizes were successfully synthesized. The structural characteristics of these cOMC microparticles indicated that the internal structures of the PC templates were faithfully replicated as single skeletal cubic networks, suggesting the importance of the selective infiltration of the precursors in the open channel network of the PC template. In particular, the cOMC microparticles with relatively large pore sizes and surface areas exhibited single cubic structures. The cOMC microparticles synthesized by templated synthesis using PC templates showed electrochemical capacitive properties and a long-term stability of the capacitance retention after 1000 cycles of charge–discharge processes (Fig. 1).
:8 v/v), was used as the carbonaceous precursor.32–35 This carbonaceous mixture could be polymerized and cross-linked in the presence of an acidic vapor at ambient temperature.36,37 The cross-linked carbonaceous materials contained in the PC templates were fully cured and carbonized under high temperatures (>900 °C), during which the PC templates were removed. Two types of cOMC microparticles with different pore and lattice sizes were successfully synthesized. The structural characteristics of these cOMC microparticles indicated that the internal structures of the PC templates were faithfully replicated as single skeletal cubic networks, suggesting the importance of the selective infiltration of the precursors in the open channel network of the PC template. In particular, the cOMC microparticles with relatively large pore sizes and surface areas exhibited single cubic structures. The cOMC microparticles synthesized by templated synthesis using PC templates showed electrochemical capacitive properties and a long-term stability of the capacitance retention after 1000 cycles of charge–discharge processes (Fig. 1).
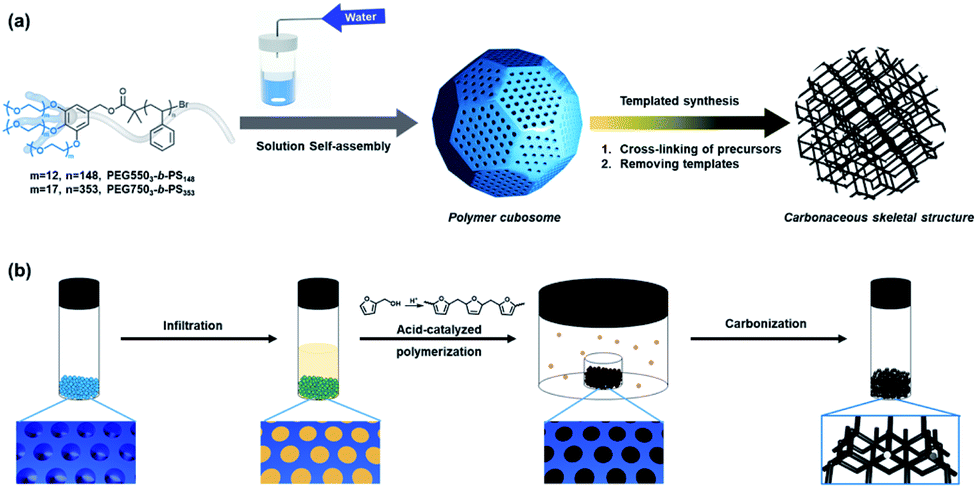 | ||
| Fig. 1 (a) Schematic representation of polymer cubosomes (PCs) formed by solution self-assembly of block copolymers and the cubic-ordered mesoporous carbon (cOMC) microparticles prepared by templated synthesis using PCs as templates. (b) The carbon precursors were infiltrated and polymerized by acid vapor inside water-channel networks of PCs. Following carbonization, cOMC microparticles were created via removing PCs. | ||
Results and discussion
Synthesis of polymer cubosomes
Block copolymers (BCPs) self-assemble into inverse mesophase structures when the block ratio (r) of the BCP is less than 10%, which warrants the critical packing parameter of the BCP to be greater than unity.23,38 We synthesized two branched-linear diblock copolymers by the atom-transfer radical polymerization (ATRP) of styrene with tri-arm poly(ethylene glycol) (PEG) macroinitiators, PEG5503 (Mn (PEG) = 1650 g mol−1) and PEG7503 (Mn (PEG) = 2250 g mol−1) (Fig. S1†). Two branched-linear BCPs, PEG5503-b-PS148 (Mn = 19000 g mol−1, Đ = 1.10, r = 9.5% (Fig. S2†)) and PEG7503-b-PS353 (Mn = 44600 g mol−1, Đ = 1.16, r = 5.4% (Fig. S3†)), were specifically synthesized to form PCs with Schwarz D surface internal structures. There branched-linear BCPs were self-assembled in the solution to form PCs with different lattice parameters and pore sizes by a co-solvent method using 1,4-dioxane and water as the common solvent and the selective solvent, respectively. The organic solvents were removed by dialysis against water. Subsequently, the PCs of the branched-linear BCPs were obtained as microparticles suspended in water. The scanning electron microscopy (SEM) analysis of these microparticles confirmed the formation of PCs with regularly arranged pores on the surface of the dried microparticles (Fig. 2c and i). Transmission electron microscopy (TEM) images of the PCs showed the presence of highly ordered porous networks within the PCs. The average diameters (d) of PEG5503-b-PS148 (PC-550) and of PEG7503-b-PS353 (PC-750) were found to be 22 ± 3 μm and 3 ± 0.5 μm, respectively. These diameters were determined using low magnification SEM images (Fig. 2b and h).
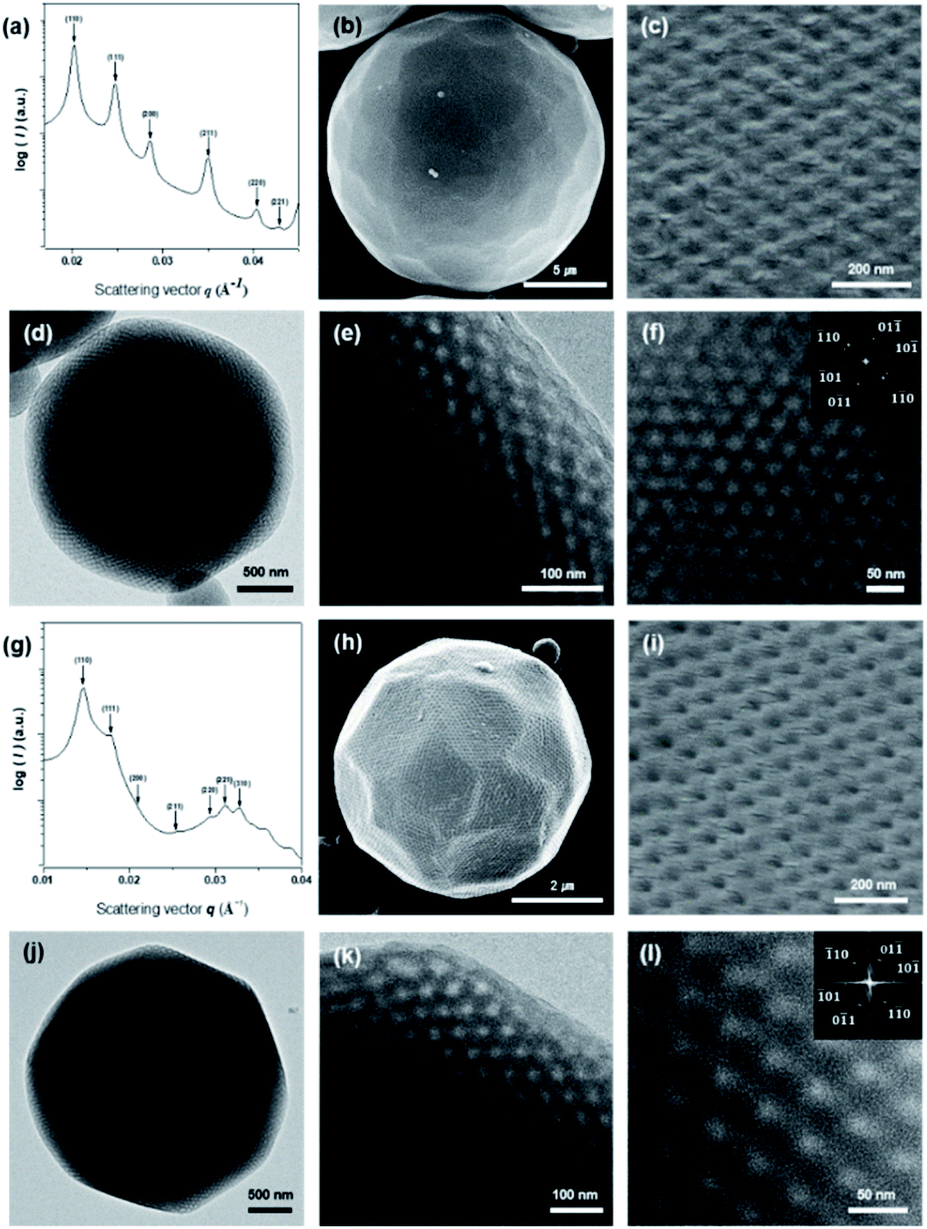 | ||
Fig. 2 Structural representation of PCs. (a and g) SAXS data of the dried PCs, PC-550 and PC-750, respectively. Both were assigned to the Pn![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and lattice constant (a) of 43.9 and 60.6 nm, respectively. (b and h) Low-resolution SEM images showing PC-550 (b) and PC-750 (h) with the spherical morphology and the average diameter (d) of 22 μm and 3 μm, respectively. (c and i) Magnified views of the perforated surface layer of PC-550 (c) and PC-750 (i) viewed along the [111] direction. (d and j) Low-magnification TEM image of PC-550 and PC-750, respectively. (e and f) TEM images showing the internal water channel network of PC-550. The magnified views of the internal network and the FFT (inset) show a Pnm lattice viewed along the [111] direction. (k and l) TEM images showing the internal network of PC-750. The magnified view of the internal network and the FFT (inset) showing a Pnm lattice viewed along the [111] direction. m space group and lattice constant (a) of 43.9 and 60.6 nm, respectively. (b and h) Low-resolution SEM images showing PC-550 (b) and PC-750 (h) with the spherical morphology and the average diameter (d) of 22 μm and 3 μm, respectively. (c and i) Magnified views of the perforated surface layer of PC-550 (c) and PC-750 (i) viewed along the [111] direction. (d and j) Low-magnification TEM image of PC-550 and PC-750, respectively. (e and f) TEM images showing the internal water channel network of PC-550. The magnified views of the internal network and the FFT (inset) show a Pnm lattice viewed along the [111] direction. (k and l) TEM images showing the internal network of PC-750. The magnified view of the internal network and the FFT (inset) showing a Pnm lattice viewed along the [111] direction. | ||
The internal bicontinuous structures of the PCs were studied by small-angle X-ray scattering (SAXS). The SAXS results of PC-550 indicated the presence of a well-defined inverse bicontinuous cubic phase, which can be assigned to the double diamond lattice (Schwarz D surface, Pnm space group) with a lattice parameter (a) of 43.9 nm (Fig. 2a). The SAXS result of PC-750 was assigned to the Schwarz D surface with a larger lattice dimension (Pnm space group, a = 60.6 nm) (Fig. 2g).
Synthesis of cOMC microparticles with PC templates
Composed of TPMSs of BCP bilayers, PCs internalize two non-intersecting water channel networks that are exposed to the surrounding through the surface pores. Particularly, the surface and internal pore sizes of the PCs are proportional to the overall molecular weight of the BCP building blocks. In this study, we used two PC templates, PC-550 and PC-750. These PC templates exhibited different pore sizes and lattice parameters due to the molecular weight differences of the BCP used for self-assembly. These PCs were collected from the aqueous suspension by centrifugation (14000 rpm for min), followed by drying of water at room temperature.
The structural integrity of the bicontinuous cubic structures of PC-550 and PC-750 could be compromised in the presence of small organic molecules used as a carbon source because of the dissolution of non-covalently held BCP bilayers. Moreover, the thermal polymerization of carbon precursors at an elevated temperature, exceeding the glass transition temperature (Tg) of polystyrene blocks (∼100 °C), could be detrimental to the structural integrity of the PCs. Therefore, we investigated a new cross-linking reaction that was compatible with the PC template for the polymerization and cross-linking of monomers without disrupting the structural order of the internal cubic nanochannel networks. We adopted furfuryl alcohol (FA) as a carbon source for the templated synthesis of the microparticles with carbonaceous skeletal structures because of its facile polymerization in the presence of acidic catalysts at room temperature. Because of the partial solubility of PS in neat FA at room temperature, the ratio of the water–FA mixture (1:4 v/v) was optimized for polymerization in the internal channel networks of PC templates at room temperature. The PC templates immersed in the water–FA mixture were collected by centrifugation and placed in the humidity chamber saturated with concentrated HCl. The acid-catalyzed polymerization of FA was conducted for 12 h, during which the progress of the polymerization in the PC template was monitored by the appearance of a dark hue. After the completion of FA polymerization, the brown colored PCs were heated at different temperatures ranging from 450 to 900 °C under N2 atmosphere for the cross-linking and carbonization of the polymerized FA. During the carbonization of FA at 900 °C, the PC templates were completely removed and resulted in the formation of cOMC microparticles. The carbonization of FA in the PC templates resulted in a ∼90% weight loss during TGA analysis, suggesting the loss of oxygen atoms from the cOMC particles and the removal of the PC templates (Fig. S4†).
Characterization of cOMC microparticles
We investigated the morphology and structural characteristics of cOMC-550 and cOMC-750 microparticles synthesized using the corresponding PCs as templates. The SEM and TEM images of cOMC-550 and cOMC-750 showed a spherical morphology, indicating the high fidelity of the synthesis of carbonaceous microparticles using the PC templates (Fig. 3b, e and 4b, e). The SEM images of these microparticles clearly showed the negative-tone replication of the PC template, which corroborated that the FA precursors were introduced in the internal channel networks through the surface pores of the templates (Fig. 3c and f). The presence of cubic crystalline skeletal networks also indicated that the polymerization of FA in the nanochannel network in a PC template stabilized the precursors, which was required to maintain the structural order during the carbonization at high temperature. The long-range internal order of cOMC microparticles was also evidenced by TEM images, indicating that all the PC templates were successfully replicated by the infiltration and carbonization of FA (Fig. 4c and f). The analysis of low magnification SEM images confirmed that the average diameter of the cOMC microparticles was reduced (14 ± 2 μm for cOMC-550 and 2 ± 0.6 μm for cOMC-750) due to the shrinkage of the cubic network structures during carbonization (Fig. 3a and d). | ||
| Fig. 3 Representative SEM images of cOMC microparticles. (a–c) Low-resolution SEM image showing the spherical morphology of cOMC-550 (d = 14 μm). Magnified views of the carbon skeletal network in the surface and the internal carbonaceous cubic structure of the fractured cOMC-550. High resolution SEM images showing the internal double diamond (Pnm) lattice of cOMC-550. (d–f) Low-resolution SEM image showing the spherical morphology of cOMC-750 (d = 2 μm). Magnified views of the carbonaceous network in the surface and the internal carbonaceous cubic mesophase of the fractured cOMC-750. High resolution SEM images showing the internal single diamond (Fdm) lattice of cOMC-550. | ||
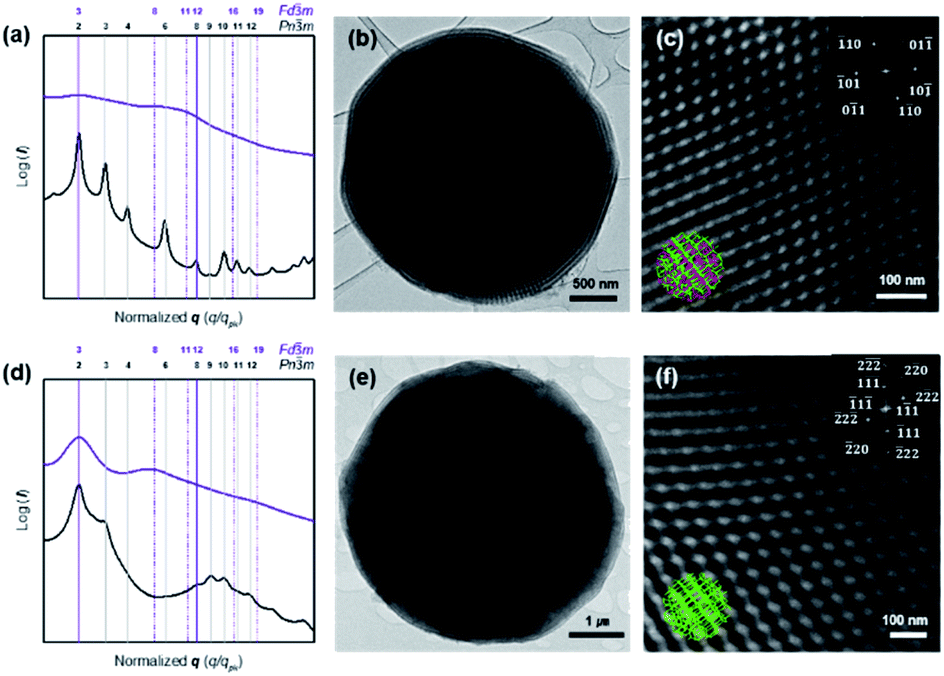 | ||
| Fig. 4 Structural analysis of carbonaceous microparticles with 3D-defined internal skeletal structures by SAXS and TEM. (a and d) SAXS results of the cOMC microparticles (violet lines) synthesized from the PCs (black lines). The symmetry transition from the corresponding double networks of the PC templates to double diamond (Pnm, a = 51.8 nm) (a) and single diamond (Fdm, a = 92.8 nm) (d). Vertical lines correspond to the expected Bragg peak positional ratios (violet; common peaks, dashed violet; Fdm only, black; PCs only). (b and c) TEM images showing the internal carbon skeletal network of cOMC-550. Magnified image of the internal network and the FFT (inset) show a Pnm lattice viewed along the [111] direction. (e and f) TEM images showing the internal skeletal network of cOMC-750. Magnified image of the internal carbonaceous cubic structure and the FFT (inset) show a Fdm lattice viewed along the [110] direction. | ||
PCs internalize two non-intersecting water channel networks. At the interface, only one of the two embedded channel networks is exposed to the surroundings in order to maintain the continuity of the TPMSs at the outermost layer of the PC. This topological feature at the interface of the PC allows the creation of single cubic networks by templated synthesis of silica, titania, and transition metal nanostructures.30,31 Therefore, we expected that the carbonaceous microparticles reported herein could be composed of the single diamond networks of nanoskeletal carbons. However, the SAXS results of the cOMC-550 were assigned to double diamond lattices (Pnm space group) with the lattice parameter of 51.8 nm (Fig. 4a). The lattice symmetry of cOMC-750 was determined to be a single diamond network (Fdm space group) with a lattice parameter of 92.8 nm by SAXS analysis (Fig. 4b). The SAXS analysis suggested that the interfacial topology of PC-550 could be compromised by the presence of FA, which could dissolve the lower molecular weight PEG5503-b-PS148 building blocks to form the BCP bilayers. In contrast, PC-750 composed of the higher molecular weight PEG7503-b-PS353 maintained the topological features at the interface, which allowed the selective infiltration of the FA–water mixture into the open channel networks of PC-750. The nature of the lattices and their dimensions were also supported by the SEM and TEM images. The lattice dimension estimated from the SEM and TEM images of the skeletal carbonaceous structure of cOMC-550 was 73 ± 2 nm, which corroborated the lattice parameter of the double diamond network determined by SAXS (Fig. 3c and 4c). The SEM and TEM images of the internal skeletal structures of cOMC-750 exhibited single diamond networks with the lattice parameter of 133 ± 1 nm, which coincided with the SAXS analysis (Fig. 3e and 4e). Cross-sectional TEM images of cOMC-550 and cOMC-750 confirmed the presence of double diamond and single diamond network structures, respectively (Fig. S5†). Considering the theoretical value of the single diamond lattice originating from the double diamond lattice of PC-750, the lattice parameter of cOMC-750 was reduced by 23%, indicating the shrinkage of the carbonaceous skeletal structures during the carbonization at high temperature.
The porosities and surface areas of the microparticles with carbonaceous nanoskeletal networks were measured by N2 adsorption–desorption experiments at 77 K. In the case of cOMC-550, the Brunauer–Emmett–Teller (BET) isotherms showed a surface area of 276 m2 g−1 and a total pore volume of 0.41 cm3 g−1. The Barrett–Joyner–Halenda (BJH) pore-size distribution analysis displayed a peak pore diameter of 10 nm (Fig. 5a and b). Moreover, cOMC-750 exhibited a BET surface area of 455 m2 g−1, pore volume of 0.92 cm3 g−1, and a peak pore diameter of 18.3 nm, which was derived from the BJH curve (Fig. 5c and d). Compared with those of the PC templates,24 the BET surface area of cOMC microparticles was dramatically increased and the peak pore diameter was diminished by ∼50%. Additionally, NLDFT pore size distribution of cOMC microparticles was measured (Fig. S6†).
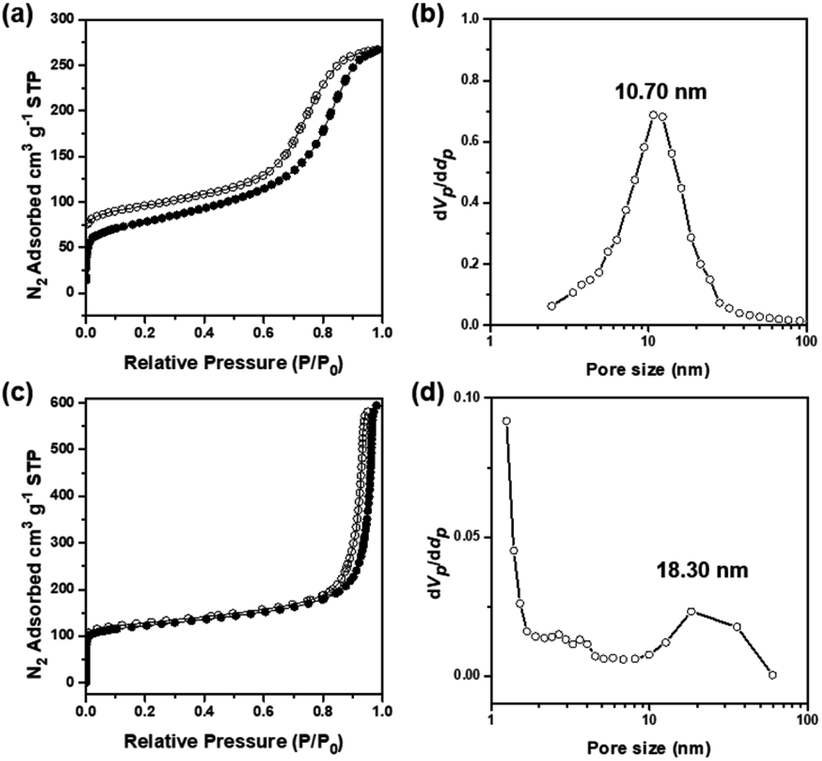 | ||
| Fig. 5 The porosity and surface area of cOMC microparticles. (a) N2 adsorption–desorption isotherms of cOMC-550 measured at 77 K (BET surface area 276 m2 g−1, pore volume 0.41 cm3 g−1). (b) The BJH pore size distribution of cOMC-550 peaked at 10.7 nm. (c) N2 adsorption–desorption isotherms of cOMC-750 measured at 77 K (BET surface area 455 m2 g−1, pore volume 0.92 cm3 g−1). (d) The BJH pore size distribution of cOMC-750 peaked at 18.3 nm. STP, standard temperature and pressure. | ||
As shown in Fig. 6a, Raman spectra of cOMC-550 and cOMC-750 revealed two distinct peaks at 1350 (D-band) and 1580 cm−1 (G-band). The D-band was induced by the defects in the sp2 carbon systems at graphite defects, and the G-band was attributed to the C–C bond stretching of the graphite sp2 hybridized carbon atoms. This result demonstrated that graphitic sp2 carbons existed in the cOMC microparticles despite the defects of disordered carbon atoms. To investigate the crystallinity of the microstructure of carbonaceous skeletal structures, X-ray diffraction (XRD) analysis of cOMC-550 and cOMC-750 was performed, as shown in Fig. 6b. The XRD peaks of 25.9° and 42.9° were designated to (002) and (100) peaks, demonstrating an interlayer stacking of the aromatic systems in the cOMC microparticles and a π-stacked in-plane graphitic structure. The two broad peaks indicated the defects in the sp2 carbon system of the cOMC microparticles. Furthermore, Field-Emission TEM (FE-TEM) images revealed the existence of ordered graphitic carbon in cOMC microparticles (Fig. S7†). The d-spacing from the lattice fringes was approximately 3.64 Å and 4.45 Å at thin-edge sites of cOMC-550 and cOMC-750, respectively. The d-spacing indicates the distance between the two nearby (002) planes in the crystalline carbon structure. These obtained values agreed with the XRD results. However, the lattice fringes in the FE-TEM image were unclear because the crystalline carbon systems were embedded in the disordered carbon structures. This observation corresponded with the results of Raman spectra and XRD patterns, which implied a low degree of graphitization during the templated synthesis of cOMC microparticles.
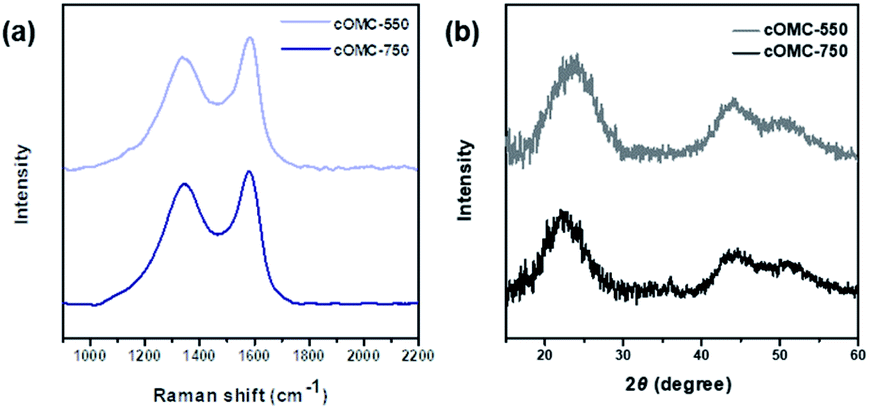 | ||
| Fig. 6 Characterization of the microstructure of the carbonaceous skeletal network of cOMC microparticles. (a) Raman spectra of cOMC-550 and cOMC-750. (b) XRD patterns of cOMC-550 and cOMC-750. | ||
Electrochemical characterization of cOMC microparticles
Electrochemical properties of cOMC microparticles were investigated in a three electrode configuration using 6 M KOH as an aqueous electrolyte. For the measurements, the working electrodes were prepared by mixing the cOMC microparticles, activated carbon, and Nafion® binder and loading onto a Ni foam.Electrochemical impedance spectroscopy (EIS) analyzes the ion and charge transport kinetics of cOMC microparticles. Nyquist plots of the cOMC microparticles show incomplete semicircles in high-frequency regions, followed by steep lines in the low-frequency region (Fig. 7c). In addition, the small x-intercept and small radius of the semi-circle corresponds to the low internal resistance and low charge transfer resistance. cOMC-750 exhibited steeper slopes in the low-frequency region than cOMC-550, indicating improved electrolytic ion diffusion. The cyclic voltammogram (CV) curves of cOMC-750 showed higher capacitor characteristics than that of cOMC-550 (Fig. 7a). cOMC microparticles exhibited nearly quasi-rectangular shapes across the scan rates of 5–100 mV s−1 in the CV profiles (Fig. S8†). The capacitive properties of cOMC microparticles linearly increased with increasing scan rates. The gravimetric capacitive performance of the cOMC microparticles was evaluated through the galvanostatic charge–discharge (GCD) measurement. The GCD curves of cOMC microparticles recorded at different current densities were almost symmetric in the wide potential window of −0.9 to 0.25 V versus Hg/HgO (Fig. S9†). Gravimetric capacitances calculated based on GCD at different current densities (Fig. 7b) were 93, 76, 65, and 56 F g−1 at the current densities of 0.5, 1, 2, and 5 A g−1, respectively, for cOMC-750. The gravimetric capacitance of cOMC-750 is higher than that of cOMC-550. Furthermore, the cycling tests were conducted at 0.5 A g−1 to assess the long-term stability of cOMC-750 (Fig. 7d) and it retained more than 90% of the initial capacitance after 1000 galvanostatic charge–discharge cycles.
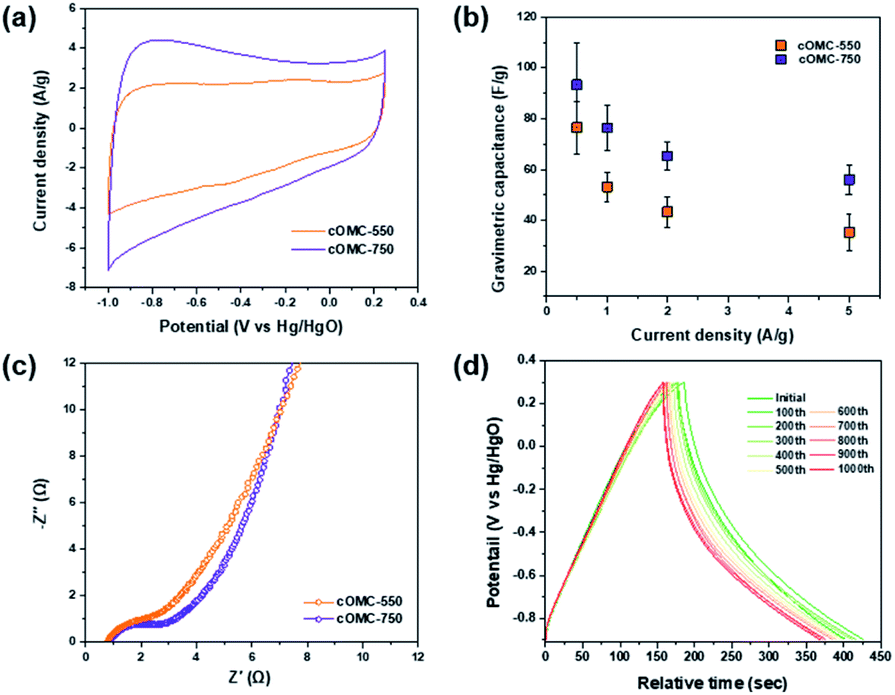 | ||
| Fig. 7 Electrochemical characterization of cOMC microparticles. (a) CV curves at a scan rate of 50 mV s−1. (b) Gravimetric capacitance versus current density ranging from 0.5 to 5 A g−1 of cOMC microparticles. (c) Nyquist plots collected at open circuit potentials with 10 mV perturbation and a frequency range from 100 kHz to 0.5 Hz. (d) Cycling stability of cOMC-750 over 1000 times charge–discharge processes at current density of 0.5 A g−1. | ||
Conclusions
In summary, we synthesized the cOMC microparticles, cOMC-550 and cOMC-750, via templated synthesis using PC-550 and PC-750 as the templates, respectively. Here, new mesoporous polymeric materials having two non-intersecting water-channel networks were used as templates. The hard-templating strategy of cOMC microparticles using PCs as templates was reported for the first time. FA was used as the carbon precursor; it was polymerized under acidic conditions inside the water channel network of the PC templates. The resulting cOMC-550 and cOMC-750 exhibited 3D crystalline skeletal structures inherited from the PCs and large BET surface areas of 276 m2 g−1 and 455 m2 g−1, respectively. cOMC microparticles showed capacitive performances in the electrochemical experiments. cOMC-750 exhibited relatively good capacitance, low resistance, and long-term durability for energy storage compared with cOMC-550. More than 90% of the initial capacitance of cOMC-750 was retained after 1000 galvanostatic charge–discharge cycles. This study suggests a new strategy for the synthesis of the microparticles with the carbonaceous interconnected networks possessing electrochemical properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Hoi Ri Moon, Hong Kyu Lee, and Sungeun Jeoung for measuring and analyzing the porosity of cOMC microparticles at Ulsan National Institute of Science and Technology (UNIST). This work was supported by National Research Foundation (NRF) of Korea (NRF-2019R1A2C3007541 and NRF-2020M3D1A1110504). K. T. K thanks Seoul National University (SNU) for the support by Creative-Pioneering Researchers Program (305-20210035).Notes and references
- A. Vinu, C. Streb, V. Murugesan and M. Hartmann, J. Phys. Chem. B, 2003, 107, 8297–8299 CrossRef CAS.
- A. Vinu, M. Miyahara and K. Ariga, J. Phys. Chem. B, 2005, 109, 6436–6441 CrossRef CAS PubMed.
- Z. Wu and D. Zhao, Chem. Commun., 2011, 47, 3332–3338 RSC.
- Y. Zhang, S. Xu, Y. Luo, S. Pan, H. Ding and G. Li, J. Mater. Chem., 2011, 21, 3664–3671 RSC.
- G. Wang, G. Gao, S. Yang, Z. Wang, P. Jin and J. Wei, Microporous Mesoporous Mater., 2021, 310, 110623 CrossRef CAS.
- G. Xu, S. Liu, H. Niu, W. Lv and R. Wu, RSC Adv., 2014, 4, 33986–33997 RSC.
- C. Karavasili, E. P. Amanatiadou, L. Sygellou, D. K. Giasafaki, T. A. Steriotis, G. C. Charalambopoulou, I. S. Vizirianakis and D. G. Fatouros, J. Mater. Chem. B, 2013, 1, 3167–3174 RSC.
- L. Miao, Z. Song, D. Zhu, L. Li, L. Gan and M. Liu, Mater. Adv., 2020, 1, 945–966 RSC.
- Y. Zhai, Y. Dou, D. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828–4850 CrossRef CAS PubMed.
- A. Eftekhari and Z. Fan, Mater. Chem. Front., 2017, 1, 1001–1027 RSC.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and T. Osamu, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- M. Kruk, M. Jaroniec, T. W. Kim and R. Ryoo, Chem. Mater., 2003, 15, 2815–2823 CrossRef CAS.
- T. Kyotani, L. F. Tsai and A. Tomita, Chem. Mater., 1995, 7, 1427–1428 CrossRef CAS.
- T. Kyotani, L. F. Tsai and A. Tomita, Chem. Mater., 1996, 8, 2109–2113 CrossRef CAS.
- T. Kyotani, T. Nagai, S. Inoue and A. Tomita, Chem. Mater., 1997, 9, 609–615 CrossRef CAS.
- J. Rodriguez-Mirasol, T. Cordero, L. R. Radovic and J. J. Rodriguez, Chem. Mater., 1998, 10, 550–558 CrossRef CAS.
- C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128(16), 5316–5317 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- T.-Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- W. C. Chu, B. P. Bastakoti, Y. V. Kaneti, J. G. Li, H. R. Alamri, Z. A. Alothman, Y. Yamauchi and S. W. Kuo, Chem.–Eur. J., 2017, 23, 13734–13741 CrossRef CAS PubMed.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed.
- S. Ha, Y. La and K. T. Kim, Acc. Chem. Res., 2020, 53, 620–631 CrossRef CAS PubMed.
- T. H. An, Y. La, A. Cho, M. G. Jeong, T. J. Shin, C. Park and K. T. Kim, ACS Nano, 2015, 9, 3084–3096 CrossRef CAS PubMed.
- A. Cho, Y. La, T. J. Shin, C. Park and K. T. Kim, Macromolecules, 2016, 49, 4510–4519 CrossRef CAS.
- J. Kim, M. Yoon, S.-M. Jin, J. Lee, Y. La, E. Lee and K. T. Kim, Polym. Chem., 2019, 10, 3778–3785 RSC.
- Y. Sun, J. Kim and K. T. Kim, RSC Adv., 2019, 9, 25423–25428 RSC.
- C. Park, Y. La, T. H. An, H. Y. Jeong, S. Kang, S. H. Joo, H. Ahn, T. J. Shin and K. T. Kim, Nat. Commun., 2015, 6, 6392 CrossRef CAS PubMed.
- Z. Lin, J. Zhou, C. Cortez-Jugo, Y. Han, Y. Ma, S. Pan, E. Hanssen, J. J. Richardson and F. Caruso, J. Am. Chem. Soc., 2020, 142, 335–341 CrossRef CAS PubMed.
- Y. La, J. Song, M. G. Jeong, A. Cho, S. M. Jin, E. Lee and K. T. Kim, Nat. Commun., 2018, 9, 5327 CrossRef CAS PubMed.
- H. Lee, D. Kim, H. Ma and K. T. Kim, Chem. Commun., 2020, 56, 14059–14062 RSC.
- P. Niebrzydowska, R. Janus, P. Kuśtrowski, S. Jarczewski, A. Wach, A. M. Silvestre-Albero and F. Rodríguez-Reinoso, Carbon, 2013, 64, 252–261 CrossRef CAS.
- K. Nanaj, A. Jyothirmayi, U. Varadaraju, T. N. Rao and S. Anandan, J. Alloys Compd., 2017, 723, 488–497 CrossRef.
- S. Inagaki, T. Nakao, T. Miki, N. Kuroda and Y. Kubota, Microporous Mesoporous Mater., 2017, 241, 123–131 CrossRef CAS.
- H. Wang and J. Yao, Ind. Eng. Chem. Res., 2006, 45, 6393–6404 CrossRef CAS.
- T. Kim, R. S. Assary, C. L. Marshall, D. J. Gosztola, L. A. Curtiss and P. C. Stair, ChemCatChem, 2011, 3, 1451–1458 CrossRef CAS.
- M. Choura, N. M. Belgacem and A. Gandini, Macromolecules, 1996, 29, 3839–3850 CrossRef CAS.
- Y. La, T. H. An, T. J. Shin, C. Park and K. T. Kim, Angew. Chem., Int. Ed., 2015, 54, 10483–10487 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00832g |
| This journal is © The Royal Society of Chemistry 2022 |