
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel method of preparing vanadium-based precursors and their enhancement mechanism in vanadium nitride preparation
Ailian Wenabcd,
Zhenlei Cai *abcd,
Yimin Zhang*abcde and
Hong Liuabcd
*abcd,
Yimin Zhang*abcde and
Hong Liuabcd
aSchool of Resource and Environmental Engineering, Wuhan University of Science and Technology, Wuhan, 430081, Hubei Province, PR China. E-mail: caizhenlei@wust.edu.cn; zym126135@126.com; Tel: +86 15271803213 Tel: +86 13907158287
bState Environmental Protection Key Laboratory of Mineral Metallurgical Resources Utilization and Pollution Control, Wuhan University of Science and Technology, Wuhan, 430081, China
cCollaborative Innovation Center of Strategic Vanadium Resources Utilization, Wuhan University of Science and Technology, Wuhan, 430081, China
dHubei Provincial Engineering Technology Research Center of High Efficient Cleaning Utilization for Shale Vanadium Resource, Wuhan University of Science and Technology, Wuhan, 430081, China
eSchool of Resource and Environmental Engineering, Wuhan University of Technology, Wuhan, 430070, China
First published on 29th April 2022
Abstract
Vanadium nitride is widely used because of its excellent properties. The existing production methods are affected by the problems of complex preparation for the vanadium source, high temperature, and low N content. In this work, a wide range of vanadium solutions were used as the vanadium source to prepare vanadium nitride with high N content. In this work, a novel precursor was prepared by a microwave-assisted precipitation process, and then the vanadium nitride was prepared by reduction and nitridation precursor at 1150 °C. The results show that in the microwave-assisted method, the particle size and structure of the precursor can be adjusted, so that the contact area of the precursor with N2 during the nitridation process becomes larger, the N2 diffusion path becomes shorter, and the formation of vanadium nitride is enhanced. The prepared product has a nitrogen content of 17.67 wt% and is composed of uniform spherical particles. The content of other chemical components and density can achieve the standard requirements specified in VN16. Meanwhile, the thermodynamic analysis showed that the NH3 generated by the thermal decomposition of the precursor can be used directly as a reducing gas to reduce V2O5, and reduced the emission of polluting gases. It is a feasible method to prepare vanadium nitride by reduction and nitridation.
1 Introduction
Transition metal nitrides are attracting increasing focus owing to their excellent physical and chemical properties.1–4 Vanadium nitride is widely used in alloy components, coatings, battery materials, supercapacitor materials, and catalytic carriers5–12 due to its high melting point, excellent wear resistance, high-temperature stability, and excellent electrical conductivity.13–17 In order to meet the ever-increasing demand for vanadium nitride and the development of high-quality vanadium nitride, new methods to prepare vanadium nitride are becoming increasingly important.Therefore, how to prepare vanadium nitride efficiently has attracted a lot of attention, and there are several methods available for the preparation of vanadium nitride. These are ammonolysis sintering,18,19 carbothermal reduction nitridation,20,21 mechanochemical synthesis,22 hydrothermal synthesis,23 deposition,24 and magnesium thermal reduction.25 However, only the carbothermic reduction of vanadium oxide is a method that can accomplish industrial production. It is characterized by co-grounding the vanadium source and reducing agent, and then prepared VN by holding the mixture above 1300–1500 °C for 4–6 h.26–28 The production of vanadium nitride using vanadium oxide exist many problems, such as high reaction temperature, long reaction time and low N content of vanadium nitride.
Therefore, researchers have improved the preparation process of vanadium nitride to make it more mature with lower temperature, shorter time, higher quality and better performance. Qin used an ultra-fast dissolution method to prepare VO2 precursor in the form of porous foam, and then the precursor was ammoniated at 700 °C for 2 h to prepare VN.18 Han reported that precursor containing vanadium source and carbon source was precipitated using vanadium-rich liquid as the vanadium source. Vanadium nitride was prepared by reduction nitride of the precursor at 1150 °C for 1 h, with a N content of 16.38 wt%. Compared with the conventional carbon thermal reduction technique, the reaction temperature can be reduced and the reaction time can be shortened.29 In addition, Huang used microwave efficient heating to promote the preparation of vanadium nitride, and the N content of the VN was 12–16 wt%.30 However, studies on the precursor properties are not common.
In the material preparation process, microwave is used to assisted in the preparation of precursors. The purpose is to reduce the time and improve the comprehensive performance. It was reported that after microwave-assisted in the hydrothermal reaction, Ce(OH)CO3 hexagonal microplate precursor were formed in a short time, and then heating decomposition the precursor can product CeO2 powder, which improved the catalytic performance and physicochemical properties of CeO2.31–33 Liu synthesized zinc layered double hydrotalcites (ZnIn-LDHs) precursor by microwave hydrothermal method and prepared zinc layered double oxides (ZnIn-LDOs) by controlled calcination method using precursor. The catalytic properties of the products were improved.34
Herein, we investigated the preparation of precursor by microwave-assisted precipitation and their enhancement mechanism on the vanadium nitride production. The thermodynamic behavior related to the precursor decomposition was analyzed. The effects of the precursor prepared by different conditions on vanadium nitride were studied. In addition, the mechanism of the enhanced properties of vanadium nitride prepared after microwave assistance was discussed by analyzing the phase, microstructure and particle size distribution of the precursor.
2 Materials and methods
2.1 Materials
The vanadium solution used was obtained by blank roasting, acid leaching, and solvent extraction from vanadium shale obtained from Tongshan, Hubei, China. The main chemical composition of the solution is shown in Table 1. Carbon black containing 94.80 wt% of carbon with a particle size of less than −0.074 mm was used as the reducing agent.| Element | V | Al | Fe | Na | K | P |
| Concentration | 25.73 | 5.78 | 0.06 | 0.42 | 0.34 | 0.10 |
2.2 Material characterization
Vanadium concentration of the solution was measured through ferrous ammonium sulfate titrimetry.35The nitrogen content (N content) was analyzed and determined by distillation–neutralization titration.
X-ray diffraction (XRD) patterns were performed using a Rigaku D/MAX-RB X-ray diffractometer (Rigaku, Akishima, Japan) with Cu Kα radiation to analyze the phase compositions in the products.
Microscopic observation and elemental analysis were performed using scanning electron microscopy (JSM-IT300, Jeol, Tokyo, Japan) equipped with X-ACT energy dispersive X-ray attachment (Oxford Instruments, Oxford, UK).
Thermogravimetric analysis (TG) experiments were performed using an STA449C analyzer (Netzsch, Germany), heated from room temperature to 1400 °C at a heating rate of 10 °C min−1, and a N2 flow rate of 50 mL min−1.
2.3 Synthesis of vanadium nitride
The process flow sheet of vanadium nitride preparation is shown in Fig. 1. Typically, vanadium(IV) was oxidized to vanadium(V) by adding an appropriate amount of NaClO3 to the vanadium solution. After vanadium(IV) complete oxidation, C was added to the solution and a mixed slurry was obtained. Adjusted the pH of the slurry to 1.9 with ammonia, and then stirred for 30 min in a microwave reactor with a power of 400 W and a temperature of 90 °C. After solid–liquid separation, all precursor were dried. Pressed into a cylindrical block with a diameter of 30 mm under a pressure of 10 KN. Put the cylindrical block into a tubular atmosphere furnace (BTF-1700C, AnHui BEQ Equipment Technology Co., Ltd) with a certain flow rate of nitrogen (99.999 wt%). The sample was calcined at 550 °C for 20 min to remove ammonia and pre-reduction at 650 °C for 120 min, and then heated to the required temperature for reduction and nitridation reaction. Subsequently, the product was cooled to room temperature in a N2 atmosphere and taken out for testing.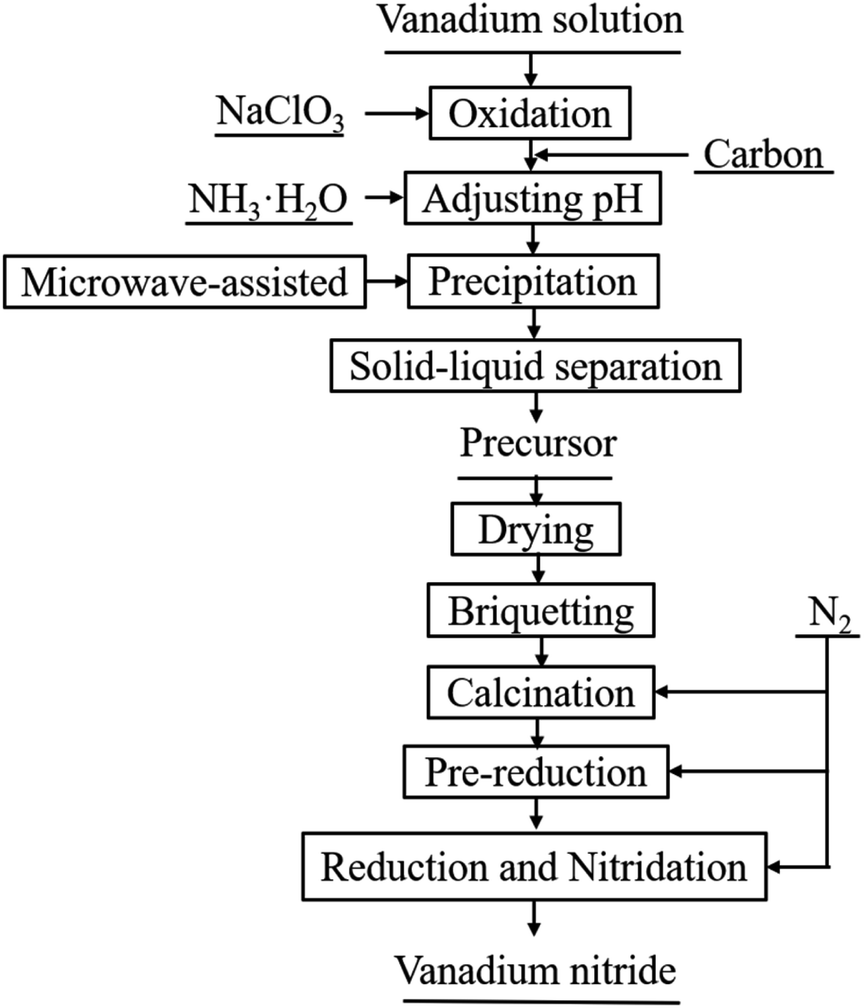 | ||
| Fig. 1 Process flow sheet for preparation of vanadium nitride. | ||
3 Thermodynamic theory of precursor decomposition
In addition to stable V2O5, VO2, V2O3, and VO, there are some vanadium oxides in other valence states, such: V3O5, V3O7, V4O9, and V6O13 in the V–O system. In this study, thermodynamic analysis showed that the existence state of V in the precursor prepared by microwave-assisted is (NH4)2V6O16·1.5H2O. (NH4)2V6O16·1.5H2O is heated and decomposed to generate V2O5 and NH3. The N2 in the tube furnace will use a protective atmosphere at low temperature, and NH3 can reduce vanadium(V) to low-value vanadium. With (NH4)2V6O16·1.5H2O as the vanadium source, the possible reaction equations and corresponding thermodynamic data of the heating deamination process areas in Table 2.| Number | Reaction | ΔGθ (T)/J mol−1 | T/K |
|---|---|---|---|
| 1 | (NH4)2V6O16·1.5H2O(s) = 3V2O5(s) + 2NH3(g) + 2.5H2O(g) | No (NH4)2V6O16·1.5H2O data | — |
| 2 | 9/10V2O5(s) + NH3(g) = 3/5V3O5(s) + 1/2N2(g) + 3/2H2O(g) | No V3O5 data | — |
| 3 | 9/2V2O5(s) + NH3(g) = 3V3O7(s) + 1/2N2(g) + 3/2H2O(g) | No V3O7 data | — |
| 4 | 3V2O5(s) + NH3(g) = 3/2V4O9(s) + 1/2N2(g) + 3H2O(g) | No V4O9 data | — |
| 5 | 9/4V2O5(s) + NH3(g) = 3/4V6O13(s) + 1/2N2(g) + 3/2H2O(g) | No V6O13 data | — |
| 6 | 3V3O7(s) + NH3(g) = 3/2V6O13(s) + 1/2N2(g) + 3/2H2O(g) | No V6O13 and V3O7 data | — |
| 7 | 3/2V3O7(s) + NH3(g) = 9/4V2O4(s) + 1/2N2(g) + 3/2H2O(g) | No V3O7 data | — |
| 8 | 2/3V6O13(s) + NH3(g) = 2V2O4(s) + 1/2N2(g) + 2/3H2O(g) | No V6O13 data | — |
| 9 | 3/2V2O5(s) + NH3(g) = 3/2V2O4(s) + 1/2N2(g) + 3/2H2O(g) | ΔGθ = −235![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 615.4−373.075T 615.4−373.075T |
631 |
| 10 | 3/4V2O5(s) + NH3(g) = 3/4V2O3(s) + 1/2N2(g) + 3/2H2O(g) | ΔGθ = −314037.7−609.024T |
516 |
| 11 | 1/2V2O5(s) + NH3(g) = VO(s) + 1/2N2(g) + 3/2H2O(g) | ΔGθ = 36534.3–304.002T |
120 |
| 12 | 3/10V2O5(s) + NH3(g) = 3/5V(s) + 1/2N2(g) + 3/2H2O(g) | ΔGθ = 1336803.0–1489.893T |
897 |
| 13 | 3/10V2O5(s) + NH3(g) = 3/5VN(s) + 1/5N2(g) + 3/2H2O(g) | ΔGθ = 77191.1–1014.606T |
76 |
Analyzed the reaction equation according to the corresponding thermodynamic data. Under the standard state, the ΔGθ − T diagram of the reaction is as Fig. 2. Combined with the calculation in Table 1 and Fig. 2, it can be seen that V2O5 is easily reduced to low-valent vanadium oxide by NH3, and may be reduced to V2O4 and V2O3 at room temperature. At higher temperatures, V2O5 is reduced to other vanadium compounds. When the temperature is higher than 897 K, V2O5 can be reduced to V. When the amount of NH3 is sufficient, V2O5 has been reduced to V2O3, so reactions (11)–(13) will not occur.
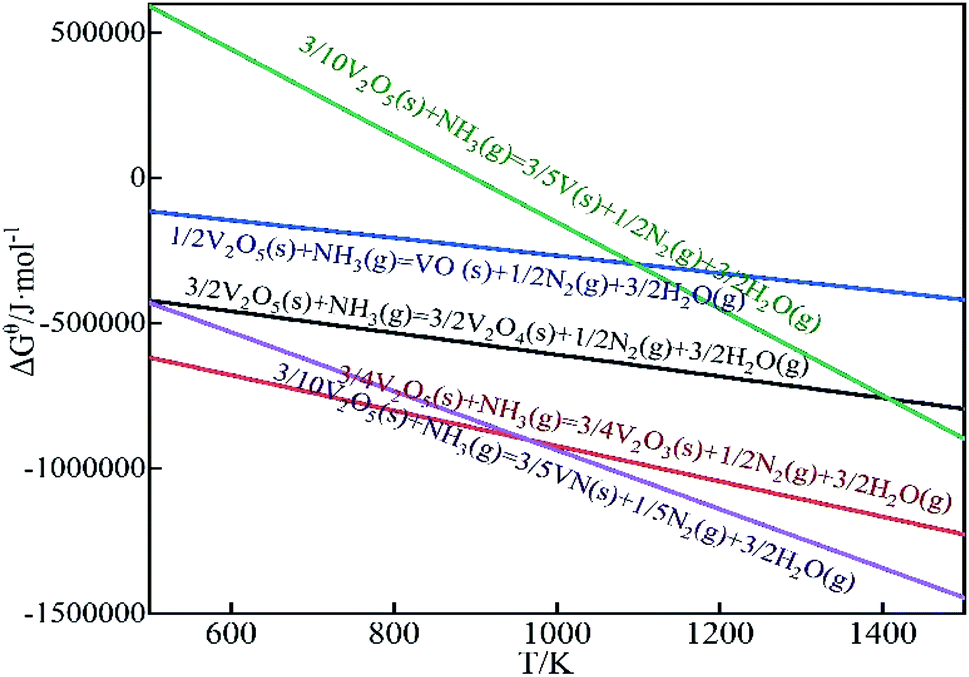 | ||
| Fig. 2 The ΔGθ − T diagram of the calcination deamination process. | ||
According to the research of scholars, the precursor (NH4)2V6O16·1.5H2O can be decomposed at a lower temperature.36 Under N2 as a protective atmosphere, low-temperature decomposition produces NH3. Due to the limited composition and quality of the raw materials, the generated NH3 is not enough to reduce it all. That is, NH3 partially reduces V2O5. Therefore, the vanadium oxides that may be generated through the calcination deamination process are V3O5, V3O7, V4O9, V6O13, V2O4, V2O3.
4 Results and discussion
4.1 Effect of main reaction conditions on N content of vanadium nitride
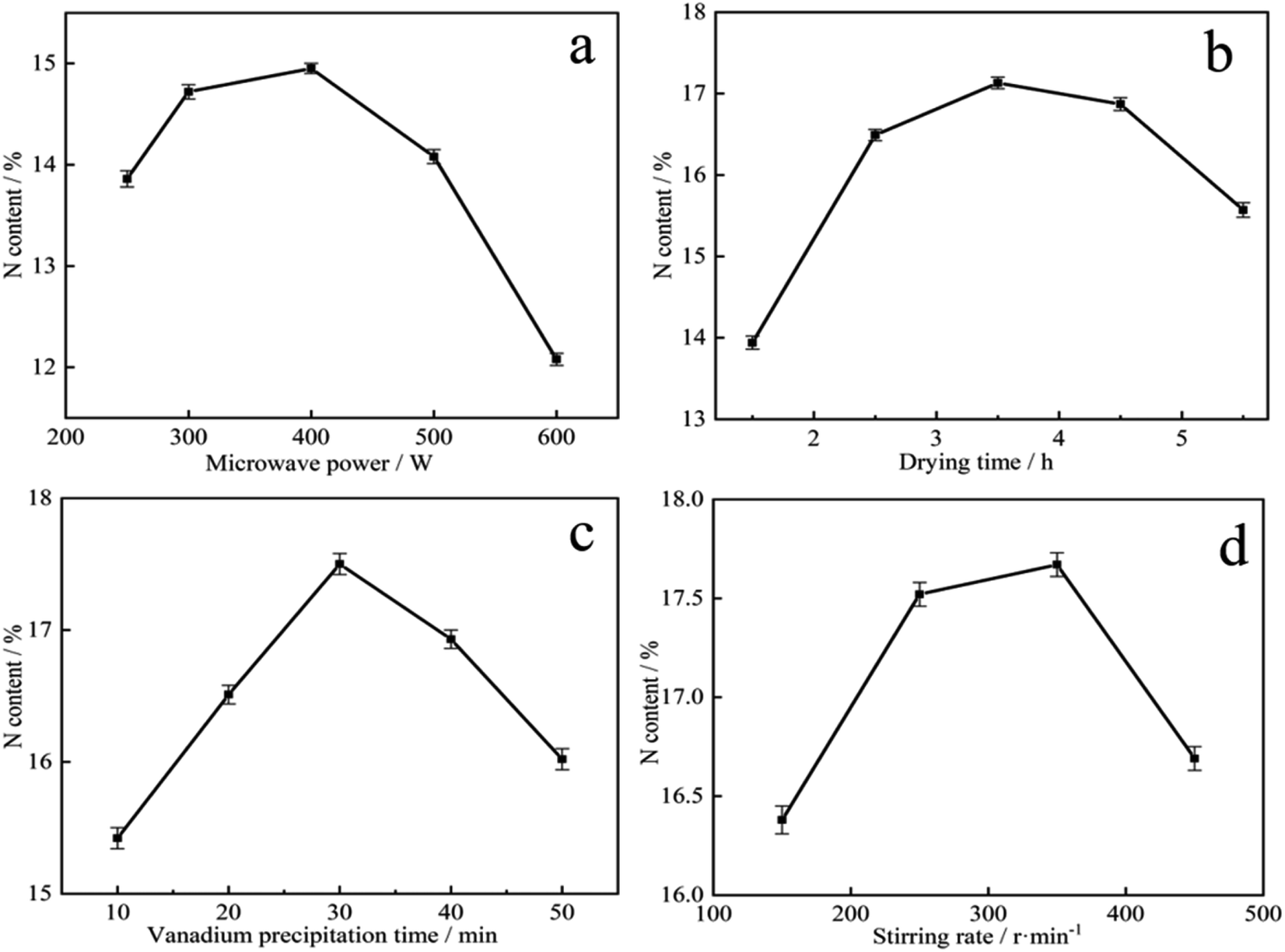 | ||
| Fig. 3 Effect of precursor preparation conditions on N content of vanadium nitride: (a) microwave power; (b) drying time; (c) vanadium precipitation time; (d) stirring speed. | ||
It is showed that microwave-assisted reaction had great potential in improving efficiency and shortening time, and high microwave power was beneficial to selective heating of solution.37 The enhanced reactivity under microwave heating was in favor of direct carbothermic reduction of ilmenite concentrate with sodium carbonate.38 Microwave-assisted hydrothermal synthesis of CuO nanostructures is an effective method to improve the gas sensitivity of materials.39 It can be seen from Fig. 3a that as the microwave power increased, the N content of vanadium nitride products increased and then decreased. When increased from 250 W to 400 W, the nitrogen content increased from 13.85 wt% to 14.95 wt%. This result may be due to the acceleration of precipitation kinetics under microwave radiation. At 600 W, the N content was only 12.08 wt%. After microwave-assisted, the vanadium precipitation rate can reach 99.3 wt%. Considering energy consumption and N content comprehensively, 400 W is determined as the best power for preparing the precursor.
As shown in Fig. 3b, the N content rapidly increased from 13.94 wt% to 17.13 wt% when the precursor drying time increased from 1.5 h to 3.5 h. After 3.5 h, the N content decreased. Drying 1.5 h is very short resulted a mass of water remains on the precursor. As the temperature is increased, the water evaporates and a large number of voids appear in the precursor, with a narrow contact area with N2 and a lack of reaction zone. When the drying time exceeds 3.5 h, there is almost no water in the precursor, and under the same pressure conditions, the material is compacted by the briquettes and N2 cannot enter the interior, resulting in a very low N content in vanadium nitride. According to Fig. 3c, the precipitation time of vanadium increased, the N content increased first and then decreased. When the precipitation time was 30 min, the maximum N content was 17.52 wt%. The increased of the N content is due to the enhance in vanadium content with the time accumulation. While exceeded 30 min, as water evaporates, other impurity ions such as SO42− and Na+ were crystallized and mixed with the vanadium precipitation product.40 The precursor has low purity, resulted a low N content in vanadium nitride. As shown in Fig. 3d, stirring rate can speed up the mass transfer process and make the reaction materials in a homogeneous system. Insufficient stirring rate, the distribution of various components is uneven and the crystal growth of the precursor is not complete. More impurities together with precipitation to form precursor because of the adsorption of C, which may reduce the purity of vanadium nitride and also make the N content lower. When the stirring speed too fast, intense mixing and shearing force destroy the precipitated crystal particles, increased surface area and adsorbed much impurities enter into precursor, resulted a decreased of the N content in the vanadium nitride. As a result, a drying time of 3.5 h, precipitation 30 min and stirring in 350 rpm was made sure as the best conditions.
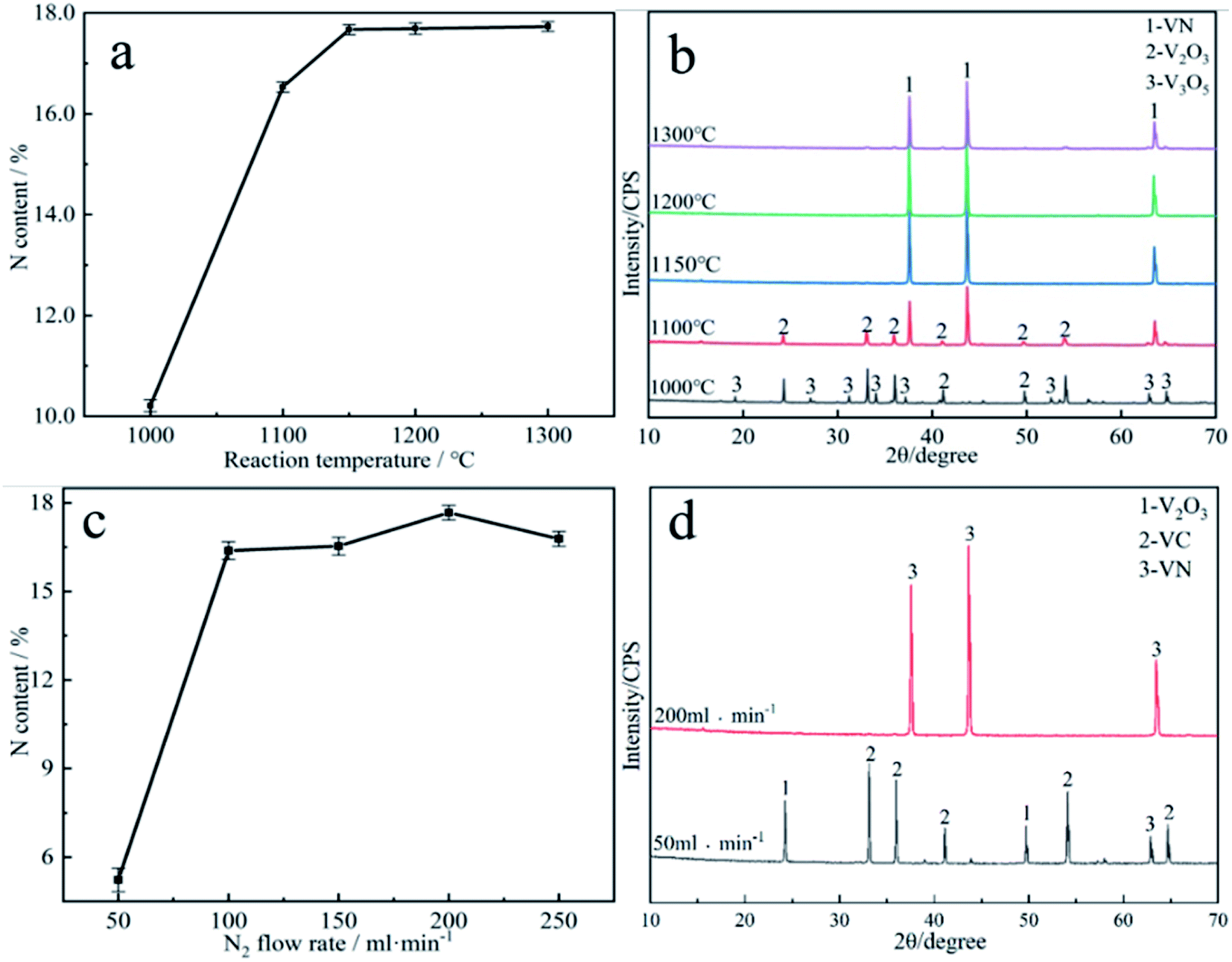 | ||
| Fig. 4 Effect of reduction nitride conditions on the N content of vanadium nitride: (a) reaction temperature; (b) the physical phases of the products at each reaction temperature condition; (c) N2 flow rate; (d) the physical phases of the products at different N2 flow rate. | ||
It can be evidently observed from Fig. 4a that the reaction temperature has a marked effect on the N content in the vanadium nitride. The N content is only 10.21 wt% at 1000 °C. The temperature rised from 1100 °C to 1150 °C as well as the N content increased from 11.47 wt% to 17.65 wt%. When the temperature exceeded 1150 °C, the N content increased slightly, indicating that higher reaction temperature had little effect on the N content of the vanadium nitride. It can be looked into from Fig. 4b that the phases are V3O5 and V2O3 at 1000 °C. The phases are VN and V2O3 at 1100 °C. When the temperature reached above 1150 °C, the phase is mainly VN. It indicated that the temperature is higher than 1150 °C, and the reduction and nitridation are fundamentally complete. Fig. 4c showed that the N content increased from 5.6 wt% to 17.67 wt%, and then dropped to 16.53 wt% as the N2 flow rate increased. The optimum flow rate of N2 was 200 ml min−1 while the N content was 17.67 wt%. The phase analysis obtained with different nitrogen flow rates is shown in Fig. 4d. A flowing N2 can increased the partial pressure of N2 and decreased the partial pressure of CO, thereby accelerating the reduction and nitridation reaction. The excessively fast of N2 flow rate results in N2 not being heated sufficiently, and the nitridation reaction cannot occur sufficiently during the fast flow, which leads to a decrease in N content. Therefore, 1150 °C and 200 ml min−1 was confirmed as the optimal reaction conditions.
4.2 Characterization of the precursor
Although the formation of vanadium nitride can be promoted by preparing precursor.29,30 However, in this study, a precursor with uniform particle size and porous surface morphology was used to prepare vanadium nitride, a fact we consider an essential difference. Therefore, we had to investigate this precursor. To understand the comparison of the precursor phase composition, and XRD analysis was performed, the results are demonstrated in Fig. 5. The phase of the product obtained by direct vanadium precipitation, the precursor obtained by adding C for vanadium precipitation and the precursor obtained by microwave-assisted were analyzed. There was no change about the phase, and both are composed of (NH4)2V6O16·1.5H2O and (NH4)Al(SO4)2·12H2O. At the same time, the precursor obtained by microwave-assisted has sharper peaks, and the product crystallinity degree is higher. The C peak was not detected in the precursor was affiliated because of the amorphous state of C.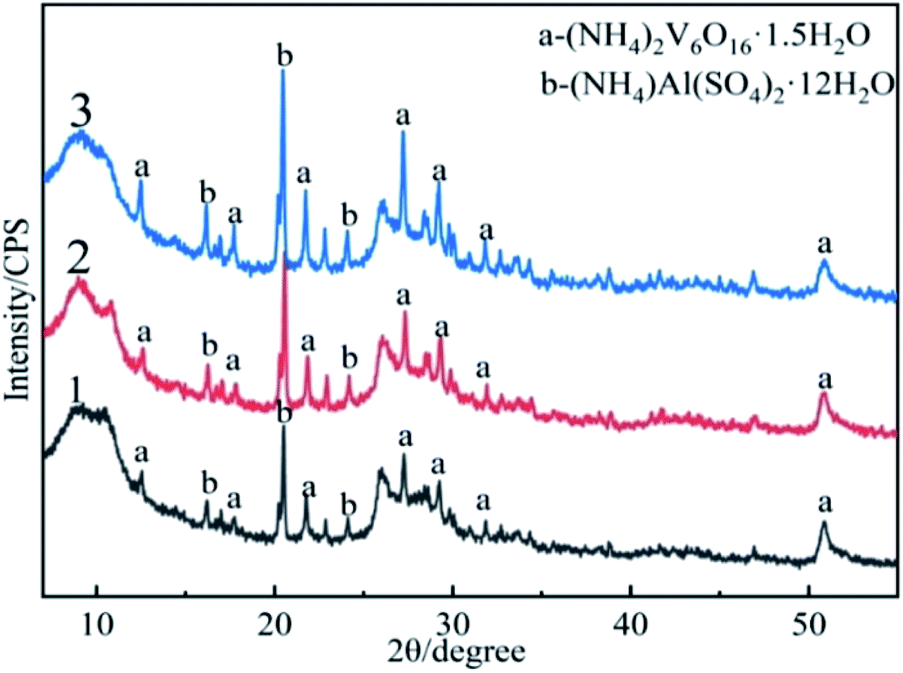 | ||
| Fig. 5 XRD patterns of the precursor: (1) the product precipitated directly from vanadium solution; (2) the precursor obtained by vanadium precipitation with adding C; (3) the precursor obtained by microwave-assisted. | ||
SEM-EDS was used to observe the microscopy of precursor, the results are shown in Fig. 6. The precursor obtained by vanadium precipitation with adding C were agglomerated and with a large particle size. After microwave-assisted, the precursor showed a good dispersion, uniform and small particle size. The structure exhibited appear porous structure in addition to the layered structure. The contact area with N2 have increased. At the same time, the residual water in the precursor evaporated during the reaction process, forming a large number of pores. The N2 is more likely to enter the material when it flowing rapidly, it can promote the formation of vanadium nitride.
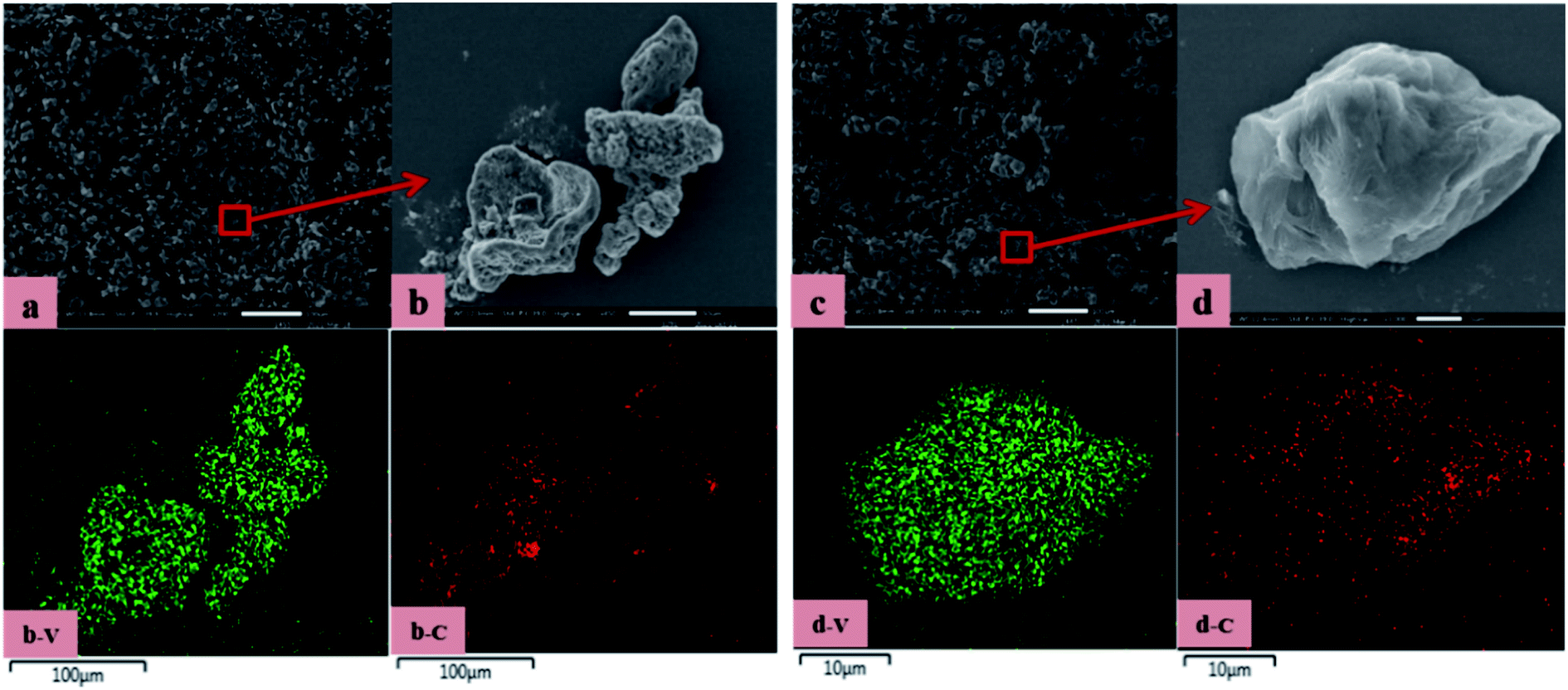 | ||
| Fig. 6 SEM-EDS: (a) he precursor obtained by microwave-assisted; (b) the partially enlarged view of the edge of the precursor obtained by microwave-assisted; (c) the precursor obtained by vanadium precipitation with adding C; (d) the partially enlarged view of the edge of the precursor obtained by vanadium precipitation with adding C. | ||
The detection and analysis results of the particle size distribution was shown in Fig. 7. According to the particle size distribution diagram A, it can be seen that the precursor particles are normally distributed and both larger than 0.283 μm. Compared the D10, D50, D90 of the two precursors in the diagram A, the particle size distribution width after microwave-assisted is more smaller. It could be seen that the particle size of the two precursor particles is dmicrowave-assisted < dwithout-microwave. It was discovered from diagram B that the cumulative distribution range of the precursor prepared by microwave-assisted is mainly 10–40 μm, and the content reached 77 wt% when the particle content without a microwave was only 66 wt%. After microwave-assisted, the precursor particles smaller than 10 μm decreased by 4.46 wt%, and those larger than 30 μm increased by 6.46 wt%. When the particle size is too small, vanadium and C do not form a package structure, the contact area decreased. When the particle size of is too large, the particle envelope formed with C as the nucleation point is too thick,29 that C in the middle of the structure cannot be fully released to participate in the response, affecting the progress of the reaction. Therefore, the decreased and uniformity of precursor particle size is the mainly reason to promote the increase of N content.
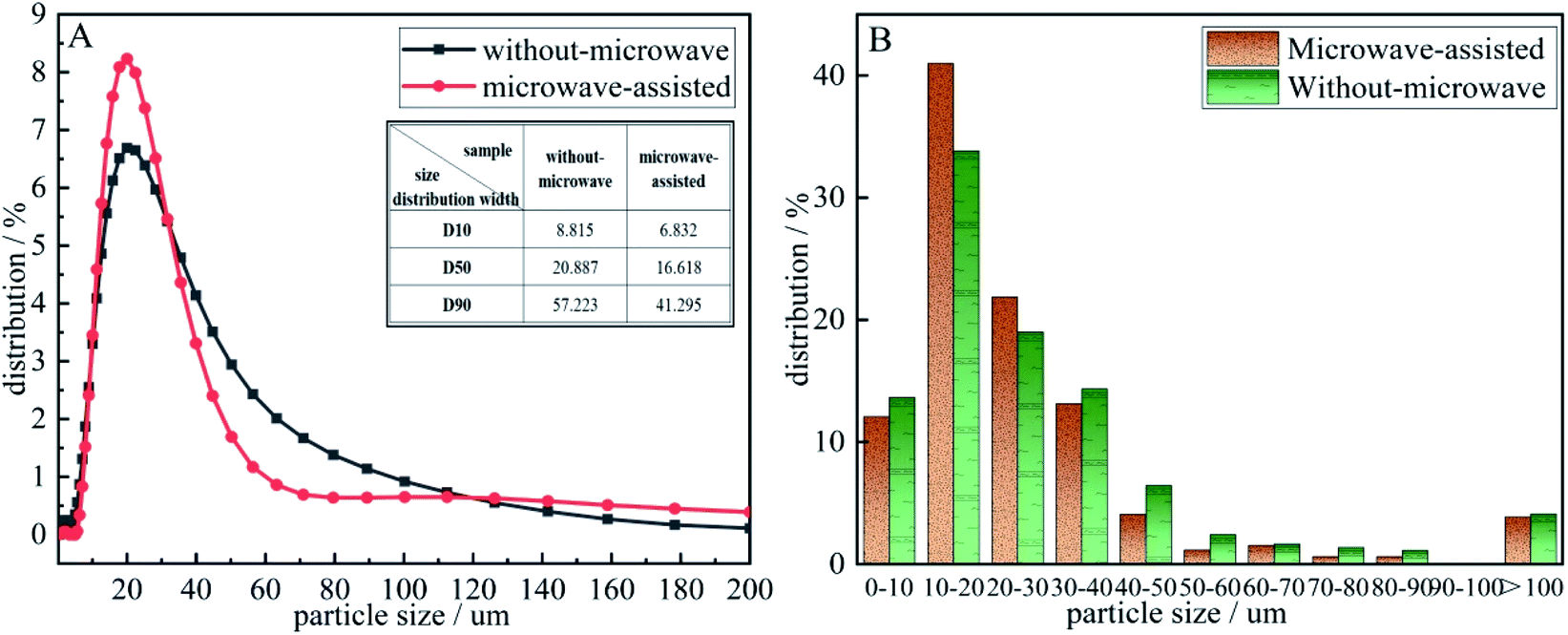 | ||
| Fig. 7 The particle size distribution of the precursor (A): precursor particle size distribution diagram. (B) accumulative distribution of precursor grain size. | ||
4.3 Characteristics of the precursor
Thermogravimetric experiments were carried out in N2 atmosphere to reflect the phenomenon of weight loss during the preparation of vanadium nitride. Fig. 8a showed that when different raw materials are used as vanadium sources, the weight loss percentage increased with the increase of temperature. When the temperature is below 600 °C, V2O5 as a vanadium source has little weight change while the precursor as a vanadium source had significant weight change, indicated that N2 plays a protective role in the early stage. Using the precursor as vanadium source, there are several obvious weight loss peaks at 230 °C, 471 °C, 682 °C and 979 °C. Four stages can be found in the whole DTG curve of the reaction. The first stage is room temperature to 304 °C, mainly caused by the evaporation of physical adsorption water and crystal water. The second stage is 304 °C to 536 °C. It is mainly the thermal decomposition of the precursor (NH4)2V6O16·1.5H2O and the initial reduction of NH3 to V2O5, corresponding reaction (1)–(13). The third stage 536–894 °C, which is caused by the carbon thermal reduction of vanadium oxide to generate V2O3. The fourth stage 894–1145 °C, which products by reduction and nitridation V2O3. Using V2O5 as vanadium source, the reaction can be divided into three stages. The first stage is from room temperature to 710 °C, which is caused by the pre-reduction of V2O5.41 The second stage is from 710–1188 °C, which is the carbonization process of low-price vanadium oxide after pre-reduction to generate vanadium carbide. The third stage 1188–1281 °C is caused by the formation of vanadium nitride from vanadium carbide nitride.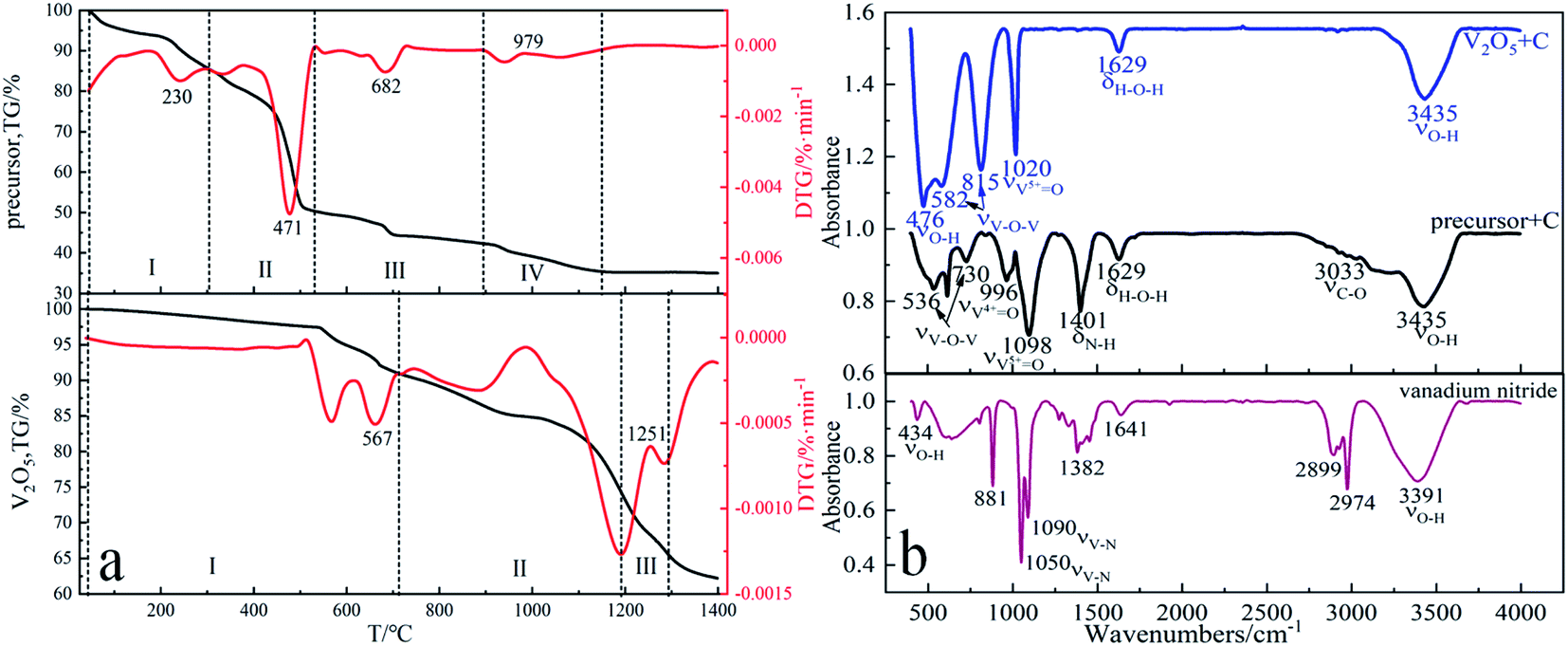 | ||
| Fig. 8 (a) TG-DTG curves of precursor and V2O5 from 20 °C to 1400 °C under N2 atmosphere; (b) FTIR analysis of precursor, V2O5 and vanadium nitride. | ||
From the FTIR curves in Fig. 8b, it can be seen that the νO–H absorption peaks of V2O5 and C as raw material are at 476 cm−1, 3435 cm−1, provided by water of crystallization, νV–O absorption peaks at 582 cm−1, 815 cm−1, 1020 cm−1, and H–O–H bond formation at 1629 cm−1, while the absorption characteristic peak of the precursor at 1401 cm−1 is νN–H, provided by the precursor 1401 cm−1 in the precursor NH4+ provided.42–44 Combined with the thermodynamic analysis and Fig. 8a TG-DTG, found NH4+ is present in the form of NH3 during reductive nitration, which can facilitate the thermal decomposition process of the precursor. From the FTIR curves of vanadium nitride, it can be seen that νV–N characteristic absorption peaks are formed at 1050 cm−1 and 1090 cm−1.
A comparative analysis of the reaction process for the preparation of vanadium nitride from two different raw materials as vanadium sources is shown in Table 3.
| Precursor | V2O5 | |
|---|---|---|
| I | Room temperature – 304 °C; the evaporation of physical adsorption water and crystal water | Room temperature – 710 °C; the pre-reduction of V2O5 |
| II | 304–536 °C; the thermal decomposition of the precursor and the initial reduction of NH3 to V2O5 | 710–1188 °C; low-price vanadium oxide is carbonized to form vanadium carbide |
| III | 536–894 °C; the carbon thermal reduction of vanadium oxide to generate V2O3 | 1188–1281 °C; the formation of VN from VC |
| IV | 894–1145 °C; V2O3 through carbonization and nitriding reaction generated VN | No response |
The formation process of vanadium nitride after microwave-assisted preparation of the precursor is shown in Fig. 9. During the vanadium precipitation process, C was used as the nucleation point and (NH4)2V6O16·1.5H2O in the slurry is adsorbed on C. With time increased, the precipitated particles increased and wrapped C. After the vanadium precipitation is completed, the precursor was prepared by filtering and drying. The precursor undergoes ammonia off, pre-reduction, reduction and nitridation to form vanadium nitride with an N2 atmosphere.
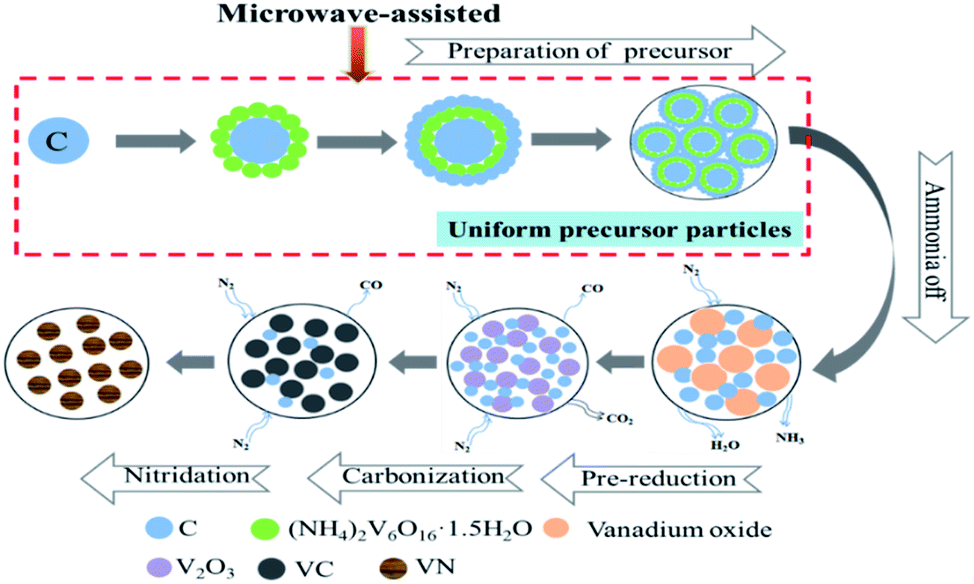 | ||
| Fig. 9 Mechanism of preparation of vanadium nitride. | ||
4.4 Technical index of vanadium nitride
The microscopic observation of vanadium nitride was performed by SEM-EDS, the results are shown in Fig. 10. The vanadium nitrogen prepared by microwave-assisted had more structurally complete and consisted of homogeneous spherical particles. Area scans were carried out to check the distribution of the elements, it found a good correlation between V and N, with complete overlap between V and N regions. A point scan was performed and found that the V and N content increased and the impurity content decreased of the vanadium nitrogen produced after microwave-assisted. The chemical element content was shown in Table 4. The V content is 79.32 wt%, the N content is 17.67 wt%, the C content is 2.11 wt%, and the content of other impurity ions also meets the requirements of VN16. After the as-prepared product was briquetted under a pressure of 40 MPa, the measured density was 3.10 g cm−3. Can be used as an additive in steelmaking.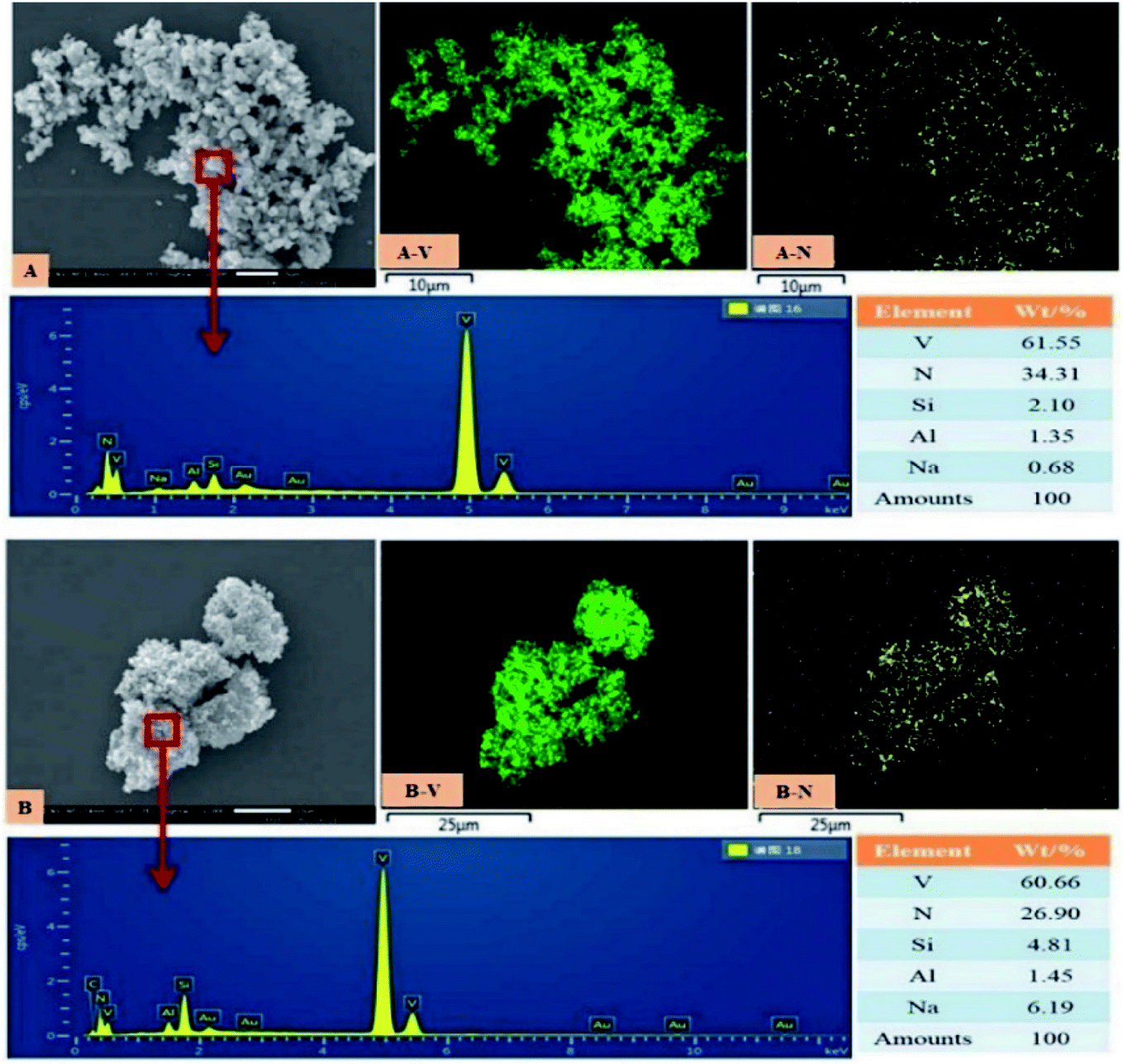 | ||
| Fig. 10 SEM-EDS (A): vanadium nitride prepared by microwave-assisted in the precursor preparation; (B): vanadium nitride prepared without microwave in the precursor preparation. | ||
| V | N | C | P | S |
|---|---|---|---|---|
| 79.32 | 17.67 | 2.11 | 0.058 | 0.10 |
5 Conclusion
Microwave-assisted synthesis of precursor for the production of vanadium nitride is a feasible method. The precursor prepared under the conditions of microwave power of 400 W, drying time of 3.5 h, vanadium precipitation time of 30 min and stirring speed of 350 rpm, and then reduction and nitridation precursor under the conditions of the reaction temperature of 1150 °C and N2 flow rate of 200 ml min−1 to production vanadium nitride. The vanadium nitride has good performance, and the content of various chemical components meets the requirements of VN16 in GB/T 20567-2020. The N content is 17.67 wt%, and V content is 79.32 wt%, which can be used as steel additives.The NH3 produced by the thermal decomposition of the precursor can be used as a reducing agent to reduce V2O5 in N2 atmosphere. The decomposition products of the precursor reduced by NH3 may be V2O5, V3O5, V3O7, V4O9, V6O13, V2O4, V2O3 and VO. ΔGθ under standard conditions was analyzed and ΔGθ − T plots were plotted.
The mechanism of microwave-assisted synthesis of precursor promoting the formation of vanadium nitride was explained by XRD, SEM-EDS and particle size distribution changed about precursor. The particle size of the precursor prepared after microwave-assisted is reduced, which effectively shorten the N2 diffusion path, and nitrogen can more easily penetrate into the material, improving the conversion efficiency of the internal raw material. At the same time, the specific surface area increased, and the reaction interface increased, which promoted the reaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (No. 2018YFC1900605), the National Natural Science Foundation of China (No. 52174259), Open Fund of State Key Laboratory of Vanadium and Titanium Resources Comprehensive Utilization (2021P4FZG03A).References
- D. Holec, M. Friák, J. Neugebauer and P. H. Mayrhofer, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 064101 CrossRef.
- F. Rovere, D. Music, S. Ershov, H.-G. Fuss, P. H. Mayrhofer and J. M. Schneider, J. Phys. D: Appl. Phys., 2010, 43, 035302 CrossRef.
- G. Li, J. Song, G. Pan and X. Gao, Energy Environ. Sci., 2011, 4, 1680–1683 RSC.
- H. Xie, F. Deng, H. Wang and S. Han, Powder Technol., 2021, 378, 639–646 CrossRef CAS.
- N. Y. Kim, J. H. Lee, J. A. Kwon, S. J. Yoo, J. H. Jang, H.-J. Kim, D.-H. Lim and J. Y. Kim, J. Ind. Eng. Chem., 2017, 46, 298–303 CrossRef CAS.
- F. Ran, Y. Wu, M. Jiang, Y. Tan, Y. Liu, L. Kong, L. Kang and S. Chen, Dalton Trans., 2018, 47, 4128–4138 RSC.
- X. Lu, M. Yu, T. Zhai, G. Wang, S. Xie, T. Liu, C. Liang, Y. Tong and Y. Li, Nano Lett., 2013, 13, 2628–2633 CrossRef CAS PubMed.
- O. Bondarchuk, A. Morel, D. Bélanger, E. Goikolea, T. Brousse and R. Mysyk, J. Power Sources, 2016, 324, 439–446 CrossRef CAS.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H.-M. Cheng and F. Li, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- Q. Sun and Z.-W. Fu, Electrochim. Acta, 2008, 54, 403–409 CrossRef CAS.
- A. Glaser, S. Surnev, F. Netzer, N. Fateh, G. Fontalvo and C. Mitterer, Surf. Sci., 2007, 601, 1153–1159 CrossRef CAS.
- M. Gao, X. Xu and H. Li, Mater. Lett., 2020, 274, 128045 CrossRef CAS.
- Z. Baochun, Z. Tan, L. Guiyan and L. Qiang, Mater. Sci. Eng., 2014, 604, 117–121 CrossRef.
- P. Maugis and M. Gouné, Acta Mater., 2005, 53, 3359–3367 CrossRef CAS.
- B. Zhao, T. Zhao, G. Li and Q. Lu, Mater. Sci. Eng., A, 2014, 604, 117–121 CrossRef CAS.
- J. Qing, L. Wang, K. Dou, B. Wang and Q. Liu, High Temp. Mater. Processes, 2016, 35, 575–582 CrossRef CAS.
- G. Maier, E. Astafurova, V. Moskvina, E. Melnikov, S. Astafurov, A. Burlachenko and N. Galchenko, Procedia Struct. Integr., 2018, 13, 1053–1058 CrossRef.
- M. Qin, H. Wu, Z. Cao, D. Zhang, B. Jia and X. Qu, J. Alloys Compd., 2019, 772, 808–813 CrossRef CAS.
- Š. Huber, O. Jankovský, D. Sedmidubský, J. Luxa, K. Klímová, J. Hejtmánek and Z. Sofer, Ceram. Int., 2016, 42, 18779–18784 CrossRef.
- X. Duan, S. C and H. Zhang, Arabian J. Sci. Eng., 2015, 40, 2133–2139 CrossRef CAS.
- A. Biswas, C. Sahoo, W.-T. Du, I.-H. Jung and M. Paliwal, Metall. Mater. Trans. B, 2021, 52, 956–967 CrossRef CAS.
- M. Roldan, V. López-Flores, M. Alcala, A. Ortega and C. Real, J. Eur. Ceram. Soc., 2010, 30, 2099–2107 CrossRef CAS.
- T. Huang, S. Mao, G. Zhou, Z. Wen, X. Huang, S. Ci and J. Chen, Nanoscale, 2014, 6, 9608–9613 RSC.
- H. Guo, C. Lu, Z. Zhang, B. Liang and J. Jia, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 1–8 CrossRef CAS.
- X. Rui, Y.-d. Wu and G.-h. Zhang, Trans. Nonferrous Met. Soc. China, 2019, 29, 1776–1783 CrossRef.
- Y. Sansan, F. Nianxin, G. Feng and S. Zhitong, J. Mater. Sci. Technol., 2007, 23, 43 Search PubMed.
- P. K. Tripathy, J. C. Sehra and A. V. Kulkarni, J. Mater. Chem., 2001, 11, 691–695 RSC.
- D. Xinhui, C. Srinivasakannan, Z. Hong and Z. Yuedan, Arabian J. Sci. Eng., 2015, 40, 2133–2139 CrossRef.
- J. Han, Y. Zhang, T. Liu, J. Huang, N. Xue and P. Hu, Metals, 2017, 7, 360 CrossRef.
- J.-W. Huang, H. Peng and G.-B. Xia, Ironmaking Steelmaking, 2009, 36, 110–114 CrossRef CAS.
- C. Riccardi, R. Lima, M. Dos Santos, P. R. Bueno, J. A. Varela and E. Longo, Solid State Ionics, 2009, 180, 288–291 CrossRef CAS.
- N. J. Vickers, Curr. Biol., 2017, 27, R713–R715 CrossRef CAS PubMed.
- M. Kopp, D. Coleman, C. Stiller, K. Scheffer, J. Aichinger and B. Scheppat, Int. J. Hydrogen Energy, 2017, 42, 13311–13320 CrossRef CAS.
- X. Liu, Q. Shao, Y. Zhang, X. Wang, J. Lin, Y. Gan, M. Dong and Z. Guo, Colloids Surf., A, 2020, 592, 124588 CrossRef CAS.
- B. Chen, S. Bao and Y. Zhang, Int. J. Min. Sci. Technol., 2021, 31, 1095–1106 CrossRef CAS.
- B. Chen, S. Bao, Y. Zhang and L. Ren, J. Cleaner Prod., 2022, 339, 130755 CrossRef CAS.
- Z. y. Ma, Y. Liu, J. k. Zhou, M. d. Liu and Z. z. Liu, Int. J. Miner., Metall. Mater., 2019, 26, 33–40 CrossRef CAS.
- Z. g. Yu, J. w. Xiao, H. y. Leng and K. c. Chou, Trans. Nonferrous Met. Soc. China, 2021, 31, 1818–1827 CrossRef CAS.
- Y. Li, Y.-L. Lu, K.-D. Wu, D.-Z. Zhang, M. Debliquy and C. Zhang, Rare Met., 2021, 40, 1477–1493 CrossRef CAS.
- J. p. Wang, Y. m. Zhang, J. Huang and T. Liu, Cryst. Res. Technol., 2017, 52, 1700104 CrossRef.
- Y. Wu, G. Zhang and K. C. Chou, J. Min. Metall., Sect. B, 2017, 53, 383–390 CrossRef CAS.
- M. Eslami, V. Vahabi and A. A. Peyghan, Phys. E, 2016, 76, 6–11 CrossRef CAS.
- X. Wang, B. Xi, Z. Feng, W. Chen, H. Li, Y. Jia, J. Feng, Y. Qian and S. Xiong, J. Mater. Chem. A, 2019, 7, 19130–19139 RSC.
- F. Meng, L. Guo, J. He, Z. Wang, Z. Ma, Y. Zeng, S. Zhang and Q. Zhong, J. Phys. Chem. Solids, 2021, 155, 110112 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |