
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of a novel heterogeneous sulfite activation with copper(I) sulfide (Cu2S) for efficient iohexol abatement
Ying Wu,
Danying Xing,
Linna Zhang,
Hualiang Suo and
Xiaodan Zhao *
*
College of Chemical Engineering, Department of Environmental Science & Engineering, Huaqiao University, Xiamen, 361021, Fujian, China. E-mail: zhaoxd_1987@163.com; Fax: +86-592-6162300; Tel: +86-592-6166216
First published on 11th March 2022
Abstract
Transition metal ion-activated sulfite autoxidation processes for the production of sulfate radicals (SO4˙−) have been widely investigated to achieve efficient abatement of recalcitrant organic pollutants. However, these homogeneous processes suffered from narrow effective pH range and metal release, thus restricting their practical application. In order to address this problem, we report a simple and efficient approach to iohexol abatement by a combined Cu2S and sulfite process (simplified as Cu2S/sulfite process) based on the superior activation performance of copper and the excellent electron donating capacity of the low-valent sulfur species. Compared with typical copper oxides, Cu2S can significantly accelerate the sulfite autoxidation to generate radicals, leading to 100% iohexol abatement in the Cu2S/sulfite process. The influence of solution pH and dissolved oxygen on iohexol abatement is also investigated. Qualitative and quantitative analysis of reactive radicals is performed by electron paramagnetic resonance (EPR) and radical quenching experiments. Generation of SO4˙− from sulfite activation with Cu2S mainly contributes to the iohexol abatement. X-ray photoelectron spectroscopy (XPS) suggests that copper is the main activation site and the reductive sulfur species can achieve the continuous regeneration of copper. Application potential of the Cu2S/sulfite process is also assessed. This study provides a new method for the treatment of water and wastewater containing organic micropollutants.
1 Introduction
Advanced oxidation processes (AOPs) based on sulfate radicals (SO4˙−) have attracted increasing research interest owing to SO4˙− possesses having high standard redox potential (2.5–3.1 V), excellent selectivity and favorably long life (τ = 30–40 μs).1,2 The generation of SO4˙− and other reactive radicals including SO3˙−, SO5˙− and HO˙ from sulfite autoxidation has been recently investigated for the abatement of organic contaminants.3–5 In comparison with other traditional SO4˙− precursors including persulfate (PS) and peroxymonosulfate (PMS), sulfite has great potential for application due to its cost-effectiveness and non-toxicity.6 However, the single sulfite process is not applicable due to its negligible autoxidation rate. Therefore, activation of sulfite with addition of the appropriate activators is necessary in the water treatment process.Homogeneous processes of sulfite activation with transition metal ions including Fe(II), Cu(II), Co(II) and Mn(II) have been demonstrated to be effective for the abatement of organic contaminants.6 In the sulfite activation process, transition metal ions (Me(II)) react with sulfite to form complexes (Me(II)–SO32−) via a rapid complexation equilibrium process. This complex is then transformed to Me(III)–SO32− complexes in the presence of dissolved oxygen, which can be decomposed to Me(II) and SO3˙−, initiating the following oxysulfur radicals chain reactions. In comparison with Fe(II), Co(II) and Mn(II), Cu(II) exhibits great capability in promoting sulfite autoxidation to generate radicals under circumneutral conditions, in which Cu(II)–SO32− complexes can directly generate Cu(I) and SO3˙−. For example, Zhao et al. reported that iohexol abatement was mainly attributed to SO4˙− produced in the Cu(II)/sulfite process under aerobic conditions.7 Yang et al. demonstrated that Cu(I) was mainly responsible for diatrizoate abatement for the Cu(II)/sulfite process under anaerobic conditions.8 However, the application of Cu(II) can be compromised by the potential toxicity and difficult operation of recycling. To address these limitations, heterogeneous activators can be an important substitute for the sulfite activation.
Recently, activation of sulfite with the heterogeneous copper-bearing nanoparticles has been investigated extensively. Zhao et al. reported that copper ferrite (CuFe2O4) significantly accelerated the sulfite autoxidation rate to generate SO4˙− for the effective removal of iohexol (80%) within 2 min at pH 8.0.3 Xiao et al. demonstrated that copper oxides with dominantly exposed (0 0 1) reactive facets exhibited an excellent sulfite activation performance with remarkable degradation of tetracycline (87%) within 5 min for the pH range of 4.5–9.0.9 Ding et al. prepared a low-leaching silica-supported copper catalyst, which exhibited excellent activation efficacy of sulfite with 90% of As(III) oxidation in neutral wastewater.10 In the sulfite activation process with copper-bearing nanoparticles, it is proposed that the decomposition of the surface >Cu(II)–SO32− complexes to Cu(I) and SO3˙− is the key step and the >Cu(II)/>Cu(I) redox cycling is critical to the acceleration of sulfite autoxidation. However, the efficiency of this >Cu(II)/>Cu(I) redox cycling is limited due to the absence of electron-donating species. Recently, sulfur, as an efficient electron donor, was reported to be favorable for the radical generation and contaminant abatement in AOPs.11,12 Especially, He et al. demonstrated that copper sulfides (Cu2S and CuS) showed good performance in the heterogeneous Fenton reactions due to the synergy between copper and S(−II).13 Therefore, it can be proposed that Cu2S and CuS could be employed as the activator to accelerate the sulfite autoxidation. Especially, Cu2S is expected to be more advantageous than CuS in maintaining stable surface functional groups via the enhanced >Cu(I)/>Cu(II) cycling by sulfur and its application in sulfite activation has never been studied previously and needs to be explored.
Herein, the Cu2S/sulfite process is studied for abatement of iohexol, which is a widely-used iodinated pharmaceutical and frequently detected in the aquatic environment. The objectives of this study are (i) to validate the feasibility of Cu2S in the sulfite activation and examine the corresponding iohexol abatement performance; (ii) to explore the dominant radicals in the iohexol abatement by EPR, fluorescence detection and radical scavenging experiments; (iii) to investigate the sulfite activation mechanism by Cu2S via XPS and (iv) to assess the application potential of the Cu2S/sulfite process in the real water matrixes.
2 Results and discussion
2.1 Characterization of the activator
Fig. 1(a) illustrates the phase information of Cu2S via X-ray diffraction (XRD) measurement. It can be seen that Cu2S exhibits the characteristic diffraction peak of the standard Cu2S spectra (JCPDS card no. 84-0208). The diffraction peaks at 2θ = 37.1°, 45.6° and 48.0° are attributable to the (1 0 2), (1 1 0) and (1 0 3) crystal planes of Cu2S. Fig. 1(b) shows the microstructure of Cu2S via transmission electron microscopy (TEM) characterization. Irregular shape of the Cu2S particles is observed with the lattice spacings of 0.243 nm corresponding to the (1 0 2) crystalline plane. | ||
| Fig. 1 (a) XRD pattern and (b) TEM images of Cu2S. | ||
2.2 Feasibility of sulfite activation by Cu2S for iohexol abatement
Fig. 2 presents a comparison of iohexol abatement efficiency by sulfite activation with the copper based activators including CuO, Cu2O, CuS and Cu2S. It can be seen that the iohexol abatement efficiency by sulfite activation with Cu2S, CuS, CuO and Cu2O is 95.6%, 88.6%, 81.0% and 15.0%, respectively. Apparently, CuS and Cu2S exhibit better iohexol abatement performance than the corresponding copper oxides. Especially, iohexol can be completely degraded within 30 s during sulfite activation with Cu2S, which is significantly higher than that of Cu2O. It is worthy to point out that the adsorption of iohexol by Cu2S is negligible. The above results suggest that the reductive sulfur might play an important role in the sulfite activation process. The rate of sulfite decay in the presence of the copper based activators follows an order of Cu2S > CuS > CuO > Cu2O (Fig. 2(b)), which also reflects that copper sulfides show superior activation performance in accelerating sulfite autoxidation.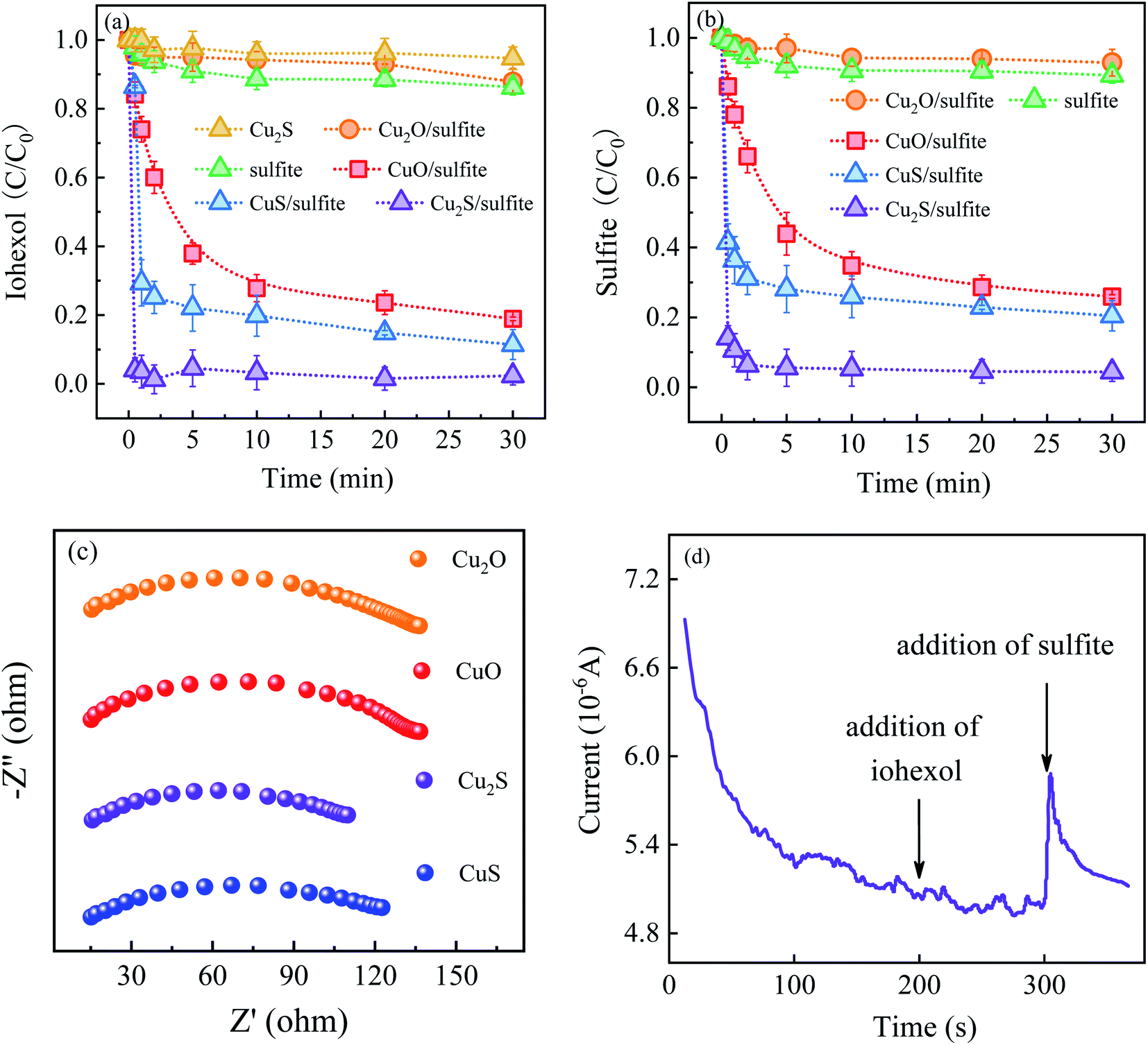 | ||
| Fig. 2 (a) Comparison of iohexol abatement by sulfite activation with CuO, Cu2O, CuS, Cu2S; (b) sulfite decay; (c) EIS Nyquist plots of above activators and (d) i–t response curve in the Cu2S/sulfite process. Experimental conditions: [sulfite] = 500 μM, [CuO]/[Cu2O]/[CuS]/[Cu2S] = 0.025 g L−1, [iohexol]0 = 10 μM, pH = 8.0. | ||
In the previous studies, Liu et al. demonstrated that basic copper(II) carbonate showed efficient activation of sulfite with 94% of As(III) removal at 20 min (pH 8.0).14 An et al. reported a hollow-structured self-floating copper-loaded catalyst for sulfite activation, which could achieve the efficient abatement of As(III) (93.7%) within 20 min at pH 7.5.15 Dong et al. prepared a zero-valent Fe–Cu bimetallic nanoparticles to facilitate sulfite activation with 87% abatement of sulfamethazine at 60 min (pH 6.0).16 Compared to the aforementioned copper-based activated sulfite processes, Cu2S exhibits excellent iohexol abatement efficiency of 95.6% within 0.5 min at pH 8.0. The rapid iohexol abatement is likely due to the electron transfer promoted by the reducing sulfur species, which is further confirmed by electrochemical characterization. As shown in Fig. 2(c), EIS Nyquist plots confirm the lower electrical impedance of the copper sulfides, which is favorable for the advanced oxidation processes. The i–t response curve in the Cu2S/sulfite process demonstrates the electron transfer between Cu2S and sulfite based on the occurrence of current response after addition of sulfite (Fig. 2(d)).
Fig. 3 shows the effect of solution pH on iohexol abatement and sulfite decay in the Cu2S/sulfite process. As illustrated in Fig. 3(a), iohexol abatement is dependent on solution pH. Lower iohexol abatement efficiencies (<30%) are observed in the pH range of 4.0–6.0. With the increase of solution pH, iohexol abatement is elevated with the efficiency being 72.0% at pH 7.0. For further increase of pH from 7.0 to 10.0, complete iohexol abatement (∼100%) can be achieved. The trend of sulfite decay, as shown in Fig. 3(b), is consistent with the iohexol abatement. This suggests that the formation of reactive radicals is corresponding to the sulfite autoxidation process, which is influenced by solution pH.
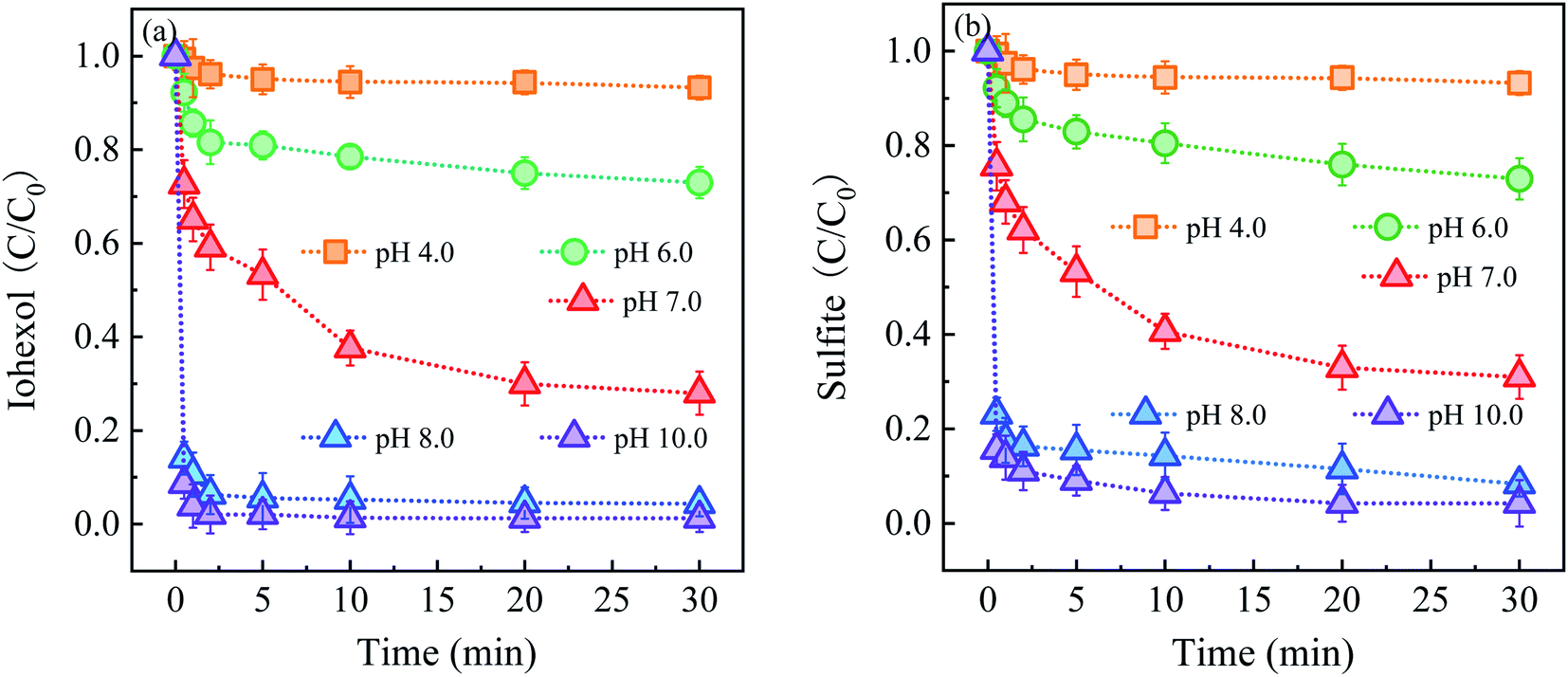 | ||
| Fig. 3 (a) Effect of solution pH on iohexol abatement and (b) sulfite decay in the Cu2S/sulfite process. Experimental conditions: [sulfite] = 500 μM, [Cu2S] = 0.025 g L−1, [iohexol]0 = 10 μM. | ||
The effect of solution pH on iohexol abatement and sulfite decay can be interpreted as the following reasons. On the one hand, the distribution of HSO3− species (HSO3−/SO32−, pKa = 7.2) is dependent on pH. HSO3− is the main species for the acidic conditions while SO32− is the dominating species for the alkaline conditions. According to a previous study, SO32− is easily activated to produce SO3˙− compared with HSO3− owing to the lower redox potential of SO3˙−/SO32− (0.63 V) than that of SO3˙−/HSO3− (0.84 V).17 On the other hand, the presence of the deprotonated copper complexes (>Cu(I)–OH and >Cu(II)–OH) under the alkaline conditions is favorable for the reaction with SO32− with the corresponding formation of the surface >Cu(I)–SO32− and >Cu(II)–SO32− complexes. This formation of complexes has been confirmed to be important for accelerating sulfite autoxidation to generate reactive radicals in the presence of the heterogeneous transition metal based activators.10 Regarding the combined effect of distribution of HSO3− species and the formation of the above surface complexes, iohexol abatement is more favorable in the Cu2S/sulfite process for the near-neutral and alkaline conditions than the acidic conditions.
2.3 Mechanistic insights
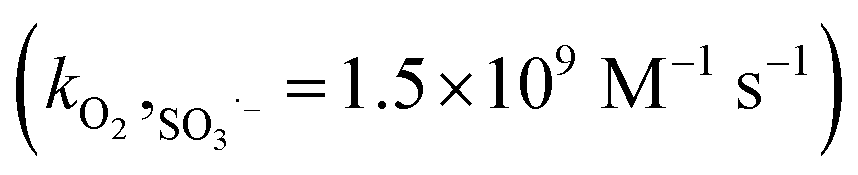 ,19 the higher dosage of DMPO (100 mM) than dissolved oxygen (0.25 mM) leads to the interruption of the subsequent chain reactions of the oxysulfur radicals. The formation of SO3˙− reflects the potential generation of the other oxysulfur radicals.
,19 the higher dosage of DMPO (100 mM) than dissolved oxygen (0.25 mM) leads to the interruption of the subsequent chain reactions of the oxysulfur radicals. The formation of SO3˙− reflects the potential generation of the other oxysulfur radicals.
 | ||
| Fig. 4 (a) Fluorescence spectra of CuO, Cu2O, CuS and Cu2S for sulfite activation, (b) effect of radical scavengers on iohexol abatement in the Cu2S/sulfite process, (c) EPR spectra of the Cu2S/sulfite process and (d) quantification of SO4˙− and HO˙ in the Cu2S/sulfite process. Experimental conditions: [sulfite] = 500 μM, [CuO]/[Cu2O]/[CuS]/[Cu2S] = 0.025 g L−1, [iohexol]0 = 10 μM, [MeOH]/[TBA] = 10 mM, [coumarin] = 5 mM, pH = 8.0. | ||
From the quantitative perspective, radical quenching experiments and direct quantification of SO4˙− and HO˙ were performed. Methanol (MeOH) and tert-butanol (TBA) are used as the radical scavengers,20,21 and the contribution of SO4˙− and HO˙ to iohexol abatement in the Cu2S/sulfite process can be differentiated, as shown in Fig. 4(c). It can be seen that TBA exerts an unnoticeable inhibitory effect on iohexol abatement, which rules out the contribution of HO˙. In comparison, iohexol abatement in the presence of MeOH is considerably suppressed with a reduction of abatement efficiency as high as 80%. Therefore, SO4˙− is the dominant radical being contributable to iohexol abatement. Then, MeOH and dimethyl sulfoxide (DMSO) are employed to quantify the production of SO4˙− and HO˙ in the Cu2S/sulfite process.22,23 Formaldehyde is quantitatively produced via the respective reactions of MeOH and DMSO with SO4˙− and HO˙. Then the formed formaldehyde can react with 2,4-dinitrophenylhydrazine to form the corresponding hydrazone, as illustrated in Fig. 4(d). It can be seen that the Cu2S/sulfite process under the studied conditions can produce 9.8 μM of SO4˙− and 1.2 μM of HO˙. This result further confirms that SO4˙− is the dominating radical contributing to iohexol abatement.
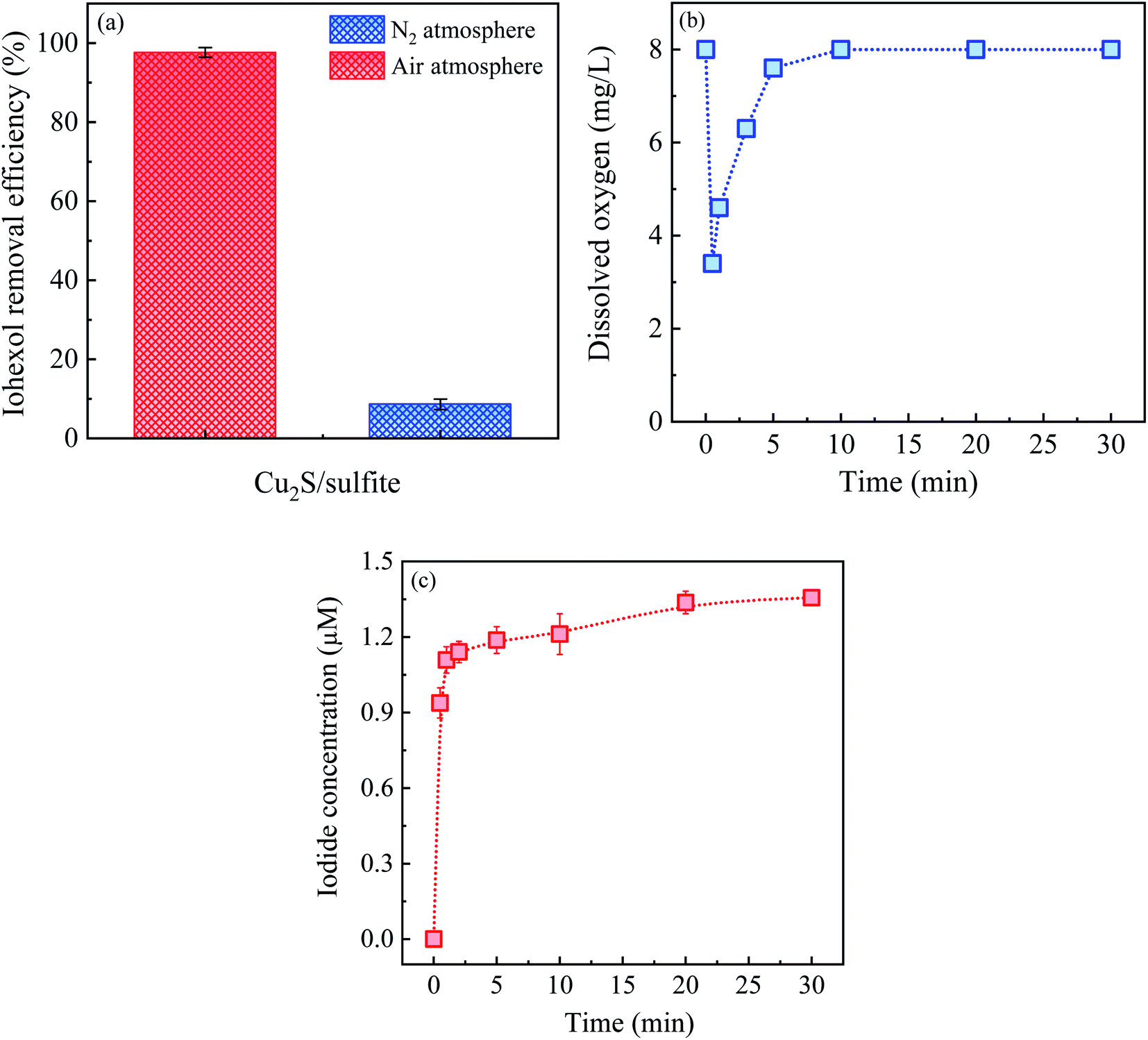 | ||
| Fig. 5 (a) Iohexol abatement under aerobic and anaerobic conditions, (b) variation of dissolved oxygen concentration in the Cu2S/sulfite process and (c) iodide release in the Cu2S/sulfite process. Experimental conditions: [sulfite] = 500 μM, [Cu2S] = 0.025 g L−1, [iohexol]0 = 10 μM, pH = 8.0. | ||
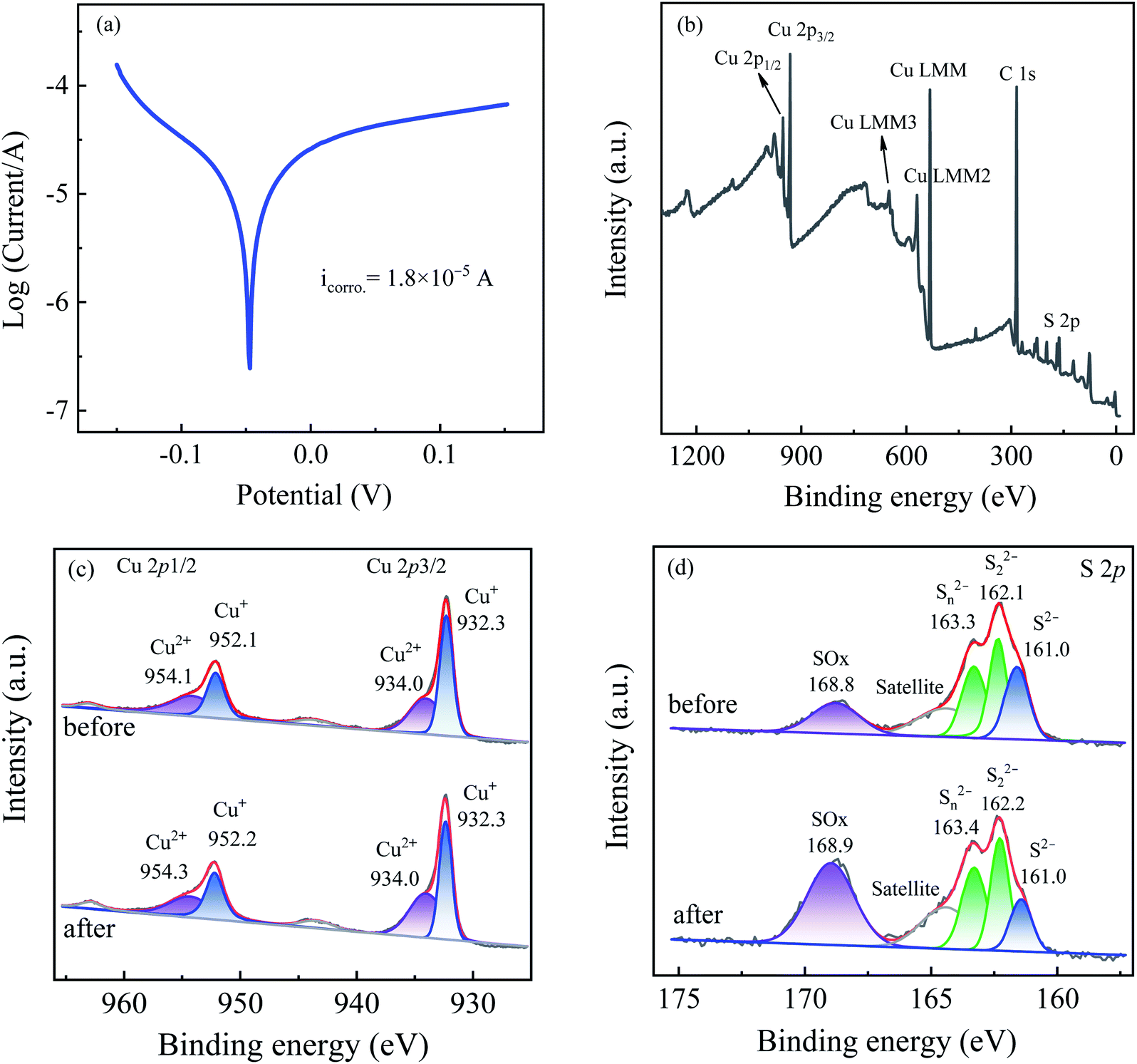 | ||
| Fig. 6 (a) Tafel scans of Cu2S electrode, (b) XPS survey spectrum, (c) Cu 2p and (d) S 2p XPS spectra before and after use in the Cu2S/sulfite process. Experimental conditions: [sulfite] = 500 μM, [Cu2S] = 0.025 g L−1, [iohexol]0 = 10 μM, pH = 8.0. | ||
The S 2p spectra displayed in Fig. 6(d) indicates the existence of monosulfide (S2−), polysulfides (S22−/Sn2−) and sulfate (SO42−) at 160.0 eV, 162.1/163.4 eV, and 168.8 eV,26 which account for 24.8%, 54.5% and 20.7% of the total sulfur species. After the reaction, the relative intensities of S2− and S22−/Sn2− are reduced to 12.3% and 45.1% with a concomitant increase of the SO42− intensity (42.6%), indicating the transformation of S2− and S22−/Sn2− to SO42−. The oxidation of S2− and S22−/Sn2− to SO42− takes a dominant role in the transformation of >Cu(II)–SO32− to >Cu(I)–SO32− and guarantees the regeneration of the >Cu(I) species, which in turn explains the negligible variation of the chemical valence of the copper species. In conclusion, the sulfur species on the Cu2S particles mainly contributes to the enhanced performance of Cu2S in accelerating the sulfite autoxidation and the corresponding radical generation.
On the basis of the above results, the sulfite activation mechanism by Cu2S for efficient iohexol abatement is illustrated in Fig. 7. The copper species on the Cu2S surface serves as the dominant active sites, which is critical for accelerating sulfite autoxidation in the presence of dissolved oxygen. The regeneration of Cu(I) by S2− is thermodynamically feasible because the standard reduction potential of sulfur (S2−/S0, −0.48 V) is much lower than that of copper (Cu(II)/Cu(I), 0.167 V).27 The consumption of sulfur species and the corresponding accumulation of SO42−, as shown in the XPS results, lead to a reduction of the sulfite activation efficiency of Cu2S, which is confirmed by the decrease of iohexol abatement efficiency during reusability tests. The reductive sulfur species of Cu2S is transformed to SO42− via a series of reaction and the generated electron could be favorable for the transformation of the >Cu(II)–SO32− complexes to the >Cu(I)–SO32− complexes with a concomitant generation of SO3˙−. This process can in turn achieve the redox cycling of >Cu(I)/>Cu(II) and maintain the stable surface copper species, which guarantees the higher overall iohexol abatement efficiency. In the presence of dissolved oxygen, the produced sulfite radical (SO3˙−) initiates the oxysulfur radical chain reactions (SO4˙−, SO5˙− and HO˙). The produced reactive radicals (SO4˙−) are the dominant species being responsible for efficient abatement of iohexol.
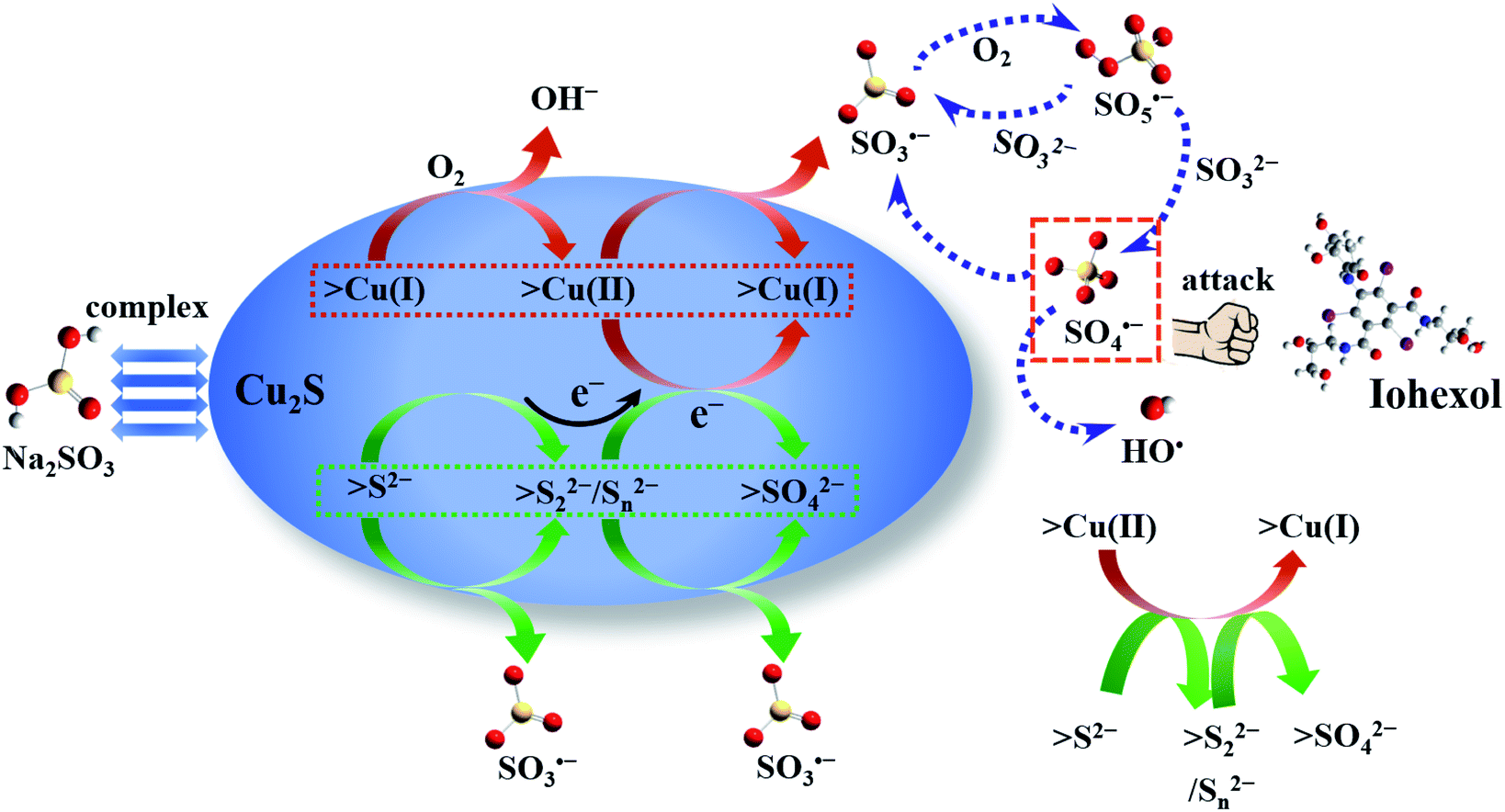 | ||
| Fig. 7 The proposed mechanism in the Cu2S/sulfite process. | ||
2.4 Application potential of the Cu2S/sulfite process in the real water matrix
Fig. 8(a) and (b) displays the effect of coexisting cations and anions on iohexol abatement in the Cu2S/sulfite process. As can be seen, the presence of cations exerts slightly inhibitory effect on iohexol abatement. This phenomenon is probably due to that the increase of ionic strength can affect the interaction between the copper and sulfite. In contrast, the anions exhibit inhibitory effect to different extent. Cl− and HPO42− exhibit considerably inhibitory effect on iohexol abatement whereas the presence of HCO3− exerts relatively lower inhibition and NO3− shows minimal effect. The increase of Cl− concentration from 0 mM to 2 mM leads to a significant reduction of iohexol abatement (∼73%), which could be ascribed to the radical quenching effect of Cl−.28 With regard to the effect of HPO42−, the iohexol abatement efficiency is reduced by 79% when the concentration of HPO42− is increased to 8 mM. This phenomenon may be due to the complexation of the active copper sites by HPO42−, which hinders the complexation reaction between copper and sulfite.29 Regarding the effect of HCO3−, iohexol abatement efficiency is reduced from 77.6% to 55.7% when the concentration of HCO3− is in the range of 2 mM to 8 mM. This inhibitory effect is caused by the radical quenching effect of HCO3−.30 The minor effect of NO3− on iohexol abatement is owing to negligible quenching effect of SO4˙−.10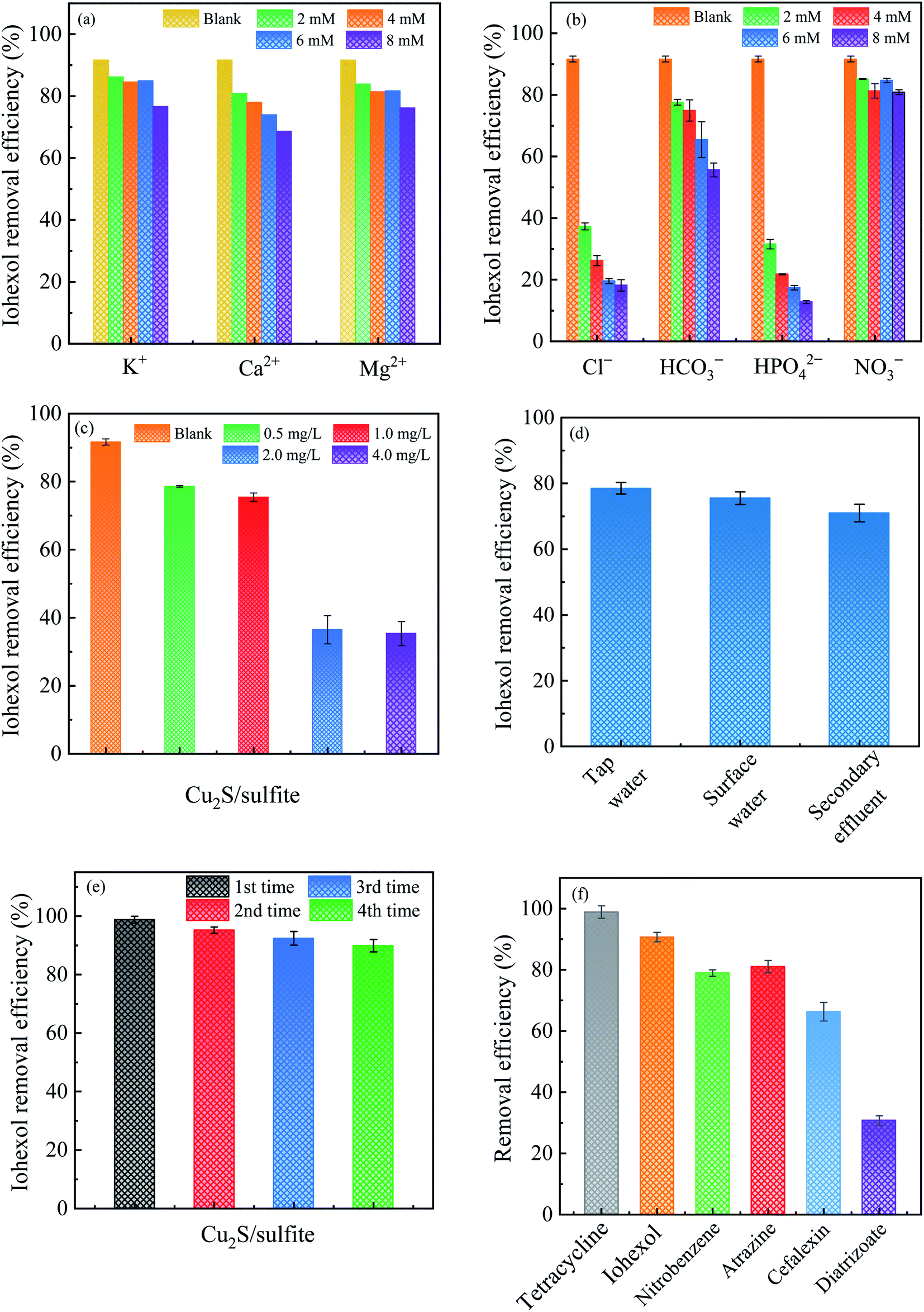 | ||
| Fig. 8 Effects of (a) cations, (b) anions, (c) humic acid, (d) different real waters on iohexol abatement, (e) iohexol abatement during four cycles and (f) removal efficacy of various pollutants in the Cu2S/sulfite process. Experimental conditions: [sulfite] = 500 μM, [Cu2S] = 0.025 g L−1, [organic pollutants]0 = 10 μM, pH = 8.0. | ||
Fig. 8(c) illustrates the effect of humic acid on iohexol abatement in the Cu2S/sulfite process. An increase of humic acid concentrations from 0 to 4.0 mg L−1 results in a decrease of iohexol abatement efficiency from 91.5% to 35.3%. This indicates that humic acid shows a significantly inhibitory effect due to its radical quenching effect with the apparent second order rate constant of 2.35 × 107 M−1 s−1 and 3.0 × 108 M−1 s−1 for SO4˙− and HO˙, respectively.31 Fig. 8(d) presents the iohexol abatement in the tap water, surface water and secondary effluent from a municipal wastewater treatment plant. The iohexol abatement in the Cu2S/sulfite process is inhibited in the real water matrixes. This can be ascribed to the coexistence of anions and humic matter. The reusable performance of Cu2S shown in Fig. 8(e) suggests that Cu2S exhibits good reusability with slight reduction of iohexol abatement efficiency after recycled for 4 times. The decrease of the activation performance may be due to the consumption of the reductive sulfur species and the accumulation of SO42− on the Cu2S surface.32,33 Fig. 8(f) gives the efficacy of the Cu2S/sulfite process towards different organic pollutants including tetracycline, cefalexin, atrazine, diatrizoate and nitrobenzene. The abatement efficiencies for the above pollutants are 95.9%, 90.9%, 78.9%, 80.3% and 62.7%, respectively. The above results demonstrate the selectivity of the Cu2S/sulfite process for the abatement of organic pollutants.
3 Conclusions
In this study, a novel Cu2S/sulfite process for production of reactive radicals is proposed to achieve effective abatement of iohexol. High abatement efficiency of iohexol is obtained for the pH range of 7.0–10.0, which is ascribed to the speciation of sulfite. The radical scavenging tests and EPR analysis demonstrate that SO4˙− and HO˙ are the dominant reactive radicals in the Cu2S/sulfite process. The coexistence of NO3− shows insignificant effect on iohexol abatement while the presence of HCO3−, HPO42−, Cl− and humic acid exerts inhibitory influence due to radical scavenging effect. Superior iohexol abatement efficiencies have been obtained for the four recycles, confirming the good reusability and satisfactory activation stability. Cu2S exhibits significant advantages of applicability in wide pH range and the elevated sulfite activation efficiency with lower dosages of the activator and thus is a promising alternative for the abatement of organic pollutants in the water and wastewater treatment.4 Experimental section
4.1 Chemicals and reagents
Iohexol (purity > 99.5%) and sodium sulfite (Na2SO3, purity > 99.0%) were obtained from Xilong Scientific Co. (China). Commercially available Cu2S (purity > 99.5%), CuS (purity > 99.5%), Cu2O (purity > 99.5%) and CuO (purity > 99.5%) were supplied from Aladdin Reagent Co., China. Ethylenediaminetetraacetic acid (EDTA, purity > 99.5%) and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB, purity > 99.0%) were supplied from Xiya Reagent Co., China. DMPO (purity > 99.0%) as the spin trapping reagent in the EPR analysis was supplied from Sigma-Aldrich Co. (China). All the reaction solutions were prepared by 18.2 MΩ cm deionized water from Milli-Q water system. All the chemicals used in this work were at least analytical reagent grade and used as received.4.2 Characterizations
The electrochemical analysis including electrochemical impedance spectroscopy Nyquist plots and i–t response curve were conducted by electrochemical workstation (CHI 760E, China). The morphological details of Cu2S were observed with transmission electron microscopy (FEI Tecnai G2 F20, USA). XRD patterns were analyzed by X-ray diffractometer (SmartLa) at room temperature with Cu-Kα radiation in the range of 10–80°. XPS of Cu2S before and after activation were obtained on a Thermo Scientific Escalab 250Xi. The obtained spectra were then analyzed by XPSPEAK software and calibrated by adventitious C 1s peak at 284.8 eV as the internal standard.4.3 Experimental procedures
Iohexol abatement reactions were carried out in a 100 mL glass reactor with Teflon-lined caps and stirred constantly with mechanical agitation (200 rpm). The desired amounts of iohexol, sulfite and Cu2S were added in the aqueous solution, which was kept at 25 °C with a thermostat. In a typical iohexol abatement experiment, the concentrations for the chemicals in the reaction solution were iohexol 10 μM, sulfite 500 μM and Cu2S 0.025 g L−1. During the reaction, 1 mL sample was withdrawn at predetermined time intervals, filtered with 0.22 μm syringe filters, and immediately terminated with methanol for HPLC analysis. Diluted NaOH and H2SO4 were used to adjust solution pH. All the experiments were performed in duplicate to calculate the average values and the error bars for reproducibility.4.4 Analytical methods
Iohexol was quantified by high performance liquid chromatography (HPLC, Agilent 1290 Infinity) equipped with a UV-vis photodiode array detector (245 nm) and Waters Sunfire C18 column (4.6 mm × 150 mm, 5 μm particle size). The acetonitrile/water mixture (5%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95%) was employed as the mobile phase at a flow rate of 1 mL min−1. The quantification limit for iohexol was 0.15 μM with a lower standard deviation (<5%). The release of iodide during the iohexol degradation process was also quantified by HPLC at the wavelength of 226 nm. Mixture of acetonitrile and monobasic potassium phosphate (2%:98%) was used as the mobile phase at a flow rate of 0.80 mL min−1. The sulfite concentrations were determined employing DTNB as chromogenic reagent. At a predetermined time interval, samples were directly added into a colorimetric tube containing 1 mM EDTA, 1 mM DTNB and phosphate buffer solution. UV-vis spectrophotometer (V-1600) was used to quantify the sulfite decay during the reaction process at the wavelength of 412 nm.34 In electrochemical analysis, activators were uniformly loaded onto the glassy carbon electrode, with platinum electrode as opposite electrode, Ag/AgCl electrode as reference electrode, and all experiments were carried out in 0.1 M sodium nitrate electrolyte. EPR signals with DMPO as the spin-trapping agent (100 mM) were recorded on Bruker EMX-10/12 spectrometer (Germany). With regard to the quantification of reactive radicals, 5 mM MeOH and dimethyl sulfoxide (DMSO) were employed to react with SO4˙− and HO˙ to generate quantitatively formaldehyde, which 1 mol formaldehyde was produced from 1 mol SO4˙− and 2.17 mol HO˙. Then, 2,4-dinitrophenylhydrazine (240 μM) reacted with formaldehyde with a 1:1 ratio to form the corresponding hydrazone, which can be measured by HPLC.22,23
:95%) was employed as the mobile phase at a flow rate of 1 mL min−1. The quantification limit for iohexol was 0.15 μM with a lower standard deviation (<5%). The release of iodide during the iohexol degradation process was also quantified by HPLC at the wavelength of 226 nm. Mixture of acetonitrile and monobasic potassium phosphate (2%:98%) was used as the mobile phase at a flow rate of 0.80 mL min−1. The sulfite concentrations were determined employing DTNB as chromogenic reagent. At a predetermined time interval, samples were directly added into a colorimetric tube containing 1 mM EDTA, 1 mM DTNB and phosphate buffer solution. UV-vis spectrophotometer (V-1600) was used to quantify the sulfite decay during the reaction process at the wavelength of 412 nm.34 In electrochemical analysis, activators were uniformly loaded onto the glassy carbon electrode, with platinum electrode as opposite electrode, Ag/AgCl electrode as reference electrode, and all experiments were carried out in 0.1 M sodium nitrate electrolyte. EPR signals with DMPO as the spin-trapping agent (100 mM) were recorded on Bruker EMX-10/12 spectrometer (Germany). With regard to the quantification of reactive radicals, 5 mM MeOH and dimethyl sulfoxide (DMSO) were employed to react with SO4˙− and HO˙ to generate quantitatively formaldehyde, which 1 mol formaldehyde was produced from 1 mol SO4˙− and 2.17 mol HO˙. Then, 2,4-dinitrophenylhydrazine (240 μM) reacted with formaldehyde with a 1:1 ratio to form the corresponding hydrazone, which can be measured by HPLC.22,23
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the National Natural Science Foundation of China (Grant No. 51808233).References
- J. Zhu, H. Li, C. Shan, S. Wang, L. Lv and B. Pan, J. Hazard. Mater., 2021, 409, 124920 CrossRef CAS PubMed.
- T. Olmez-Hanci and I. Arslan-Alaton, Chem. Eng. J., 2013, 224, 10–16 CrossRef CAS.
- X. Zhao, W. Wu, G. Jing and Z. Zhou, Environ. Pollut., 2020, 260, 114038 CrossRef CAS PubMed.
- T. Sun, Y. Su, H. Liu, H. Song and Y. Lv, Chem. Commun., 2020, 56, 6993–6996 RSC.
- H. Dong, J. Chen, L. Feng, W. Zhang, X. Guan and T. J. Strathmann, Chem. Eng. J., 2019, 357, 328–336 CrossRef CAS.
- D. Zhou, L. Chen, J. Li and F. Wu, Chem. Eng. J., 2018, 346, 726–738 CrossRef CAS.
- X. Zhao, W. Wu and Y. Yan, Environ. Sci. Pollut. Res., 2019, 26, 24707–24719 CrossRef CAS.
- J. Yang, Y. Luo, X. Fu, Z. Dong, C. Wang, H. Liu and C. Jiang, Water Res., 2021, 19, 117137 CrossRef PubMed.
- B. Xiao, M. Wu, Y. Wang, R. Chen and H. Liu, Chem. Eng. J., 2021, 406, 126693 CrossRef CAS.
- W. Ding, X. Huang, W. Zhang, F. Wu and J. Li, Chem. Eng. J., 2019, 359, 1518–1526 CrossRef CAS.
- Y. Zhang, H. P. Tran, X. Du, I. Hussain, S. Huang, S. Zhou and W. Wen, Chem. Eng. J., 2017, 308, 1112–1119 CrossRef CAS.
- M. Xing, W. Xu, C. Dong, Y. Bai, J. Zeng, Y. Zhou, J. Zhang and Y. Yin, Chem, 2018, 4, 1359–1372 CAS.
- H. He, M. Sun, D. Wu, G. Di and X. Fei, J. Clean. Prod., 2021, 278, 123572 CrossRef CAS.
- Z. Liu, J. Liu, H. Yang, J. Guo, Y. Chen, Q. Shao, S. Gao and L. Luo, Chem. Eng. J., 2022, 433, 133662 CrossRef CAS.
- Y. An, M. Han, H. Zheng, W. Ding, Q. Sun, C. Hu and L. Zheng, Chem. Eng. J., 2021, 407, 127193 CrossRef CAS.
- Q. Dong, H. Dong, Y. Li, J. Xiao, S. Xiang, X. Hou and D. Chu, J. Hazard. Mater., 2022, 431, 128601 CrossRef CAS PubMed.
- W. Ding, W. Xiao, H. Zheng, S. Zhang, H. Liu, Y. An and R. Zhao, Chem. Eng. J., 2020, 402, 126168 CrossRef CAS.
- L. Chen, M. Tang, C. Chen, M. Chen, K. Luo, J. Xu, D. Zhou and F. Wu, Environ. Sci. Technol., 2017, 51, 12663–12671 CrossRef CAS PubMed.
- K. Ranguelova, S. Chatterjee, M. Ehrenshaft, D. C. Ramirez, F. A. Summers, M. B. Kadiiska and R. P. Mason, J. Biol. Chem., 2010, 285, 24195–24205 CrossRef CAS.
- C. Chen, L. Liu, J. Guo, L. Zhou and Y. Lan, Chem. Eng. J., 2019, 361, 1304–1316 CrossRef CAS.
- J. Hu, X. Zeng, G. Wang, B. Qian, Y. Liu, X. Hu, B. He, L. Zhang and X. Zhang, Chem. Eng. J., 2020, 400, 125869 CrossRef CAS.
- J. Qiao, L. Feng, H. Dong, Z. Zhao and X. Guan, Environ. Sci. Technol., 2019, 53, 10320–10328 CrossRef CAS PubMed.
- C. Tai, J. F. Peng, J. F. Liu, G. B. Jiang and H. Zou, Anal. Chim. Acta, 2004, 527, 73–80 CrossRef CAS.
- P. Xie, Y. Guo, Y. Chen, Z. Wang, R. Shang, S. Wang, J. Ding, Y. Wan, W. Jiang and J. Ma, Chem. Eng. J., 2017, 314, 240–248 CrossRef CAS.
- M. H. Conkiln and M. R. Hoffmann, Environ. Sci. Technol., 1988, 22, 8 CrossRef.
- J. Fan, L. Gu, D. Wu and Z. Liu, Chem. Eng. J., 2018, 333, 657–664 CrossRef CAS.
- T. V. Popova and N. V. Aksenova, Russ. J. Inorg. Chem., 2003, 29, 743–765 CAS.
- Y. Qi, J. Wei, R. Qu, A. B. Gadah, X. Pan, A. D. Afzal, S. Asam, D. Zhou and Z. Wang, Chem. Eng. J., 2021, 403, 126396 CrossRef CAS.
- Y. Wu, Y. Xing, X. Zhao, Z. Zhou and G. Jing, Chem. Eng. J., 2022, 429, 132404 CrossRef CAS.
- L. Zhou, C. Yan, M. Sleiman, C. Ferronato, J. M. Chovelon, X. Wang and C. Richard, Chem. Eng. J., 2019, 368, 252–260 CrossRef CAS.
- C. Jiang, Y. Ji, Y. Shi, J. Chen and T. Cai, Water Res., 2016, 106, 507–517 CrossRef CAS PubMed.
- A. Jawad, W. Lei, S. Ajmal, I. Jerosha, G. A. Gebremedhin, I. S. Irshad, E. Zouhair, Z. Chen and Z. Chen, Water Res., 2020, 181, 115862 CrossRef PubMed.
- J. Peng, X. Lu, X. Jiang, Y. Zhang, Q. Chen, B. Lai and G. Yao, Chem. Eng. J., 2018, 354, 740–752 CrossRef CAS.
- R. Humphrey, M. Ward and W. Hinze, Anal. Chem., 1970, 42, 698–702 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |