
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSteered polymorphic nanodomains in TiO2 to boost visible-light photocatalytic oxidation†
Zeju Zhang‡
a,
Mang Niu‡a,
Wei Lia,
Chenfeng Ding*be,
Peitao Xiea,
Yongxin Li a,
Lili Chena,
Xiaopeng Lana,
Chunlei Liua,
Xiaodong Yanc,
Xuewei Fud,
Yaochun Liue,
Yuan Liu*ae,
Dapeng Caof,
Jingjie Daig,
Xiaofen Hongh and
Chunzhao Liu*a
a,
Lili Chena,
Xiaopeng Lana,
Chunlei Liua,
Xiaodong Yanc,
Xuewei Fud,
Yaochun Liue,
Yuan Liu*ae,
Dapeng Caof,
Jingjie Daig,
Xiaofen Hongh and
Chunzhao Liu*a
aState Key Laboratory of Bio-fibers and Eco-textiles, Institute of Biochemical Engineering, The Affiliated Qingdao Central Hospital of Qingdao University, College of Materials Science and Engineering, Qingdao University, Qingdao 266071, China. E-mail: liuyuan@qdu.edu.cn; czliu@qdu.edu.cn
bEnergy Materials and Surface Sciences Unit (EMSSU), Okinawa Institute of Science and Technology Graduate University (OIST), 1919-1 Tancha Kunigami-gun, Onna-son, Okinawa 904-0495, Japan. E-mail: chenfeng.ding@oist.jp
cKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, China
dCollege of Polymer Science and Engineering, State Key Laboratory of Polymer Materials Engineering, Sichuan University, Chengdu 610065, Sichuan, China
eFoshan (Southern China) Institute for New Materials, Foshan 528200, China
fBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China
gSchool of Mechanical and Electronic Engineering, Qingdao Binhai University, Qingdao 266555, Shandong, China
hZhejiang Rich Environmental Protection Technology Co., Ltd, Hangzhou 310000, China
First published on 28th March 2022
Abstract
A breakthrough in enhancing visible-light photocatalysis of wide-bandgap semiconductors such as prototypical titania (TiO2) via cocatalyst decoration is still challenged by insufficient heterojunctions and inevitable interfacial transport issues. Herein, we report a novel TiO2-based composite material composed of in situ generated polymorphic nanodomains including carbon nitride (C3N4) and (001)/(101)-faceted anatase nanocrystals. The introduction of ultrafine C3N4 results in the generation of many oxygen vacancies in the TiO2 lattice, and simultaneously induces the exposure and growth of anatase TiO2(001) facets with high surface energy. The photocatalytic performance of C3N4-induced TiO2 for degradation of 2,4-dichlorophenol under visible-light irradiation was tested, its apparent rate being up to 1.49 × 10−2 min−1, almost 3.8 times as high as that for the pure TiO2 nanofibers. More significantly, even under low operation temperature and after a long-term photocatalytic process, the composite still exhibits exceptional degradation efficiency and stability. The normalized degradation efficiency and effective lifespan of the composite photocatalyst are far superior to other reported modified photocatalysts.
1. Introduction
Design of suitable photocatalytic semiconductors is regarded as one of the most promising solutions to effectively utilize the sustainable solar energy to tackle the challenges of energy exhaustion and environmental destruction.1–3 Titania (TiO2) has been proved as the most versatile photocatalyst among semiconductors owing to its cost-effectiveness and high stability,.4 However, TiO2 as a photocatalyst suffers from large band gap (>3 eV) and a lack of suitable electronic/surface structures, resulting in limited visible-light (400–700 nm) absorption ability and charge carrier separation efficiency.5–7 Developing high-performance TiO2 photocatalysts with visible-light response is needed in order to fully utilize and convert the visible solar light.To overcome these bottlenecks, many strategies have been adopted to strengthen visible light absorption and realize effective conversion. The strategies involve doping/co-doping transition metal ions (e.g. copper, vanadium, and iron, etc.)8–10 and/or electronegative anions (e.g. boron, carbon, and nitrogen, etc.) as well as introducing oxygen/nitrogen vacancies11–14 into the lattice of photocatalysts. These approaches effectively narrow the original band gap via forming impurity energy levels or elevating the original valence band of oxygen.15,16 However, these dopants thermodynamically and easily agglomerate and locate at the surface/interface layers: a low density of localized states is introduced into the TiO2 bandgap, resulting in unsatisfactory visible-light absorption and conversion.13 In addition to the above strategies, optimization of high-surface-energy defects and facets of TiO2 such as anatase TiO2(001) represents an effective way for enhancing the photocatalytic activity and efficiency.17–19 Compared with the dominant and thermodynamically stable TiO2(101) facet, the TiO2(001) facet exhibits a high density of unsaturated titanium atoms and surface oxygen atoms for enhanced photocatalytic activity.20–22 However, according to the Wulff construction and surface energy, only 6% (001) facets can be exposed in the stable anatase TiO2.23 More significantly, these methods have limited accommodation of adsorption of low-energy protons and restraint on recombination of electron and holes, leading to the poor visible light photocatalytic performance. Thus, there is a long-sought-after demand for further improving both the absorption and conversion of visible light for TiO2 photocatalysts.
To design advanced photocatalysts should integrate three elements, that is, good photochemical properties, high photocatalytic activity, and excellent long-term stability/capacity.24,25 Designing composite photocatalysts with heterogeneous junctions is an effective approach for tuning the electronic and interface structure,26,27 that are beneficial for the fast charge transfer.28,29 Thus far, manifold heterogenous components such as noble metal,30,31 transition metal oxide/sulfide,32,33 and metal–organic framework materials,34,35 etc. have been intensively studied and demonstrated to show satisfactory improvement of visible light absorption and photochemical activity. As the most promising alternative photocatalyst to metal-based ones, carbon nitride (C3N4) possesses suitable valence energy and conduction band, good chemical/thermal stability, and visible-light harvesting capability.36–38 After C3N4-decoration, the TiO2-based composites exhibit visible-light absorption and enhanced photocatalytic activity.39,40 However, the insufficient heterojunctions and inherent poor interfacial compatibility between C3N4 and TiO2 matrix inevitably lead to energy loss during the light transportation and inhibition of charge migration. Therefore, to overcome these issues, more critical efforts on TiO2-based photocatalysts that can simultaneously realize visible-light harvesting, high photocatalytic activity, and good long-term life span as well as stability are highly needed.
Nanodomains is a nanometer-sized protein found in a natural cell membrane.41 Constructing suitable biomimetic nanodomains can realize the rapid transportation of ions/electrons in composite media.42,43 Similarly, we report a polymorphic TiO2-based composite material formed by in situ diverse nanodomains including C3N4 and (001)/(101)-faceted anatase nanocrystals. The unique composite structure is fabricated by calcinating the nanofibrous composite precursor of thiourea and tetrabutyl titanate in the air. Induced by in situ formation of heterogeneous ultrafine C3N4 crystals, oxygen vacancy occurs in the lattice structure and high-surface-energy anatase TiO2(001) facets are exposed. The resulting composite simultaneously presents excellent photocatalytic activity and a relatively narrow bandgap. As a result, the composite exhibits excellent degradation efficiency of 2,4-dichlorophenol (2,4-DCP) and various organic dyes under visible light with high kinetic rates. Additionally, even under low operation temperature or after a long-term photocatalytic process, the composite still exhibits desirable degradation efficiency and stability.
2. Experimental section
2.1 Materials and chemicals
Tetrabutyl titanate (C16H36O4Ti, CP) was provided by Sinopharm Chemical Reagent Co., China. The sodium hydroxide (KOH, AR), polyvinylpyrrolidone (PVP, Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000, AR), and thiourea (CN2H4S, AR), silver nitrate (AgNO3, AR), benzoquinone (BQ, AR), tertiary butanol (TBA, AR), and ethylene diamine tetraacetic acid (EDTA, AR) were purchased from Aladdin Chemical Co., China. Tertiary butanol (TBA). N,N-dimethylformamide (DMF, AR) and hydrochloric acid (HCl, AR) were purchased from Sinopharm Chemical Reagent Co. (Shanghai), China. 2,4-DCP was purchased from Macklin Co., China. CR and methylene blue (MB) were bought from the National Pharmaceutical Co., China. Rhodamine-b (Rh-b) was obtained from Tianjin Tianxin Chemical Co., China. All chemicals were used without further purification.
300000, AR), and thiourea (CN2H4S, AR), silver nitrate (AgNO3, AR), benzoquinone (BQ, AR), tertiary butanol (TBA, AR), and ethylene diamine tetraacetic acid (EDTA, AR) were purchased from Aladdin Chemical Co., China. Tertiary butanol (TBA). N,N-dimethylformamide (DMF, AR) and hydrochloric acid (HCl, AR) were purchased from Sinopharm Chemical Reagent Co. (Shanghai), China. 2,4-DCP was purchased from Macklin Co., China. CR and methylene blue (MB) were bought from the National Pharmaceutical Co., China. Rhodamine-b (Rh-b) was obtained from Tianjin Tianxin Chemical Co., China. All chemicals were used without further purification.
2.2 Preparation of C3N4-induced TiO2 nanofibers and counterparts
As shown in Scheme 1, the TiO2-based composite nanofibers were prepared by an electrospinning technology and subsequent thermal treatment. The detailed synthesized method is as follows. First, 1 g of PVP was dissolved in anhydrous ethanol (4.5 mL) and N–N dimethylformamide (4.5 mL) to achieve a homogeneous solution. Then thiourea were added into the mixture solvent with various mass (0.1, 0.5, and 1 g), separately. Then 6 mmol tetrabutyl titanate was gradually added into the pre-solution. After full homogenization, the mixture was used as the electrospinning solution. During the electrospinning process, the precursor solution was loaded into a 20 mL plastic syringe pump and injected through a stainless-steel needle with a flow rate of 1 mL h−1. A high positive charge of 16 kV was applied between the needle tip and the collector. The receiving speed of the collector is around 400 rpm. Then, the fibrous membranes were dried at 60 °C in a vacuum oven for 12 hours and calcined in air at 500 °C with an increasing rate of 3 °C min−1. The fibrous membranes were maintained at 500 °C for 2 h to obtain C3N4-induced TiO2 nanofibers. TiO2 nanofibers were prepared by the same procedures above, except that thiourea was not added in the prepared electrospinning solution. | ||
| Scheme 1 Flowchart for the synthesis of TiO2-based composite nanofibers. | ||
Similarly, the obtained precursor nanofibers were mixed with ethanol solution of thiourea with a mass ratio of 1:1. Then composite was dried at 60 °C in a vacuum oven for 12 hours and calcined in air at the temperature of 500 °C with an increasing rate of 3 °C min−1 and maintain at 500 °C for 2 h. The obtained sample is denoted as the C3N4/TiO2.
2.3 Surface and structure characterization
An ultra-micro balance (Mettler Toledo) with an accuracy of 10 μg was used to weigh the samples and chemicals. The microscopic architecture and chemical structure of samples were characterized by SEM (FE-SEM, Supra55, Carl Zeiss) with energy dispersive X-ray (EDX) spectroscopy, a high-resolution transmission electron microscope (HR-TEM, JEM-3010, JEOL), UV-visible spectrophotometer (UV-2700, Shimadzu), Raman spectroscopy (Ar laser, wavelength: 532 nm, RM2000, Renishaw), XPS (ThermoFisher scientific), and a D8 Advance Diffractometer (Bruker) with a Cu Kα source. Nitrogen adsorption/desorption isotherms were collected at 77 K on a Micromeritics ASAP 2020 instrument. The specific surface area was obtained by the Brunauer–Emmett–Teller (BET) method. The pore size distribution was obtained from the density functional theory (DFT) method. The wetting behaviors were characterized by a contact angle analyzer (SL-200KB, KINO). The tap densities of the powders were measured as follows. A certain quantity of powders was added to a dry quartz tube and was then vacuumed until the volume of the powders did not change to measure the volume of the tapped powders. Then the mass and the measured volume of the tapped powders were used to calculate the tap density.2.4 Evaluation on photocatalytic activity
The photocatalytic activity of the samples was evaluated via the photocatalytic degradation of Rh-b and other pollutants under UV/visible-light irradiation. These experiments were conducted in a photocatalytic reactor. The light source was a 230 W xenon lamp with a UV-light filter from 300 to 400 nm and a visible-light filter from 420 to 780 nm, respectively. 50 mg of the photocatalytic material was added to a 50 mL pollutants reaction mixture (10 mg L−1), including Rh-b, 2,4-DCP, CR, and MB, in a quartz tube. The pH values were controlled by the addition of sodium hydroxide and hydrochloric acid. For 2,4-DCP degradation, the photocatalyst powder and 2,4-DCP solution were mixed in a photochemical glass reactor under magnetic stirring at the room temperature and standard pressure. Prior to irradiation, the reactive system was stirred magnetically in dark for 30 min to reach the adsorption–desorption equilibrium. During the photoreaction process, 3 mL of the solution was collected every 30 min and then centrifuged for 5 min with a rate of 5000 rpm to remove the material. The concentration variation of 2,4-DCP under the UV light was determined by High-performance liquid chromatography (HPLC, Agilent 1260). Before determination, the samples were filtered by 0.22 μm membrane, and then the filtrates were performed using HPLC with the UV detector at 290 nm. The HPLC was equipped with a Kromasil C18 column and worked at 25 °C. The mobile phase was 85:15 (v/v) methanol (CH3OH) and ultrapure water, and the flow rate was 1 mL min−1. The compounds were identified by comparing retention times with standard samples and then quantified with calibration curve method. In addition, for the photocatalytic degradation of Rh-b, CR, and MB, 3 mL of aliquots were collected and sampled at certain time interval of 10 min for subsequent analysis after centrifugation and filtration to remove the photocatalyst. The real time concentrations of dye were determined on UV-visible spectrophotometer at λ = 555 nm, λ = 497 nm and λ = 664 nm for Rh-b, CR and MB, respectively. Usually, it is ascertained that the removal of pollutants by photocatalysis is related to several reactive oxygen species (ROS) including hydroxyl radical (˙OH), superoxide radical (˙O2−) and so on. In order to verify the dominated ROS of 2,4-DCP or Rh-b degradation, the radicals trapping experiments were conducted with the addition of different scavenger agents. EDTA, AgNO3, TBA, and benzo-quinone (BQ) were adopted to quench photo-generated hole (h+), photo-generated electron (e−), OH−, and O2−, respectively. Typically, C3N4-induced TiO2 is added into the 2,4-DCP mixture and stirred for 0.5 h in the dark. After that, the scavengers (5 mM) are added into the liquor and irradiated under visible light for 3 h with continuous stirring.
The first-order kinetic equation during photocatalytic degradation is shown as follows:
| ln(C/C0) = −Ka × t | (1) |
The normalized photocatalytic rates are calculated using the equation as follows:
| Kn = m1 × ε/t × m2 | (2) |
2.5 Calculation method
The DFT calculations were carried out using the Vienna Ab initio Simulation Package (VASP) with the frozen-core all-electron projector-augment-wave (PAW) method. The Perdew–Burke–Ernzerhof (PBE) of generalized gradient approximation (GGA) was adopted to describe the exchange and correlation potential. The cutoff energy for the plane-wave basis set was set to 450 eV. The geometry optimizations were performed until the forces on each ion was reduced below 0.01 eV Å−1. The resulting structures were then used to calculate the electronic structures. Here, we adopted DFT + U method for geometry optimization and electronic structure calculation, the effective on-site coulomb interaction parameter of 6 eV was applied to the Ti-3d electrons.The thiourea and tetrabutyl titanate were placed in a 20 × 20 × 20 Å3 vacuum box to minimize the interactions between neighboring systems, and gamma k-point sampling of the Brillouin zone was used. The adsorption energy, Eads, is calculated using the equation as follows:
| Eads = Ethiourea+tetrabutyl-titanate − Etetrabutyl-titanate − Ethiourea | (3) |
A vacuum region of 15 Å was added above monolayer TiO2(101) plane (1 × 3) and tri-layer TiO2(001) planes (3 × 3) to minimize the interactions between neighboring systems. The climbing image nudged elastic band (CI-NEB) method was adopted to calculate free radical (˙OH) generation process on the TiO2(101) plane. An anatase TiO2 supercell (2 × 2 × 1) was used to simulate pure TiO2, and the model of TiO2−X was built by deleting a O atom in pure anatase TiO2 supercell. The structure of C3N4/TiO2 composite was built by placing a monolayer C3N4 on double-layer TiO2(101) plane (2 × 3), and a vacuum region of 15 Å was also added above the C3N4/TiO2 slab model. For geometry optimization, the bottom-layer atoms of TiO2(101) plane were fixed in their bulk positions.
3. Result and discussion
3.1 Nanoarchitecture of C3N4-induced TiO2
Tuning the interaction and localized homogeneity of molecules in precursors is quite significant for the construction of continuous heterogeneous interfaces in the composite photocatalytic materials.44 As illustrated in Fig. 1a, there is a strong electrostatic interaction between thiourea and tetrabutyl titanate precursors. Specifically, based on the density factor calculation, the charge density differences between thiourea and tetrabutyl titanate (e.g. NH2–Ti and S–Ti sites) are shown in Fig. S1a and b.† The amino groups on the thiourea exhibit a lower stable distance (∼2.83 Å), indicating a stronger interaction with central titanium atom on tetrabutyl titanate than that between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and central titanium atoms. In addition, the amino group on thiourea presents an absorption energy of ∼1.86 eV (Fig. S1c†), which is much higher than that of CS (∼1.54 eV). In comparison with original TiO2 (Fig. 1b), C3N4/TiO2 composite (details in ESI†) in Fig. 1c shows a narrow bandgap and enhanced photocatalytic activity due to the introduction of localized heterogeneous nanodomains. Furthermore, via regulation of the precursor, C3N4-induced TiO2 (Fig. 1d) displays a narrow bandgap and high photocatalytic activity, which are attributed to the heterogenous junctions and high-surface-energy facets.
S and central titanium atoms. In addition, the amino group on thiourea presents an absorption energy of ∼1.86 eV (Fig. S1c†), which is much higher than that of CS (∼1.54 eV). In comparison with original TiO2 (Fig. 1b), C3N4/TiO2 composite (details in ESI†) in Fig. 1c shows a narrow bandgap and enhanced photocatalytic activity due to the introduction of localized heterogeneous nanodomains. Furthermore, via regulation of the precursor, C3N4-induced TiO2 (Fig. 1d) displays a narrow bandgap and high photocatalytic activity, which are attributed to the heterogenous junctions and high-surface-energy facets.
 | ||
| Fig. 1 Schematic illustration and morphologies of C3N4-induced TiO2: (a) schematic illustration of the electrostatic interaction between thiourea and tetrabutyl titanate. (b)–(d) Comparison of constituent and contribution of TiO2, C3N4/TiO2, and C3N4-induced TiO2. (e) Scanning electron microscope (SEM) of C3N4-induced TiO2 nanofibers. (f) Transmission electron microscope (TEM) images of single nanofiber. (g)–(j) Energy dispersive spectrometer (EDS) elemental mappings of partial C3N4-induced TiO2 nanofiber. (k) High-resolution TEM image of constituents in C3N4-induced TiO2, and (l) selected area electron diffraction. (m) X-ray diffraction spectra of C3N4-induced TiO2 as compared with counterparts. | ||
Construction of suitable nanostructures with the continuous heterogeneous interface is quite vital for charge separation and migration in composite photocatalytic materials.25 Specifically, C3N4-induced TiO2 materials exhibit a hierarchical nanofibrous structure (Fig. 1e) with a uniform diameter. As shown in Fig. 1f and S2a,† C3N4-induced TiO2 nanofiber exhibits a grainy surface with an ultrafine diameter of only around 120 nm. The ultrafine nanofibrous structure with the porous surface similar to the schematic illustration in Fig. S2b† is highly beneficial to light transmission, scattering, and absorption.45 With increasing thiourea in the precursor, nanofibers present curved morphology and smooth surface with an average diameter increasing from 102 to 257 nm (Fig. S3†). Furthermore, the spatial distribution of each element, including Ti, C, O, and N in a single nanofiber, is investigated by energy dispersive spectrometer (EDS) elemental mapping (Fig. 1g–j). As compared with the elemental mapping images of intrinsic Ti and O elements (Fig. 1g and h), the mapping of C and N suggests the uniform distribution of both C and N dopants in the nanofiber. The homogeneous distribution of the substitutional element is highly favorable to the generation of numerous heterogeneous junctions in the nanofibers.46
Pore structure and size distribution are crucial structural parameters for spatial mass transfer. The pore structure of C3N4-induced TiO2 is measured using the nitrogen-sorption isothermal analysis with the Barrett–Joyner–Halenda (BJH) model. The C3N4-induced TiO2 nanofibers exhibit a similar specific surface area (22 m2 g−1) to the counterpart C3N4/TiO2 composite (25 m2 g−1). However, C3N4-induced TiO2 presents a higher tapping density (∼0.11 g cm−3) (Fig. S4†), which is close to that of commercial TiO2–P25 (∼0.13 g cm−3). As shown in Fig. S5a,† the nitrogen adsorption/desorption curves display a type II/IV isotherm curve, which interaction demonstrates the macropore-dominant structure.47 Furthermore, the pore size distribution of C3N4-induced TiO2 nanofibers is characterized by the density functional theory (DFT) method. The C3N4-induced TiO2 in Fig. S5b† exhibits micropores at around 1.3 nm. The developed microporous structure is highly beneficial to the absorption of small molecules for photocatalysis.
Furthermore, the polymorphic nature of C3N4-induced TiO2 is confirmed by high-resolution transmission electron microscopy (HR-TEM) images (Fig. 1k). The lattice spacing of 0.35 and 0.24 nm are assigned to the stable low-index (101) facet and high-surface-energy (004) facet of anatase TiO2, respectively.48,49 Additionally, the lattice spacing of 0.32 nm is attributed to the (002) plane of C3N4.50 There are some disordered microregions in C3N4-induced TiO2 due to the introduction of N/C heteroatoms and O vacancies. The crystalline composition of C3N4-induced TiO2 can be further investigated by selected area electron diffraction (Fig. 1l). Corresponding diffraction rings of anatase TiO2 and C3N4 can be found, demonstrating the well-crystalline structure. The crystal structures of all the three samples are further confirmed by X-ray diffraction spectra (Fig. 1m). All the three diffraction patterns show six typical and strong diffraction peaks that locate at 25.3°, 37.8°, 48.1°, 53.9°, 62.69° and 75.01°, which are corresponded to the (101), (004), (200), (105), (204) and (215) crystal planes of anatase TiO2 (JCPDS no. 21-1272), respectively.51,52 According to the relative (004)/(200) peak intensity ratio of in Fig. S6,† C3N4-induced TiO2 exhibits a higher ratio of ∼0.69 than that of TiO2 (∼0.65) and C3N4/TiO2 (∼0.67), indicating a better crystal orientation growth along the [001] direction.53–55 Furthermore, C3N4/TiO2 composite and C3N4-induced TiO2 exhibit additional peaks at around 26.8°, which are assigned to the (002) plane of g-C3N4 (JCPDS no. 87-1526) in the composite constituent.56–58 Besides, as illustrated in Table S1,† according to the Debye–Scherrer formula, C3N4-induced TiO2 exhibits a lower average crystallite size of 9.5 nm in comparison with that of TiO2 nanofiber and C3N4/TiO2 composite. In a word, well-connected nanosized polymorphic facets of C3N4 and anatase TiO2 are believed to promote the vectorial transfer of photogenerated electrons, thus improving the charge separation and photocatalytic efficiency.48
3.2 Catalytic mechanisms and performances of TiO2(101) and TiO2(001) plane
It is generally believed that oxidizing radicals in water can oxidize organic pollutants. In order to understand the differences in the photocatalytic process between TiO2(101) and TiO2(001) planes, we calculated the free radical (˙OH) generation process by using DFT calculations. Specifically, the C3N4/TiO2 composite in Fig. 2a is dominated by exposed anatase TiO2(101) with a lattice fringe of 0.35 nm.48 As shown in Fig. 2b, when H2O approaches the TiO2(101) plane, the O atoms in H2O are adsorbed on 5-coordinated Ti atoms of the TiO2(101) plane. Then, one of the H atoms in adsorbed H2O splits to bridge the O atom of the TiO2(101) plane. The splitting process of H2O needs to overcome an energy barrier of 0.32 eV. Finally, the free ˙OH is generated after the desorption of adsorbed OH. It is found that ˙OH generation process on the TiO2(001) plane is much easier than that on the (101) plane. As compared with C3N4/TiO2 in Fig. 2a, C3N4-induced TiO2 in Fig. 2c displays emerging high-energy-surface anatase TiO2(004) with a lattice fringe of 0.24 nm corresponding to the low-index anatase TiO2(001) facet.49 As shown in Fig. 2d, the H2O on the TiO2(001) plane is directly split into OH and H. The OH is adsorbed on 5-coordinated Ti atoms, and the H forms a new OH with the bridge O atom of the TiO2(001) plane. The desorption of OH can generate a free ˙OH. Therefore, the free radical generation process on the TiO2(001) plane is more energetically favorable than that on the TiO2(101) plane. | ||
| Fig. 2 Photocatalytic activity of samples with/without TiO2(001) plane: HR-TEM images of each plane in (a) C3N4/TiO2 and (c) C3N4-induced TiO2. Simulated free radical generation process on (b) TiO2(101) plane and (d) TiO2(001) plane. Calculated band structures of (e) TiO2(101) and (f) TiO2(001) plane. (g) Comparative photocatalytic rates between C3N4/TiO2 and C3N4-induced TiO2 under the UV light. | ||
Furthermore, the energy band structures of the TiO2(101) and TiO2(001) planes are also calculated in Fig. 2e and f. Energy zero is taken as Fermi level and displayed with a red dashed line. It is found that both TiO2(101) and TiO2(001) planes are indirect bandgap semiconductors, and their conduction band minimum (CBM) and valence band maximum (VBM) locate at Γ and S point, respectively. The calculated band gap of TiO2(001) plane is 2.38 eV, which is 0.53 eV smaller than that of TiO2(101) plane, indicating higher photocatalytic activity under the same light irradiation. Additionally, the comparison of photocatalytic activity with/without TiO2(001) facets is further demonstrated by photocatalytic degradation of Rh-b under UV light in Fig. 2g. The C3N4-induced TiO2 with high-surface-energy TiO2(001) facets exhibits a high kinetic rate of 0.16 min−1, which is higher than that of C3N4/TiO2 (∼0.11 min−1). Even at a low operating temperature of 5 °C, C3N4-induced TiO2 still displays a higher photocatalytic activity as compared with C3N4/TiO2 composite. It is further established that the (001) facets of TiO2 are highly favorable to the enhancement of photocatalytic activity.
3.3 Constituent and electronic structure of C3N4-induced TiO2
The analysis of crystal constituents and structure is quite vital for the investigation of enhancement mechanisms of photocatalytic performance. As shown in Fig. S7a and b,† exposed (101) facets are the most stable and dominant planes in TiO2 nanofibers. In contrast, the HR-TEM images of C3N4-induced TiO2 indicate the coexistence of various interfaces between C3N4 and TiO2 nanocrystals as well as (101) and (001) facets of anatase TiO2 (Fig. 3a and b).50 Raman scattering measurements are employed to characterize the structural properties of C3N4-induced TiO2 (Fig. 3c). The six Raman modes of the anatase phase were detected in both C3N4-induced TiO2 and counterparts, which are assigned to the peaks of 143.6 cm−1 (Eg(1)), 198.1 cm−1 (Eg(2)), 398.1 cm−1 (B1g(1)), 519.5 cm−1 (A1g/B1g(2)), and 637.5 cm−1 (Eg(3)).59,60 As compared with anatase TiO2, C3N4-induced TiO2 presents a broad Eg(1) at 143.6 cm−1 with an obvious blueshift (Fig. 3d). The blueshift and broadening of the Eg peak are attributed to the nano-sized grain (<10 nm) and/or the presence of defects such as (Ti3+ and oxygen vacancy).61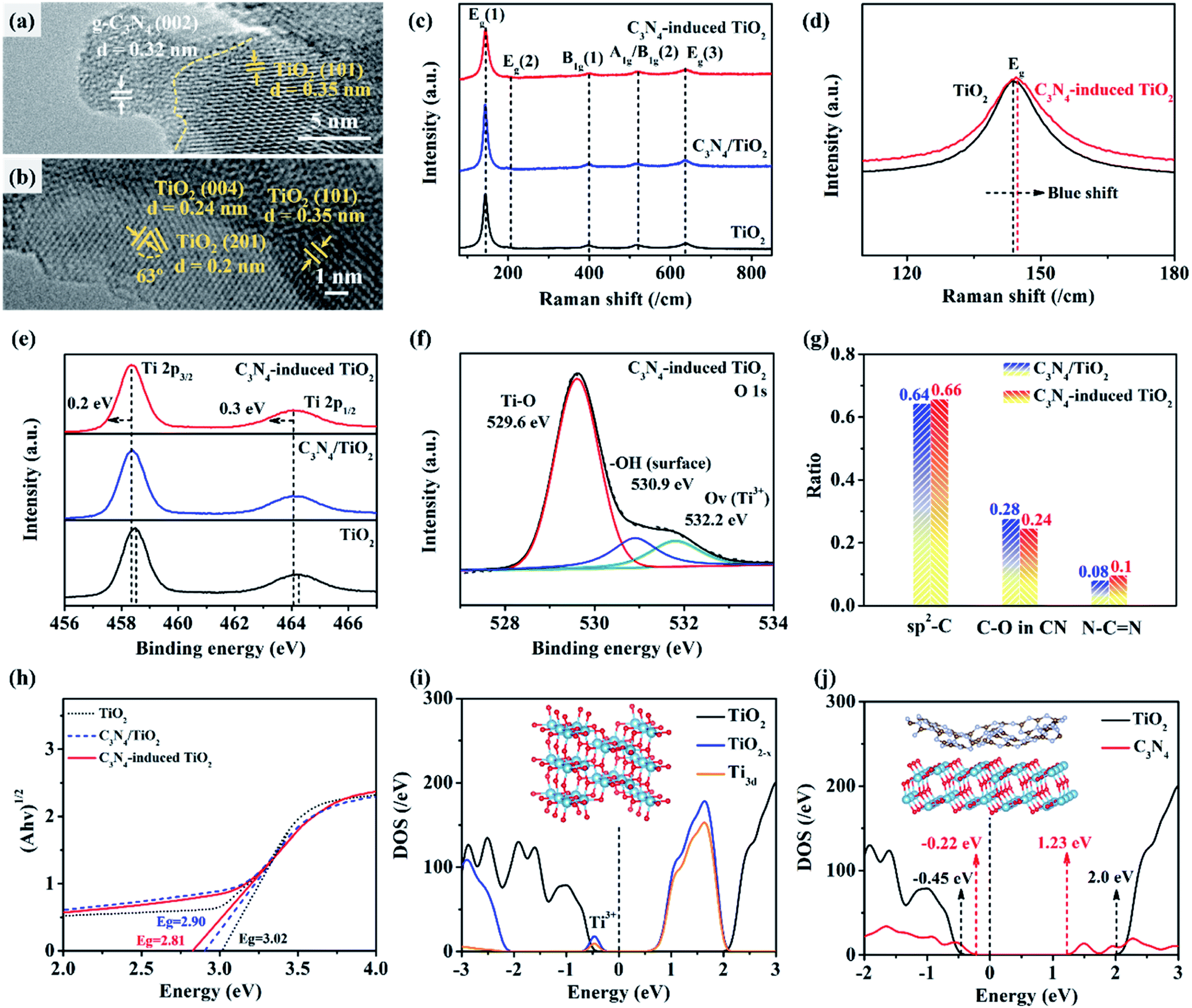 | ||
| Fig. 3 Characterization on constituent and band gap of C3N4-induced TiO2 as compared with counterparts: (a) and (b) are HR-TEM images of each interface in C3N4-induced TiO2. (c) Raman spectra of C3N4-induced TiO2, C3N4/TiO2, and TiO2. (d) Magnified partial spectra of C3N4-induced TiO2 and TiO2. (e) XPS spectra of C3N4-induced TiO2, C3N4/TiO2, and TiO2. (f) XPS spectra of O 1s in C3N4-induced TiO2. (g) Comparative carbonaceous constituents of C3N4/TiO2 and C3N4-induced TiO2. (h) Kubelka–Munk-transformed reflectance of TiO2, C3N4/TiO2, and C3N4-induced TiO2. (i) Calculated density of states (DOS) of pure TiO2 and TiO2−x in C3N4-induced TiO2. (j) Calculated DOS of TiO2 and C3N4 in C3N4/TiO2 and C3N4-induced TiO2. | ||
Furthermore, X-ray photoelectron spectroscopy (XPS) spectra are used to further demonstrate the element constituent in C3N4-induced TiO2. As exhibited in Fig. 3e, the typical peaks of Ti 2p3/2 and Ti 2p1/2 in pure TiO2 are located at 458.5 and 464.3 eV, respectively. Among them, Ti 2p3/2 peak is used to demonstrate Ti element with different valence states by change of the binding energy. Interestingly, with the introduction of C3N4 into TiO2, both C3N4/TiO2 and C3N4-induced TiO2 exhibit the shift of the total binding energy of Ti element to the low-energy direction, indicating the existence of Ti3+. The increasing Ti3+ is attributed to the electron transfer in the coordination environment of O and Ti atoms due to the incorporation of trace amounts of carbon and nitrogen.62 Additionally, despite similar Ti–O (529.6 eV) and –OH (530.9 eV) peaks to C3N4/TiO2 (Fig. S8†), C3N4-induced TiO2 shows additional oxygen vacancy (Ov) at 532.2 eV, which is consistent with the previous result in Fig. 3d. The carbonaceous constituents in C3N4-induced TiO2 and its counterpart are further illustrated in Fig. 3f. Notably, based on the fitted carbon curves in Fig. S9,† C3N4-induced TiO2 shows a higher ratio of sp2-C with lower oxygen content than that of C3N4/TiO2 (Fig. 3g), which is favorable for the electronic conduction and transfer in polymorphic structure.63 Moreover, more nitrogen-containing functional groups are found in the carbonaceous structure of C3N4-induced TiO2, which is beneficial to amelioration of surface polarity. Accordingly, as shown in Fig. S10,† C3N4-induced TiO2 presents the improved water permeability with a two times lower contacting angle than that of pure TiO2. The good water wettability and permeability promote the mass transfer between C3N4-induced TiO2 and H2O as well as targeted molecules, thus improving the photocatalytic efficiency.
Additionally, according to the transformed Kubelka–Munk-transformed reflectance in Fig. 3h, C3N4-induced TiO2 presents a lower bandgap of 2.81 eV than that of TiO2 nanofibers (∼3.02 eV) and C3N4/TiO2 composite (∼2.90 eV), respectively. Among that, the bandgap of TiO2 nanofibers in the study is lower than the theoretical one (∼3.2 eV) due to the presence of amorphous constituents and incorporation of trace carbon derived from precursor during calculation. To further reveal the mechanism for reduction of bandgap, the theoretical simulation and calculation in terms of DFT are operated based on previous observed crystal constituents and structure. Specifically, DFT calculated DOS of pure TiO2 and O-deficient TiO2 (TiO2−x) is shown in Fig. 3i, in which the energy zero is denoted as Fermi level with a black dashed line. It is found that the Fermi level of pure TiO2 locates above the VBM, indicating intrinsic semiconductive property. In contrast, for TiO2−x, the Fermi level locates below the CBM, demonstrating n-type semiconductive characteristics.
However, there is an appearance of defect state below the CBM of TiO2−x, which mainly consists of the Ti-3d orbital. The defect state is mainly caused by the reduction of Ti4+ to Ti3+ in O-deficient TiO2. The defect state induced by O-vacancy can improve the visible-light absorption of TiO2−x to some extent. However, these defects often act as recombination centers of photogenerated electron–hole pairs, which limits the further improvement of photocatalytic performance of TiO2−x due to the relatively high recombination rate. Moreover, as displayed in Fig. 3j, the Fermi level (energy zero) of C3N4 and TiO2 locate at 0.22 eV and 0.45 eV above each VBM value. Benefiting from the unique combination of Fermi level, the C3N4/TiO2 composite presents a bandgap of 1.45 eV as an intrinsic semiconductor, demonstrating the high potential for visible-light absorption and conversion.
3.4 Photocatalytic performances of C3N4-induced TiO2
The excitation processes of C3N4-induced TiO2 and counterparts under visible light are demonstrated by the photocatalytic oxidation of 2,4-DCP and other various organic dyes. According to the standard plots in Fig. S11,† the concentration of 2,4-DCP as a function of illumination intensity is plotted in Fig. 4a. C3N4-induced TiO2 displays a degradation efficiency of 94.6%, which is much higher than that of TiO2 (∼59.2%) and C3N4/TiO2 (75.5%). Simultaneously, the corresponding plots are in accordance with the first-order kinetic eqn (1). It is found that C3N4-induced TiO2 presents a Ka of 0.015 min−1 that is up to two times higher than that of TiO2 and C3N4/TiO2 (Fig. 4b), demonstrating the enhanced photocatalytic efficiency at room temperature. Furthermore, the photocatalytic activity of C3N4-induced TiO2 is further demonstrated by the photodegradation of different organic dyes. According to standard plots of Rh-b in Fig. S12,† corresponding photodegradation curve and Ka at different operation temperatures are calculated and summarized in Fig. 4c. As compared with TiO2 and C3N4/TiO2, C3N4-induced TiO2 exhibits a higher photodegradation kinetic rate of 0.092 min−1. As known, low operation temperature has a negative effect on molecular dynamics of the reactant surface, thereby restricts the mass transfer and diffusion process during the photocatalytic reaction. However, even at a low operation temperature (5 °C) in Fig. 4c, C3N4-induced TiO2 still shows a higher Ka of 0.038 min−1 based on the degradation process (Fig. S13†) than that of counterparts. As shown in Fig. S14,† the photocatalytic efficiency of C3N4-induced TiO2 decreases with increasing pH value, demonstrating good stability in acidic condition. Additionally, according to the standard plots in Fig. S15† and photocatalytic degradation curve in Fig. S16,† C3N4-induced TiO2 simultaneously displays the good photodegradation ability of CR and MB, which further demonstrates its good universality in degrading various organic dyes. | ||
| Fig. 4 Photocatalytic activity of C3N4-induced TiO2 under visible light (λ > 420 nm) as compared with counterparts: (a) photocatalytic degradation curve of 2,4-DCP. (b) Pseudo-first-order kinetics curves. (c) Photodegradation kinetic number of Rh-b at various temperatures, CR, and MB. (d) Photocatalytic degradation efficiency with the introduction of EDTA (h+), TBA (˙OH), and BQ (O2−) as scavengers during the degradation of 2,4-DCP. (e) Kinetic number of C3N4-induced TiO2 with various concentrations of TBA. (f) Long-term photocatalytic degradation of C3N4-induced TiO2. (g) Comparison of final degradation efficiency as a function of maximum degradation capacity between C3N4-induced TiO2 and other reported work. | ||
To explore the main reactive species of C3N4-induced TiO2 during the photodegradation of 2,4-DCP, the trapping experiments were implemented with TBA, EDTA, and BQ as the ˙OH, h+, and ˙O2− scavengers, respectively. As shown in Fig. 4d, the initial degradation efficiency of C3N4-induced TiO2 is regarded as 100%. The relative photodegradation efficiency of C3N4-induced TiO2 are generally decreased with introduction of scavengers into the systems, indicating all active species are all participated into the photodegradation process. But with introduction of TBA as ˙OH scavenger, C3N4-induced TiO2 exhibits a limited photodegradation efficiency of 12%. The sudden drop of photodegradation efficiency demonstrates that the key active specie is ˙OH. With increasing of TBA concentration in Fig. 4e, the Ka of photodegradation process exhibits the nonlinear decreasing relationship, further indicating coexistence of ˙OH as the key active specie other active species during photodegradation process. As for the degradation of organic dyes such as Rh-b, ˙OH also plays the major role to degrade the dyes as demonstrated by Fig. S17.† Moreover, the durability and stability of C3N4-induced TiO2 are further investigated by cyclic photocatalytic oxidation reactions of 2,4-DCP. After 20 cycles in Fig. 4f, the photocatalytic efficiency under visible light still retains 90.7%. Simultaneously, C3N4-induced TiO2 maintains an inherent nanofibrous structure with a rough surface (Fig. S18†). In contrast to the reported modified photocatalytic materials (Fig. 4g and Table 1), C3N4-induced TiO2 in this study simultaneously presents a higher degradation capacity and satisfactory final efficiency. The excellent photocatalytic performances are attributed to stable nanostructure and polymorphic nanodomains/interfaces, which facilitate electron separation and extend the lifetime of the separated electron and hole.
| Sample | Light source | Rate constants (min−1) | Final efficiency (%) | Capacity (mg) | Ref. |
|---|---|---|---|---|---|
| C3N4-Induced TiO2 | 230 W xenon lamp (420 nm–780 nm) | 0.015 | 90.76 | 10.86 | This work |
| TiO2/FeOX/POM | 150 W xenon lamp (>400 nm) | Not available | 58 | 2.03 | 64 |
| CeO2/g-C3N4 | 150 W xenon lamp (>420 nm) | Not available | 78 | 1.54 | 65 |
| In2O3/ZnIn2S4 | 300 W xenon lamp (>420 nm) | 0.0246 | 95.8 | 4.75 | 66 |
| TiO2/g-C3N4 | 150 W xenon lamp (>400 nm) | 0.012 | 97 | 3.9 | 67 |
| Ag2CrO4/Ag/g-C3N4 | 500 W xenon lamp (>400 nm) | 0.015 | 80 | 2.2 | 68 |
| N-Carbon@ZnO | 300 W xenon lamp (380 nm–800 nm) | 0.028 | 72 | 5.7 | 69 |
| SnO2/Pt/In2O3 | 500 W xenon lamp (>420 nm) | 0.023 | 80 | 8.45 | 70 |
| Ag3PO4/Fe3O4 | 250 W LED lamp (>420 nm) | 0.056 | 74 | 1.34 | 71 |
| SnO2/Ag/MoS2 | 150 W xenon lamp (>400 nm) | Not available | 90 | 3.89 | 72 |
| P–g-C3N4 | 230 W xenon lamp (420 nm–780 nm) | Not available | 84 | 2.1 | 73 |
| BiO(OH)xI1−x–AgI | 150 W xenon lamp (>400 nm) | 0.16 | 85 | 2.622 | 74 |
| 3S-BN | 300 W xenon lamp (380 nm–800 nm) | Not available | 63 | 1.12 | 75 |
4. Conclusion
In summary, we fabricate a polymorphic TiO2 composite composed of in situ diverse nanodomains including C3N4 nanocrystals and (001)-/(101)-faceted anatase. The unique structure is fabricated by calcinating the composite nanofibrous precursor of thiourea and tetrabutyl titanate in air. Induced by in situ formation of heterogeneous ultrafine C3N4 crystals, more oxygen vacancies can be generated in the TiO2 lattice structure. And high-surface-energy anatase TiO2(001) facets are simultaneously exposed. The resulting composite simultaneously presents excellent photocatalytic activity and relatively narrow bandgap. The composite exhibits a degradation efficiency of 94.6% in 2 h under visible light with a high kinetic rate of 0.015 min−1. Additionally, the composite still exhibits excellent durability even under extreme conditions such as low-temperature and acidic environment, and desirable long-term stability with high degradation efficiency. More significantly, the normalized degradation rate and effective durability of the composite photocatalyst are far superior to other reported modified TiO2 photocatalysts. This work not only leads to a novel TiO2 composite material, but also provides a new strategy for the design of advanced photochemical semiconductors.Author contributions
Chunzhao Liu supervised the project. Chenfeng Ding and Yuan Liu conceived the idea, designed the experiments, and participated in the data analysis. Zeju Zhang carried out the fabrication and characterizations. Mang Niu provided support for the DFT stimulation and corresponding analysis. Wei Li, Peitao Xie, Yongxin Li, Xiaodong Yan, Xuewei Fu, Yaochun Liu, Lili Chen, Xiaodong Lan, Chunlei Liu, Jingjie Dai, Xiaofei Hong, and Dapeng Cao provided valuable suggestions for the manuscript. All authors contributed to writing the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Shandong Province (ZR2020QE060), the Key Research and Development Project of Shandong Province (2019GSF109079), the Guangdong Basic and Applied Basic Research Foundation (2019A1515110504, 2020A1515110830), the Medical Science and Technology Development Plan Project of Shandong Province (202104050762), the Qingdao Key Health Discipline Development Fund, the Medical Science and Technology Development Plan Project of Qingdao City (2021-WJZD072, 2021-WJZD069), and the State Key Laboratory of Bio-Fibers and Eco-Textiles (Qingdao University). We also acknowledge the support from the Organizing Committee of the 19th Asian Games in 2022 and Zhejiang province indoor environment depuration industry association.References
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- C. Y. Toe, Z. Zheng, H. Wu, J. Scott, R. Amal and Y. H. Ng, Angew. Chem., Int. Ed., 2018, 57, 13613–13617 CrossRef CAS PubMed.
- Q. Sun, N. Wang, J. Yu and J. C. Yu, Adv. Mater., 2018, 30, 1804368 CrossRef PubMed.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913–17918 CrossRef CAS PubMed.
- Z. Xing, J. Zhang, J. Cui, J. Yin, T. Zhao, J. Kuang, Z. Xiu, N. Wan and W. Zhou, Appl. Catal., B, 2018, 225, 452–467 CrossRef CAS.
- T. Wei, Y.-N. Zhu, X. An, L.-M. Liu, X. Cao, H. Liu and J. Qu, ACS Catal., 2019, 9, 8346–8354 CrossRef CAS.
- R. Jaiswal, J. Bharambe, N. Patel, A. Dashora, D. Kothari and A. Miotello, Appl. Catal., B, 2015, 168, 333–341 CrossRef.
- G. Rossi, L. Pasquini, D. Catone, A. Piccioni, N. Patelli, A. Paladini, A. Molinari, S. Caramori, P. O'Keeffe and F. Boscherini, Appl. Catal., B, 2018, 237, 603–612 CrossRef CAS.
- M. Surówka, M. Kobielusz, M. Trochowski, M. Buchalska, K. Kruczała, P. Broś and W. Macyk, Appl. Catal., B, 2019, 247, 173–181 CrossRef.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, science, 2001, 293, 269–271 CrossRef CAS PubMed.
- J. H. Lee, D. F. Hevia and A. Selloni, Phys. Rev. Lett., 2013, 110, 016101 CrossRef PubMed.
- T. Wu, P. Niu, Y. Yang, L. C. Yin, J. Tan, H. Zhu, J. T. Irvine, L. Wang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2019, 29, 1901943 CrossRef.
- Y. Lin, C. Yang, S. Wu, X. Li, Y. Chen and W. L. Yang, Adv. Funct. Mater., 2020, 30, 2002918 CrossRef CAS.
- N. Nishiyama, Y. Fujiwara, K. Adachi, K. Inumaru and S. Yamazaki, Appl. Catal., B, 2015, 176, 347–353 CrossRef.
- T. M. Breault and B. M. Bartlett, J. Phys. Chem. C, 2012, 116, 5986–5994 CrossRef CAS.
- S. Liu, J. Yu and M. Jaroniec, Chem. Mater., 2011, 23, 4085–4093 CrossRef CAS.
- G. Liu, H. G. Yang, X. Wang, L. Cheng, J. Pan, G. Q. Lu and H.-M. Cheng, J. Am. Chem. Soc., 2009, 131, 12868–12869 CrossRef CAS PubMed.
- Y. Gong, X. Quan, H. Yu and S. Chen, Appl. Catal., B, 2017, 219, 439–449 CrossRef CAS.
- L. Ye, J. Mao, J. Liu, Z. Jiang, T. Peng and L. Zan, J. Mater. Chem. A, 2013, 1, 10532–10537 RSC.
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CrossRef CAS.
- M.-V. Sofianou, C. Trapalis, V. Psycharis, N. Boukos, T. Vaimakis, J. Yu and W. Wang, Environ. Sci. Pollut. Res., 2012, 19, 3719–3726 CrossRef CAS PubMed.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B, 2001, 63, 155409 CrossRef.
- Y. Wei, F. Zhang, J. Hao, Y. Ling, Y. Gong, S. Wang, J. Wei and Z. Yang, Appl. Catal., B, 2020, 272, 119035 CrossRef CAS.
- Y. Wei, N. Yang, K. Huang, J. Wan, F. You, R. Yu, S. Feng and D. Wang, Adv. Mater., 2020, 32, 2002556 CrossRef CAS PubMed.
- C.-C. Wang, X. Wang and W. Liu, Chem. Eng. J., 2020, 391, 123601 CrossRef CAS.
- S. You, Y. Hu, X. Liu and C. Wei, Appl. Catal., B, 2018, 232, 288–298 CrossRef CAS.
- Y. Lin, H. Liu, C. Yang, X. Wu, C. Du, L. Jiang and Y. Zhong, Appl. Catal., B, 2020, 264, 118479 CrossRef.
- Y. Lin, C. Yang, Q. Niu and S. Luo, Adv. Funct. Mater., 2022, 32, 2108814 CrossRef CAS.
- W. Zhang, G. Li, H. Liu, J. Chen, S. Ma, M. Wen, J. Kong and T. An, Appl. Catal., B, 2020, 272, 118969 CrossRef CAS.
- Y. Zou, B. Yang, Y. Liu, Y. Ren, J. Ma, X. Zhou, X. Cheng and Y. Deng, Adv. Funct. Mater., 2018, 28, 1806214 CrossRef.
- H. Yan, L. Liu, R. Wang, W. Zhu, X. Ren, L. Luo, X. Zhang, S. Luo, X. Ai and J. Wang, Chem. Eng. J., 2020, 401, 126052 CrossRef CAS.
- G. Malekshoar, K. Pal, Q. He, A. Yu and A. K. Ray, Ind. Eng. Chem. Res., 2014, 53, 18824–18832 CrossRef CAS.
- J. Qin, J. Wang, J. Yang, Y. Hu, M. Fu and D. Ye, Appl. Catal., B, 2020, 267, 118667 CrossRef CAS.
- M. Valero-Romero, J. Santaclara, L. Oar-Arteta, L. Van Koppen, D. Osadchii, J. Gascon and F. Kapteijn, Chem. Eng. J., 2019, 360, 75–88 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- C. Huang, Y. Wen, J. Ma, D. Dong, Y. Shen, S. Liu, H. Ma and Y. Zhang, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- Q. Zhang, Y. Peng, Y. Lin, S. Wu, X. Yu and C. Yang, Chem. Eng. J., 2021, 405, 126661 CrossRef CAS.
- Q. Sun, K. Lv, Z. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420–427 CrossRef.
- Y. Wang, W. Yang, X. Chen, J. Wang and Y. Zhu, Appl. Catal., B, 2018, 220, 337–347 CrossRef CAS.
- J. F. McKenna, D. Rolfe, S. E. Webb, A. F. Tolmie, S. W. Botchway, M. L. Martin-Fernandez, C. Hawes and J. Runions, Proc. Natl. Acad. Sci., 2019, 116, 12857–12862 CrossRef CAS PubMed.
- P. Maksymovych, A. N. Morozovska, P. Yu, E. A. Eliseev, Y.-H. Chu, R. Ramesh, A. P. Baddorf and S. V. Kalinin, Nano Lett., 2012, 12, 209–213 CrossRef CAS PubMed.
- W. Wang, Y. Zhang, G. Xu, L. Peng, B. Ding, Y. Wang, Z. Hou, X. Zhang, X. Li and E. Liu, Adv. Mater., 2016, 28, 6887–6893 CrossRef CAS PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- Y. Gao, N. Yan, C. Jiang, C. Xu, S. Yu, P. Liang, X. Zhang, S. Liang and X. Huang, Appl. Catal., B, 2020, 268, 118737 CrossRef CAS.
- G. Liu, L.-C. Yin, J. Wang, P. Niu, C. Zhen, Y. Xie and H.-M. Cheng, Energy Environ. Sci., 2012, 5, 9603–9610 RSC.
- Y. Liu, C. Ding, P. Xie, X. Yan, M. Feng, Y. Liu, C. Liu, Y. Yu and Y. Lin, Mater. Chem. Front., 2021, 5, 3216–3225 RSC.
- Z. Tong, D. Yang, T. Xiao, Y. Tian and Z. Jiang, Chem. Eng. J., 2015, 260, 117–125 CrossRef CAS.
- L. Gu, J. Wang, Z. Zou and X. Han, J. Hazard. Mater., 2014, 268, 216–223 CrossRef CAS PubMed.
- Y. Tan, Z. Shu, J. Zhou, T. Li, W. Wang and Z. Zhao, Appl. Catal., B, 2018, 230, 260–268 CrossRef CAS.
- X. He, M. Wu, Z. Ao, B. Lai, Y. Zhou, T. An and S. Wang, J. Hazard. Mater., 2021, 403, 124048 CrossRef CAS PubMed.
- E. Kusiak-Nejman, A. Wanag, J. Kapica-Kozar, Ł. Kowalczyk, M. Zgrzebnicki, B. Tryba, J. Przepiórski and A. W. Morawski, Catal. Today, 2020, 357, 630–637 CrossRef CAS.
- M. Liu, L. Piao, W. Lu, S. Ju, L. Zhao, C. Zhou, H. Li and W. Wang, Nanoscale, 2010, 2, 1115–1117 RSC.
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed.
- F. Tian, Y. Zhang, J. Zhang and C. Pan, J. Phys. Chem. C, 2012, 116, 7515–7519 CrossRef CAS.
- L. Ge, Mater. Lett., 2011, 65, 2652–2654 CrossRef CAS.
- L. Ye, J. Liu, Z. Jiang, T. Peng and L. Zan, Appl. Catal., B, 2013, 142, 1–7 Search PubMed.
- M. Zhang, N. Han, Y. Fei, J. Liu, L. Xing, A. Núñez-Delgado, M. Jiang and S. Liu, J. Environ. Manage., 2021, 297, 113311 CrossRef CAS PubMed.
- J. A. Rengifo-Herrera, M. Blanco, J. Wist, P. Florian and L. R. Pizzio, Appl. Catal., B, 2016, 189, 99–109 CrossRef CAS.
- Y. Liu, C. Ding, X. Yan, P. Xie, B. Xu, L. Chen, Y. Liu, C. Liu, Y. Yu and Y. Lin, Chem. Eng. J., 2021, 420, 129894 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- H. Qian, Q. Hou, E. Duan, J. Niu, Y. Nie, C. Bai, X. Bai and M. Ju, J. Hazard. Mater., 2020, 391, 122246 CrossRef CAS PubMed.
- C. Ding, L. Huang, X. Yan, F. Dunne, S. Hong, J. Lan, Y. Yu, W. H. Zhong and X. Yang, Adv. Funct. Mater., 2020, 30, 1907486 CrossRef CAS.
- J. Yu, T. Wang and S. Rtimi, Appl. Catal., B, 2019, 254, 66–75 CrossRef CAS.
- M. Humayun, Z. Hu, A. Khan, W. Cheng, Y. Yuan, Z. Zheng, Q. Fu and W. Luo, J. Hazard. Mater., 2019, 364, 635–644 CrossRef CAS PubMed.
- Q. Zhu, Y. Sun, S. Xu, Y. Li, X. Lin and Y. Qin, J. Hazard. Mater., 2020, 382, 121098 CrossRef CAS PubMed.
- A. Zada, Y. Qu, S. Ali, N. Sun, H. Lu, R. Yan, X. Zhang and L. Jing, J. Hazard. Mater., 2018, 342, 715–723 CrossRef CAS PubMed.
- C. Feng, Y. Deng, L. Tang, G. Zeng, J. Wang, J. Yu, Y. Liu, B. Peng, H. Feng and J. Wang, Appl. Catal., B, 2018, 239, 525–536 CrossRef CAS.
- X. Chen, X. Xu, J. Cui, C. Chen, X. Zhu, D. Sun and J. Qian, J. Hazard. Mater., 2020, 392, 122331 CrossRef CAS PubMed.
- Y. Sun, Q. Zhu, B. Bai, Y. Li and C. He, Chem. Eng. J., 2020, 390, 124518 CrossRef CAS.
- K. Talukdar, B.-M. Jun, Y. Yoon, Y. Kim, A. Fayyaz and C. M. Park, J. Hazard. Mater., 2020, 398, 123025 CrossRef CAS PubMed.
- B. Khan, F. Raziq, M. Bilal Faheem, M. Umar Farooq, S. Hussain, F. Ali, A. Ullah, A. Mavlonov, Y. Zhao, Z. Liu, H. Tian, H. Shen, X. Zu, S. Li, H. Xiao, X. Xiang and L. Qiao, J. Hazard. Mater., 2020, 381, 120972 CrossRef CAS PubMed.
- J. Ma, K. Wang, C. Wang, X. Chen, W. Zhu, G. Zhu, W. Yao and Y. Zhu, Appl. Catal., B, 2020, 276, 119150 CrossRef CAS.
- H. Ji, L. Zhang and C. Hu, Appl. Catal., B, 2017, 218, 443–451 CrossRef CAS.
- C. Feng, L. Tang, Y. Deng, G. Zeng, J. Wang, Y. Liu, Z. Chen, J. Yu and J. Wang, Appl. Catal., B, 2019, 256, 117827 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00782g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |