
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequential olefination–dimerisation of benzylic dithioacetals by the nickel-catalysed reaction with methyl Grignard or zinc reagent†
Lei Yua,
Guo-Qiao Lai‡
*a,
Pinglu Zhang*a,
Ze Li*a and
Tien-Yau Luh‡ *b
*b
aKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, College of Material, Chemistry, and Chemical Engineering, Hangzhou Normal University, Hangzhou 311121, China. E-mail: laiguoqiao@aliyun.com; zeli@hznu.edu.cn
bDepartment of Chemistry, National Taiwan University, Taipei, Taiwan 10617. E-mail: tyluh@ntu.edu.tw; zhangpinglu@hotmail.com
First published on 28th March 2022
Abstract
Products of main group elements from cross-coupling reactions have been shown to serve as Lewis acids, mediating further reactions of organic coupling products. Thus, the nickel-catalysed olefination of benzylic dithioacetal with MeMgI in benzene in a sealed Schlenk tube at 130 °C generates magnesium mercaptide which regioselectively converts 2-arylpropene into a dimer in good yield. Aryl iodide reacts with 2-propenylmagnesium bromide in the presence of 1,2-ethanedithiol and NiCl2(PPh3)2 to yield the same dimer. Replacement of the Grignard reagent by an organozinc reagent gives the dimers in a better yield.
Introduction
Transition metal-catalysed cross coupling reactions have significantly contributed to the formation of carbon–carbon bonds.1–3 In general, these reactions require an organic electrophile, a transition-metal catalyst, and a main group organometallic compound as a nucleophile. After the reaction is complete, the main group element is converted into the corresponding inorganic salt or equivalent. To illustrate this, Mg(II) and Zn(II) are produced from Kumada–Corriu–Tamao2 and Negishi3 reactions, respectively. These in situ generated inorganic salts can serve as a Lewis acid catalyst4 or a reagent to mediate further reactions of the cross-coupling product. In particular, these Lewis catalysed reactions can even take place in strong basic conditions. For example, the regioselective ring-opening of acetals of contiguous polyols, such as monosaccharides or inositols, by a Grignard regent is well documented (eqn (1)).5
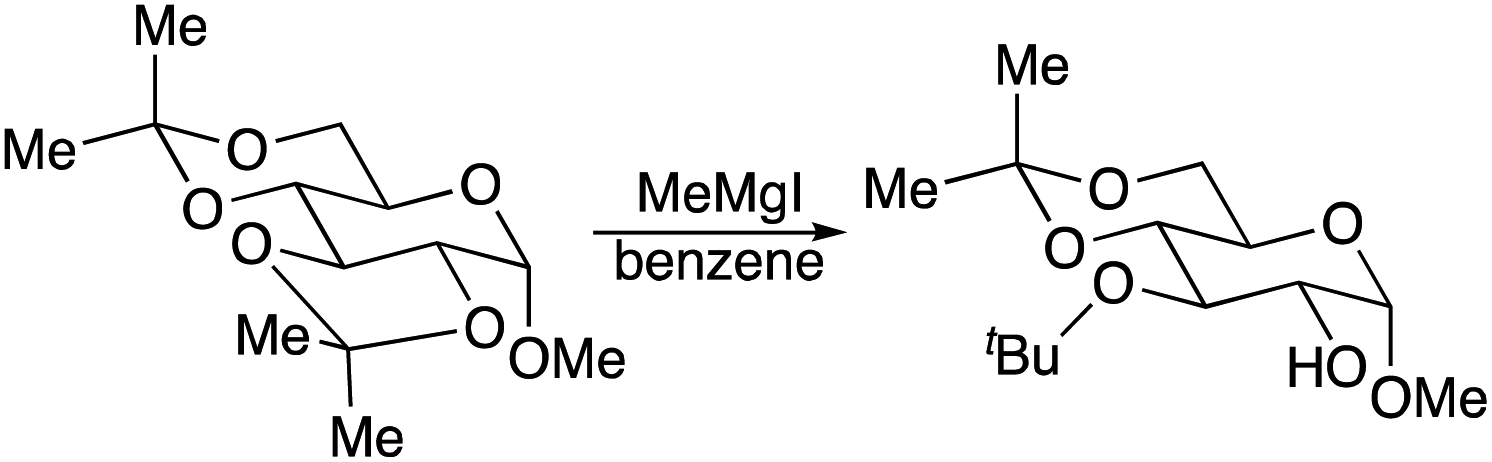 | (1) |
Dimerisation of α-methylstyrene 1 under various conditions, leading to different kinds of products 2–4, has been extensively studied (eqn (2)).6–8 Under strong acidic conditions or longer reaction times, indane derivative 2 is obtained predominantly, if not exclusively.6 Regioisomers 3 and 4 are occasionally isolated as a mixture under acid-catalysed conditions.6a–c,7 Notably, these pentene derivatives are industrially important to serve as chain-transfer agents or molecular weight regulators for styrene-based polymers or copolymers.6–8
 | (2) |
Compound 1 can readily be obtained by the nickel-catalysed olefination9,10 of benzylic dithioacetals 5 with excess MeMgI under conditions similar to those of the Kumada–Corriu–Tamao reaction (eqn (3)).2 As previously mentioned, Mg(II) salt will be generated from this reaction. Compound 1 may be vulnerable to further transformation under these reaction conditions. Since Mg(II) can be considered as a mild Lewis acid, it is envisaged that dimerisation might take place to give 3 and/or 4, but not the Friedel–Crafts product 2. In this paper, we wish to report for the first time the direct conversion of 5 to 6 regioselectively under cross-coupling reaction conditions.
 | (3) |
Results and discussion
NiCl2(PPh3)2MeMgI catalysed olefination–dimerisation reactions with 5a
Treatment of 5a with MeMgI in the presence of 10 mol% of NiCl2(PPh3)2 in refluxing benzene gave 67% of the corresponding dimer 6a. The isolation of 6a from the reaction under the conditions of the Kumada–Tamao reaction2a is striking. The reaction mixture may contain a Grignard reagent or a Mg(II), Ni(II) or Ni(0) catalyst, Ph3P, and −SCH2CH2S−. It seems likely that some of these components might be responsible for promoting the formation of 6 from 1. Our initial aim was to find suitable conditions to give 6 predominantly, if not exclusively.Effect of halide X in MeMgX on the dimerisation step
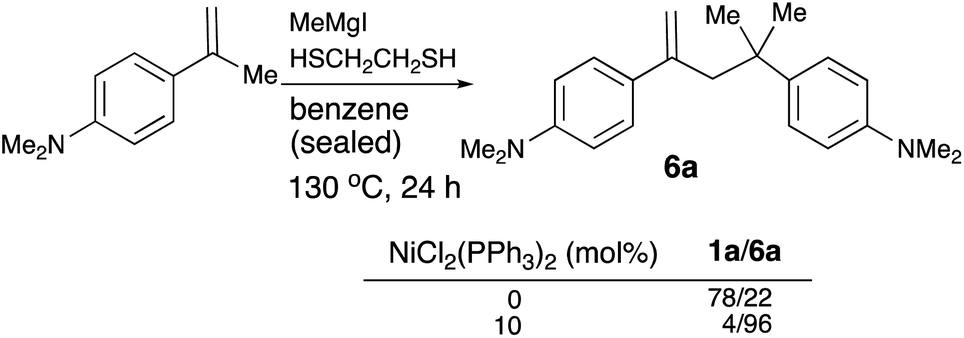
The halogen atom in the Grignard reagent has been shown to play an important role in the reactivity of the reaction (eqn (4)). A sealed Schlenk tube was chosen for the reaction of 5a with the Grignard reagent in benzene and the reaction tube was immersed in an oil bath at 130 °C for 24 h. When MeMgCl was employed, the ratio of 1a to 6a was 87/13. The ratio changed to 37/63 if MeMgBr was used. MeMgI, on the other hand, gave excellent selectivity with 1a/6a = 4/96. The magnesium ion liberated during the course of the cross-coupling reaction would interact with the negatively charged halide and/or sulfide moieties. Apparently, when these softer anions are associated with magnesium ion, the reactivity of the magnesium ion would be enhanced and dimer 6a would be formed preferentially.
 | (4) |
Effect of −SCH2CH2S− on the reactivity of dimerisation of 1a
Since −SCH2CH2S− would be generated from the olefination of dithiolanes, it seems likely that the sulfide moiety might also bind to the magnesium ion that might promote the formation of dimer 6. Dimerisation of 1a to 6a has been carefully examined. Notably, there is no sulfur atom in 1a. In the absence of 1,2-ethanedithiol, no dimerisation took place. Interestingly, a benzene solution of 1a, MeMgI, and HSCH2CH2SH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1) in a sealed Schlenk tube was heated to 130 °C for 24 h. Only 23% of conversion of 1a was observed and the ratio of dimer 6a versus unreacted 1a was 22 to 78. When 10 mol% of NiCl2(PPh3)2 was added to the reaction mixture, only 3% of the starting 1a was recovered and the yield of 6a was improved to 67%. It is worth mentioning that no dimer 6a was detected from the mixture without MeMgI. In other words, NiCl2(PPh3)2-HSCH2CH2SH alone does not catalyse the dimerisation of 1a at all. Presumably, the direct displacement of the chloride by a mercaptan would generate HCl that would not be a favored process.
:3:1) in a sealed Schlenk tube was heated to 130 °C for 24 h. Only 23% of conversion of 1a was observed and the ratio of dimer 6a versus unreacted 1a was 22 to 78. When 10 mol% of NiCl2(PPh3)2 was added to the reaction mixture, only 3% of the starting 1a was recovered and the yield of 6a was improved to 67%. It is worth mentioning that no dimer 6a was detected from the mixture without MeMgI. In other words, NiCl2(PPh3)2-HSCH2CH2SH alone does not catalyse the dimerisation of 1a at all. Presumably, the direct displacement of the chloride by a mercaptan would generate HCl that would not be a favored process.
 | (5) |
Examples of sequential olefination–dimerisation reactions
Being successful in this transformation shown in eqn (5), we tested the generality of this catalyst system for sequential olefination–dimerisation. The reaction mixture consists of excess Grignard reagent and the medium should, therefore, be basic. The Lewis acidity of magnesium may not be active enough to promote the dimerisation process. It is envisaged that zinc might be a stronger Lewis acid. Accordingly, the reactivity of the organozinc reagent obtained by the addition of ZnI2 to the above Grignard reagent was also examined and the results are compared in Table 1.| Ar in dithioacetal 5 | % yield of 6 (without ZnI2) | % yield of 6 (With ZnI2) |
|---|---|---|
| a Conditions: a sealed Schlenk tube containing 3, NiCl2(PPh3)2 (10 mol%) and MeMgI (4 equiv.), with or without ZnI2 (4 equiv.), was heated at 130 °C for 24 h, unless otherwise specified.b Reaction for 48 h. | ||
| 4-Me2NC6H4 (5a) | 88 | 76 |
| 4-HOC6H4 (5b) | 82 | 89 |
| 4-MeOC6H4 (5c) | 52b | 81 |
| 4-tBuC6H4 (5d) | 48b | 67 |
| 4-iPrC6H4 (5e) | 43b | 78 |
| 4-MeC6H4 (5f) | 21 | 74 |
| C6H5 (5g) | 10 | 64 |
| 4-CF3C6H4 (5h) | — | — |
Substrates with highly electron-donating substituents gave the corresponding 6 in a much better yield than those with alkyl substituents. The compound with an electron withdrawing group, such as the CF3 group, gave the corresponding olefination product 1h only. These results imply that the dimerisation reaction may proceed via a carbocation-like mechanism. Notably, organozinc reagent appears to be more potent than the corresponding Grignard reagent in these sequential olefination–dimerisation reactions.
Nickel-catalysed sequential cross coupling-dimerisation of aryl iodide 7 with H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) C(Me)MgBr/ZnI2
C(Me)MgBr/ZnI2
Dimer 6a could also be obtained from the reaction of 7a with 7 equiv. each of the 2-propenyl Grignard reagent and ZnI2 in the presence of 10 mol% of NiCl2(PPh3)2, and 1 equiv. of 1,2-ethanedithiol at rt for 6 h. The temperature was then raised to 130 °C for 42 h to give, after workup, 6a in 72% yield (eqn (6)). When 1,2-ethanedithiol was replaced by 2 equiv. of propanethiol, otherwise under the same conditions, dimer 6a and monomer 1a were obtained in 15 and 44% yield, respectively.
Without 1,2-ethanedithiol, the reaction stopped at the 1a stage and no dimer 6a was detected. These results suggest that the presence of bismercaptan is essential to facilitate the formation of dimer 6a. Representative examples are tabulated in Table 2.
 | (6) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)MgBr, ZnI2 and HSCH2CH2SH in benzenea
C(Me)MgBr, ZnI2 and HSCH2CH2SH in benzenea
| Aryl halide | 6 (% yield) | 1 (% yield) |
|---|---|---|
| a Conditions: in a sealed Schlenk tube, a mixture of 7, NiCl2(PPh3)2 (10 mol%), H2CC(Me)MgBr (7 equiv.), ZnI2 (7 equiv.) and HSCH2CH2SH (1 equiv.) was stirred at rt for 6 h and then heated at 130 °C for 42 h. |
||
| 7a | 6a (72) | 1a (7) |
| 7c | 6c (47) | 1c (16) |
| 7d | 6d (37) | 1d (30) |
| 8a | 6a (56) | 1a (17) |
| 8b | 6e (55) | 1e (34) |
As can be seen from Table 2, the yield of 6 is no more than moderate, a significant amount of the intermediate product 1 being obtained. In addition, the reaction is much longer than those starting with dithioacetals shown in Table 1. As shown in eqn (4), the nature of the halide in the Grignard reagent may exhibit different reactivity in the overall sequential reaction.
Possible nature of the Lewis acid catalyst for the dimerisation step
In the reaction mixture, there are two (Column 2 of Table 1 and eqn (6)) or three (Column 3 of Table 1) kinds of divalent metallic species that can serve as a Lewis acid catalyst for the dimerisation step. They are Mg(II), Ni(II) and/or Zn(II). As can be seen from Table 1, the yields of reactions in the presence of ZnI2 (Column 3) are, in general, much higher than those without the ZnI2 additive (Column 2). It seems likely that Zn(II) might be a more active catalyst than Mg(II) for the dimerisation step.As shown in eqn (5), the addition of NiCl2(PPh3)2 to the MeMgI–HSCH2CH2SH mixture will significantly accelerate the dimerisation of 1a. Grignard reagent is known to reduce Ni(II) to Ni(0). The role of the nickel species rendering the dimerisation of 1a is not clear.
Another interesting feature in this sequential reaction is the regioselectivity for the formation of the double bond in 6. In other words, no double bond migration (like 4) would take place under the reaction conditions. Whether the nickel catalyst would be involved in the regioselective formation of the double bond in 6 remains to be clarified.
Conclusions
In summary, we have demonstrated the first example of using the main-group by-product from the transition-metal catalysed cross-coupling reaction as the second catalyst to mediate further reaction of the main coupling product. Thus, under Kumada–Corriu–Tamao reaction conditions,2 the cascade olefination–dimerisation of benzylic dithioacetals derived from acetophenones affords the corresponding dimers 6 via 2-arylpropenes 1. Incorporation of ZnI2 has been shown to significantly improve the yield of the reaction. An extension of this reaction to aryl iodide 7 by applying the catalytic system NiCl2(PPh3)2-H2CC(Me)MgBr–ZnI2–HSCH2CH2SH to promote the cross-coupling and dimerisation leads to the same dimeric products 6. The Lewis acid character of the product of the main group element [Mg(II) or Zn(II)] from the transition metal catalysed cross-coupling reaction may play a critical role for the dimerisation process of the organic coupling products. The presence of 1,2-ethanedithiol is also crucial for the success of these sequential reactions. Presumably, chelation of the sulfur moiety to Mg(II) or Zn(II) may tune the catalytic activity for the dimerisation step. Indeed, the involvement of this dithiol for the dimerisation of 2-arylpropenes can be understood within this framework. The application of this strategy to other cross-coupling reactions is in progress in our laboratories.
Experimental section
General information
All melting points were measured on an SGW X-4A apparatus and were uncorrected. 1H and 13C NMR nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Advance 400 MHz spectrometer at ambient temperature. Chemical shifts (δ) and coupling constants (J) were expressed in units of ppm and Hz, respectively. Samples for measurements were dissolved in CDCl3. The residual peak of CHCl3 at δ 7.26 was used as the reference for 1H NMR chemical shifts and the central peak at δ 77.16 of CDCl3 was used as the reference for 13C NMR chemical shifts. Infrared spectra were taken on a Nicolet 700 FT infrared spectrophotometer using a KBr plate for solid samples or forming a film on an NaCl window.Materials
Compounds 5b, 5e, 5f and 5g were prepared according to a literature procedure.7General procedure for the synthesis of dithioacetals
To a chloroform solution (50 mL) of acetophenone derivative (20 mmol) and 1,2-ethanedithiol (density = 1.123 g cm−3, 22–44 mmol), boron trifluoride etherate (density = 1.15 g cm−3, 14–34 mmol) was added. The mixture was stirred at rt overnight. The solution was washed twice with 10% aqueous sodium hydroxide (30 mL) and the aqueous layer was extracted twice with DCM (20 mL). The combined organic layers were washed twice with water (50 mL), dried (Na2SO4) and filtered. The solvent was removed in vacuo and the residue was chromatographed on silica gel using petroleum ether and ethyl acetate as the eluent to give the corresponding dithioacetals.General procedure for the NiCl2(PPh3)2-catalysed reaction of dithioacetal 5 with MeMgX
In a sealed Schlenk tube, a benzene solution of dithioacetal (0.5 mmol), NiCl2(PPh3)2 (0.05 mmol), and MeMgI (3 M in ether, 2–3 mmol) was immersed in an oil bath at 130 °C for 24 or 48 h. The mixture was then poured into saturated Na2CO3 solution. The organic layer was separated and the aqueous layer was extracted twice (10 mL) with diethyl ether. The combined organic portions were washed twice each (10 mL) with 10% NaOH and brine (10 mL), and dried (Na2SO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel (petroleum ether and EtOAc) to give the product.General procedure for the nickel-catalysed reaction of dithioacetal with organozinc reagent
In a manner of the same procedure as described above except ZnI2 (0–3 mmol) was added to the reaction mixture, which was heated at 130 °C for 24 h.General procedure for the reaction of aryl halides with 2-propenylmagnesium bromide in the presence of NiCl2(PPh3)2, ZnI2 and 1,2-ethanedithiol
THF was removed from the solution of 2-propenylmagnesium bromide (3.5 mL, 1 M in THF, 3.5 mmol) and benzene (1 mL) was then added. To a Schlenk tube containing aryl halide (0.5 mmol), NiCl2(PPh3)2 (0.05 mmol), ZnI2 (3.5 mmol) and 1,2-ethanedithiol (0.5 mmol) in benzene (1 mL), the benzene solution of 2-propenyl Grignard reagent was added. The mixture was then poured into saturated Na2CO3 solution. The organic layer was separated and the aqueous layer was extracted twice (10 mL) with diethyl ether. The combined organic portions were washed twice each (10 mL) with 10% NaOH and brine (10 mL), and dried (Na2SO4). The solvent was removed in vacuo and the residue was chromatographed on silica gel (petroleum ether and EtOAc) to give the product.:EtOAc = 9:1) to give 1a (6 mg, 7%) and 6a (58 mg, 72%).:EtOAc = 9:1) to give 1a (14 mg, 17%) and 6a (45 mg, 56%).Author's contributions
T.-Y. L. and G.-Q. L. contributed to the conception and design of this study in addition to the data analysis. P. Z. and Z. L. contributed intellectually and took the responsibility to supervise L. Y., a graduate student, working on this project in the laboratory. All co-authors actively participated in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T.-Y. L. thanks the Ministry of Science and Technology (Taipei) for partial support. P. Z. thanks the Hangzhou Normal University for a start-up research fund.Notes and references
-
(a) A. de Meijere and F. Diederich, Metal-catalyzed cross coupling reactions; 2nd completely revised and enlarged edition, vol. 1 and 2, Wiley-VCH, Mannheim, 2004 Search PubMed
; (b) Y. Nishihara, Applied cross coupling reactions, Springe, 2013 Search PubMed
-
(a) K. Tamao, K. Sumotani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374 CrossRef CAS
-
(a) E.-i. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2017 CrossRef CAS PubMed
- Y. Motoyama and H. Nishiyama, in Lewis Acids in Organic Synthesis, ed., H. Yamamoto, Wiley-VCH, 2000, ch. 3 Search PubMed
-
(a) Y.-H. Chen, T.-Y. Luh, G.-H. Lee and S.-M. Peng, J. Chem. Soc., Chem. Commun., 1994, 2369 RSC
-
(a) C. Peppe, E. S. Lang, F. M. de Andrade and L. B. de Castro, Synlett, 2004, 10, 1723 CrossRef
-
(a) H. M. Wang, P. Cui, G. Zou, F. Yang and J. Tang, Tetrahedron, 2006, 62, 3985 CrossRef CAS
-
(a) E. Bergmann, H. Taubadel and H. Weiss, Ber., 1931, 64, 1493 Search PubMed
-
(a) T.-Y. Luh, Acc. Chem. Res., 1991, 24, 257 CrossRef CAS
-
(a) Z.-J. Ni and T.-Y. Luh, J. Chem. Soc., Chem. Commun., 1987, 1515 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ra00841f |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |