
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of multi-substituted pyrazoles via potassium carbonate-mediated [3 + 2] cycloaddition of in situ generated nitrile imines with cinnamic aldehydes†
Mei-Mei Li‡
 a,
Hui Huang‡a,
Wanrong Tiana,
Yiru Pua,
Chaozheng Zhanga,
Jirui Yanga,
Qing Rend,
Feiyan Tao*c,
Yun Deng*a and
Jun Lu*ab
a,
Hui Huang‡a,
Wanrong Tiana,
Yiru Pua,
Chaozheng Zhanga,
Jirui Yanga,
Qing Rend,
Feiyan Tao*c,
Yun Deng*a and
Jun Lu*ab
aState Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu, 611137, China. E-mail: ljaaa111@163.com; dengyun@cdutcm.edu.cn; taofeiyan@163.com
bInstitute for Advancing Translational Medicine in Bone & Joint Diseases, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong SAR 999077, China
cResearch and Development Centre, China Tobacco Sichuan Industrial Co., Ltd, Chengdu, 610066, China
dSchool of Chinese Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR 999077, China
First published on 29th April 2022
Abstract
A highly efficient potassium carbonate-mediated [3 + 2] cycloaddition reaction of hydrazonoyl chlorides with cinnamic aldehydes to furnish multi-substituted pyrazoles under nontoxic and mild conditions has been developed. A plausible stepwise cycloaddition reaction mechanism is proposed. This protocol featured broad substrate coverage, good functional group tolerance, wide scalability, and operational simplicity, as well as conveniently constructed pyrazole scaffolds.
Pyrazole, as a ubiquitous structural motif found in many natural products and biomolecules possessing significant biological and therapeutic effects, is utilized in various areas of pharmaceutical, agrochemical, and biological research.1 The multi-substituted pyrazoles, especially 1,3,4-tri, 1,3,5-tri and 1,3,4,5-tetrasubstituted pyrazoles, are the structural basis for a wide range of commercially available drugs, such as celecoxib, used as a cyclooxygenase-2 inhibitor for the treatment of rheumatoid arthritis,2 CDPPB, described as the first positive allosteric modulator of human metabotropic glutamate receptor,3 Lonazolac, regarded as an anti-inflammatory and anti-hyperglycemic drug,4 and PNU-32945 employed as a non-nucleoside reverse transcriptase inhibitor for the treatment of HIV (Fig. 1).5
 | ||
| Fig. 1 Example of pyrazole-containing bioactive compounds. | ||
Considering the therapeutic potential of these moieties, the development of new methods towards their synthesis has attracted substantial interest. The past decades have witnessed the development of a large number of simple and efficient methods for synthesis of various functionalized pyrazoles, including the addition condensation of hydrazines with carbonyl derivatives,6 the annulation reaction of hydrazones with nitroolefins,7 cycloadditions of diazo compounds with alkenes, alkynes or 1,3-dicarbonyl compounds,8 the synthesis of 5-formyl-1,3,4,5-tetrasubstituted pyrazoles by using alkyne surrogates and the preparation of 4-formyl-1,3,4,5-tetrasubstituted pyrazoles by Pd-catalyzed direct arylation at C-5 position (Scheme 1).9
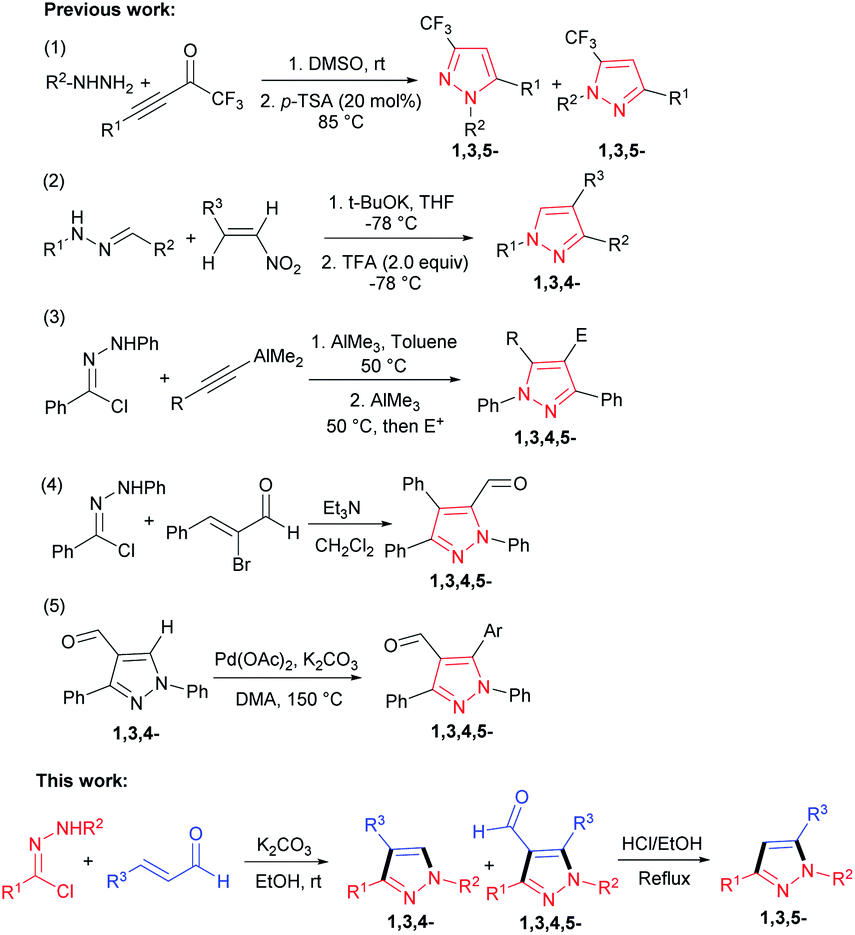 | ||
| Scheme 1 Different strategies for synthesis of pyrazoles. | ||
In addition, the cascade reaction of hydrazines to acyl chlorides was catalyzed by palladium via Sonogashira coupling reaction in situ to prepare pyrazoles.10 Moreover, TsOH-catalyzed Mannich-type cyclization–oxidation of arylhydrazines, aromatic aldehydes, and terminal arylalkynes has been reported to provide multi-substituted pyrazoles.11 Later, the condensation of α,β-unsaturated ketones with aryl hydrazines afforded the functionalized pyrazoles under the presence of water-soluble metal ion catalyst.12
Recently, several approaches of preparing pyrazoles by the [3 + 2] cycloaddition reaction of hydrazonoyl chlorides with active alkynes, bromoalkenes, or ketones bearing an active methylene group at the α-position were developed. For example, the 1,3-dipolar cycloaddition of hydrazonoyl chloride with o-(trimethyl-silyl)phenyl triflate to access multi-substituted pyrazoles has been developed by Spiteri et al.13 Then, the regioselective [3 + 2] cycloaddition of in situ formed nitrile imine with 2-bromo-3,3,3-trifluoropropene was successfully demonstrated for the construction of functionated pyrazoles in simple and mild reaction conditions.14 Moreover, nitrile imines, generally in situ generated by a base-induced dehydrodechlorination of corresponding hydrazonoyl chlorides, are also known for their ability to serve as the prominent [3 + 2] cycloaddition synthons.15 Herein, we report the first [3 + 2] cycloaddition reaction of in situ generated nitrile imines and cinnamic aldehydes under mild conditions, conveniently delivering various multi-substituted pyrazoles in good yields.
With encouragement from the preliminary results, we commenced our exploration using the readily accessible hydrazonoyl chloride and cinnamic aldehyde to investigate the feasibility of the designed [3 + 2] cycloaddition reaction for preparing 1,3,4,5-tetrasubstituted pyrazole. However, our surprise was the achievement of the compound beyond our expectation, the 1,3,4-trisubstituted pyrazole, which was prepared by extremely limited methods, was also presented as a minor substance (Scheme 1). As the potential benefits of this transformation seemed to be promising, we decided to engage in a more thorough investigation.
Subsequently, 1a and 2a were used as model substrates to examine the optimal conditions of the annulation reaction in this study. We were delighted to observe that when the reaction was conducted in THF with K2CO3 as the base, two products as 3aa and 4aa were isolated in 55% and 27% yields, respectively (Table 1, entry 1). As a result of the solvent screening, it was found that ethanol was the most effective solvent with a drastic increase in total yield (95%) (Table 1, entry 3), and oxygenated solvent, such as tetrahydrofuran, 1,4-dioxane or ethanol, could have a detrimental effect on the reaction, affording moderate to excellent yields of the two products (Table 1, entries 1–3). Upon further screening of other solvents, additional improvements in reaction efficiency were not obtained even with prolonged time (Table 1, entries 4–9). Accordingly, different types of bases were screened in order to enhance the yields (Table 1, entries 10–16). To our delight, K2CO3, Na2CO3, KHCO3, DIPEA, Et3N could provide moderate to excellent yields (81–95%) (Table 1, entries 3, 10–13). Unfortunately, those bases, such as Cs2CO3, NaOH or DBU, displayed no efficacies for this reaction (Table 1, entries 14–16). An obvious decrease in yield has been observed with a decrease in the amount of K2CO3 (Table 1, entry 17), while the doubled amount of the base resulted in a slight reduction of the total yield (Table 1, entry 18).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yieldb (%) (3aa) | Yieldb (%) (4aa) |
| a The reactions were performed with 1a (0.2 mmol), 2a (1.2 equiv.), and base (2.5 equiv.) in the solvent (2.5 mL) at room temperature.b Isolated yields.c K2CO3 was pre-dried.d With 4 Å MS in anhydrous toluene.e K2CO3 (1.0 equiv.) was used.f K2CO3 (5.0 equiv.) was used.g The reaction was carried out at 0 °C.h The reaction was carried out at 60 °C.i DDQ (2.5 equiv.) was added.j Under the protection of N2. | |||||
| 1 | K2CO3 | THF | 12 | 55 | 27 |
| 2 | K2CO3 | 1,4-Dioxane | 12 | 51 | 29 |
| 3 | K2CO3 | EtOH | 12 | 53 | 42 |
| 4 | K2CO3 | DCM | 12 | 39 | 14 |
| 5 | K2CO3 | CH3CN | >72 | 47 | 26 |
| 6 | K2CO3 | CHCl3 | 12 | 34 | 22 |
| 7 | K2CO3 | Toluene | 12 | 48 | 17 |
| 8c | K2CO3 | Toluene | 12 | 45 | 16 |
| 9d | K2CO3 | Toluene | 12 | 43 | 16 |
| 10 | Na2CO3 | EtOH | 12 | 47 | 39 |
| 11 | KHCO3 | EtOH | 12 | 49 | 33 |
| 12 | DIPEA | EtOH | 12 | 45 | 36 |
| 13 | Et3N | EtOH | 12 | 49 | 32 |
| 14 | Cs2CO3 | EtOH | 12 | Trace | Trace |
| 15 | NaOH | EtOH | 12 | Trace | Trace |
| 16 | DBU | EtOH | 12 | Trace | Trace |
| 17e | K2CO3 | EtOH | >72 | 39 | 20 |
| 18f | K2CO3 | EtOH | 12 | 49 | 39 |
| 19g | K2CO3 | EtOH | 12 | 37 | 27 |
| 20h | K2CO3 | EtOH | 12 | 50 | 39 |
| 21i | K2CO3 | EtOH | 12 | 51 | 42 |
| 22j | K2CO3 | EtOH | 12 | 52 | 40 |

Furthermore, we attempted to conduct the reaction in a precooled ice bath at 0 °C with a considerably decreased total yield (Table 1, entry 19) and in a preheated oil bath at 60 °C with a slightly decreased total yield (Table 1, entry 20). Finally, a combination of an oxidant, such as DDQ, or protection under inert gas, such as N2, failed to provide better results (Table 1, entries 21 and 22).
Subsequently, we conducted a systematic evaluation of the substrate scope of nitrile imines in this [3 + 2] annulation reaction under optimal reaction conditions (Scheme 2). Generally, various hydrazonoyl chlorides exhibited good functional group tolerance, and the desired multi-substituted pyrazole derivatives were synthesized in moderate to excellent yields (51–96%). The hydrazonoyl chlorides bearing none or electron-rich substituents on the phenyl ring of the R1 moiety reacted with 2a and provided higher total yields (3aa/4aa, 3ha/4ha and 3ia/4ia), compared with that bearing electron-deficient groups, such as fluoro, chloro and bromo at para-, meta-, and ortho-positions of the phenyl ring R1 (3da/4da, 3ea/4ea and 3fa/4fa). The para-position with electron-withdrawing substituents on R1 displayed a subtler negative effect on the total yields compared to the meta-position (3ea/4ea to 3fa/4fa, 3oa/4oa to 3pa/4pa). Substrates with electron-donating groups in the aryl ring at R2, such as methoxy- and methyl-, were all directly converted into desired products in excellent yields (3ia/4ia, 3ma/4ma, 3na/4na, 3oa/4oa and 3pa/4pa). Although the electron-withdrawing substituents of R2 on hydrazonoyl chlorides can also transform efficiently, an overall slight decline in yields of the corresponding products was observed (3ba/4ba, 3ca/4ca, 3ja/4ja, 3ka/4ka, 3la/4la and 3qa/4qa, 3sa/4sa). Interestingly, no matter what type of electron properties a substituent on the R1 has, the electron-donating substituents on the R2 could contribute to an increase in the yields of products. Moreover, the optimal condition was also applicable for heteroaryl hydrazonoyl chlorides, such as N-phenylthiophene-2-carbohydrazonoyl chloride, affording the desired multi-substituted pyrazoles 3ga and 4ga in 45% and 33% yields, respectively.
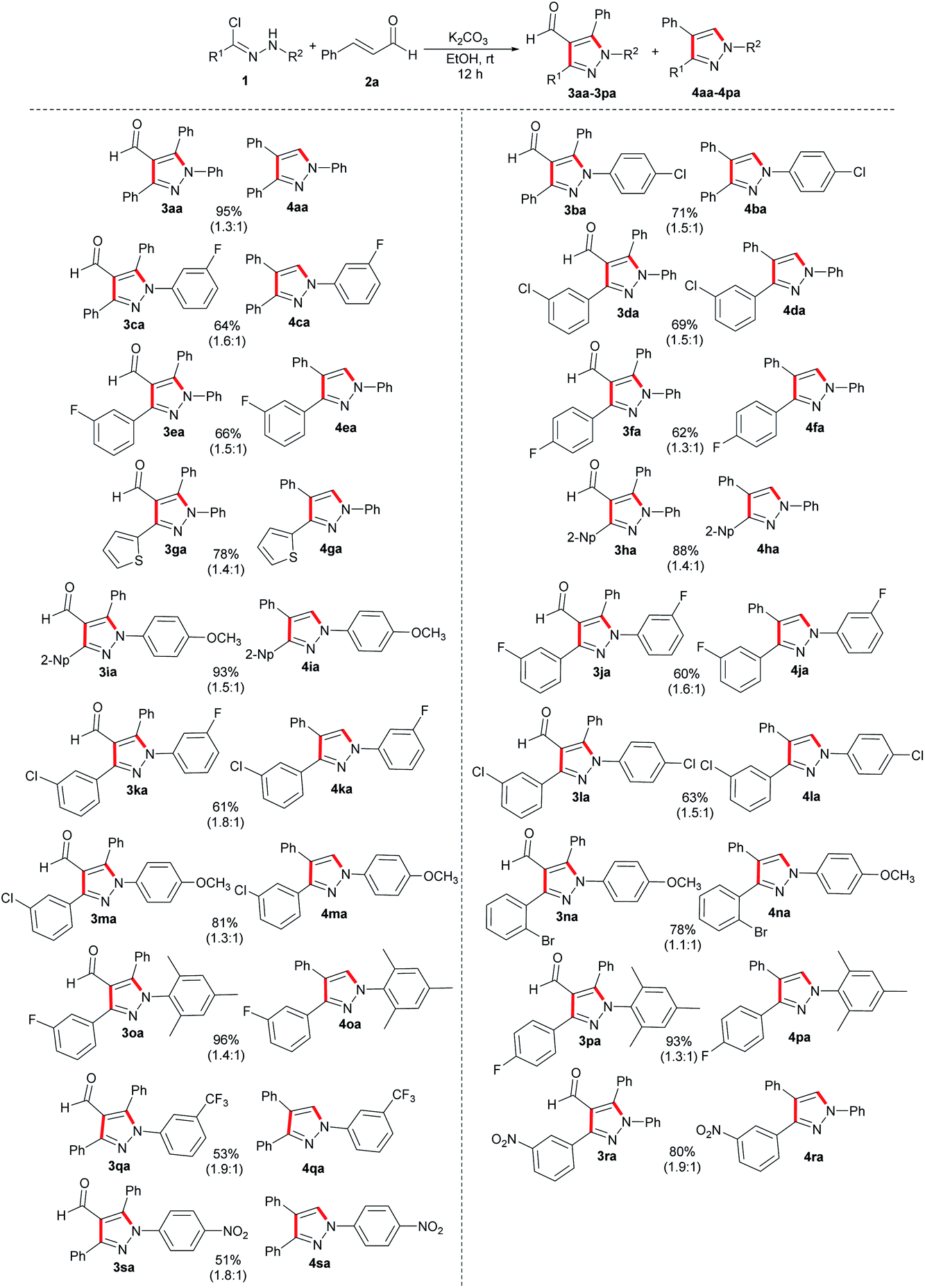 | ||
| Scheme 2 Substrate scope of hydrazonoyl chlorides. | ||
In addition, we investigated how the substituted cinnamic aldehydes could be employed as substrates for the optimum conditions established previously (Table 2). The protocol worked efficiently regardless of the electronic nature of substituted groups on cinnamic aldehydes. The cinnamic aldehyde derivatives with electron-withdrawing groups, such as nitro, chloro and bromo, or electron-donating groups, such as methoxy, on the phenyl ring could react with a variety of hydrazonoyl chlorides to afford the desired products in good to excellent yields (73–96%) (entries 1–21). Interestingly, a systematic comparison showed the electron-donating group on cinnamic aldehyde has a slightly detrimental effect on the reaction efficiency with a lessened yield compared to electron-withdrawing groups (entries 1–3, 4–6, 13–14, 17–18 and 20). In addition, the reaction of p-chlorocinnamaldehyde with 1n afforded an equal proportion of the annulation products (3l/4l = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (entry 12). During the reaction with p-chlorocinnam-aldehyde, it was found that when 1g was substituted for 1o, this optimal condition resulted in the highest total yield (96%) (entry 16). Furthermore, the positions of the substituent on the phenyl ring R1 exhibited no apparent influence on the total yields (entries 4 and 8, 5 and 7, 16–17).
:1) (entry 12). During the reaction with p-chlorocinnam-aldehyde, it was found that when 1g was substituted for 1o, this optimal condition resulted in the highest total yield (96%) (entry 16). Furthermore, the positions of the substituent on the phenyl ring R1 exhibited no apparent influence on the total yields (entries 4 and 8, 5 and 7, 16–17).
|
|||
|---|---|---|---|
| Entry | 1 (R1, R2) | 2 (Ar) | Product, yieldb |
| a The reaction of 1 (0.2 mmol) with 2 (0.24 mmol) was carried out in the presence of K2CO3 (0.5 mmol) in EtOH (2.5 mL) at room temperature for 12 h.b Isolated yield. | |||
| 1 | Ph, Ph | 4-BrC6H4 | 3a, 54%; 4a, 29% |
| 2 | Ph, Ph | 4-ClC6H4 | 3b, 59%; 4b, 28% |
| 3 | Ph, Ph | 4-OMeC6H4 | 3c, 48%; 4c, 27% |
| 4 | 3-FC6H4, Ph | 4-BrC6H4 | 3d, 57%; 4d, 24% |
| 5 | 3-FC6H4, Ph | 4-ClC6H4 | 3e, 59%; 4e, 22% |
| 6 | 3-FC6H4, Ph | 4-OMeC6H4 | 3f, 50%; 4f, 25% |
| 7 | 4-FC6H4, Ph | 4-ClC6H4 | 3g, 55%; 4g, 27% |
| 8 | 4-FC6H4, Ph | 4-BrC6H4 | 3h, 50%; 4h, 27% |
| 9 | 3-ClC6H4, Ph | 4-ClC6H4 | 3i, 54%; 4i, 27% |
| 10 | 3-ClC6H4, Ph | 4-BrC6H4 | 3j, 44%; 4j, 29% |
| 11 | 3-ClC6H4, 4-OMeC6H4 | 4-OMeC6H4 | 3k, 52%; 4k, 29% |
| 12 | 2-BrC6H4, 4-OMeC6H4 | 4-ClC6H4 | 3l, 48%; 4l, 47% |
| 13 | 2-Np, 4-OMeC6H4 | 4-ClC6H4 | 3m, 56%; 4m, 30% |
| 14 | 2-Np, 4-OMeC6H4 | 4-OMeC6H4 | 3n, 54%; 4n, 32% |
| 15 | 2-Np, Ph | 4-ClC6H4 | 3o, 54%; 4o, 31% |
| 16 | 3-FC6H4, 2,4,6-triMeC6H2 | 4-ClC6H4 | 3p, 67%; 4p, 29% |
| 17 | 4-FC6H4, 2,4,6-triMeC6H2 | 4-ClC6H4 | 3q, 65%; 4q, 30% |
| 18 | 4-FC6H4, 2,4,6-triMeC6H2 | 4-OMeC6H4 | 3r, 60%; 4r, 28% |
| 19 | 2-Thiophenyl, Ph | 4-ClC6H4 | 3s, 59%; 4s, 21% |
| 20 | Ph, Ph | 4-NO2C6H4 | 3t, 52%; 4t, 24% |
| 21 | Ph, 3-CF3C6H4 | 4-NO2C6H4 | 3u, 45%; 4u, 29% |

To determine the possibility of using the optimal reaction condition to produce multi-substituted pyrazoles on a gram scale, we carried out the cycloaddition reaction of 1p and 2a, which proceeded smoothly and gave the corresponding products of 3pa and 4pa in 60% yield and 22% yield, respectively (Scheme 3). The result indicated that the total yield was slightly decreased compared to the milligram-scale reaction. Furthermore, we demonstrated the synthetic utility by transforming trifluoromethyl-substituted nitrile imine 1q in two steps to the fructose 1,6-bisphosphatase (FBPase) inhibitor 7,16 which could decrease high blood glucose levels. In order to explore the utility of the final compounds, the aldehyde group on the product 3aa was transformed into hydroxyl, hydrogen and imine via reduction reaction, elimination reaction and aldimine condensation, respectively, to provide the corresponding multi-substituted pyrazole derivatives 8, 9 and 10.
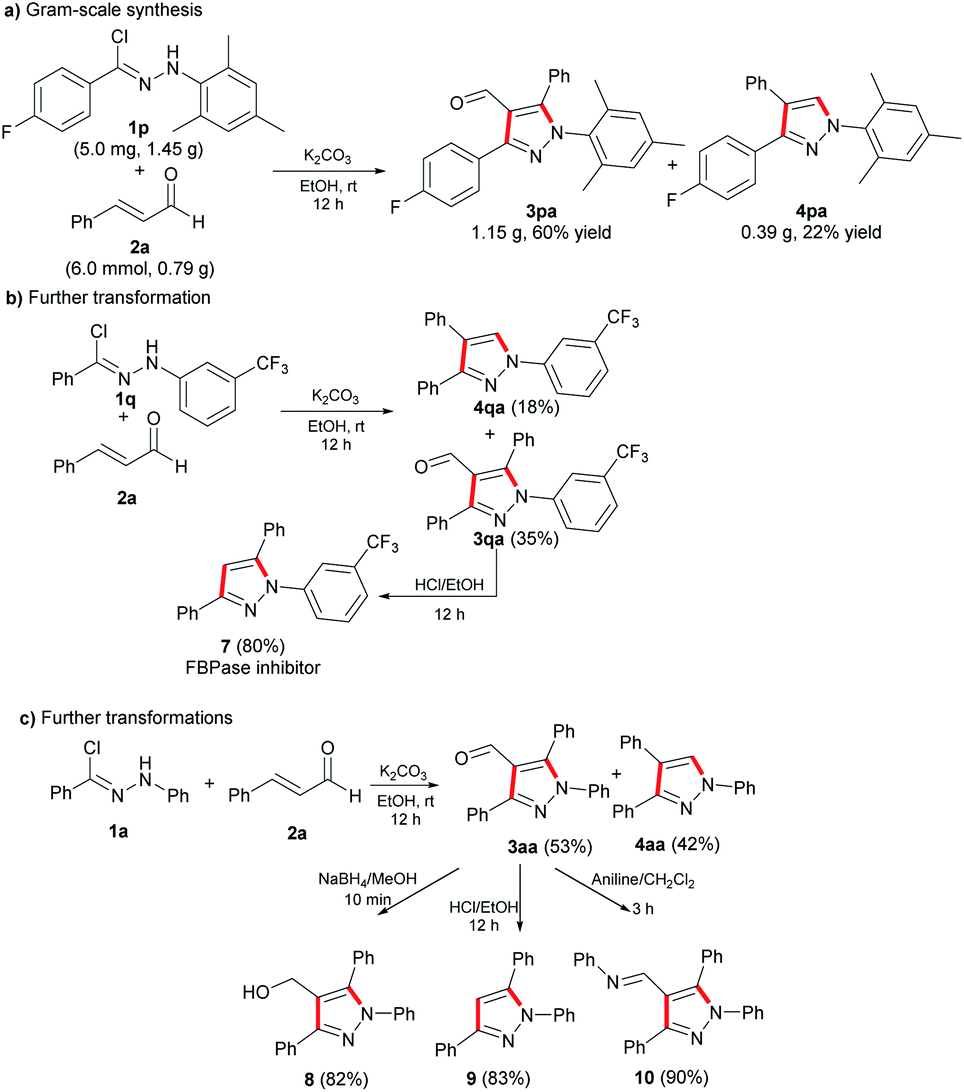 | ||
| Scheme 3 Gram-scale synthesis and further transformations. | ||
Although inconclusive, these results indicated that when a strong electron-withdrawing group was attached to the olefins, the stable aromatization products were obtained after the rapid elimination of small molecules, without 1,3-dipolar cycloaddition products being observed. A tentative reaction pathway is outlined in Scheme 4. Initially, nitrile imine specie Int-1 was generated from hydrazonoyl chloride 1a in situ in the presence of K2CO3. Subsequently, Michael addition reaction of nitrile imine to cinnamic aldehyde 2a led to the formation of intermediate Int-2 and Int-3. Int-2 underwent oxidative dehydrogenation to generate fully substituted pyrazole 3aa, and Int-3 underwent an elimination of HCHO to give 1,3,4-trisubstituted pyrazole 4aa, driven by the strongly electron-withdrawing groups such as CHO and NO2. For the reaction intermediates Int-2 and Int-3, whether the elimination of formaldehyde or hydrogen is the priority, the main consideration may be the stability of the elimination product. In order to prove this proposed mechanism, when we applied nitrile imine 1a and nitroolefin 5 as the raw materials, which successfully underwent the previous process of our optimal cycloaddition condition to afford the desired 4-nitro-pyrazole 6 and 1,3,4-trisubstituted pyrazole 4aa in a good total yield (90%).
 | ||
| Scheme 4 Plausible reaction mechanism for two multi-substituted pyrazoles. | ||
Conclusions
In summary, we have developed an efficient cycloaddition reaction of nitrile imine and cinnamic aldehyde, which allow expedient access to 1,3,4,5-tetra and 1,3,4-trisubstituted pyrazoles under mild conditions without transition metals. In addition, the products could be efficiently converted to other different types of pyrazoles via the reduction, oxidation, or elimination reaction. Furthermore, we have analyzed the possible mechanism of this [3 + 2] cycloaddition. The further exploration for the construction of pyrazole scaffold and the potential application of these compounds are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 81803374), Xinglin Scholar Research Promotion Project of Chengdu University of Traditional Chinese Medicine (No. BJRC2020002, No. BSH2019027), China Postdoctoral Science Foundation (2020M673565XB) and Scientific Research Foundation of the Science & Technology Department of Sichuan Province (Grant No. 2018JY0599).Notes and references
- (a) M. F. Khan, M. M. Alam, G. Verma, W. Akhtar, M. Akhter and M. Shaquiquzzaman, Eur. J. Med. Chem., 2016, 120, 170 CrossRef CAS PubMed; (b) Ş. G. Küçükgüzel and S. Şenkardeş, Eur. J. Med. Chem., 2015, 97, 786 CrossRef PubMed; (c) M. Laavola, R. Haavikko, M. Hämäläinen, T. Leppänen, R. Nieminen, S. Alakurtti, V. n. M. Moreira, J. Yli-Kauhaluoma and E. Moilanen, J. Nat. Prod., 2016, 79, 274 CrossRef CAS PubMed; (d) J. K. Williams, D. Tietze, J. Wang, Y. Wu, W. F. DeGrado and M. Hong, J. Am. Chem. Soc., 2013, 135, 9885 CrossRef CAS PubMed; (e) J. J. Talley, D. L. Brown, J. S. Carter, M. J. Graneto, C. M. Koboldt, J. L. Masferrer, W. E. Perkins, R. S. Rogers, A. F. Shaffer and Y. Y. Zhang, J. Med. Chem., 2000, 43, 775 CrossRef CAS PubMed; (f) H.-Z. Zhang, Z.-L. Zhao and C.-H. Zhou, Eur. J. Med. Chem., 2018, 144, 444 CrossRef CAS; (g) M.-M. Li, X. Chen, Y. Deng and J. Lu, RSC Adv., 2021, 11, 38060 RSC.
- (a) T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha and J. M. Miyashiro, J. Med. Chem., 1997, 40, 1347 CrossRef CAS PubMed; (b) R. F. Jacoby, K. Seibert, C. E. Cole, G. Kelloff and R. A. Lubet, Cancer Res., 2000, 60, 5040 CAS.
- (a) T. De Paulis, K. Hemstapat, Y. Chen, Y. Zhang, S. Saleh, D. Alagille, R. M. Baldwin, G. D. Tamagnan and P. J. Conn, J. Med. Chem., 2006, 49, 3332 CrossRef CAS PubMed; (b) S. Fowler, J. Walker, D. Klakotskaia, M. Will, P. Serfozo, A. Simonyi and T. Schachtman, Neurobiol. Learn. Mem., 2013, 99, 25 CrossRef CAS PubMed.
- G. R. Bebernitz, G. Argentieri, B. Battle, C. Brennan, B. Balkan, B. F. Burkey, M. Eckhardt, J. Gao, P. Kapa and R. J. Strohschein, J. Med. Chem., 2001, 44, 2601 CrossRef CAS PubMed.
- M. J. Genin, C. Biles, B. J. Keiser, S. M. Poppe, S. M. Swaney, W. G. Tarpley, Y. Yagi and D. L. Romero, J. Med. Chem., 2000, 43, 1034 CrossRef CAS PubMed.
- (a) A. Schmidt and A. Dreger, Curr. Org. Chem., 2011, 15, 1423 CrossRef CAS; (b) X. Zhang, J. Kang, P. Niu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2014, 79, 10170 CrossRef CAS PubMed; (c) V. Lellek, C.-y. Chen, W. Yang, J. Liu, X. Ji and R. Faessler, Synlett, 2018, 29, 1071 CrossRef CAS.
- (a) X. Deng and N. S. Mani, Org. Lett., 2006, 8, 3505 CrossRef CAS PubMed; (b) X. Deng and N. S. Mani, Org. Lett., 2008, 10, 1307 CrossRef CAS PubMed; (c) C. Shi, C. Ma, H. Ma, X. Zhou, J. Cao, Y. Fan and G. Huang, Tetrahedron, 2016, 72, 4055 CrossRef CAS.
- (a) D. L. Browne and J. P. Harrity, Tetrahedron, 2010, 66, 553 CrossRef CAS; (b) V. K. Aggarwal, J. de Vicente and R. V. Bonnert, J. Org. Chem., 2003, 68, 5381 CrossRef CAS PubMed; (c) Y. Shao, J. Tong, Y. Zhao, H. Zheng, L. Ma, M. Ma and X. Wan, Org. Biomol. Chem., 2016, 14, 8486 RSC; (d) T. Baiju and I. N. Namboothiri, Chem. Rec., 2017, 17, 939 CrossRef CAS PubMed; (e) Y. Shao, H. Zheng, J. Qian and X. Wan, Org. Lett., 2018, 20, 2412 CrossRef CAS PubMed.
- (a) S. Dadiboyena, E. J. Valente and A. T. Hamme, Tetrahedron Lett., 2010, 51, 1341 CrossRef CAS PubMed; (b) I. Smari, C. Youssef, K. Yuan, A. Beladhria, H. Ben Ammar, B. Ben Hassine and H. Doucet, Eur. J. Org. Chem., 2014, 2014, 1778 CrossRef CAS.
- A. C. Götzinger, F. A. Theßeling, C. Hoppe and T. J. Müller, J. Org. Chem., 2016, 81, 10328 CrossRef PubMed.
- P. Liu, Y.-M. Pan, Y.-L. Xu and H.-S. Wang, Org. Biomol. Chem., 2012, 10, 4696 RSC.
- (a) G. Bharathiraja, M. Sengoden, M. Kannan and T. Punniyamurthy, Org. Biomol. Chem., 2015, 13, 2786 RSC; (b) G. S. Ananthnag, A. Adhikari and M. S. Balakrishna, Catal. Commun., 2014, 43, 240 CrossRef CAS; (c) T. Zhang and W. Bao, J. Org. Chem., 2013, 78, 1317 CrossRef CAS PubMed; (d) X. Li, L. He, H. Chen, W. Wu and H. Jiang, J. Org. Chem., 2013, 78, 3636 CrossRef CAS PubMed; (e) J. J. Neumann, M. Suri and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7790 CrossRef CAS PubMed.
- C. Spiteri, S. Keeling and J. E. Moses, Org. Lett., 2010, 12, 3368 CrossRef CAS PubMed.
- H. Zeng, X. Fang, Z. Yang, C. Zhu and H. Jiang, J. Org. Chem., 2021, 86, 2810 CrossRef CAS PubMed.
- (a) A. Sami Shawali and A. Osman Abdelhamid, Curr. Org. Chem., 2012, 16, 2673 CrossRef; (b) M. Breugst and H. U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293 CrossRef CAS PubMed; (c) L. K. Garve, M. Petzold, P. G. Jones and D. B. Werz, Org. Lett., 2016, 18, 564 CrossRef CAS PubMed; (d) Y. Ohshiro, M. Komatsu, M. Uesaka and T. Agawa, Heterocycles, 1984, 22, 549 CrossRef CAS; (e) A. Kowalczyk, G. Utecht-Jarzynska, G. Mloston and M. Jasinski, Org. Lett., 2022, 24, 2499 CrossRef CAS PubMed.
- A. Rudnitskaya, K. Huynh, B. Torok and K. Stieglitz, J. Med. Chem., 2009, 52, 878 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ra00331g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |